

Abstract
Genetically engineered T cell immunotherapies have provided remarkable clinical success to treat B cell acute lymphoblastic leukaemia by harnessing a patient’s own T cells to kill cancer, and these approaches have the potential to provide therapeutic benefit for numerous other cancers, infectious diseases and autoimmunity. By introduction of either a transgenic T cell receptor or a chimeric antigen receptor, T cells can be programmed to target cancer cells. However, initial studies have made it clear that the field will need to implement more complex levels of genetic regulation of engineered T cells to ensure both safety and efficacy. Here, we review the principles by which our knowledge of genetics and genome engineering will drive the next generation of adoptive T cell therapies.
Through natural selection, our immune system has evolved the ability to recognize and eliminate a wide array of pathogens and tumours while sparing healthy tissue. Advances in gene and cell engineering mean that one no longer has to wait for natural selection to devise ways to successfully control pathogens and tumours. Rather, immune interventions, often called ‘immunotherapies’, have been developed that collaborate with the immune system to reverse or prevent a disease state. Vaccines were the earliest immunotherapy and have been by far the most impactful, saving millions of lives1. Additionally, infusion of antibodies, the product of B cells, can block undesired immune interactions, flag the immune system to remove cells coated with particular antibodies or enhance effector responses by blocking negative regulators of T cell activation (checkpoint therapies), a particularly effective therapeutic for a wide spectrum of diseases spanning autoimmunity and cancer2–4. The newest form of immunotherapy is cell and gene therapy, whereby immune cells are modified to express synthetic molecules and are genetically altered to generate a tailored immune response for a specific disease (FIG. 1). This is an emerging field in which all immune cells could be utilized, although T cells have been the most studied to date.
Fig. 1 |. Autologous and allogeneic T cell immunotherapy.
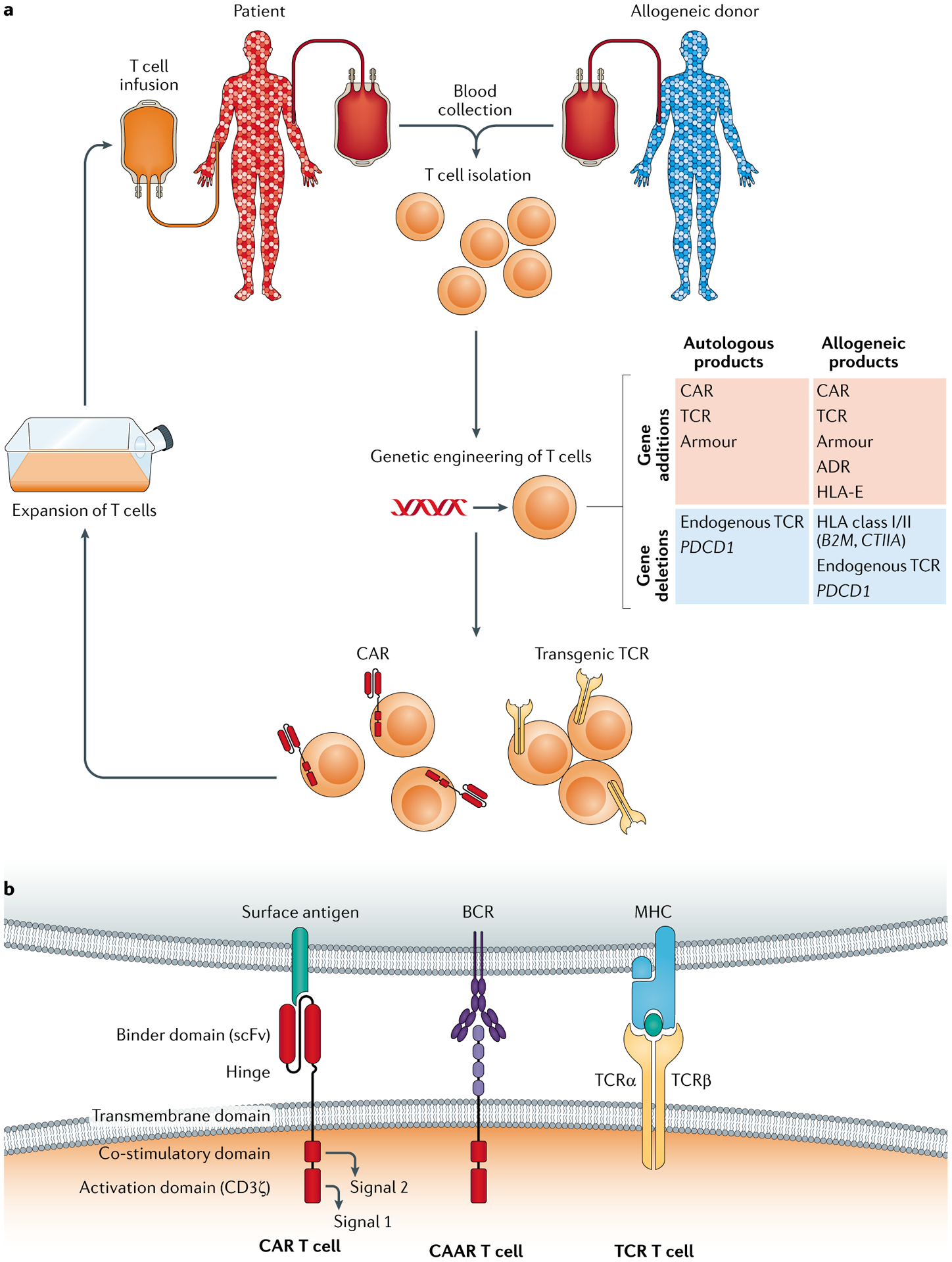
a | Manufacturing process. Blood is collected by venipuncture or apheresis, and T cells are isolated either from the patient (autologous donor) or from an allogeneic donor. Purified T cells undergo engineering to introduce a transgene using viral vectors or non-viral strategies and/or genome editing to eliminate protein expression. Following stimulation, engineered T cells are expanded to increase cellular dosage and infused into the patient. Site-specific transgene insertion may be preferred for allogeneic products to reduce batch-to-batch variation (not shown). T cells can undergo genetic engineering (middle right), for example, to endow antigen specificity, evade the host immune response or confer resistance to the tumour microenvironment. From an efficacy perspective, allogeneic T cell products are vulnerable to immune-mediated rejection34. This can be ameliorated by genetic deletion of both HLA class I and HLA class II expression by targeting B2M and CIITA, respectively, which extends the survival of chimeric antigen receptor (CAR) T cells in preclinical models27. However, allogeneic CAR T cells without HLA molecules can be killed by the recipient’s natural killer cells, which may necessitate the overexpression of HLA-E or other non-classical major histocompatibility complex (MHC) molecules as a remedy226. Deleting multiple genes and adding HLA-E in addition to a CAR or T cell receptor (TCR) is fairly complex, and thus recently an alloimmune defence receptor (ADR) approach was described whereby a CAR is used to trigger cytotoxicity of alloreactive host T cells and natural killer cells227. b | Composition of CARs, chimeric autoantibody receptors (CAARs) and transgenic TCRs. CARs and CAARs recognize antigen via a binder domain — typically a single-chain variable fragment (scFv) or a protein target of a B cell receptor (BCR), respectively. Each also has a flexible hinge domain, a transmembrane domain and a CD3ζ activation domain (signal 1), along with a choice of co-stimulatory domains (signal 2) tailored to fit the therapeutic task. Alternatively, a pair of TCRα and TCRβ chains dimerize to form a transgenic TCR, which recognizes MHC-presented antigens. Wild-type chains can be engineered to facilitate their dimerization preferentially over cross-pairing with the T cell’s endogenous chains.
T cells are a key component of the adaptive immune system and can be subdivided on the basis of function, co-receptor expression (CD4 or CD8), trafficking, metabolism and lifespan5,6. T cells arise from haematopoietic stem cells and mature in the thymus. During this process of development, T cells undergo a complex gene-editing programme by which gene segments within the T cell receptor (TCR) locus are randomly rearranged to generate a TCR of unique specificity that enables a population of T cells to recognize a wide array of peptides presented by major histocompatibility complex (MHC) molecules. When a TCR recognizes cognate peptide presented via an MHC molecule on an antigen-presenting cell, a signalling complex is recruited to the TCR (signal 1), which, when coupled with one or more co-stimulatory signals (signal 2), initiates a second phase of development that leads to the expansion of T cell numbers, differentiation and, ultimately, the development a memory pool of cells poised to eliminate the pathogen or tumour on secondary exposure.
Some pathogens and tumours deftly avoid the T cell response; for example, Ebola virus sterically interferes with T cell–MHC interactions7, and some tumours overexpress the immune regulator PDL1, resulting in a higher signalling threshold to induce the full complement of T cell effector functions8. By using genetic tools, investigators can take advantage of what is known about a particular pathogen or tumour to generate synthetic approaches to sidestep evasion mechanisms and redirect T cells towards a desired target. This approach has led to durable, complete cures in the case of CD19-expressing tumours9 (BOX 1), begetting many other potential therapies that are in development for infectious disease, autoimmunity and cancer.
Box 1 |. The evolution of CAR T cell therapies.
Some of the first iterations of engineered T cells were designed to combat viral infections. Chimeric antigen receptor (CAR) T cells expressing a CD4 extracellular binder domain and the CD3ζ signalling domain (signal 1 alone) capitalized on HIV’s dependency on CD4 for viral entry229. Although these CD4ζ CAR T cell clinical trials were unable to show long-term clinical benefit, they were able to demonstrate the long-term safety and feasibility of engineered adoptive T cell transfer19. The addition of co-stimulatory signalling domains (signal 2) to CARs enhanced in vivo function70,230 and persistence109, and has enabled T cell engineers to design collections of co-stimulatory signalling domains that tailor CAR T cells to a particular malady53,54. A major clinical breakthrough for engineered T cell therapeutics came in 2011, when a CAR T cell therapy targeting CD19 achieved durable remission of paediatric B cell acute lymphoblastic leukaemia231. This report was swiftly followed by other reports of clinical activity of CD19 CAR T cells in B cell malignancies232,233, and together these findings contributed to the regulatory approval of Kymriah (Novartis) and Yescarta (Kite) and the near-term approval of lisocabtagene maraleucel (Bristol Myers Squibb), all of which target the CD19 lineage antigen on B cells. The resulting proliferation of transgenic T cell receptor T cell123 and CAR T cell234 trials for other haematologic malignancies has been met with enthusiasm. However, due to chemical and mechanical barriers in the tumour microenvironment, T cells engineered for solid malignancies have been less effective and require further engineering to overcome the tumour microenvironment and tumour immune evasion mechanisms.
From lessons learned from cancer CAR therapy235, a new generation of HIV-specific CAR T cells have been generated81,107,236–239, some of which are currently being tested in phase I clinical trials (https://clinicaltrials.gov/ct2/show/NCT03617198 and https://clinicaltrials.gov/ct2/show/NCT03240328). Preclinical models have shown that CAR T cells can be developed to oppose other infectious diseases, including hepatitis B virus infection240, hepatitis C virus infection241 and opportunistic fungal infections242, suggesting that CAR T cells could be an important therapeutic for many infectious diseases.
A host of strategies have been developed to prolong T cell function, protect allogeneic T cells from immune rejection, thwart viruses from infecting the T cells programmed to recognize and eliminate them, and augment effector functions to clear a particular tumour or pathogen. However, until recently, the field lacked the tools to implement complex gene therapy approaches that would take full advantage of our knowledge of T cell function. Only single genetic alterations of T cells had been tested, and the only FDA-approved therapies to date, which treat some leukaemias and lymphomas, use T cells engineered to harbour randomly placed genomic insertions of a single synthetic gene that redirects T cells to kill. Additional engineering, beyond redirecting the T cell to a target, is likely required to increase the number of indications for which genetically modified T cells provide durable cures. Recently, results from a phase I clinical trial of TCR transgenic triple-gene-edited T cells were reported10, demonstrating feasibility and preliminary safety, ushering in a new phase of adoptive T cell therapy involving multiple genetic manipulations. While this initial trial found off-target gene edits and chromosomal translocations, a long-term in vitro culture assay used before infusion did not find T cell transformation10. To minimize off-target gene editing, a variety of improvements to CRISPR gene-editing systems, including base editing, are being developed (reviewed in REF.11). With an ever-expanding and ever more precise toolbox with which to engineer T cells, investigators are now broadening the scope of T cell alterations to cure a variety of disease states.
Here, we review why T cells are attractive cells to genetically engineer, approaches to engineer T cells, how these engineered T cells are being used to combat chronic disease and outline how future multifaceted genetic engineering strategies can increase safety and efficacy. We focus on cancer therapy and refer readers interested in engineering T cells for autoimmunity and infectious disease to other in-depth reviews12,13.
Why are T cells attractive to engineer?
Although cell types such as haematopoietic stem cells, B cells and macrophages have unique properties that make them attractive for immune-based therapies14–16, T cells are on the leading edge of this exciting new pillar of medicine. They are readily available via a simple venipuncture; culture and delivery systems compatible with good manufacturing practice (GMP) have been devised to expand T cells ex vivo and safely reinfuse them into patients17; lessons learned from HIV have generated high-efficiency vectors that readily transduce T cells; T cells are fully mature cells that resist oncogenic transformation18; and engineered T cells have the potential to be very long-lived, with some studies showing decade-long persistance19. Moreover, owing to natural genomic rearrangement and editing that occurs to generate a unique TCR, it is tempting to speculate that they are well equipped to perform and survive all forms of genome engineering. Lastly, the mobility and functional range of T cells make them excellent agents for cellular engineering.
Sources of T cells
The first wave of approved gene-engineered adoptive T cell therapies uses bulk autologous T cells harvested by apheresis20,21, which minimizes the risk of graft-versus-host disease (GVHD) and avoids immune clearance of therapeutic T cells22–25. This approach has notable disadvantages, including the turnaround time needed for the individualized manufacturing and logistics process (known as the ‘vein-to-vein’ time) during which a patient’s disease may progress23,25, the risk of manufacturing failure23,25 and high cost26. For these reasons, significant effort is being expended to develop gene-engineered products made from allogeneic T cells as ‘off-the-shelf’ therapies. Three sources of allogeneic gene-edited T cells are being explored: healthy adult donors27–29, umbilical cord blood30,31 and induced pluripotent stem cells32,33.
Allogeneic genetically engineered T cells pose unique risks and drawbacks that are not encountered with autologous T cell products. To counter these challenges, the optimal use of third-party T cells may require additional gene engineering steps that are not required for autologous products (FIG. 1a). From a safety perspective, allogeneic T cells can mediate serious or fatal GVHD34, so either they need to be obtained from low-risk sources such as partially or fully HLA-matched donors29,35, virus-specific T cells28 or umbilical cord blood36,37, or the endogenous TCR must be deleted27,38,39. Finding the optimal combination of gene engineering approaches to enable allogeneic T cells to avoid GVHD and survive in patients of different HLA types will be key to establishing off-the-shelf replacements for autologous cell therapies.
Engineered T cell types
Transgenic TCR T cells and CAR T cells.
The most common approaches to T cell engineering are the introduction of either a transgenic TCR or a chimeric antigen receptor (CAR), which enable the T cell to recognize a new target (FIG. 1b). Expansion and adoptive transfer of these cells enables the killing of cancerous or infectious cells en masse. Both approaches have their strengths and weaknesses. TCRs are able to access the full proteome of the cell, but are confined by the requirement for antigen processing and presentation of peptide targets17. CARs bind their antigens directly, and are not limited to the proteome but expand to other macromolecules such as glycans, which can differ markedly between normal and tumour cells40. However, only surface-accessible or secreted antigens can act as targets for CARs17.
TCR T cells are predominantly engineered to express one transgenic TCRα chain and one transgenic TCRβ chain in addition to or in place of their endogenous chains. The concept stemmed from cloning TCRs from tumour-infiltrating lymphocytes, which could redirect bulk T cells to have tumour specificity41. On binding of transgenic TCR to a therapeutically relevant peptide–MHC of interest — for example, the 9-mer peptide derived from amino acids 157–165 of the highly immunogenic cancer testis antigen NY-ESO-1, which is presented by HLA-A*02:01 (REF.42) — natural TCR signalling activates T cell function and expansion.
The endogenous TCR can limit the efficacy of TCR T cells in two major ways: through competition for the CD3 complex, reducing transgenic surface TCR expression43, and through mispairing between endogenous and transgenic TCR chains43. TCR chain mispairing can generate novel TCRs with autoreactivity and alloreactivity44, reduces the amount of the desired transgenic receptor, limiting potency45,46, and poses a risk of autoimmune toxicity, which fortunately so far has been observed only in mice47. Numerous strategies have been used to avoid mispairing between the endogenous and transgenic TCR chains as well as to increase expression of the transgenic TCR (reviewed in REF.48) and include equimolar expression of chains via 2A ribosomal skip sequences49; modifications facilitating intramolecular bonding between transgenic α and β chains, fusing the chains to CD3ζ; and knockout of endogenous TCR expression via nuclease editing50 or small interfering RNAs51. Recently, the first clinical trial of adoptively transferred TCR T cells with edited endogenous TCR α and β chains demonstrated safety and feasibility of the approach10, building upon a decade of work following the first TCR T cell trial52.
CAR T cells harbour a customizable transgenic receptor that is a chimera of a ‘binder domain’, typically derived from an antibody, with T cell-derived transmembrane and intracellular signalling domains (FIG. 1b). CARs harness the exquisite MHC-independent binding specificity of antibodies to activate T cells via TCR (signal 1) and co-stimulatory signalling domains (signal 2)53–55. Affinity tuning of binder domains and selection of signalling domains are important considerations in CAR T cell therapy. High-affinity binder domains prevent tumour escape by antigen-low cancer cells56, whereas lower-affinity domains confer increased cytotoxicity and proliferation57 and can spare normal cells expressing physiological levels of target antigen56. The choice of intracellular signalling domain should match the optimal character of the desired immune response. For example, CAR T cells expressing the ICOS co-stimulatory domain secrete more T helper 17 cell cytokines compared with the CD28 or 4–1BB domains54.
Chimeric autoantibody receptor (CAAR) T cells (FIG. 1b) are a subset of CAR T cells that assuage antibody-mediated autoimmune diseases by reversing the antibody–protein binding paradigm of CAR T cells. CAARs contain autoantigen as their extracellular binder domain, eliminating autoantibody-secreting B cells by binding cognate B cell receptors. CAAR T cells with a desmoglein 3 binder domain kill pathogenic B cells to ameliorate pemphigus in animal models of disease58, which became the foundation for a phase I clinical trial (https://clinicaltrials.gov/ct2/show/NCT04422912).
Regulatory T cells.
Regulatory T cells (Treg cells) are anti-inflammatory CD4+ T cells tasked with maintenance of immunological homeostasis with self and commensal antigens. Adoptive transfer of Treg cells is envisioned to ameliorate the scores of autoimmune diseases in which Treg cell paucity or dysfunction contributes to the cause59–61 or to prevent disease where dominant tolerance fails to block the initiation of an unwanted immune response12. Clinical-scale adoptive transfer of in vitro expanded, unmodified Treg cells was safe and decreased the incidence of GVHD following allogeneic bone marrow transplantation62, paving the way for engineered Treg cells. Treg cells are broadly suppressive; stimulation via transgenic TCR or CAR can simultaneously suppress an array of inflammatory cell types targeting multiple autoantigens. Treg cells seem to have a lower threshold for signal 1, as CD8-derived TCRs readily work in CD4+ Treg cells but not effector CD4+ T cells63. CAR Treg cells that recognize MHC class I are protective in animal models of allotransplantation and GVHD64–67, whereas transgenic TCR Treg cells have been designed to recognize pancreatic β-cell antigens for the treatment of type 1 diabetes mellitus68,69.
Treg cell engineering has benefited from progress in effector T cell engineering, yet fundamental differences in biology between the two cell types present unique design and manufacturing challenges. Even the most successful T cell modifications must be assessed for translation to Treg cells both in vitro and in vivo. For example, the 4–1BB and CD28 co-stimulatory domains, which are major contributors to the efficacy of Kymriah and Yescarta CAR T cells, signal differently in Treg cells; 4–1BB co-stimulation causes Treg cells to lose suppressive function70,71. Moreover, the relative paucity of Treg cells in peripheral blood necessitates GMP sorting followed by serial stimulation ex vivo, neither of which is part of the proinflammatory CAR T cell manufacturing process72,73. The major safety concern of engineered Treg cells is the loss of Treg cell identity by conversion into IL-17-secreting or interferon-γ-secreting ‘ex-FOXP3 cells’ within inflammatory milieus, which hastens disease progression74. Mitigating strategies include stringent isolation of Treg cells72, reinforcement of Treg cell identity by overexpressing FOXP3, or redirecting effector T cells into suppressor ‘edited Treg cells’ by placing endogenous FOXP3 under the control of an active promoter75.
Genome engineering approaches
At their core, T cell therapeutics rely on the ability to engineer the genome. The strategies used can be broadly stratified into viral or non-viral types and can have non-specific or targeted effects (FIG. 2). As we discuss the pros and cons of putative approaches, it is important to realize that one size will not fit all. Disease cause, severity, corporeal location and pathophysiology should inform the design and approach of the intended knock-ins, knockouts or other genetic mutations. As the toolbox of methods to engineer DNA expands, the fidelity and efficiency of each method must be thoroughly characterized in preclinical and clinical studies to inform the optimal approach for each type of therapeutic challenge.
Fig. 2 |. The genetic outcome of modifying the T cell genome.
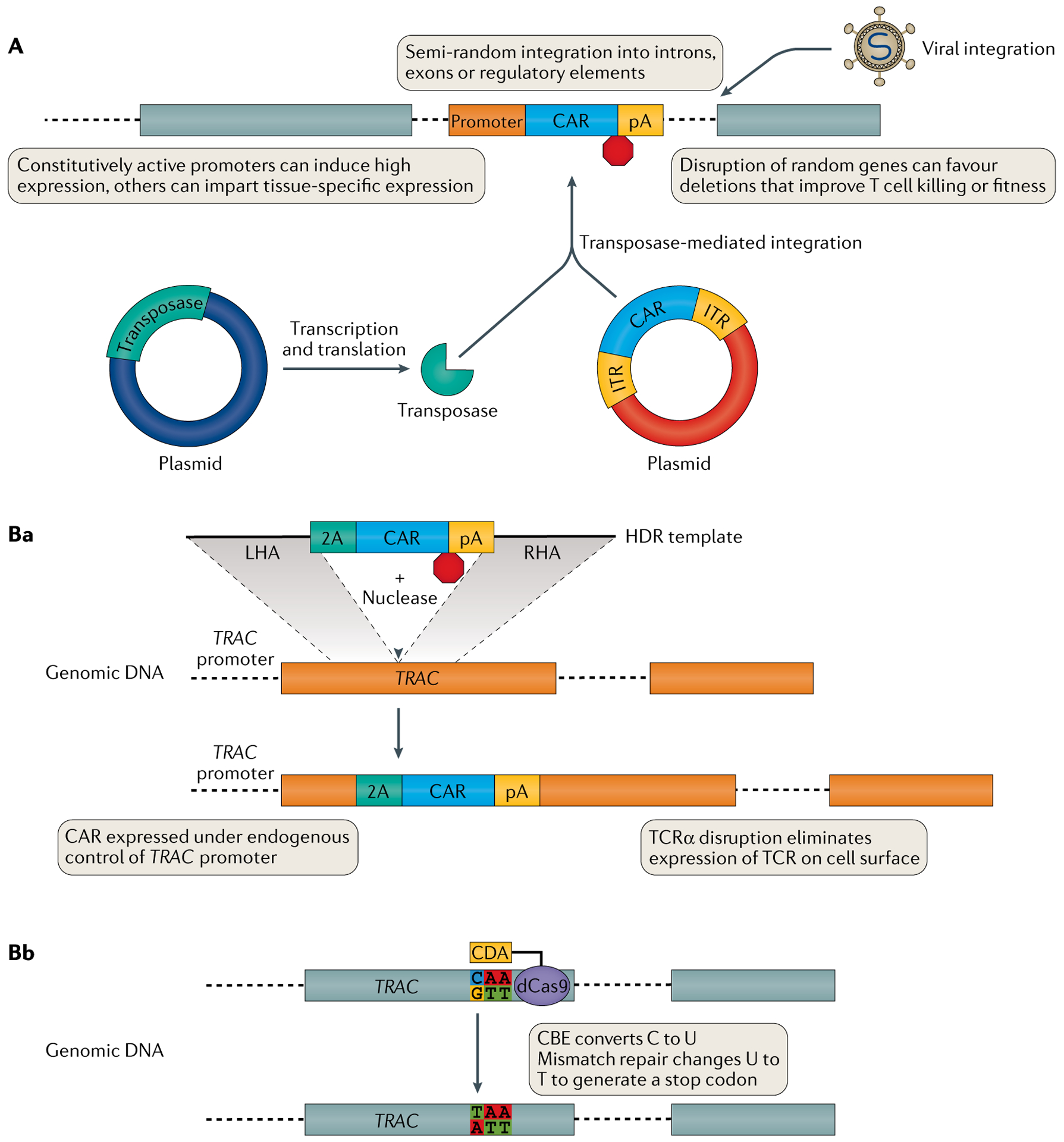
A | Viral vectors (top) or co-delivery of transposase and transposon (bottom) can integrate transgenes into the genome in a non-targeted fashion. Ba | Nuclease-targeted DNA double-strand breaks in the presence of homology directed repair (HDR) template DNA facilitates integration of template DNA at a specific genomic locus. Depending on the design of the template and the location of the DNA double-strand breaks, the transgene can be placed under the control of an endogenous promoter and/or can knock out expression of a gene into which it integrates. For example, integrating a transgene expressing a chimeric antigen receptor (CAR) into the TRAC locus, which encodes the T cell receptor (TCR) α-chain constant region, eliminates endogenous TCR expression. Bb | Cytidine deaminase (CDA) or adenine deaminase tethered to catalytically inactive (‘dead’) Cas9 (dCas9) forms a cytosine base editor (CBE) or adenosine base editor, respectively. Base editing can repair damaged genes or generate functional knockouts through introduction of a novel stop codon (shown) or by disrupting RNA splice acceptor–donor pairs. 2A, ribosomal skip peptide; ITR, inverted terminal repeat; LHA, left homology arm; pA, poly(A) tail; RHA, right homology arm; TCR, T cell receptor.
Non-targeted genome editing
Viral gene delivery.
Viral vector transduction is a reliable means of integrating a transgene into the T cell genome (FIG. 2a). The large packaging size enables delivery of CARs or TCRs, in addition to multicistronic suites containing additional transgenes to enhance the behaviour of the T cell product. However, larger provirus inversely correlates with titre, imposing an upper limit to cargo size76. Integration is semi-random, with Moloney murine leukaemia virus-based gammaretroviral integration clustering near transcriptional start sites77, and HIV-based lentiviral integration showing a propensity for active genes. Insertional mutagenesis of oncogenes or tumour suppressor genes has the potential to cause oncogenic transformation via dysregulation of these genes. Although gammaretroviral vectors were used for the first CAR T cell trials in the late 1990s78–80, lentiviral vectors have since become the virus of choice, owing to their higher transduction efficiency and transgene expression in T cells81,82.
Tuning of viral transgene expression can be modulated via the promoter and 3′ untranslated region sequences accompanying the coding region. Often, low-affinity CAR or TCR and/or low expression of target antigen can render some cancer cells invisible, necessitating maximum receptor expression for sufficient therapeutic effect. Constitutively active promoters combined with optimized 3′ untranslated regions83 endow stable, high-level transgene expression in primary human T cells81,84. However, overexpression of CAR can lead to T cell exhaustion85 and may render the CAR more reactive against low-expressing, healthy tissue targets. Thus, more regulated kinetics may be favourable that allow the CAR T cell to eliminate tumours, ignore healthy tissue and have physiological activation strength and kinetics. Incorporation of activation-dependent or tissue-specific regulatory elements may provide an additional layer of genetic control86.
In contrast to oncogenic gene integration events, insertional mutagenesis causing overexpression or knockout of other proteins in a therapeutic T cell product is a phenomenon being actively exploited. Analysis of virally transduced T cells that are selected by their thriving in the hypoglycaemic, hypoxic, cytokine-laden tumour microenvironment could identify genes that are vital for survival and therapeutic function among such pressures. Since each T cell’s unique TCR acts as a barcode, deep sequencing of the TCR locus can characterize the therapeutic response as polyclonal, pauciclonal or monoclonal and track a functional clonal population from the infusion product through the peak of response to long-term maintenance. In a patient with chronic lymphocytic leukaemia participating in a CD19 CAR T cell clinical trial, TCR deep sequencing revealed that a clonal population derived from a single CAR T cell was responsible for remission, constituting 94% of all peripheral blood T cells at the peak of response87. Integration site analysis revealed insertion of the CAR transgene into the methylcytosine dioxygenase TET2 locus, causing loss of gene function. CAR T cells with knockdown of TET2 showed a skewing towards a central memory phenotype, increased growth in vitro and perturbed cytokine secretion. Fortunately, this massive clonal expansion of TET2CAR/− cells retracted to set point levels, absent of oncogenic transformation87, cementing TET2 knockdown as a testable candidate for intentional knockdown in future CAR T cell therapy. Similar molecular analyses of responders and non-responders alike will unearth other processes fundamental to clinical efficacy.
Non-viral gene delivery.
Non-viral gene delivery offers an alternative method of T cell engineering free from the high cost of production and safety testing of cells modified using viral vectors. T cells are readily electroporated, allowing DNA, RNA, proteins and ribonucleoprotein complexes to cross the plasma and nuclear membranes. In vitro transcribed mRNA can be introduced into both resting and activated T cells, inciting transient expression of a protein of interest88. The magnitude and persistence of transgene expression can be regulated by varying the amount of RNA electroporated88, the length of the poly(A) tail89 and the inclusion of a 5′ cap.
Co-transfection of piggyBac or Sleeping Beauty transposase with transgene-containing DNA leverages ‘jumping genes’ to non-virally integrate genetic information into engineered T cells90,91 (FIG. 2a). As with viral vectors, piggyBac prefers integration into genes and thus carries the risk and reward of insertional mutagenesis92,93. By contrast, Sleeping Beauty integrates genes favourably for therapeutic application in a close-to-random fashion. However, toxicity associated with template DNA delivery and lower transduction efficiency compared with lentiviral vectors limits engineered T cell expansion in vitro94, and care must be taken to avoid integration of the transposase itself, leading to unconstrained remobilization of the transgene. The development of novel forms of Sleeping Beauty95,96 along with methods for delivery of both transposase and transgene template97 continue to support the clinical translation of this therapeutic modality. Clinical trials of transposon-based CD19-specific CAR T cell adoptive therapy demonstrated safety and feasibility of this approach98,99.
Targeted genome editing
A new crop of site-specific nucleases grants the ability to select where a construct integrates into the genome (BOX 2). Each method in this class shares an underlying tenet — the ability to shepherd a DNA nuclease to a predetermined region of interest. DNA double-strand breaks can be repaired by non-homologous end joining or homology directed repair (HDR) processes. Non-homologous end joining repairs DNA double-strand breaks without end processing, occasionally inducing insertion and deletion (indel) frameshift mutations, leading to premature stop codons and engineered knockout of unwanted protein expression.
Box 2 |. Genome-editing technologies.
ZFNs, TALENs and megaTALs
Zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) are chimeric proteins that fuse site-specific DNA binding with the non-specific cleavage domain of the endonuclease FokI. The obligate dimer character of FokI243 necessitates ZFN or TALEN binding to both plus and minus strands of DNA, decreasing off-target cleavage at the expense of on-target efficiency. The ease in generating TALENs for virtually any genomic sequence is an advantage over ZFNs, and a collection of TALENs targeting every protein in the human genome has been created244. However, TALENs are about threefold larger than ZFNs, making them more difficult to package in viral vectors. When TALEN and ZFN editing were directly compared at the CCR5 and IL2RG loci, TALENs were as specific as ZFNs and provoked less cytotoxicity220. Infusion of ZFN-modified and TALEN-modified T cells into patients has demonstrated safety and efficacy, including a lack of product tumorigenesis, despite the presence of rearranged chromosomes generated by DNA double-strand breaks (DSBs)212,218.
MegaTAL nucleases share DNA-binding domains with TALENs but have a meganuclease replacing FokI, allowing cutting with a single molecule rather than a pair245. In comparison with TALENs, megaTALs induced more non-homologous end joining insertions and deletions (indels) at TRAC246 and fewer at CCR5 (REF.103).
CRISPR–Cas9
CRISPR–Cas9 uses a guide RNA to ferry the nuclease Cas9 to a region of interest to induce gene knockout by DSB and indel formation following non-homologous end joining repair. dCas9 is an enzymatically inactive variant that retains its ability to be guided by RNA247 and can be tethered to transcriptional activators248, repressors249 or methyltransferases250 to influence transcription of DNA, often in non-coding regions. Other Cas molecules are in development that have different protospacer adjacent motifs, enabling CRISPR-mediated DSBs in areas inaccessible to Cas9 (REF.251). A clinical trial of multiplex CRISPR T cell receptor-engineered T cell adoptive therapy demonstrated feasibility and persistence of cells for up to 9 months, albeit with chromosomal translocations between CRISPR sites10.
Base editing
Base editing uses a fusion protein consisting of either a cytidine deaminase or an adenosine deaminase and dCas9 to impart RNA-guided transition point mutations without DSBs252, reducing the chance of chromosomal translocation. Mutations induced by base editing can correct genes that contribute to a disease phenotype or knock out protein expression by alteration of RNA splice donor–acceptor pairs or introduction of a premature stop codon. In T cells, triplex base editing of B2M, CIITA and TRAC occurred at more than 98% of sequencing reads and eliminated surface expression of β2M and HLA-DR225. CD3 expression due to TRAC editing was downregulated but not extinguished, emphasizing that nonsense-mediated decay is not sufficient to knock out protein expression for some loci225. Additionally, ribosomal stop codon readthrough can circumvent novel stop codon mutations.
HDR exploits homology found on undamaged sister chromatids to resolve damage. This pathway can be commandeered by cellular engineers to deliver transgenes to a specific locus by delivering DNA repair templates containing the transgene enveloped by homology arms specific for either side of the DNA lesion (FIG. 2Ba). In contrast to semi-random integration with viral vectors or transposases, the integration of a transgene at a precise genomic location via HDR allows avoidance of integration near an oncogene or tumour suppressor gene, finer control of transgene by an endogenous promoter and/or simultaneous knockout of another protein. Since the only prerequisite for HDR is a DNA double-strand break, multiple nucleases85,100–102 have been used for HDR-mediated knock-ins in T cells. Donor DNA can be delivered to T cells by either adeno-associated virus serotype 6 (REFS100,103) or DNA electroporation104,105. To increase safety, DNA can be inserted into a ‘safe harbour’ site identified as unlikely to cause insertional mutagenesis106. Alternatively, the HDR template can be targeted to a gene, generating a two-for-one benefit: knockout of an undesired protein of interest and control of transgenic protein expression under a novel promoter. For example, in TCR T cells or CAR T cells, integration of a transgene into the TCRα-encoding TRAC locus concurrently eliminates TCR expression, increasing efficacy by decreasing crosspairing with endogenous TCRα, and places the transgenic receptor under endogenous control of the TRAC promoter85 (FIG. 2Ba). Such regulation improves function versus constitutive expression by reducing T cell exhaustion in the absence of tonic signalling85. This approach highlights that controlling expression can be as important as the character of the transgene. Along the same lines, targeted integrations can help customize a cell product for a particular disease indication. For example, targeted insertion of HIV-specific CARs into the CCR5 locus renders the resulting HIV CAR T cells HIV resistant as well by eliminating a viral co-receptor107.
Each method of engineering has its own benefits and limitations (TABLE 1). Because many are in the nascent stages of characterization, the overall safety profile of each is still being delineated. A balance between on-target and off-target editing must fall within the therapeutic range whereby enough cells are engineered correctly to have an impact on disease without causing oncogenesis or dysfunction due to editing of an unintended locus.
Table 1 |.
Advantages and limitations of technologies for genome editing of therapeutic T cell products
| Technology | Mechanism | On-target efficacy | Limitations | Benefits | Targets | Rationale |
|---|---|---|---|---|---|---|
| ZFN | DSB; ZFN DBD + FokI endonuclease domain; operates as dimer | Up to 28% in a clinical trial212; up to 90% on a non-clinical scale213 | ZFN specificity designed by screening then tested empirically; requires pair of ZFNs | Low off-target editing | CCR5 (REFS100,212,214) | Resistance to HIV by knockout of viral co-receptor |
| CXCL4 (REFS215,216) | ||||||
| TRAC213 | Knockout of endogenous TCRa | |||||
| TRBC217 | ||||||
| TALEN | DSB; TALE DBD + FokI endonuclease domain; operates as dimer | Up to 64% in a clinical trial218; up to 80% on a non-clinical scale219 | Large protein size; can induce translocations218 | Low off-target editing | CD52 (REF.218) | Resistance to anti-CD52 antibody therapy |
| CD40L102 | Repair of damaged gene causing hyper-IgM syndrome | |||||
| CCR5 (REF.220) | Resistance to HIV by knockout of viral co-receptor | |||||
| DCK (encodes deoxycytidine kinase)219 | Resistance to purine nucleotide analogue drugs | |||||
| PDCD1 (encodes PD1)221 | Reduces T cell exhaustion | |||||
| TRAC219,221 | Knockout of endogenous TCRa | |||||
| MegaTAL | DSB; TALE DBD + meganuclease | Up to 61% on a non-clinical scale101 | Off-target editing103 | Small protein size | CCR5 (REF.103) | Resistance to HIV by knockout of viral co-receptor |
| CRISPR-Cas9 | DSB; gRNA + Endonuclease Cas9 | Up to 45% in a clinical trial10; up to 95% on a non-clinical scale222 | Target must be adjacent to PAM; high degree of off-target editing or translocations | Easy to design and screen guides, high efficiency | B2M222 | Protects universal T cells from alloreactive rejection by host |
| CCR5 (REF.223) | Resistance to HIV by knockout of viral co-receptor | |||||
| PDCD1 (REF.222) | Reduces T cell exhaustion | |||||
| RNF20 (REF.159) | Stabilizes FOXP3 in Treg cells | |||||
| TRAC85,222 | Knockout of endogenous TCRa | |||||
| TRBC222 | ||||||
| Nuclease + HDR | DSB in presence of ssDNA or dsDNA delivered by AAV or electroporation | Non-viral: up to 61% on a non-clinical scale105 Viral: 40–60% on a non-clinical scale85,224 |
Immunogenicity of DNA or AAV, off-target integration; dependent on presence of adjacent PAM | Integration of large cargoes | AAVS1 (REF.100) | Safe harbour site for genome integration |
| CD40L102 | Repair of damaged gene causing hyper-IgM syndrome | |||||
| FOXP3 (REF.75) | Converts Teff cells into Treg-like cells | |||||
| IL2RA104 | Repair of damaged gene causing autoimmunity | |||||
| IL2RG220 | Repair of damaged gene causing SCID | |||||
| TRAC85,104 | Puts CAR or other transgenes under control of endogenous promoter | |||||
| Base editing | Nucleotide deaminase + dCas9 + uracil DNA glycosylase inhibitor + single gRNA | >95% on a non-clinical scale225 | Transition mutations only, no deletions or insertions; bystander edits in editing window, dependent on adjacent PAM; may not eliminate protein expression | No DSB, no detectable translocations in triple editing | TRAC225 | Knockout of endogenous TCRa |
| CIITA225 | Protects universal T cells from alloreactive rejection by host | |||||
| B2M225 |
AAV, adeno-associated virus; CAR, chimeric antigen receptor; DBD, DNA-binding domain; dCas9, dead (catalytically inactive) Cas9; DSB, DNA double-strand break; dsDNA, double-stranded DNA; gRNA, guide RNA; HDR, homology directed repair; PAM, protospacer adjacent motif; SCID, severe combined immunodeficiency; ssDNA, single-stranded DNA; TALE, transcription activator-like effector; TALEN, transcription activator-like effector nuclease; TCR, T cell receptor; Teff cell; effector T cell; Treg cell; regulatory T cell; ZFN, zinc-finger nuclease.
Knockout of endogenous TCR improves surface expression of transgenic TCR or generates universal T cells.
Engineering approaches to increase efficacy
How a T cell is engineered alters its trafficking, function, survival and, ultimately, ability to cure disease. For example, within a single CAR framework, constructs using the CD28 or 4–1BB co-stimulatory domains have marked differences, with CD28-based CARs displaying enhanced effector function and 4–1BB-based CARs having enhanced survival and expansion24,81,108,109. Given that even subtle changes to how T cells are programmed can have dramatic effects on function and persistence, correctly programming a T cell to meet the challenges of a given disease setting is a daunting task. In the oncology field, where the exploration of gene-engineered T cells is concentrated, no two cases of a given cancer histology are identical in their genetic and epigenetic aberrations, and tumours acquire further heterogeneity within an individual patient as they evolve, further complicating the engineering challenge8. In keeping with the heterogeneity of tumours, preclinical models and early clinical studies of gene-engineered T cell therapies have revealed a panoply of mechanisms that potentially contribute to insufficient or transient efficacy. Such insights provide a premise for the rational design of ‘next-generation’ gene-engineered T cell therapies equipped either through addition (‘armoured’) or deletion of specific regulatory factors to overcome the principal immune resistance mechanisms of tumours while avoiding toxicity (see the section entitled Engineering approaches to increase safety). As more advanced forms of genetic engineering emerge, the ultimate goal is to programme T cells to act in a more discriminating and context-dependent fashion to optimally harness both the potency and the selectivity of T cells against cancer.
Defining the problem
While barriers to the efficacy of engineered T cell therapies can be categorized in numerous ways, here we consider them in the oncology setting using a well-known framework from the broader drug development field, the ‘three pillars’ of survival of candidate medicines in mid-phase clinical testing110: (1) exposure of a drug at the site of action, which for T cell therapies refers to the active process of trafficking to the tumour; (2) target engagement, which for T cell therapies can be inferred to mean addressing diversity in target antigen expression levels or antigen loss as a result of immune editing; and (3) expression of functional pharmacological activity, which in this context equates to the sustained function of the therapeutic T cell once it has engaged its target, which is strongly influenced by the tumour microenvironment. The three-pillar framework may be especially useful given that even phase I trials of CAR T or TCR T therapies are closely scrutinized for signs of clinical benefit, and regulatory approval has been based on single-arm studies of fewer than 100 patients20,21, akin to the typical phase II study size for traditional pharmaceuticals. FIGURE 3 summarizes some of the specific strategies that have been used to enhance the efficacy of engineered T cells that are discussed in the following sections.
Fig. 3 |. Gene-engineered T cell products to enhance efficacy.
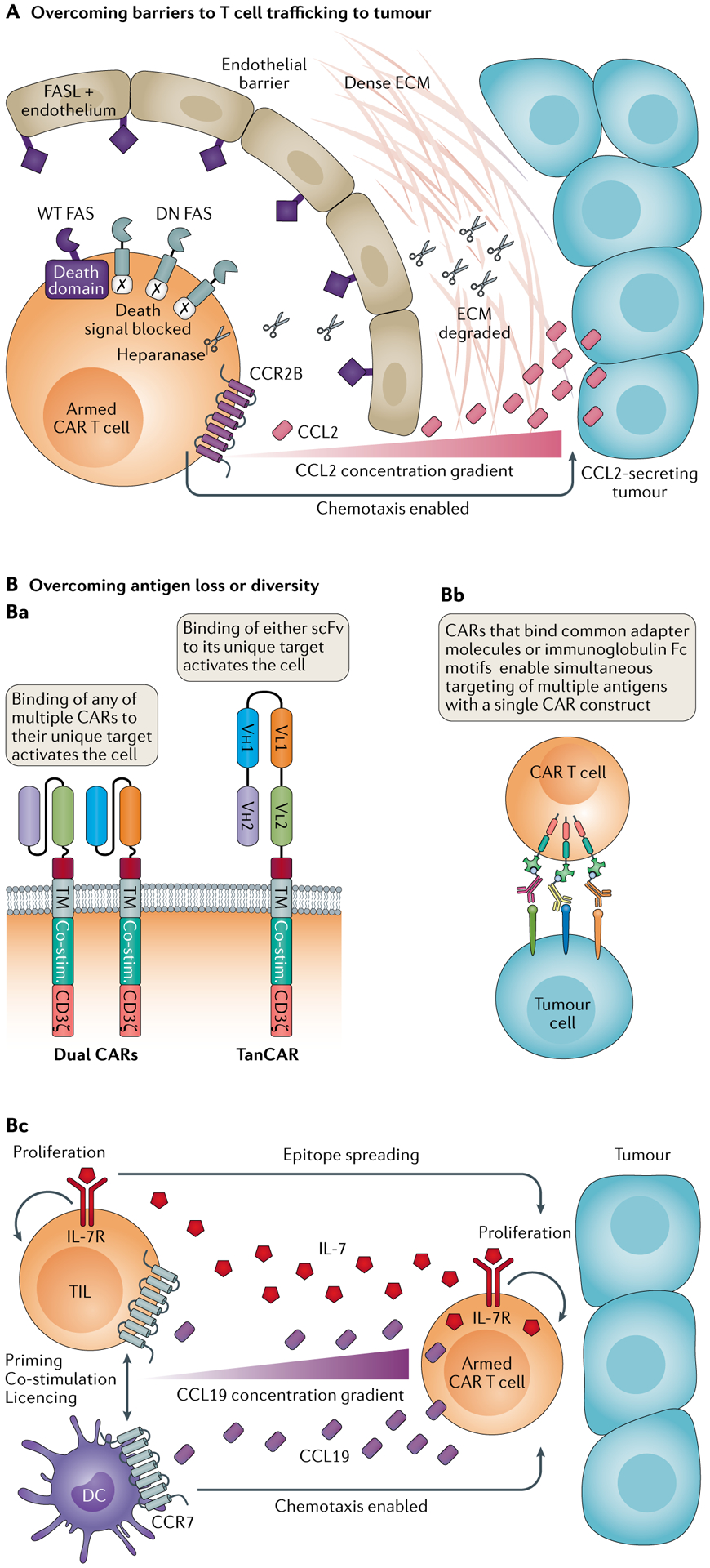
A | Overcoming barriers to T cell trafficking to tumour. The addition of an appropriate chemokine receptor matched to sense chemokines released by the target tumour has increased T cell infiltration in animal models. Addition of a dominant negative (DN) FAS receptor has enabled engineered T cells to avoid FAS ligand (FASL)-mediated apoptosis induction. Equipping T cells to secrete heparanase has enabled them to counter the dense extracellular matrix (ECM) of tumours in preclinical models. B | Overcoming antigen loss or diversity can be achieved by transduction with two or more chimeric antigen receptors (CARs) (OR gate logic) or via tandem CARs (TanCARs) (panel Ba), using a universal CAR and separate tumour-targeting ligands (panel Bb) or equipping T cells to express both growth factors and chemokines to enhance infiltration of tumours by T cells and dendritic cells (DCs) (panel Bc). Co-stim., co-stimulatory domain; scFv, single-chain variable fragment; TIL, tumour-infiltrating lymphocyte; TM, transmembrane domain; WT, wild type.
Exposure at the site of action.
T cell trafficking is a complex, active, multistep process that requires the engagement of chemokine receptors on T cells by chemokines released in the tumour microenvironment; the attachment, rolling and arrest of T cells on activated endothelium; and subsequent extravasation and migration through the extracellular matrix of the tissue stroma111. There is often a mismatch between the types of chemokines released by tumours and the chemokine receptors expressed by effector T cells, resulting in diminished chemoattraction111. Furthermore, the endothelium of tumour-associated blood vessels is often anergic to activation by inflammatory stimuli111, poorly conducive to T cell attachment and extravasation112, and tends to overexpress FAS ligand (FASL), triggering apoptosis in effector T cells113,114. Even when effector T cells overcome these hurdles and make it into the tumour stroma, they typically face a dense extracellular matrix as an additional physical barrier through which they must migrate115. The culmination of the various barriers results in fewer than 1% of adoptively transferred T cells successfully arriving at the sites of action to exert a pharmacological effect116,117.
To improve the trafficking of gene-engineered T cells to tumours, numerous strategies have been demonstrated in preclinical models (FIG. 3A). Engineering T cells to express a chemokine receptor that responds to the chemokines produced by the tumour facilitates T cell infiltration of tumour lesions118. T cells equipped with a dominant negative version of FAS were able to resist FASL-induced apoptosis114. Those armoured to overexpress heparanase were able to digest the dense extracellular matrix of solid tumours119. Each of these approaches showed benefit in preclinical models, resulting in improved tumour control, which warrants potential exploration in patients. However, differences between patients and even between different sites of tumour metastasis may be a key hurdle for implementation.
Target engagement.
To address the intrinsic antigen diversity of tumours120 and the tendency of both haematological tumours121 and solid tumours10,122,123 to undergo antigen loss on treatment with engineered T cells, efforts to target multiple antigens or to enhance the endogenous immune response by promoting epitope spreading have been proposed (FIG. 3B).
Conceptually, perhaps the most straightforward approach to address antigen diversity or escape is a cocktail or ‘CAR pool’ in which traditional CAR T cells of distinct specificity are mixed124. However, to meet GMP standards, CAR pools will likely require the manufacture of each component separately before mixing at defined ratios, multiplying cost considerations. Alternatively, T cells can be engineered to express two kinds of CAR molecule125; that is, a dual CAR, or both a CAR and a TCR126. Either approach imparts on T cells the function of a classical OR Boolean logic gate, such that encountering either antigen is sufficient to activate the T cell. Dual CARs were found to have superior activity compared with CAR pools of the same individual specificities as well as being more straightforward to prepare125. In addition, our group recently demonstrated that dual CAR T cells transduced with two separate CARs containing a CD28/CD3ζ endodomain or a 4–1BB/CD3ζ endodomain had improved effector function, increased survival and increased expansion versus CAR T cells containing a single CD28/4–1BB/CD3ζ CAR53.
An alternative to expressing two separate CAR molecules is the bispecific or tandem CAR (TanCAR), in which both binders are engineered into a single CAR molecule127,128. When both antigens are present, this approach has the advantage of superior activity compared with dual CARs owing to enhanced immune synapse formation, and TanCARs require less nucleotide coding capacity than dual CARs127,128. However, TanCARs are inferior to CAR pools or dual CARs when antigen escape at one antigen has already occurred129. They also have the disadvantage of needing to determine the optimal order and spacing of the binders, and the potential of single-chain variable fragment (scFv) binders to mispair130. Expanding on the dual CAR concept, a trispecific CAR T design has also been reported in which three separate CAR constructs are delivered via one lentivirus and expressed in a single T cell; the trivalent CAR T cells were able to recognize any of three independent antigens131. While even greater valency may be possible with future designs using gene delivery systems with sufficient nucleotide coding capacity, a significant limitation to increased valency may be the issue of compounding the on-target off-tumour toxicity (discussed in the section entitled Engineering approaches to increase safety) to unacceptable levels.
Two alternatives to address antigen diversity beyond merely increasing the valency of gene-engineered T cells have been proposed: the ‘universal’ CAR T cell paired with a library of targeting modules that can be infused separately, and engineering T cells to optimize the induction and/or recruitment of an endogenous immune response, resulting in epitope spreading against the tumour.
Universal CAR T cells make use of a single T cell product that can be infused in multiple disease settings, followed by infusion of distinct targeting moieties where the variable end recognizes the selected tumour antigen and the constant end is recognized by the universal CAR. The simplest form of this platform borrows the CD16-dependent antibody-dependent cellular cytotoxicity mechanism from natural killer cells, allowing T cells expressing a CD16-based CAR to be redirected by existing therapeutic monoclonal antibodies (mAbs)132. Alternative universal CARs can be redirected using tagged or hapten-conjugated mAbs or antibody fragments, bispecific adaptors133 or natural binding partners such as streptavidin–biotin (reviewed in REF.134) or SpyTag-SpyCatcher135. To implement universal CAR systems clinically, each targeting ligand needs to demonstrate adequate pharmacokinetics, safety and low immunogenicity, and the degree to which universal CAR T cells can persist in the absence of targeting a ligand will need to be determined.
Knowledge of the antigen landscape in a given patient’s tumour, especially with respect to the variation between metastatic sites, is likely to remain limited. Hence, the concept of engineering T cells to promote the induction of endogenous immune responses is a worthy one. To this end, CAR T cells engineered to secrete IL-7 together with either CCL19 (known as 7×19 CAR T cells)136 or CCL21 (known as 7×21 CAR T cells)137 have been reported (FIG. 3B). IL-7 and CCL19 were selected on the basis of their importance in maintaining T cell zones in lymphoid organs136, whereas CCL21 was selected owing to its ability to attract naive T cells and dendritic cells to T cell zones, as well as for its antiangiogenic effects137. Mice treated with 7×19 CAR T cells demonstrated an increase in dendritic cell and T cell tumour infiltration with improved resolution of large established tumours compared with CAR T cells alone136. Efficacy was reduced if host T cells were eliminated before 7×19 CAR T cell therapy, and endogenous CAR-negative T cells formed memory responses to tumour136. Meanwhile, 7×21 CAR T cells outperformed both 7×19 CAR T cells and CAR T cells in several mouse models, with evidence of enhanced dendritic cell and T cell infiltration, reduced angiogenesis and activity against tumours composed of a mixed population of cells that were positive or negative for the target antigen137.
With a number of clinical trials under way, or already starting to produce results138–140, testing various strategies to counteract antigen diversity and escape, we should soon learn which approaches are most appropriate in given settings. Many B cell malignancies have fairly low mutational burdens141 and high expression of lineage-specific markers, such as CD19, CD20 and CD22, which may be ideal indications for multitargeting CAR T cells. At the other end of the spectrum, tumours with high mutational burdens such as non-small-cell lung carcinoma and melanoma141 might be better substrates for approaches that seek to break tolerance and induce epitope spreading.
Achieving sustained pharmacological activity.
The suppressive tumour microenvironment is a key barrier to immunotherapy in general and a challenge to study using engineered human T cells142. Numerous mechanisms are involved, often with significant crosstalk, which include a lack of nutrients, hypoxia, the presence of toxic metabolites, suppressive cytokines and immune checkpoints, and an abundance of suppressive stromal, myeloid and lymphoid cells (reviewed in REF.143). FIGURE 4 provides examples of some of the notable approaches that have been deployed to enable gene-engineered T cells to resist the effects of the suppressive tumour microenvironment and thereby sustain their pharmacological activity.
Fig. 4 |. Addition of armour or subtraction of suppressive genes or their transcripts enables TME resistance.
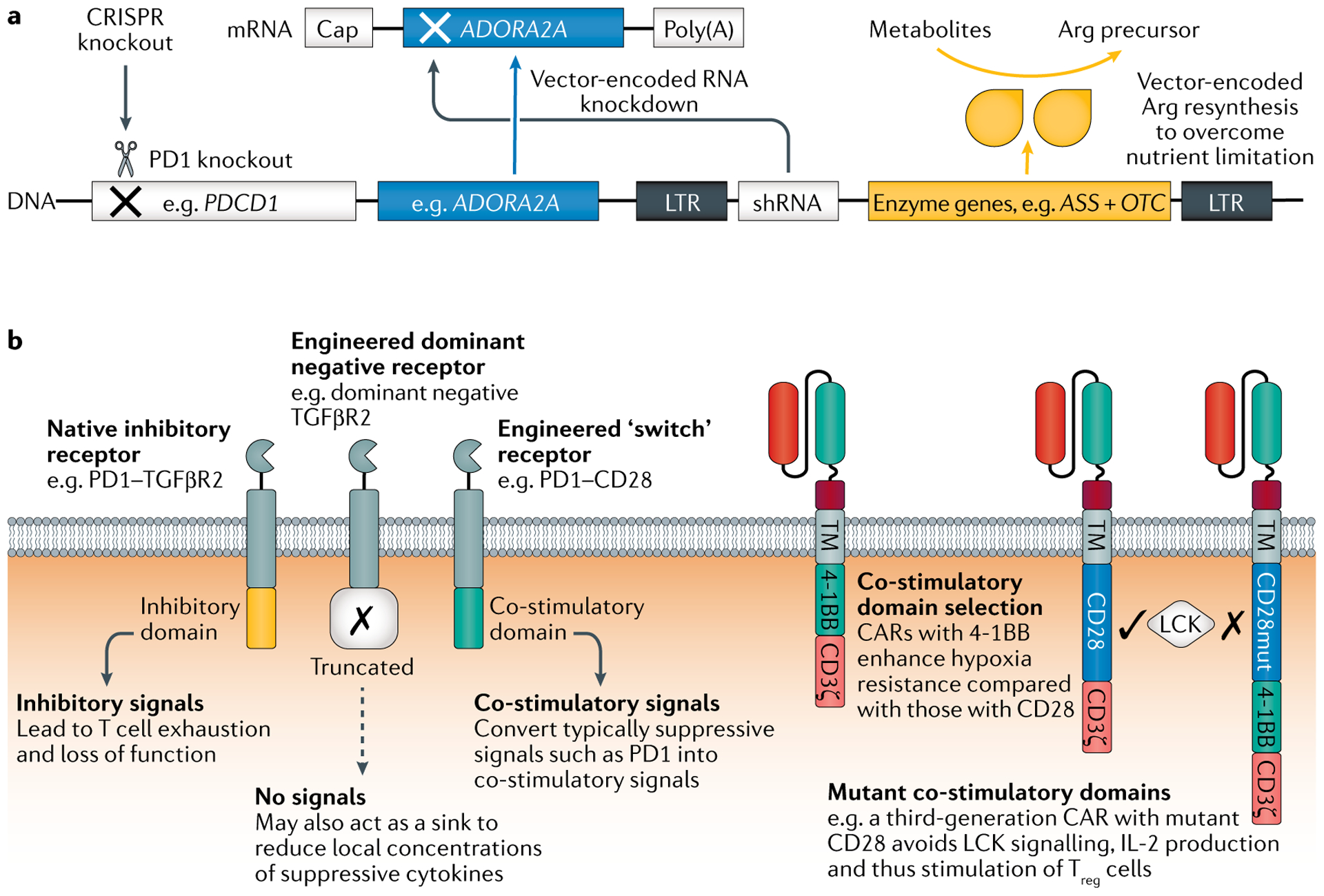
a | Many potential design solutions are available, including gene deletion (for example, using CRISPR–Cas9 genome editing to knock out PDCD1), RNA interference (for example, knockdown of the adenosine A2A receptor gene ADORA2A to confer adenosine resistance) and addition of genes to enable metabolic self-sufficiency (for example, addition of ASS and OTC, which encode the enzymes argininosuccinate synthetase and ornithine transcarbamylase, respectively) or that counter specific checkpoints and suppressive cytokines. b | Receptor-based approaches to resist tumour microenvironment (TME) suppression include expression of a dominant negative form of an inhibitory receptor to act as a sink. Alternatively, the receptor’s inhibitory domain can be switched to a co-stimulatory domain to change the inhibitory signal into an activating one. Chimeric antigen receptor (CAR) design considerations include the choice of co-stimulatory domains; both natural and mutant co-stimulatory domains may enhance the ability of the engineered T cell to survive in the suppressive TME. Multiple strategies may be used in parallel and should be customized to the specific TME roadblocks encountered. LTR, long terminal repeat; shRNA, short hairpin RNA; TM, transmembrane domain; Treg cell, regulatory T cell.
The emergence of PD1–PDL1 checkpoint antagonists as a major class of immunotherapies has stimulated a number of approaches to tackle this pathway and similar immune checkpoints in the setting of gene-engineered T cells (see the section entitled Challenges in implementing solutions). Notably, these approaches have included the first published examples of CRISPR-engineered T cells in patients, in which the gene for PD1, PDCD1, was deleted in NY-ESO-1-specific TCR T cells10 or in bulk T cells144 (FIG. 4a).
TGFβ is also emerging as a key immunosuppressive factor in many tumours145. To counter this pathway, the dominant negative receptor approach has been used (FIG. 4b), for example, in CAR T cells targeting prostate-specific membrane antigen (PSMA), where it enhanced their persistence and function in vitro and in vivo146. The dominant negative receptor lacks an endogenous signalling domain and thus fails to propagate the typical signalling pathways downstream of the receptor complex, acting as a sink for the cytokine and reducing the amount of suppressive TGFβ signalling for bystander T cells as well as the gene-engineered T cells147. In a clinical study of Epstein–Barr virus (EBV)-specific cytotoxic T lymphocytes for the treatment of EBV-positive Hodgkin lymphoma, a patient whose tumour resisted therapy with non-engineered cytotoxic T lymphocytes achieved an objective response on subsequent therapy with cytotoxic T lymphocytes bearing the dominant negative TGFβ receptor148. With numerous trials of TGFβ-resistant gene-engineered T cells in progress, we may soon learn the degree to which efficacy can be safely enhanced using this approach.
Beyond the plethora of suppressive checkpoints and cytokines, metabolites such as adenosine, acting via adenosine A2A receptors, suppress T cell activity. To counter this challenge, CAR T cells were engineered to express a short hairpin RNA molecule to trigger the degradation of the mRNA for the adenosine receptor (FIG. 4a). This approach succeeded in reducing expression of the adenosine receptor by CAR T cells and improved their in vitro and in vivo function in preclinical models149.
Suppressive cells, including Treg cells, M2-type macrophages and myeloid-derived suppressor cells, are key regulators of effector T cell function in the tumour microenvironment143. Treg cells are an especially pernicious challenge since activated effector T cells secrete IL-2, promoting Treg cell expansion150. To counter Treg cells, CAR T cells with both CD28 and 4–1BB as co-stimulatory molecules were engineered with a mutation in the CD28 element preventing the recruitment of the SRC kinase LCK, thus avoiding IL-2 induction (FIG. 4b). In mouse models, CAR T cells engineered in this fashion were resistant to the effects of Treg cells151. Since second-generation CARs with 4–1BB and CD3ζ alone were not tested, the study primarily supports a method to improve on CD28-containing CARs rather than demonstrating superiority to constructs utilizing alternative co-stimulatory domains.
Aside from the numerous immunosuppressive cells, checkpoints and cytokines in the tumour microenvironment, the metabolic demand of the tumour cells themselves, coupled with inadequate perfusion, contributes to creating an environment that is poor in many essential nutrients as well as oxygen, representing significant challenges to the sustained effector function of gene-engineered T cells152. While many design solutions are possible that may help circumvent these challenges, the choice of co-stimulatory domains for CAR T cells is a basic and impactful one. Compared with CD28 co-stimulation, 4–1BB co-stimulation results in enhanced mitochondrial spare respiratory capacity in gene-engineered T cells and thus an enhanced capability to function in hypoxic microenvironments153 (FIG. 4b). More complex mechanisms to enhance the function of gene-engineered T cells in the tumour microenvironment include equipping T cells to express enzymes involved in the resynthesis of the critical amino acid arginine154 (FIG. 4a). CAR T cells engineered to express the enzymes argininosuccinate synthase and ornithine transcarbamylase were self-sufficient in arginine and maintained function in the tumour microenvironment, showing enhanced activity154.
As the above examples show, countermeasures that have been deployed in the preclinical or clinical setting have largely been achieved via the simple addition or deletion of specific regulatory elements. However, given that specific countermeasures may increase the potency of gene-engineered T cells to potentially toxic levels, scientists are increasingly seeking more complex engineering approaches to counter immune suppression in specific contexts. For example, the cytokine IL-12 is a potent immunostimulatory cytokine that is fairly toxic when administered systemically155. When constitutively expressed from engineered T cells, it prevents the manufacture of an adequate product by dysregulating T cell expansion and promoting apoptosis156. In an attempt to effectively harness IL-12, a copy of the encoding gene was added to TCR T cells under the control of an NFAT-inducible promoter to link expression to tumour antigen recognition. While this approach worked well in the preclinical setting156, a clinical trial testing the approach using tumour-infiltrating lymphocytes (https://clinicaltrials.gov/ct2/show/NCT01236573) was terminated due to toxicity, suggesting that either the NFAT system was too leaky or the on-target activity of the tumour-infiltrating lymphocytes and their expansion in patients resulted in intolerable levels of IL-12 production. Such a result underscores the need for improved control of gene-engineered T cells especially when armoured to increase their potency.
Challenges in implementing solutions
The challenges for successful clinical application of efficacy enhancement strategies include finding an indication where the resistance mechanism being addressed is a primary barrier to efficacy, selecting an optimally designed countermeasure and establishing that the enhanced T cell is sufficiently safe.
While much of our existing knowledge of bypassing resistance mechanisms comes from preclinical models and interrogation of patient biopsy samples from early clinical studies, these sources suffer from the potential bias in what the investigators chose to study and how they measured it. CRISPR screens have been used as a non-biased way to uncover pathways involved in enhanced function, stability or persistence of engineered T cells. In this method, libraries of guide RNAs targeting the whole of the genome or curated subsets of the genome are incorporated into T cells along with Cas9 or a catalytically inactive (‘dead’) Cas9 (dCas9)157. After application of a relevant method of selection — high proliferation158, low FOXP3 expression159 or presence in the tumour microenvironment160, for example — proviral guide RNAs are amplified and sequenced to uncover targets that represent important nodes in pathways relevant to the selected process. Incorporation of more than one guide RNA per cell allows the identification of collections of knockout targets which would otherwise be masked by epistasis or complementarity. Alternatively, CRISPR knock-in in the presence of a pool of HDR templates can rapidly select for transgenes that improve CAR T cell and TCR T cell function in a high-throughput manner. Via this method, a TGFβ receptor–4–1BB switch receptor was found to enhance TCR T cell-mediated clearance of melanoma161. By discovering functional T cells that endure the selective pressures of the tumour microenvironment, engineered T cells can be designed to better match that tumour microenvironment.
Once the major immune-resistance pathway relevant to a given gene-engineered T cell therapy in a specific disease setting has been selected, the appropriate countermeasure is required. In general, numerous design solutions can be proposed to address any given challenge. Taking the PD1 pathway as an example, one may focus at the genetic level on knocking out the PDCD1 gene10 or knocking down its expression via vector-encoded RNA interference162. Alternatively, the endogenous PD1 can be left untouched and its effects eliminated by overexpressing a dominant-negative version of PD1 (REF.163), overexpressing a ‘switch’ receptor that converts the signal to a co-stimulatory signal164 (FIG. 4b), disabling downstream signalling pathways triggered by PD1 ligation165 or equipping the T cell to secrete its own PD1 pathway-blocking agents such as antibodies166 or soluble PD1 (REF.167). Moreover, beyond these mechanisms directly targeting PD1, the sensitivity of T cells to PD1-mediated dysfunction depends on many other factors, which can also be modulated. For example, specific T cell subsets seem to be intrinsically more resistant to PD1-mediated suppression168, and exhaustion in general resulting from any checkpoint pathway might be targeted via countering the transcription factors involved in establishing or maintaining T cell exhaustion, such as TOX169, TCF1 (REF.170) and NR4A family members171.
Once the design of the countermeasure to overcome the immune resistance of the tumour has been settled upon, the next hurdle is to establish that the resulting gene-engineered T cell with its enhanced potency will be sufficiently safe and will function as anticipated. In the PD1 example, a T cell intrinsically resistant to PD1 might arguably be safer than one that secretes a PD1 antagonist, as the latter would also block PD1 on bystander T cells. It might also be safer than the use of an approved PD1 checkpoint agent in combination, again due to the restriction of PD1 resistance to the therapeutic T cell. However, the use of a PD1 checkpoint antibody in combination with a therapeutic T cell could be controlled via dosing and withdrawal. Moreover, in a recent clinical trial of PD1-knockout TCR T cells, those lacking PD1 did not persist well10, in keeping with earlier findings that some level of PD1 signalling may be required to establish T cell memory172.
As discussed above, thus far the engineering of T cells for enhanced efficacy involves the fairly straightforward deletion or addition of specific genes to counteract the identified obstacles to achieving deep and sustained clinical responses. The difficulty has largely been in determining which of the barriers are paramount and which are secondary. Much emphasis in the field is on deciphering which changes have the biggest impact for a particular disease. As the ability to perform multiple genetic alterations becomes more mainstream, emphasis will shift to which changes synergize to provide greater therapeutic benefit.
Engineering approaches to increase safety
The ability of T cells to proliferate, act as serial killers of antigen-positive target cells and orchestrate a broader immune response via secreting cytokines and chemokines is a double-edged sword. These mechanisms make T cells a suitable adversary against cancer but can also result in severe or fatal toxic effects173,174. The toxic effects associated with gene-engineered T cells have been investigated extensively173,175–182 and fall into four categories: (1) product risks, (2) on-target toxicity, (3) on-target off-tumour toxicity and (4) off-target toxicity (FIG. 5). Significant toxic effects can also result from drugs that are part of the CAR T cell or TCR T cell regimen173, but these are beyond the scope of this Review. The potential to achieve safety by design via enhanced genetic engineering of T cell products is promising. The various solutions fall into two categories depending on whether they allow exogenous control (that is, physician-mediated control) or are endogenously programmed, enabling the T cell to respond autonomously to avoid or reduce toxicity (FIG. 6).
Fig. 5 |. Toxicity risks associated with gene-engineered T cells.
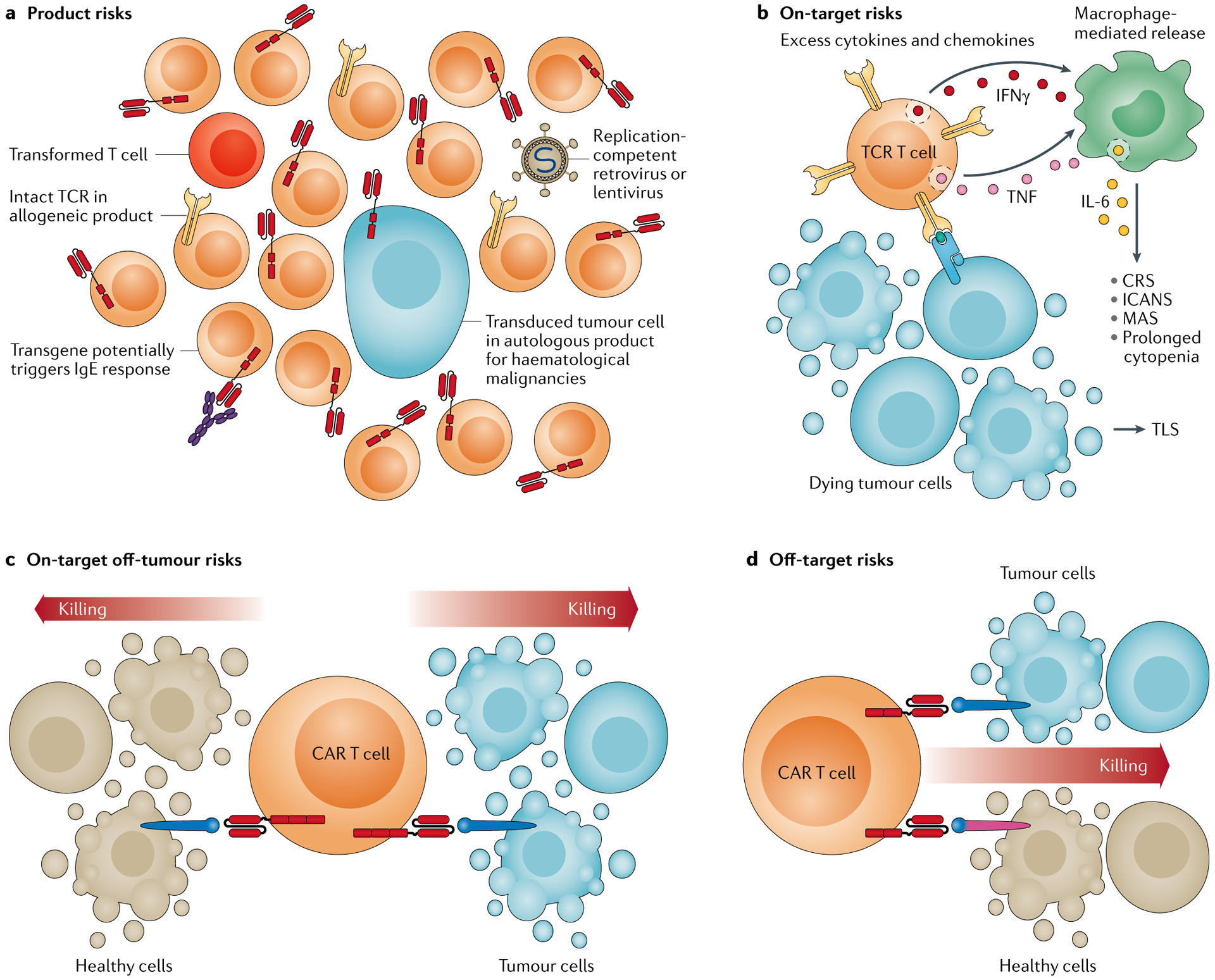
a | Product risks are the inherent liabilities associated with the manufactured product such as unwanted contaminants or by-products of the process, including replication competent vectors, chimeric antigen receptor (CAR) transformed residual tumour cells, T cells with DNA damage that may become cancerous themselves and allogeneic T cells that have escaped T cell receptor (TCR) ablation and thus pose a graft-versus-host disease risk. These risks also include the potential immunogenicity of the transgenic construct, which may provoke infusion reactions. b | On-target toxicity derives from a surplus of target cell killing resulting in tumour lysis syndrome (TLS) or a surplus of cytokine and chemokine production by the therapeutic T cells alone or in concert with myeloid cells that results in cytokine release syndrome (CRS), immune effector cell-associated neurotoxicity syndrome (ICANS), macrophage activation syndrome (MAS) or prolonged cytopenia. c | On-target off-tumour toxicity occurs if the target antigen is also found on healthy cells at levels sufficient to trigger the therapeutic T cells. d | Off-target toxicity occurs if the transgenic immune receptor cross-reacts with an antigen found on healthy tissues. IFNγ interferon-γ; TNF, tumour necrosis factor.
Fig. 6 |. Gene-engineered T cell products for enhanced safety.
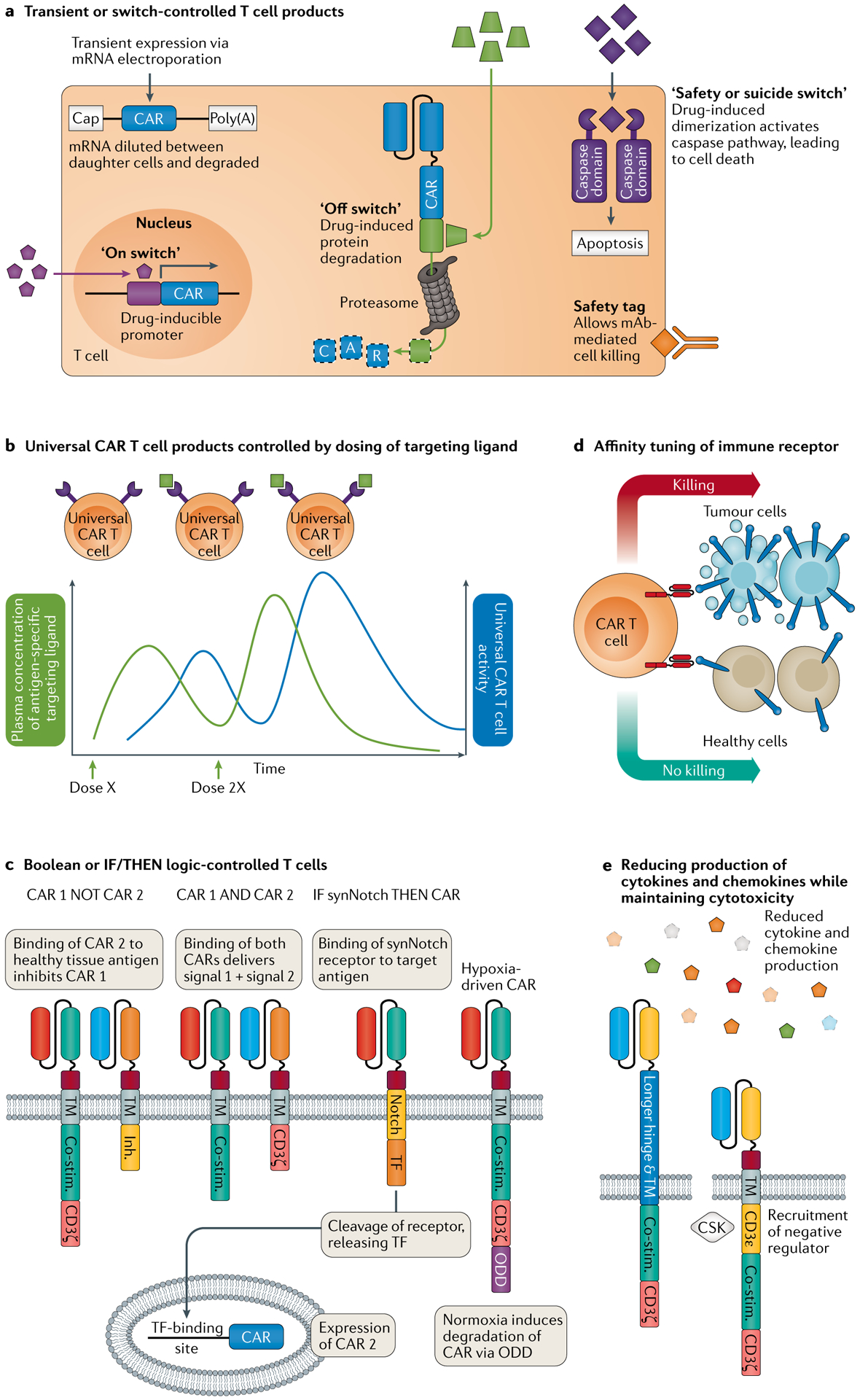
a | Exogenous control of gene-engineered T cells can be achieved via transient delivery of the transgene using mRNA electroporation or via application of a variety of on, off or suicide switches. The transgene may be switched on or off at the level of transcription or via induced protein degradation, or the therapeutic T cell can be eliminated either by a caspase-activation system or via the inclusion of specific tags recognized by approved therapeutic monoclonal antibodies (mAbs). b | Exogenous control can also be achieved via a ‘universal’ chimeric antigen receptor (CAR) system in which infusion of separate antigen-specific targeting ligands redirects the CAR T cells to the target, while the dose and schedule of the antigen-specific targeting ligand can be modified as required. c | Endogenous control can be achieved via Boolean logic-type CAR designs that require CAR T cells to recognize two antigens for full activation (AND gate, via a split CAR) or inhibit CAR T cell activation via a second inhibitory CAR (NOT gate). The synthetic Notch (synNotch) and hypoxia-inducible systems provide conditional ‘IF/THEN’ logic for endogenous control of T cell activity. In the synNotch system, the first CAR is an artificial Notch-type receptor wherein antigen recognition stimulates proteolytic cleavage of a transcription factor (TF), which in turn triggers expression of a second ‘traditional’ CAR recognizing a separate antigen, making sensing of the second antigen dependent on first sensing the first antigen. In the hypoxia-inducible system, the CAR includes oxygen-sensing elements of hypoxia inducible factor 1α, which promote its degradation via the ubiquitin pathway under normoxia but allow full expression in hypoxic conditions such as those found in solid tumours. d | Affinity tuning of the immune receptor provides another form of endogenous control whereby the affinity is set to enable the therapeutic T cell to be activated by tumours expressing high levels of the antigen but not by healthy tissues that express low levels. e | Extending the hinge and transmembrane (TM) domain of a CAR enabled maintenance of cytotoxicity but a significant reduction in the production of cytokines and chemokines, and subsequent cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome210. Similarly, engineering CARs to express the CD3ε domain recruited the negative regulator CSK and resulted in more transient signalling with maintained cytotoxicity but reduced cytokine and chemokine expression228. Co-stim., co-stimulatory domain; Inh., inhibitory domain; ODD, oxygen-dependent degradation domain.
Exogenously controlled T cells
Transient or readily depleted products.
A simple method to limit the duration of activity of gene-engineered cells is to deliver the transgene using mRNA electroporation183 (FIG. 6a). This method limits activity due to the finite half-life of the mRNA transcript and its dilution between daughter cells, with repeated infusions given to overcome the transient persistence183. However, this approach may require the production of large quantities of therapeutic T cells to meet the demands of repeated infusions. Moreover, the repeated delivery of a mRNA CAR T cell product based on a murine scFv provoked anaphylaxis owing to the emergence of human anti-murine antibodies of the IgE class184, thus creating a new product risk.
As an alternative to engineered T cell products with short half-lives, safety or suicide tags and switches are a class of engineering solution intended to enable the rapid depletion of the gene-engineered T cell in the event of unacceptable toxicity (FIG. 6a). Safety tags comprise whole185 or truncated186 antigens encoded alongside the CAR or TCR that are recognized by approved mAb therapies, allowing depletion by Fc-dependent mechanisms. However, as mAbs generally do not penetrate the central nervous system, where therapeutic T cells might need to be depleted187, and Fc-dependent mechanisms might act too slowly, especially in patients with lymphopenia, the exploration of suicide switches is warranted. The inducible caspase 9 (iCasp9) safety switch is activated by a small molecule, rimiducid, that dimerizes the domains of the iCasp9 switch, leading to rapid induction of apoptosis188. While it may be possible to partially activate the suicide switch or tag and thereby avoid total elimination of the gene-engineered T cell, an emergent severe toxicity will likely necessitate stringent intervention and thus depletion of the expensive engineered T cells, likely leaving the patient to face disease progression should they recover from the acute toxicity. For these reasons, alternatives to safety switches are being explored.
On and off switches.
On and off switches act by regulating the transcription, translation or protein stability of the CAR or TCR or, alternatively, by regulating the availability of co-stimulation to promote T cell activation and survival (FIG. 6a).
To regulate activity of the transgene at the transcription level, drug-inducible switches such as the tetracycline-inducible (Tet-On) system189–191 and the doxycycline-inducible system192 have been demonstrated. These systems are somewhat limited by ‘leaky’ expression of the transgene, the fairly slow loss of expression on drug withdrawal189 and immunogenicity of a bacterial-derived antibiotic regulatory system.
Rimiducid-inducible protein dimerization has also been used to create an ‘on switch’ for therapeutic T cells by dimerizing a ‘signal 2’ construct comprising MYD88 and CD40 to provide potent co-stimulation for CAR T cells that were designed to be minimally active without co-stimulation193. To enable both an on switch and a safety switch in the same therapeutic cell, an iCasp9 switch using rapamycin as a dimerizer was reported194. An alternative on switch design makes use of a split CAR, where dimerization is triggered by a rapamycin analogue acting on rapamycin-binding domains195. Aside from small-molecule sensing on switches, a light-responsive element has been demonstrated in the CAR T cell setting, whereby a blue light stimulates expression of the CAR196. So far, this system has been demonstrated to work only for locally delivered CAR T cells in subcutaneous tumours in mice and required cycles of 12 h of light induction, and thus applicability to visceral masses and shorter induction periods remains to be demonstrated.
To regulate activity of the transgene at the level of translation and/or protein turnover, ‘off switch’ systems have been developed. In these systems, the transgene is tagged in ways that allow drug-induced degradation via the ubiquitin–proteasome system. This is achieved by dosing with a proteolysis-targeting chimera (PROTAC) molecule197 or by activating self-degradation domains with small-molecule compounds198,199.
As an alternative to inbuilt off switches, the approved tyrosine kinase inhibitor dasatinib can switch off CAR T cells by inhibiting LCK-mediated signalling200. Indeed, the favourable pharmacokinetic profile of dasatinib supported dose titration to moderate the activity of CAR T cells, and the inhibitory effects were rapidly reversed in vitro and in vivo when the drug was withdrawn200. A potential drawback of using a non-selective off switch if applied regularly or for longer durations might be the suppression of epitope spreading, which requires the endogenous immune system and is a desired factor in immunotherapy generally.
Universal CAR T cells.
An alternative approach to enable exogenous control over gene-engineered T cells and simultaneously address the issue of antigen loss is the aforementioned universal CAR that binds to a separately infused targeting moiety. To optimize safety, the targeting moiety should have a limited half-life to enable control of universal CAR T cell activity by the dose and dosing schedule of the targeting ligand (FIG. 6b). This requirement may disfavour the CD16 CAR plus mAb system132 given the long half-life of mAbs.
A barrier for the application of all exogenously controlled gene-engineered T cell therapy platforms will be in developing the clinical guidelines for regulating the activity of the therapeutic T cells in the patient. This is especially challenging given the lack of methods for real-time analysis of biomarkers, such as cytokine and T cell levels, leaving the physician to react to data that are out of date.
Endogenously programmed T cells
Boolean and IF/THEN logic gates.
Over the past decade, the biological equivalents of the OR, AND and NOT Boolean logic gates have emerged (reviewed in REF.201). The OR gate is designed to prevent antigen escape rather than to increase specificity202 and was discussed previously, but the AND and NOT gates are both designed with increased tumour specificity in mind (FIG. 6c). However, these T cell Boolean logic gates have their drawbacks. For the AND gate, in which ‘signal 1’ (CD3ζ) and ‘signal 2’ are split onto separate CARs, signal 1 alone is sufficient to trigger activation of the memory and effector T cell subsets in the gene-engineered product, while addition of signal 2 boosts the response and enables activation of naive T cells. The loss of either target antigen will diminish the activity of the product, increasing the risk of tumour escape. For the NOT gate, in which a second CAR provides a suppressive signal, it is challenging to find an ideal target associated with healthy tissues that is absent on tumours, and to design a sufficiently potent inhibitory signal for the NOT CAR203. Such limitations have stimulated the design of a novel protein logic system and more advanced genetic circuits such as synthetic Notch (synNotch) and hypoxia-inducible systems.
The protein logic system is a true Boolean logic system that combines the combinatorial antigen requirements of the aforementioned Boolean logic CARs with the concept of a universal CAR T cell system recognizing a separately infused logic-programmed targeting ligand. The system relies on synthetically designed colocalization-dependent latching orthogonal cage–key protein (co-LOCKR) switches204. These are designed with combinatorial AND, OR and NOT logic in mind, such that after binding to a target cell, they undergo a conformational change to reveal a tag to be recognized by a tag-specific CAR T cell only if the required logic conditions are met. Precise control of CAR T cell activity has been shown in vitro with this system204, but it will be vital to establish whether co-LOCKR switches, which are non-human synthetic proteins, can be designed with the necessary pharmacokinetics and low immunogenicity to work in patients, and it will remain a challenge to find appropriate antigen targets for use in NOT logic components just as it was for the earlier NOT CARs203.
The synNotch and hypoxia-inducible CAR systems are ‘IF/THEN’-type logic systems where an encounter with a specific antigen or condition induces functional expression of a CAR to a second antigen that is capable of mediating full T cell activation. The objective with these systems is to compartmentalize activity of gene-engineered T cells to the tumour while reducing or preferably eliminating the activity in healthy tissues.
For the synNotch system, it was shown that on-target off-tumour toxicity could be avoided in animal models as long as healthy cells expressing the target of the second ‘traditional’ CAR were not co-located with the tumour205,206. Where healthy and tumour cells were co-located, once primed by the synNotch CAR, gene-engineered T cells were able to kill antigen-positive healthy and tumour tissues alike205, which is a drawback of this type of system that will need further attention. Moreover, current synNotch systems are limited by the use of immunogenic sequences of non-human origin, potentially limiting product persistence in the patient.
An alternative fully human conditional expression system links CAR expression to hypoxia by including elements of the oxygen-sensitive subdomain of HIF1α in the CAR molecule207. With use of this system, it was found that CAR expression could be induced by hypoxia and dropped by 80% within 2 h when normoxia was restored. The resulting CAR showed a significant hypoxia-dependent differential in cytotoxicity towards target cells, although this shift was not binary207. Future studies may determine whether such designs sufficiently limit on-target off-tumour toxicity by focusing T cell activation to hypoxic environments common to solid tumours.
Alternative approaches.
Alternative approaches to logic-gated or inducible systems include the tuning of the expression or affinity of the transgenic receptor, or modulating its function by altering the linker length between the antigen-binding and transmembrane domains (FIG. 6d,e). For approaches that modulate the expression or affinity of the immune receptor, the ultimate goal is to calibrate the immune receptor to recognize antigen densities typical on tumour cells while sparing healthy cells, with the possibility of also reducing or avoiding tonic signalling, which is detrimental to T cell function and persistence85. The expression of transgenes can be regulated at the level of mRNA transcript stability via inclusion of microRNA-response elements208 or at the protein level by inclusion of destabilizing tags known as ‘degrons’209. So far, working model systems for both approaches have been described, and their application to CAR T cells or TCR T cells is pending. Affinity tuning of the antigen-binding domain has been shown to enable gene-engineered T cells to avoid activation by low antigen levels on healthy cells while retaining anti-tumour activity56. Finally, changing the hinge length in a CD19 CAR T cell was shown both in preclinical models and in the clinic to uncouple efficacy from cytokine release syndrome and immune effector cell-associated neurotoxicity syndrome by reducing cytokine production while maintaining cytotoxicity210. Iterations and improvements to immune receptor design using the concepts of logic-gating, conditional induction and/or fine-tuned expression or binding affinity will likely increase the inherent safety of gene-engineered T cell therapies, thereby simplifying toxicity management.
Concluding remarks
Genetic engineering of T cells is opening new avenues to treat a wide array of disease states. Successful approaches to date rely on the constitutive expression of a single protein. To reach the full potential of engineered T cells, numerous genetic alterations will likely be necessary. Moreover, engineering approaches that allow T cells to adapt to multiple challenges simultaneously, such as adapting to antigen escape, poor nutrient environments and T cell exhaustion, will likely be required to enable durable successful clinical outcomes. The ease of engineering and the impressive array of both pro-immune and anti-immune response functions of T cells opens seemingly limitless possibilities for how T cells can be altered to fight a particular disease. However, numerous examples of engineered T cells having caused significant morbidity and in some cases death have been reported. Thus, toxicity remains the biggest barrier to widespread use of genetically engineered T cells. Improved tools such as prime editing211, controlled expression of immune modulators and safety switches will hopefully allow both effective and safe engineered T cell-based therapies.
Acknowledgements
The authors are grateful to the members of the Center for Cellular Immunotherapies for in-depth discussions regarding the topics discussed in this Review and apologize to the many investigators whose work they were unable to cite. The authors gratefully acknowledge funding from the NIH (U19AI117950, U19AI149680 UM1AI126620 and UG3DK122644), Helmsley Charitable Trust and Tmunity Therapeutics.
Competing interests
J.L.R. is a co-founder and shareholder of Tmunity Therapeutics. He has been in receipt of research funding from Tmunity Therapeutics. N.C.S. is a shareholder of Tmunity Therapeutics and Fate Therapeutics. G.I.E. declares no competing interests.
Glossary
- Allogeneic
A term to denote a genetically different individual of the same species
- Antigen loss
A mechanism of cancer or pathogen recurrence in which the target (tumour or pathogen) escapes engineered T cell recognition, typically via exon skipping, downregulation or alteration of the target antigen
- Epitope spreading
Diversification of the immune response by endogenous immune cells against new targets following engineered T cell therapy.
- Boolean logic gate
AND, OR and NOT functions, which can describe the response of combinations of immune receptors to multiple antigens
- Immune synapse
The location of physical interaction between an immune cell and its activator, either another immune cell or a non-haematopoietic target cell. The strength and the type of signals exchanged at the immune synapse can modulate an immune response
- Single-chain variable fragment
(scFv). A linear transposition of an antibody’s heavy and light chains separated by a flexible linker, which retains the antigen binding capacity of the original antibody and can be used as the binder domain of a chimeric antigen receptor molecule
- Monoclonal antibodies
(mAbs). Antibodies purified from a single B cell hybridoma that can be used as a diagnostic or therapeutic product
- Fc-dependent mechanisms
A series of effector modalities by which targets bound by antibodies are depleted via recognition of the constant region of the antibody molecule.
Footnotes
Peer review information
Nature Reviews Genetics thanks M. K. Levings, Z. Li, E. L. Smith, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
References
- 1.Rappuoli R, Mandl CW, Black S & De Gregorio E Vaccines for the twenty-first century society. Nat. Rev. Immunol 11, 865–872 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sliwkowski MX & Mellman I Antibody therapeutics in cancer. Science 341, 1192–1198 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldmann M & Maini RN Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu. Rev. Immunol 19, 163–196 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Kumar BV, Connors TJ & Farber DL Human T cell development, localization, and function throughout life. Immunity 48, 202–213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jameson SC & Masopust D Understanding subset diversity in T cell memory. Immunity 48, 214–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Francica JR et al. Steric shielding of surface epitopes and impaired immune recognition induced by the ebola virus glycoprotein. PLoS Pathog. 6, e1001098 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei F et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl Acad. Sci. USA 110, E2480–E2489 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JH, Geyer MB & Brentjens RJ CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood 127, 3312–3320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first report on the in vivo fate and genomic ramifications of CRISPR-engineered TCR T cells in a phase I clinical trial.
- 11.Li J, Hong S, Chen W, Zuo E & Yang H Advances in detecting and reducing off-target effects generated by CRISPR-mediated genome editing. J. Genet. Genomics 46, 513–521 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Maldini CR, Ellis GI & Riley JL CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol 18, 605–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosado-Sanchez I & Levings MK Building a CAR-Treg: going from the basic to the luxury model. Cell Immunol. 358, 104220 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Klichinsky M et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol 38, 947–953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimbrel EA & Lanza R Next-generation stem cells - ushering in a new era of cell-based therapies. Nat. Rev. Drug Discov 19, 463–479 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Hartweger H et al. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med 216, 1301–1310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fesnak AD, June CH & Levine BL Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newrzela S et al. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia 26, 2499–2507 (2012). [DOI] [PubMed] [Google Scholar]
- 19.Scholler J et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl Med 4, 132ra153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Leary MC et al. FDA approval summary: tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin. Cancer Res 25, 1142–1146 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Bouchkouj N et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin. Cancer Res 25, 1702–1708 (2019). [DOI] [PubMed] [Google Scholar]
- 22.Hirayama AV et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood 134, 636–640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuster SJ et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N. Engl. J. Med 380, 45–56 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Locke FL et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 20, 31–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiorenza S, Ritchie DS, Ramsey SD, Turtle CJ & Roth JA Value and affordability of CAR T-cell therapy in the United States. Bone Marrow Transplant. 55, 1706–1715 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Kagoya Y et al. Genetic ablation of HLA class I, class II, and the T-cell receptor enables allogeneic T cells to be used for adoptive T-cell therapy. Cancer Immunol. Res 8, 926–936 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Cruz CR et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 122, 2965–2973 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kochenderfer JN et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122, 4129–4139 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huls MH et al. Clinical application of Sleeping Beauty and artificial antigen presenting cells to genetically modify T cells from peripheral and umbilical cord blood. J. Vis. Exp 10.3791/50070 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pegram HJ et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 29, 415–422 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ueda T & Kaneko S In vitro differentiation of T cell: from CAR-modified T-iPSC. Methods Mol. Biol 2048, 85–91 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Themeli M et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol 31, 928–933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan LZ et al. Allogeneic CAR-T for treatment of relapsed and/or refractory multiple myeloma: four cases report and literatures review. Zhonghua Xue Ye Xue Za Zhi 40, 650–655 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brudno JN et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J. Clin. Oncol 34, 1112–1121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eapen M et al. Effect of graft source on unrelated donor haemopoietic stem-cell transplantation in adults with acute leukaemia: a retrospective analysis. Lancet Oncol. 11, 653–660 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwoczek J et al. Cord blood-derived T cells allow the generation of a more naive tumor-reactive cytotoxic T-cell phenotype. Transfusion 58, 88–99 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Torikai H et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119, 5697–5705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stenger D et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood 136, 1407–1418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steentoft C et al. Glycan-directed CAR-T cells. Glycobiology 28, 656–669 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Hughes MS et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene Ther 16, 457–472 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao Y et al. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J. Immunol 174, 4415–4423 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmadi M et al. CD3 limits the efficacy of TCR gene therapy in vivo. Blood 118, 3528–3537 (2011). [DOI] [PubMed] [Google Scholar]
- 44.van Loenen MM et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl Acad. Sci. USA 107, 10972–10977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shao H et al. TCR mispairing in genetically modified T cells was detected by fluorescence resonance energy transfer. Mol. Biol. Rep 37, 3951–3956 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Cohen CJ et al. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 67, 3898–3903 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bendle GM et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med 16, 565–570 (2010). [DOI] [PubMed] [Google Scholar]
- 48.Govers C, Sebestyen Z, Coccoris M, Willemsen RA & Debets R T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol. Med 16, 77–87 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Yang S et al. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther. 15, 1411–1423 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Legut M, Dolton G, Mian AA, Ottmann OG & Sewell AK CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 131, 311–322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okamoto S et al. Improved expression and reactivity of transduced tumor-specific TCRs in human lymphocytes by specific silencing of endogenous TCR. Cancer Res. 69, 9003–9011 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Robbins PF et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol 29, 917–924 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maldini CR et al. Dual CD4-based CAR T cells with distinct costimulatory domains mitigate HIV pathogenesis in vivo. Nat. Med 26, 1776–1787 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guedan S et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 124, 1070–1080 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kawalekar OU et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Liu X et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghorashian S et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med 25, 1408–1414 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Ellebrecht CT et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 353, 179–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA & Atkinson MA Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes 54, 1407–1414 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Thiruppathi M et al. Functional defect in regulatory T cells in myasthenia gravis. Ann. N. Y. Acad. Sci 1274, 68–76 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Viglietta V, Baecher-Allan C, Weiner HL & Hafler DA Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J. Exp. Med 199, 971–979 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brunstein CG et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood 127, 1044–1051 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plesa G et al. TCR affinity and specificity requirements for human regulatory T-cell function. Blood 119, 3420–3430 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boardman DA et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am. J. Transpl 17, 931–943 (2017). [DOI] [PubMed] [Google Scholar]
- 65.MacDonald KG et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J. Clin. Invest 126, 1413–1424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that human CAR Treg cells can prevent GVHD in vivo.
- 66.Noyan F et al. Prevention of allograft rejection by use of regulatory T cells with an MHC-specific chimeric antigen receptor. Am. J. Transpl 17, 917–930 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Dawson NA et al. Systematic testing and specificity mapping of alloantigen-specific chimeric antigen receptors in regulatory T cells. JCI Insight 4, e123672 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yeh WI et al. Avidity and bystander suppressive capacity of human regulatory T cells expressing de novo autoreactive T-cell receptors in type 1 diabetes. Front. Immunol 8, 1313 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hull CM et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J. Autoimmun 79, 63–73 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Dawson NAJ et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci. Transl Med 12, eaaz3866 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Boroughs AC et al. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight 5, e126194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hippen KL et al. Massive ex vivo expansion of human natural regulatory T cells (Tregs) with minimal loss of in vivo functional activity. Sci. Transl Med 3, 83ra41 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McKenna DH Jr. et al. Optimization of cGMP purification and expansion of umbilical cord blood-derived T-regulatory cells in support of first-in-human clinical trials. Cytotherapy 19, 250–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou X et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol 10, 1000–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Honaker Y et al. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci. Transl Med 12, eaay6422 (2020). [DOI] [PubMed] [Google Scholar]
- 76.Kumar M, Keller B, Makalou N & Sutton RE Systematic determination of the packaging limit of lentiviral vectors. Hum. Gene Ther 12, 1893–1905 (2001). [DOI] [PubMed] [Google Scholar]
- 77.Mitchell RS et al. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2, E234 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang OO et al. Lysis of HIV-1-infected cells and inhibition of viral replication by universal receptor T cells. Proc. Natl Acad. Sci. USA 94, 11478–11483 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deeks SG et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol. Ther 5, 788–797 (2002). [DOI] [PubMed] [Google Scholar]
- 80.Mitsuyasu RT et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus-infected subjects. Blood 96, 785–793 (2000). [PubMed] [Google Scholar]
- 81.Leibman RS et al. Supraphysiologic control over HIV-1 replication mediated by CD8 T cells expressing a re-engineered CD4-based chimeric antigen receptor. PLoS Pathog. 13, e1006613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bobisse S et al. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res. 69, 9385–9394 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Zufferey R, Donello JE, Trono D & Hope TJ Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol 73, 2886–2892 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Norrman K et al. Quantitative comparison of constitutive promoters in human ES cells. PLoS ONE 5, e12413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors improve CAR function by CRISPR-mediated insertion of CAR into the TRAC gene, improving expression kinetics and decreasing exhaustion.
- 86.Masiuk KE, Laborada J, Roncarolo MG, Hollis RP & Kohn DB Lentiviral gene therapy in HSCs restores lineage-specific Foxp3 expression and suppresses autoimmunity in a mouse model of IPEX syndrome. Cell Stem Cell 24, 309–317.e7 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Random insertion of CAR into the TET2 locus highlights the potential for targeting the epigenome to improve CAR T cell function and the value of analysis of the clinical product.
- 88.Zhao Y et al. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther 13, 151–159 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao Y et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 70, 9053–9061 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin Z et al. The hyperactive sleeping beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 18, 849–856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chicaybam L et al. Transposon-mediated generation of CAR-T cells shows efficient anti B-cell leukemia response after ex vivo expansion. Gene Ther. 27, 85–95 (2020). [DOI] [PubMed] [Google Scholar]
- 92.Gogol-Doring A et al. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the Piggybac transposon in primary human CD4+ T cells. Mol. Ther 24, 592–606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Berry C, Hannenhalli S, Leipzig J & Bushman FD Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol 2, e157 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chicaybam L et al. CAR T cells generated using Sleeping Beauty transposon vectors and expanded with an EBV-transformed lymphoblastoid cell line display antitumor activity in vitro and in vivo. Hum. Gene Ther 30, 511–522 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Voigt F et al. Sleeping Beauty transposase structure allows rational design of hyperactive variants for genetic engineering. Nat. Commun 7, 11126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Querques I et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol 37, 1502–1512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Monjezi R et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 31, 186–194 (2017). [DOI] [PubMed] [Google Scholar]
- 98.Kebriaei P et al. Phase I trials using sleeping beauty to generate CD19-specific CAR T cells. J. Clin. Invest 126, 3363–3376 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Magnani CF et al. Sleeping Beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J. Clin. Invest 130, 6021–6033 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang J et al. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 44, e30 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.MacLeod DT et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol. Ther 25, 949–961 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hubbard N et al. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood 127, 2513–2522 (2016). [DOI] [PubMed] [Google Scholar]
- 103.Sather BD et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl Med 7, 307ra156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roth TL et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nguyen DN et al. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol 38, 44–49 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sadelain M, Papapetrou EP & Bushman FD Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer 12, 51–58 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Hale M et al. Engineering HIV-resistant, anti-HIV chimeric antigen receptor T cells. Mol. Ther 25, 570–579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Park JH et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Milone MC et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Morgan P et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov. Today 17, 419–424 (2012). [DOI] [PubMed] [Google Scholar]
- 111.Ager A, Watson HA, Wehenkel SC & Mohammed RN Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem. Soc. Trans 44, 377–385 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Buckanovich RJ et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat. Med 14, 28–36 (2008). [DOI] [PubMed] [Google Scholar]
- 113.Motz GT et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med 20, 607–615 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yamamoto TN et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest 129, 1551–1565 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lu P, Weaver VM & Werb Z The extracellular matrix: a dynamic niche in cancer progression. J. Cell Biol 196, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Griffith KD et al. In vivo distribution of adoptively transferred indium-111-labeled tumor infiltrating lymphocytes and peripheral blood lymphocytes in patients with metastatic melanoma. J. Natl Cancer Inst 81, 1709–1717 (1989). [DOI] [PubMed] [Google Scholar]
- 117.Moon EK et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res 20, 4262–4273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Moon EK et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res 17, 4719–4730 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Caruana I et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med 21, 524–529 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Swanton C Intratumor heterogeneity: evolution through space and time. Cancer Res. 72, 4875–4882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sotillo E et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 5, 1282–1295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.O’Rourke DM et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl Med 9, eaaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rapoport AP et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med 21, 914–921 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Feng KC et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol 10, 4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ruella M et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest 126, 3814–3826 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Simon B, Harrer DC, Schuler-Thurner B, Schuler G & Uslu U Arming T cells with a gp100-specific TCR and a CSPG4-specific CAR using combined DNA- and RNA-based receptor transfer. Cancers 11, 696 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hegde M et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J. Clin. Invest 126, 3036–3052 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zah E et al. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun 11, 2283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.de Larrea CF et al. Defining an optimal dual-targeted CAR T-cell therapy approach simultaneously targeting BCMA and GPRC5D to prevent BCMA escape-driven relapse in multiple myeloma. Blood Cancer Discov. 1, 146–154 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Han X, Wang Y, Wei J & Han W Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol 12, 128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bielamowicz K et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 20, 506–518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kudo K et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 74, 93–103 (2014). [DOI] [PubMed] [Google Scholar]
- 133.Landgraf KE et al. ConvertibleCARs: a chimeric antigen receptor system for flexible control of activity and antigen targeting. Commun. Biol 3, 296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Minutolo NG, Hollander EE & Powell DJ Jr. The emergence of universal immune receptor T cell therapy for cancer. Front. Oncol 9, 176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Minutolo NG et al. Quantitative control of gene-engineered T-cell activity through the covalent attachment of targeting ligands to a universal immune receptor. J. Am. Chem. Soc 142, 6554–6568 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Adachi K et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol 36, 346–351 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Luo H et al. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin. Cancer Res 26, 5494–5505 (2020). [DOI] [PubMed] [Google Scholar]
- 138.Wang N et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed B-cell malignancies. Blood 135, 17–27 (2020). [DOI] [PubMed] [Google Scholar]
- 139.Yan Z et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol. 6, e521–e529 (2019). [DOI] [PubMed] [Google Scholar]
- 140.Zhao WH et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol 11, 141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lawrence MS et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ecker C & Riley JL Translating in vitro T cell metabolic findings to in vivo tumor models of nutrient competition. Cell Metab. 28, 190–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Munn DH & Bronte V Immune suppressive mechanisms in the tumor microenvironment. Curr. Opin. Immunol 39, 1–6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lu Y et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med 26, 732–740 (2020). [DOI] [PubMed] [Google Scholar]
- 145.Neuzillet C et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther 147, 22–31 (2015). [DOI] [PubMed] [Google Scholar]
- 146.Kloss CC et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther 26, 1855–1866 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Powell AB et al. Medulloblastoma rendered susceptible to NK-cell attack by TGFbeta neutralization. J. Transl. Med 17, 321 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bollard CM et al. Tumor-specific T-cells engineered to overcome tumor immune evasion induce clinical responses in patients with relapsed hodgkin lymphoma. J. Clin. Oncol 36, 1128–1139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Beavis PA et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Invest 127, 929–941 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sim GC et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J. Clin. Invest 124, 99–110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Suryadevara CM et al. Preventing Lck activation in CAR T cells confers Treg resistance but requires 4–1BB signaling for them to persist and treat solid tumors in nonlymphodepleted hosts. Clin. Cancer Res 25, 358–368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Franchina DG, He F & Brenner D Survival of the fittest: cancer challenges T cell metabolism. Cancer Lett. 412, 216–223 (2018). [DOI] [PubMed] [Google Scholar]
- 153.Kawalekar OU et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T Cells. Immunity 44, 712 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Fultang L et al. Metabolic engineering against the arginine microenvironment enhances CAR-T cell proliferation and therapeutic activity. Blood 136, 1155–1160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leonard JP et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood 90, 2541–2548 (1997). [PubMed] [Google Scholar]
- 156.Zhang L et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol. Ther 19, 751–759 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Joung J et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc 12, 828–863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shifrut E et al. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell 175, 1958–1971.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study uses a genome-wide CRISPR screen to unearth proteins that limit T cell proliferation and killing in the tumour microenvironment.
- 159.Cortez JT et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 582, 416–420 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Dong MB et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell 178, 1189–1204.e23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Roth TL et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell 181, 728–744.e21 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wei J et al. PD-1 silencing impairs the anti-tumor function of chimeric antigen receptor modified T cells by inhibiting proliferation activity. J. Immunother. Cancer 7, 209 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cherkassky L et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest 126, 3130–3144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ankri C, Shamalov K, Horovitz-Fried M, Mauer S & Cohen CJ Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J. Immunol 191, 4121–4129 (2013). [DOI] [PubMed] [Google Scholar]
- 165.Parry RV et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol 25, 9543–9553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rafiq S et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol 36, 847–856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhang A et al. Secretion of human soluble programmed cell death protein 1 by chimeric antigen receptor-modified T cells enhances anti-tumor efficacy. Cytotherapy 22, 734–743 (2020). [DOI] [PubMed] [Google Scholar]
- 168.Chang ZL, Silver PA & Chen YY Identification and selective expansion of functionally superior T cells expressing chimeric antigen receptors. J. Transl. Med 13, 161 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Khan O et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Chen Z et al. TCF-1-centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity 51, 840–855.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Chen J et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Odorizzi PM, Pauken KE, Paley MA, Sharpe A & Wherry EJ Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J. Exp. Med 212, 1125–1137 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Greenbaum U et al. Chimeric antigen receptor T-cell therapy toxicities. Br. J. Clin. Pharmacol 10.1111/bcp.14403 (2020). [DOI] [PubMed] [Google Scholar]
- 174.Curran KJ et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 134, 2361–2368 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Neelapu SS et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol 15, 47–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Howard SC, Trifilio S, Gregory TK, Baxter N & McBride A Tumor lysis syndrome in the era of novel and targeted agents in patients with hematologic malignancies: a systematic review. Ann. Hematol 95, 563–573 (2016). [DOI] [PubMed] [Google Scholar]
- 177.Fried S et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transpl. 54, 1643–1650 (2019). [DOI] [PubMed] [Google Scholar]
- 178.Teachey DT et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 6, 664–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ruella M et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med 24, 1499–1503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hacein-Bey-Abina S et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419 (2003). [DOI] [PubMed] [Google Scholar]
- 181.Marcucci KT et al. Retroviral and lentiviral safety analysis of gene-modified T cell products and infused HIV and oncology patients. Mol. Ther 26, 269–279 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Morgan RA et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother 36, 133–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Beatty GL et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 155, 29–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Maus MV et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res 1, 26–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Tasian SK et al. Optimized depletion of chimeric antigen receptor T cells in murine xenograft models of human acute myeloid leukemia. Blood 129, 2395–2407 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Jonnalagadda M et al. Efficient selection of genetically modified human T cells using methotrexate-resistant human dihydrofolate reductase. Gene Ther. 20, 853–860 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Cavaco M, Gaspar D, Arb Castanho M & Neves V Antibodies for the treatment of brain metastases, a dream or a reality? Pharmaceutics 12, 62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gargett T & Brown MP The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front. Pharmacol 5, 235 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gu X, He D, Li C, Wang H & Yang G Development of inducible CD19-CAR T cells with a Tet-on system for controlled activity and enhanced clinical safety. Int. J. Mol. Sci 19, 3455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Drent E et al. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS ONE 13, e0197349 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sakemura R et al. A Tet-on inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol. Res 4, 658–668 (2016). [DOI] [PubMed] [Google Scholar]
- 192.Zhang RY et al. Doxycycline inducible chimeric antigen receptor T cells targeting CD147 for hepatocellular carcinoma therapy. Front. Cell Dev. Biol 7, 233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Foster AE et al. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol. Ther 25, 2176–2188 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Duong MT et al. Two-dimensional regulation of CAR-T cell therapy with orthogonal switches. Mol. Ther. Oncolytics 12, 124–137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wu CY, Roybal KT, Puchner EM, Onuffer J & Lim WA Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huang Z et al. Engineering light-controllable CAR T cells for cancer immunotherapy. Sci. Adv 6, eaay9209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lee SM et al. A Chemical switch system to modulate chimeric antigen receptor T cell activity through proteolysis-targeting chimaera technology. ACS Synth. Biol 9, 987–992 (2020). [DOI] [PubMed] [Google Scholar]
- 198.Juillerat A et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 19, 44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Richman SA et al. Ligand-induced degradation of a CAR permits reversible remote control of CAR T cell activity in vitro and in vivo. Mol. Ther 28, 1600–1613 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Mestermann K et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl Med 11, eaau5907 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reports a pharmacological approach to modulating T cell function in vivo.
- 201.Ebert LM, Yu W, Gargett T & Brown MP Logic-gated approaches to extend the utility of chimeric antigen receptor T-cell technology. Biochem. Soc. Trans 46, 391–401 (2018). [DOI] [PubMed] [Google Scholar]
- 202.Zah E, Lin MY, Silva-Benedict A, Jensen MC & Chen YY T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B Cells. Cancer Immunol. Res 4, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Fedorov VD, Themeli M & Sadelain M PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl Med 5, 215ra172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lajoie MJ et al. Designed protein logic to target cells with precise combinations of surface antigens. Science 369, 1637–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Srivastava S et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503 e488 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Roybal KT et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the development of synNotch CAR T cells operating on IF/THEN logic.
- 207.Juillerat A et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep 7, 39833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Michaels YS et al. Precise tuning of gene expression levels in mammalian cells. Nat. Commun 10, 818 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chassin H et al. A modular degron library for synthetic circuits in mammalian cells. Nat. Commun 10, 2013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ying Z et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med 25, 947–953 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study describes the development of prime editing for genome editing in the absence of DNA double-strand breaks.
- 212.Tebas P et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med 370, 901–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Paschon DE et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun 10, 1133 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Perez EE et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol 26, 808–816 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Wilen CB et al. Engineering HIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 7, e1002020 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Yuan J et al. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4+ T cell resistance and enrichment. Mol. Ther 20, 849–859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Provasi E et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med 18, 807–815 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Qasim W et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl Med 9, eaaj2013 (2017). [DOI] [PubMed] [Google Scholar]
- 219.Valton J et al. A multidrug-resistant engineered CAR T cell for allogeneic combination immunotherapy. Mol. Ther 23, 1507–1518 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Mussolino C et al. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 39, 9283–9293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sachdeva M et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat. Commun 10, 5100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Ren J et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res 23, 2255–2266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors describe a CRISPR–Cas9-based triple knockout of genes encoding TCR, β2M and PD1 in primary human T cells.
- 223.Liu Z et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4+ T cells from HIV-1 infection. Cell Biosci. 7, 47 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Anderson W, Thorpe J, Long SA & Rawlings DJ Efficient CRISPR/Cas9 disruption of autoimmune-associated genes reveals key signaling programs in primary human T cells. J. Immunol 203, 3166–3178 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gaudelli NM et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol 38, 892–900 (2020). [DOI] [PubMed] [Google Scholar]
- 226.Gornalusse GG et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol 35, 765–772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Mo F et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat. Biotechnol (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wu W et al. Multiple signaling roles of CD3epsilon and its application in CAR-T cell therapy. Cell 182, 855–871.e23 (2020). [DOI] [PubMed] [Google Scholar]
- 229.Roberts MR et al. Targeting of human immunodeficiency virus-infected cells by CD8+ T lymphocytes armed with universal T-cell receptors. Blood 84, 2878–2889 (1994). [PubMed] [Google Scholar]
- 230.Maher J, Brentjens RJ, Gunset G, Riviere I & Sadelain M Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta / CD28 receptor. Nat. Biotechnol 20, 70–75 (2002). [DOI] [PubMed] [Google Scholar]
- 231.Porter DL, Levine BL, Kalos M, Bagg A & June CH Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med 365, 725–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kochenderfer JN et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119, 2709–2720 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Brentjens RJ et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl Med 5, 177ra138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl Med 7, 303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Kim GB, Hege K & Riley JL CAR talk: how cancer-specific CAR T cells can instruct how to build CAR T cells to cure HIV. Front. Immunol 10, 2310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Maldini CR et al. HIV-resistant and HIV-specific CAR-modified CD4+ T cells mitigate HIV disease progression and confer CD4+ T cell help in vivo. Mol. Ther 28, 1585–1599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Herzig E et al. Attacking latent HIV with convertible CAR-T cells, a highly adaptable killing platform. Cell 179, 880–894.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Anthony-Gonda K et al. Multispecific anti-HIV duoCAR-T cells display broad in vitro antiviral activity and potent in vivo elimination of HIV-infected cells in a humanized mouse model. Sci. Transl Med 11, eaav5685 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ali A et al. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J. Virol 90, 6999–7006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Festag MM et al. Evaluation of a fully human, hepatitis b virus-specific chimeric antigen receptor in an immunocompetent mouse model. Mol. Ther 27, 947–959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sautto GA et al. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut 65, 512–523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Kumaresan PR et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc. Natl Acad. Sci. USA 111, 10660–10665 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Bitinaite J, Wah DA, Aggarwal AK & Schildkraut I FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA 95, 10570–10575 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Kim Y et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol 31, 251–258 (2013). [DOI] [PubMed] [Google Scholar]
- 245.Boissel S et al. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 42, 2591–2601 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Osborn MJ et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol. Ther 24, 570–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Qi LS et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Konermann S et al. Optical control of mammalian endogenous transcription and epigenetic states. Nature 500, 472–476 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yeo NC et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 15, 611–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Huang YH et al. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 18, 176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Koonin EV, Makarova KS & Zhang F Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37, 67–78 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Komor AC, Badran AH & Liu DR CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 168, 20–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]