

Abstract
ApoE4 enhances Tau neurotoxicity and promotes the early onset of AD. Pretangle Tau in the noradrenergic locus coeruleus (LC) is the earliest detectable AD-like pathology in the human brain. However, a direct relationship between ApoE4 and Tau in the LC has not been identified. Here we show that ApoE4 selectively binds to the vesicular monoamine transporter 2 (VMAT2) and inhibits neurotransmitter uptake. The exclusion of norepinephrine (NE) from synaptic vesicles leads to its oxidation into the toxic metabolite 3,4-dihydroxyphenyl glycolaldehyde (DOPEGAL), which subsequently activates cleavage of Tau at N368 by asparagine endopeptidase (AEP) and triggers LC neurodegeneration. Our data reveal that ApoE4 boosts Tau neurotoxicity via VMAT2 inhibition, reduces hippocampal volume, and induces cognitive dysfunction in an AEP- and Tau N368- dependent manner, while conversely ApoE3 binds Tau and protects it from cleavage. Thus, ApoE4 exacerbates Tau neurotoxicity by increasing VMAT2 vesicle leakage and facilitating AEP-mediated Tau proteolytic cleavage in the LC via DOPEGAL.
Keywords: ApoE, VMAT2, DOPEGAL, norepinephrine, neurofibrillary tangle, Locus Coeruleus
Introduction
Amyloid-β (Aβ) plaques and tau neurofibrillary tangles (NFT), as well as neuronal degeneration, are the pathologic hallmarks of Alzheimer’s disease (AD). In humans, the Apolipoprotein E (ApoE) gene has three polymorphic alleles (ε2, ε3 and ε4), with the ε3 allele being the most common (77%) and ε2 allele the least common (8%) [36]. The ε4 allele increases the risk for both familial and sporadic AD [16, 27]; up to 60% of people with AD carry at least one ε4 allele, compared to only 15% of healthy controls. ApoE is an apolipoprotein abundant in cholesterol- and triglyceride-rich plasma lipoproteins [37], and is essential for Aβ deposition in amyloid precursor protein (APP) transgenic mice. When ApoE knockout (KO) mice are crossed with PDAPP or Tg2576 mice, Aβ deposition in amyloid plaques and cerebral amyloid angiopathy (CAA) is dramatically reduced [3, 31]. Moreover, the PDAPP/TRE mouse model of β-amyloidosis that overexpresses human APP carrying an autosomal dominant familial AD-linked mutation (V717F) also expresses each of the human ApoE isoforms under the control of mouse ApoE regulatory elements. Notably, Aβ clearance is slower in ApoE4-TR mice compared with ApoE3-TR mice [13]. Although accumulating clinical and preclinical studies suggest that ApoE isoforms impact AD pathogenesis by driving Aβ pathology, ApoE4 also impacts neurodegeneration in the context of Tau pathology, independently of amyloid-β. The presence of ApoE4 exacerbates tau-mediated neurodegeneration, whereas the absence of ApoE is neuroprotective [43].
Hyperphosphorylated Tau is prone to aggregation, and this “pretangle” form of the protein appears in the locus coeruleus (LC), the major central source of norepinephrine (NE), before any other area of the brain, making it the earliest detectable AD-like neuropathology [5–7, 24]. Hyperphosphorylated Tau appears exclusively in the LC in Braak stage 0 (a-c) stages, spreads to the entorhinal cortex (EC) in Braak stages I-II, then propagates to the hippocampus and frontal cortex in stages III-VI. Because the LC sends dense projections to other vulnerable brain regions that display early Tau pathology, the LC might be one of the origins of Tau neuropathology in AD [6, 19, 45]. Injection of Tau preformed filaments (PFFs) or Tau virus into the LC leads to progressive loss of LC neurons and the appearance of Tau pathology in the forebrain [21, 29, 30, 33]. However, a direct relationship between ApoE and Tau pathology in the LC has not been described.
Active transport of neurotransmitters into synaptic vesicles is required for their subsequent exocytotic release. In monoaminergic neurons, this process is carried out by the vesicular monoamine transporters (VMAT1 and VMAT2) [25]. These transporters exchange intra-vesicular protons for extravesicular neurotransmitters [4], and are essential for monoaminergic neurotransmission, which requires the sequestration of transmitter into synaptic vesicles for subsequent Ca2+-stimulated exocytotic release [47]. Cytoplasmic NE is converted into 3,4-dihydroxyphenyl-glycolaldehyde (DOPEGAL) by monoamine oxidase A (MAO-A) during normal catecholamine metabolism, but DOPEGAL production is increased in the degenerating LC neurons of AD patients [10–12]. Injection of DOPEGAL into rodent brains elicits adrenergic neuronal loss, which is likely due to generation of free radicals and activation of mitochondrial permeability transition [9]. VMAT2 LO mice, which have a 95% genetic reduction of VMAT2, exhibit age-dependent loss of noradrenergic innervation in the cortex and progressive degeneration in the LC, demonstrating that insufficient catecholamine storage can cause spontaneous degeneration in susceptible neurons, underscoring cytosolic catecholamine catabolism as a determinant of neuronal susceptibility [14, 49].
Asparagine endopeptidase (AEP, gene name LGMN) is an endo-lysosomal cysteine protease that cleaves its substrates after an asparagine (N) residue. We previously reported that AEP acts as delta-secretase (δ-secretase), which cleaves both APP and Tau in the aged mouse brains and human AD brains [54, 55]. δ-secretase predominantly cleaves APP at the N585 residue, escalating Aβ production and senile plaque deposition. It also cuts Tau at the N255 and 368 sites, elevating NFT pathology and neuronal cell death. Notably, AEP is upregulated and activated in AD mouse models and human AD brains in an age-dependent manner, tightly coupled to APP and Tau fragmentation. Depletion of AEP abolishes senile plaques in AD mouse models and NFTs in Tau P301S mice, respectively, leading to restoration of synaptic plasticity and cognitive function [54, 55]. Consequently, a small molecule inhibitor of AEP via blocks AD-like pathology and rescues cognitive deficits [53]. Recently, we showed that DOPEGAL, which is exclusively produced in noradrenergic neurons, activates AEP, leading to Tau N368 cleavage, LC degeneration, and spread of Tau pathology to the forebrain [33].
In the current study, we sought to link ApoE4 to Tau pathology in the LC via VMAT2 and DOPEGAL production. We show that ApoE4 selectively binds to VMAT2 and inhibits vesicular NE uptake, leading to increased cytosolic NE that gets oxidized into DOPEGAL and activation of AEP. Knockout of AEP in MAPT transgenic mice that express wild-type human Tau abolishes ApoE4-induced Tau pathology and hippocampal atrophy, and restores normal cognitive function. By contrast, ApoE3 binds Tau and protects it from AEP cleavage, decreasing neurotoxicity. Blocking Tau N368 cleavage by AEP or knockout of Tau abrogates ApoE4-provoked neurotoxicity, leading to attenuation of cognitive dysfunction. Finally, deletion of VMAT2 in the LC of MAPT mice enhances DOPEGAL production and facilitates ApoE4-induced pathology, toxicity, and cognitive impairment, all of which are ameliorated in Tau knockout mice.
Materials and methods
Animals.
Tau P301S mice (line PS19), wild-type human Tau mice (line MAPT), and wild-type mice (all on a C57BL/6J background) were obtained from the Jackson Laboratory (stock numbers 008169, 005491, and 000664, respectively). ApoE3 (B6.129P2-Apoetm2(APOE*3)MaeN8) and ApoE4 (B6.129P2-Apoetm3(APOE*4)MaeN8) targeted replacement mice were purchased from Taconic. AEP −/− mice were maintained on a mixed 129/Ola and C57BL/6 background. VMAT2 LO mice were generated and provided by Dr. Gary W. Miller [49]. VMAT2 LO mice were maintained on a mixed 129 and C57BL/6 background. MAPT mice were crossed with AEP −/− mice to generate MAPT/AEP −/− mice. 6-9 female mice per group were used for these studies. We used female mice because (1) epidemiological studies show that women are at higher risk for AD than men [40], and (2) in many AD mouse models, AD-like neuropathology is more prominent in females [18]. Investigators were blinded to the group allocation during the animal experiments. Animal care and procedures were conducted according to the National Institutes of Health Guide for Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Emory University.
Human tissue samples.
Paraffin sections containing the LC from 10 postmortem AD cases of Braak stages IV - VI (5 males and 5 females, age 62 ± 10.9 years, mean ± SD) and 10 cognitively normal controls (6 males and 4 females, age 59.4 ± 8.8 years) were obtained from the Emory Goizueta Alzheimer’s Disease Research Center. AD was diagnosed according to the criteria of the Consortium to Establish a Registry for AD and the National Institute on Aging. Informed consent was obtained from the subjects prior to death. Diagnoses were confirmed by the presence of amyloid plaques and neurofibrillary tangles in formalin-fixed tissue.
Transfection and viral infection of cells.
Primary cortical neurons were cultured as previously described [35]. Human iPS cells were obtained from a donor; ax0111 from AD patients with ApoE4/4 genetype (Axo1 Bioscience). All rats were purchased from the Jackson Laboratory. HEK293 cells and SH-SY5Y cells were transfected with plasmids encoding Tau, ApoE3, ApoE4, or VMAT2 using polyethylenimine (PEI) or Lipofectamine 3000 (Thermo Fisher Scientific). AAV-Tau, AAV-AEP, AAV-AEP C189S, AAV Tau N225/368A, AAV-ApoE3, and AAV-ApoE4 were used for infection in primary neuron and SH-SY5Y (ATCC® CRL-2266™) neuroblastoma cells. Neurotoxicity was analyzed using LDH assay (CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega).
Western blot analysis.
Cultured cells or mouse brain tissue samples were lysed in lysis buffer (50 mM Tns, pH 7.4, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate, supplemented with a cocktail of protease inhibitors), and centrifuged for 15 min at 16,000 g. The supernatant was boiled in SDS loading buffer. The samples were transferred to a nitrocellulose membrane, after SDS-PAGE. Primary antibodies to the following targets were used: Tau5 (Santa Cruz, SC-58860), AT-8 (Thermo Fisher Scientific, MN1020), beta-actin (Sigma-Aldrich, A5316), AEP (Cell signaling, 93627S), T22 (Millipore, ABN454), ApoE (Thermo Fisher Scientific, 701241), and Tau N368 (custom antibody [54]).
Vesicular [3H] norepinephrine (NE) uptake assay.
Mice brains were homogenized in buffer (4 mM HEPES, 0.32 M sucrose, pH 7.4) and centrifuged at 1000 g for 10 min. The supernatant was centrifuged at 20,000 g for 20 min and the pellet was resuspended in 1.6 ml of 0.32 M sucrose. After homogenization in 6.4 ml of the cold distilled water by a glass/Teflon homogenizer, 1 ml of buffer containing 250 nM HEPES and 1 M potassium tartrate was added and the mixture was centrifuged at 20,000 g for 20 min. The supernatant was centrifuged again at 120,000 g for 2 h and the vesicles were resuspended in 1 ml of buffer (100 mM potassium tartrate, 25 mM HEPES, 0.1 mM EDTA, 0.05 mM EGTA, and 1.7 mM ascorbate, pH 7.4). 300 ml of vesicle solution including NE was incubated for 30 min at 30°C with APOE3 or APOE4 recombinant proteins, and then a tracer of 2% [3H] NE was added and incubated for 5 min at 30°C. After termination of the reaction by addition of 5 ml of cold assay buffer, the vesicles were filtered through the membrane filters (Millipore). The filters were placed in scintillation fluid and counted by Beckman scintillation counter (Beckman LS6500).
AEP activity assay.
Assay buffer (20 mM citric acid, 60 mM Na2HPO4, 1 mM EDTA, 0.1% CHAPS and 1 mM DTT, pH 6.0) of 100 μl including 100 μM AEP substrate Z-Ala-Ala-Asn-AMC (ABchem) was added to culture cell or mouse brain tissue lysates. AMC released by substrate cleavage was quantified using a fluorescence plate reader at 460 nm for 1 h in kinetic mode.
MAO-A activity assay.
Cultured cell or mouse brain tissue lysates (20 μg) were incubated with a working solution of 100 μl containing 400 μM Amplex® Red reagent, 2 U/ml HRP, and 2 mM p-tyramine substrate with MAO-B inhibitor, pargyline (Molecular Probes). The fluorescence of MAO-A activity was measured in a fluorescence plate reader using excitation in the range of 530-560 nm and emission at 590 ±10 nm at 37°C for 2 h in kinetic mode.
ELISA analysis.
Cultured cell or mouse brain tissue lysates were diluted with the reaction buffer (PBS with 5% BSA, 0.03% Tween-20, and protease inhibitor mixture) and analyzed with mouse Aβ1-40 (Invitrogen, KMB3481) and mouse Aβ1-42 (Invitrogen, KMB3441) ELISA kits according to the manufacturer’s instruction. For the analysis of inflammatory cytokine, lysates were analyzed with mouse IL-6 (Abcam, ab222503), mouse IL-1β (Invitrogen, 88-7013-88), and mouse TNFβ (Invitrogen, 88-7324-88) ELISA kits according to the manufacturer’s instruction. The sample concentrations were determined by comparison with the standard curve.
HPLC analysis.
LC tissue from mouse brain was resuspended in 150 βL ice cold 0.1 M PCA containing 0.1 mM EDTA and sonicated on ice by probe sonication on setting 3 with a 30% duty cycle. Homogenates were centrifuged at 10,000 × g for 10 min at 4°C, and supernatants were transferred into 0.22 μM PVDF microcentrifuge filter tubes to remove any remaining particulate matters at 5000 × rpm for 5 min at 4°C. Monoamine concentrations were determined by reverse phase HPLC with electrochemical detection. Tissue protein pellets were dissolved in 500 μL 2% sodium dodecyl sulfate (SDS). Protein quantification was carried out in triplicate in 96-well microplates with SpectraMax M5e spectrophotometer (Molecular Devices, Sunnyvale CA) using the BCA method (Piere BCA Protein Assay Kit, Thermo Scientific). For HPLC, an ESA 5600A CoulArray detection system, equipped with an ESA Model 584 pump and an ESA 542 refrigerated autosampler (ESA, Bedford, MA) was used. Separations were performed at 23°C using an Xterra RP 4.6 × 250 mm, 3.5 μM particle size, C18 column (Waters, Milford, MA). The mobile phase consisted of 72 mM sodium hydrogen phosphate, 28 mM citric acid, 0.6 mM 1-octanesulfonic acid sodium, 0.2 mM EDTA at pH 5.35. A 20 μl of sample was injected. The samples were eluted isocratically at 0.5 mL/min and detected using a 6210 electrochemical cell. Analytical cell potentials were −175, 100, 350 mV. The analytes were identified by the matching criteria of retention time to known standards. Compounds were quantified by comparing peak areas to those of standards on the dominant sensor.
Thioflavin S staining.
Tau fibrils were stained with Thioflavin S solution (Sigma-Aldrich. Cat#T1892). The brain sections were incubated in 0.25% potassium permanganate solution for 20 min, followed by incubation in bleaching solution containing 2% oxalic acid and 1% potassium metabisulfite for 2 min. Then, the sections were transferred to blocking solution containing 1% sodium hydroxide and 0.9% hydrogen peroxide and incubated for 20 min. The sections were stained with solution of 0.0125% Thioflavin S in 50% ethanol
Immunostaining.
Paraffin-embedded human brain sections or free-floating mouse brain sections sliced by cryotome were treated with 0.3% H2O2 for 10 min. Sections were washed three times in PBS and blocked in 1% BSA, 0.3% Triton X-100, for 1 h, followed by overnight incubation with a tyrosine hydroxylase (TH) antibody (Abcam, AB112; 1: 1000), anti-T22, anti-Tau N368, anti-ApoE, anti-AEP, anti-VMAT2 (Novus, NBP1-69750), or AT-8 antibody (1: 500) at 4°C. Then, the sections were incubated with the matched fluoro-conjugated secondary antibody for 2 h at room temperature, followed by three washes in PBS. The slides were washed three times in PBS and covered with a glass using mounting solution, after DAPI staining for 5 min. Human brain sections were treated with 10 mM cupric sulfate in ammonium acetate buffer (50 mM) for 15 min to eliminate lipofuscin-like auto fluorescence.
Proximity ligation assay.
The fluorescence of proximity ligation of anti-VMAT2 (rabbit, NBP1-69750) and anti-ApoE (Goat, Millipore, AB947) was detected in LC sections of ApoE TR mice by Duolink PLA in situ detection kit (Sigma, DUO92105). All procedures were followed by Duolink PLA protocol and reagents. After blocking for 1 h at 37 °C, sections were incubated with ApoE and VMAT2 antibodies, following PLA plus and minus probes incubation for 1 h. Sections were washed three times in wash buffer and incubated in ligation buffer for 30 min and in amplification buffer for 2 h, respectively. Then, sections were washed three times in wash buffer and mounted using mounting solution, after DAPI staining for 10 min.
Electron microscopy.
Mice were perfused transcardially and fixed with 2% glutaraldehyde and 3% paraformaldehyde dissolved in PBS. Hippocampal tissues were post-fixed in 1% OsO4 for 1 h. Ultrathin sections of 90 nm were stained with uranyl acetate (Sigma-Aldrich. Germany) and lead acetate, and viewed at 100 kV in a JEOL 200CX electron microscope. Synapses were identified and quantified by the presence of synaptic vesicles and postsynaptic densities.
Golgi staining.
Mouse brains were perfused and fixed by 10% formalin and post-fixed for 24 h. Then the brains were immersed in 3% potassium bichromate for 3 d in the dark, transferred into 2% silver nitrate solution, and incubated in the dark at room temperature. Hippocampal sections were cut at 60 mm by vibratome. After mounting, the sections were air-dried, dehydrated through 70%, 95%, and 100% ethanol, and cleared in xylene and coverslipped.
Stereotaxic injection.
AAV-ApoE3 (GeneCopoeia, AA02-CS-C0189-0, >5X10^12 GC/ml), AAV-ApoE4 (GeneCopoeia, AA02-CS-C0189-02), or Lentiviral ShRNA VMAT2 (ABM, iV042581) were injected into the LC or HC region of mice. 3-month-old mice (20 - 25 g) of each group were anesthetized with isoflurane (Piramal Healthcare). Meloxicam (5 mg/kg) was injected subcutaneously for analgesia (Loxicom, Norbrook). Unilateral intracerebral injection of virus was performed stereotaxically at the following coordinates: anteroposterior (AP) −5.4 mm and mediolateral (ML) −1.2 mm relative to bregma, and dorsoventral (DV) −3.7 mm from the dural surface for LC; AP −2.1, ML −1.8, DV −1.5 for HC. 2 μl of viral suspension was injected into each site using a 10-μ1 Hamilton syringe with a fixed needle at a rate of 0.25 μl/min. The needle remained in place for 5 min after the viral suspension was completely injected, and then was slowly removed over 2 min. The mice were placed on a heating pad until recovery from the anesthesia.
Behavioral testing.
Morris water maze.
Mice were trained in a round, water-filled tub (52” diameter) in an environment with extra maze cues. Each subject was given 4 trials/d for 5 consecutive d, with a 15-min intertrial interval. The maximum trial length was 60 s, and if subjects did not reach the platform in the allotted time, they were manually guided to it. Following the 5 d of task acquisition, a probe trial was presented, during which time the platform was removed and the percentage of time spent in the quadrant that previously contained the escape platform during task acquisition was measured over 60 s. All trials were analyzed for latency and swim speed by MazeScan (CleverSys, Inc.). We monitored body weight, visual ability, and swim speed during behavioral test to verify that the deficits did not result from motor dysfunction.
Fear conditioning.
The ability to form and retain an association between an aversive experience and environmental cues was tested with a standard fear conditioning paradigm occurring over a period of 3 d. Mice were placed in the fear conditioning apparatus (7″ W, 7″ D X 12″ H, Coulbourn Instruments), composed of Plexiglas with a metal shock grid floor, and allowed to explore the enclosure for 3 min. Following this habituation period, 3 conditioned stimulus (CS)-unconditioned stimulus (US) pairings were presented with a 1-min intertrial interval. The CS consisted of a 20-s 85-db tone, and the US consisted of 2 s of a 0.5-mA foot shock, which was co-terminated with each CS presentation. One min following the last CS-US presentation, mice were returned to their home cage. On day 2, the mice were presented with a context test, during which subjects were placed in the same chamber used during conditioning on Day 1, and the amount of freezing was recorded via camera and the software provided by Colbourn. No shocks were given during the context test. On day 3, a tone test was presented, during which time subjects were exposed to the CS in a novel compartment. Initially, animals were allowed to explore the novel context for 2 min. Then the 85-db tone was presented for 6 min, and the amount of freezing behavior was recorded.
Stereological cell quantification.
Unbiased stereological cell counting was performed using the Stereo Investigator software (MBF Bioscience, Williston, VT) and Olympus microscope. LC sections of 30 μm at every 6th section were stained with TH antibody and analyzed under the randomly placed counting frames (50 × 50 μm) on a counting grid (120 × 120 μm). Optical dissector of 22 μm with 2 μm upper and lower guard zones was used. The total number of TH+ neurons in the LC was estimated using the optical fractionator method. TH-positive axon terminals in the striatum were estimated using fluorescence intensity by Image J software. For quantification of positive cells and image analysis, stained colour was selected and set the proper threshold for the binarization of the selected colour image by Image J software. The total number and intensity of immune-reactive neurons were analysed using the same threshold. The conditions of the analysis were blinded to the investigator.
Quantification and statistical analysis.
All cell culture data are expressed as mean ± S.E.M. from 3 or more independent experiments, and the level of significance between 2 groups was assessed with Student’s t-test. For experiments consisting of more than 2 groups, one-way or two-way ANOVA followed by Tukey’s post-hoc test was applied. A value of p < 0.05 was considered to be statistically significant. Sample sizes were determined by Power and Precision (Biostat).
Results
VMAT2 is lost in LC neurons in an age-dependent manner in Tau P301S mice and AD brains
We recently reported that the NE metabolite, DOPEGAL, activates AEP, triggers Tau hyperphosphorylation and aggregation, and provokes LC neuron degeneration in various AD mouse models [33]. Reduced vesicular storage of catecholamines in VMAT2-deficient (VMAT2 LO) mice causes progressive noradrenergic neuronal degeneration in LC, associated with increased levels of deaminated metabolites [14, 49]. To explore whether VMAT2 is reduced and associated with LC neuronal loss in AD, we conducted immunofluorescence (IF) co-staining on human AD brains with antibodies against VMAT2 and the catecholaminergic cell marker TH. All information concerning the postmortem human cases are presented in Supplementary Table 1. Consistent with many studies, the number of TH-positive noradrenergic neurons was prominently decreased, and we observed a concomitant deduction in VMAT2 abundance and inverse p-Tau (AT8) elevation (Fig. 1a). Quantification of double labeled LC neurons showed that the percentage of TH-positive cells that were also VMAT2-positive was significantly reduced in human AD brains compared with control brains (Fig. 1b). We also observed an age-dependent LC neuronal loss, as we previously reported [15], correlated with progressive VMAT2 signal reduction in Tau P301S transgenic mice, which express human Tau with a frontotemporal dementia-causing mutation, from 3 to 9 months of age. Concurrently, 4-HNE, a biomarker for oxidative stress, gradually increased (Fig. 1c & d). Quantification of VMAT2 and fluorescence intensities of TH and 4-HNE activities are summarized in Fig. 1f & g, indicating that oxidative stress escalation was correlated with VMAT2 decrease and loss of LC neurons. However, significant neuronal loss and the reduction of VMAT2 were not observed in 9-month-old MAPT mice, which express wild-type human Tau (Fig. 1e & f). Finally, there was an inverse relationship between NE and DOPEGAL concentration as Tau P301S mice aged (Fig. 1h), suggesting that NE metabolism into DOPEGAL was contributing to the oxidative stress and LC degeneration. By contrast, DOPEGAL was barely detectable in the LC of 9-month old wild-type mice.
Fig. 1. VMAT2 is reduced in degenerated LC neurons in Tau P301S mice and AD brains.
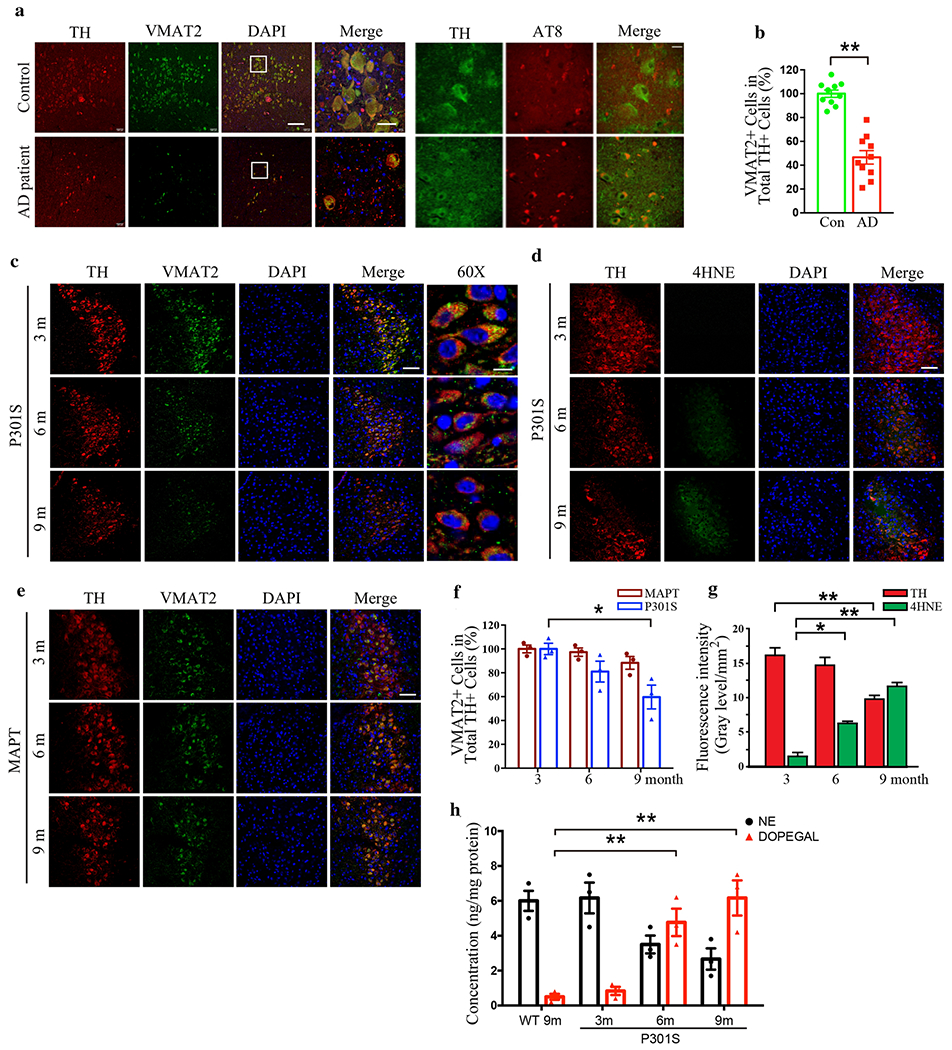
LC neurodegeneration and the reduction of VMAT2 in postmortem human AD brains and P301S mouse brains of AD mice model were examined by immunofluorescence staining of tyrosine hydroxylase (TH) and VMAT2. a. Representative images of staining in postmortem LC sections of healthy controls and AD patients by TH (red), VMAT2 (green), DAPI (blue), and AT8 (red). Scale bar is 100 μm and 60X panel is 20 μm. b. Quantification of VMAT2 + cells in TH+ LC neurons. Data are shown as mean ± SEM. N=10 per group. ** p<0.01. c. Representative images of staining by TH (red), VMAT2 (green), and DAPI (blue) in LC sections of P301S mice at 3 months, 6 months, and 9 months old. Scale bar is 100 μm and 60X panel is 20 μm. d. Representative images of staining by TH (red), 4-HNE (green), and DAPI (blue) for the oxidative stress in the LC of P301S mice. Scale bar is 100 μM. e. Representative images of staining by TH (red), VMAT2 (green), and DAPI (blue) in LC sections of MAPT mice at 3 months, 6 months, and 9 months old. Scale bar is 100 μm. f & g. Quantification of VMAT2 + cells in TH+ LC neurons and fluorescence intensity of TH (red) and 4-HNE (green) show that oxidative stress is increased with LC neurodegeneration in P301S mice in an age-dependent manner. Data are shown as mean ± SEM. N=6 per group. * p<0.05, ** p<0.01. h. Measurement of DOPEGAL escalation in the LC of P301S mice via HPLC analysis. Data were analyzed using two-way ANOVA and shown as mean ± SEM, N = 3 per group. ** p < 0.01.
ApoE4 selectively binds to VMAT2 and inhibits its NE uptake activity
To examine whether ApoE plays a role in LC neuronal degeneration, we assessed biochemical interactions between ApoEs and VMAT2. Co-immunoprecipitation (Co-IP) showed that GFP-tagged ApoE4, but not E3, specifically associated with VMAT2 in transfected SH-SY5Y cells (Fig. 2a, Supplementary Fig. 1a). Enzymatic assays revealed that MAO-A, MAO-B, and AEP activities were significantly enhanced by ApoE4 as compared to ApoE3 and GFP control (Fig. 2b–d). Next, we explored the functional consequence of VMAT2 binding to ApoE4. [3H]NE uptake assay demonstrated that the NE transport velocity and uptake were significantly repressed by the VMAT inhibitor reserpine or recombinant ApoE4 protein (rApoE4), but not rApoE3 (Fig. 2e & f). To further explore the relationship between VMAT2 and ApoE, we performed IF co-staining on the brain sections from ApoE4 and ApoE3 knockin mice (ApoE-TR target replacement mice, 6 months old), and found that ApoE4 but not ApoE3 tightly co-localized with VMAT2 in LC neurons (Fig. 2g & h). AEP, AEP-truncated Tau N368, 4-HNE, and DOPEGAL were also noticeably augmented in TH-positive LC neurons from ApoE4-TR mice compared to ApoE3-TR mice (Fig. 2g–i), suggesting that DOPEGAL might be elevated due to VMAT2 blockage by ApoE4 and triggering AEP activity (Fig. 2g & h). Further, the number of TH-positive LC neurons was significantly reduced in ApoE4-TR as compared to ApoE3-TR mice in an age-dependent way, tightly correlated with DOPEGAL levels (Fig. 2i & j). The specific physiological interaction between ApoE4 and VMAT2 was confirmed by co-staining human iPS neurons from ApoE4 or ApoE3 homozygous individuals (Fig. 2k). Co-IP validated that endogenous VMAT2 robustly associated with ApoE4 but not ApoE3 (Fig. 2l, Supplementary Fig. 1b). Moreover, proximity ligation assay with VMTA2 and ApoE antibodies verified the interaction between ApoE4 and VMAT2 in the LC of ApoE4-TR mice (Supplementary Fig. 1c). Hence, ApoE4, but not ApoE3, binds and inhibits VMAT2, escalating DOPEGAL production and provoking LC degeneration.
Fig. 2. ApoE4 selectively interacts with VMAT2 and inhibits NE uptake.
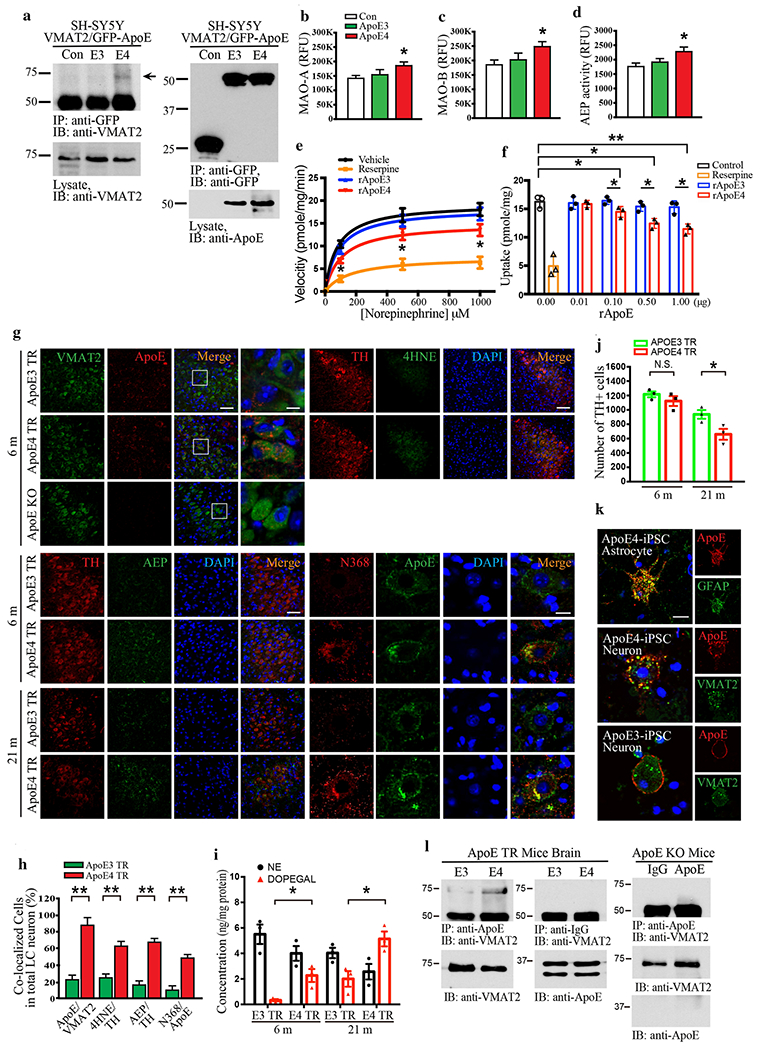
a. The binding between ApoE4 and VMAT2 was investigated in SH-SY5Y cells, which were co-transfected with GFP-APOE3, GFP-APOE4, and VMAT2. Transfected cell lysates were immunoprecipitated with anti-GFP and the precipitated proteins were analyzed by immunoblotting with anti-VMAT2. b-d. Cell lysates were analyzed by enzymatic assay of MAO-A (b), MAO-B (c), and AEP (d). Data are shown as mean ± SEM. N=3 per group. * p<0.05. e & f. [3H] NE uptake was analyzed in the purified vesicles isolated from mouse brainstem including the LC region in the presence of rApoE3 or rApoE4. The measured results were analyzed as velocity (pmole/mg/min) (e) and uptake (pmole/mg) (f). The interaction between ApoE4 and VMAT2 in the LC was analyzed using 6-month-old Target replacement ApoE3 TR and ApoE4 TR mice. g. Representative images of double immunofluorescence staining in the LC section by VMAT2 (green)/ApoE (red), TH (red)/AEP (green), TH (red)/4HNE (green), and Tau N368 (red)/ApoE (green). Scale bar is 100 μm and 60X panel is 20 μm. h. Quantification of co-localized cells in the LC staining of ApoE TR mice showed that ApoE4 induced AEP activation, oxidative stress of 4-HNE, and upregulation of Tau N368 as well as the interaction of ApoE4 and VMAT2. i. Measurement of DOPEGAL activated in the LC of ApoE4 TR mice via HPLC analysis, j. Stereological counting of TH+ cells in LC of ApoE3 TR and ApoE4 TR mice brain. All data shown in H-J were analyzed using two-way ANOVA and shown as mean ± SEM, N = 3 per group. ** p < 0.01, * p < 0.05, N.S. not significant. The co-localization and binding between ApoE4 and VMAT2 were verified in neurons and astrocytes induced from human iPS cells (ApoE4 homozygous) and brain tissue from ApoE TR mice. k. Representative images of double immunofluorescence staining in ApoE4 or ApoE3 human iPS cells by ApoE (red)/VMAT2 (green) and ApoE (red)/GFAP (green). Scale bar is 20 μm. 1. Mice brain cell lysates were immunoprecipitated with anti-ApoE, and the precipitated proteins were analyzed by immunoblotting with anti-VMAT2.
ApoE4 enhancement of Tau neurotoxicity is dependent on AEP cleavage of Tau at N368
While the AD risk associated with ApoE4 has been attributed to effects on Aβ, it was recently reported that ApoE4 also exacerbates Tau neurotoxicity [43]. To determine whether AEP contributes to this process, we infected primary cortical neuronal cultures with various viruses expressing human Tau and ApoE3 or ApoE4 in the presence of wild-type or enzymatically-dead C189S mutant AEP. As shown by immunoblotting in Fig. 3a and Supplementary Fig. 2a, Tau was strongly cleaved at position N368 by active AEP in neurons expressing ApoE4. In contrast, Tau N368 cleavage was blunted by ApoE3, even though AEP was strongly activated. Subsequently, Tau was robustly phosphorylated in ApoE4- but not ApoE3-expressing neurons as revealed by AT8 immunoreactivity. These effects were reduced in cells expressing inactive AEP C189S, indicating that active AEP is required for ApoE4 to provoke Tau pathology. LDH assay revealed that ApoE4 stimulated significantly more cell death than ApoE3 (Fig. 3b). Overexpression of wild-type AEP further augmented this effect, which was significantly reduced by C189S mutant. Enzymatic assay showed that AEP activity was much higher in neurons overexpressing wild-type AEP compared to control plasmid, and C189S mutant diminished enzymatic activity in ApoE3- and ApoE4-expressing neurons (Fig. 3c). Next, we employed the small molecule AEP inhibitor #11 to further assess the role of AEP in this process [53]. As expected, #11 potently blocked AEP proteolytic cleavage of Tau N368, and also attenuated Tau hyperphosphorylation and cell death that was more abundant in the presence of ApoE4 than ApoE3 (Fig. 3d–f, Supplementary Fig. 2b). These results demonstrate that AEP is required for ApoE4-triggered neuronal cell death, accompanied with Tau proteolytic cleavage and hyperphosphorylation.
Fig. 3. ApoE4-elicited neurotoxicity is dependent on AEP cleavage of Tau N368.
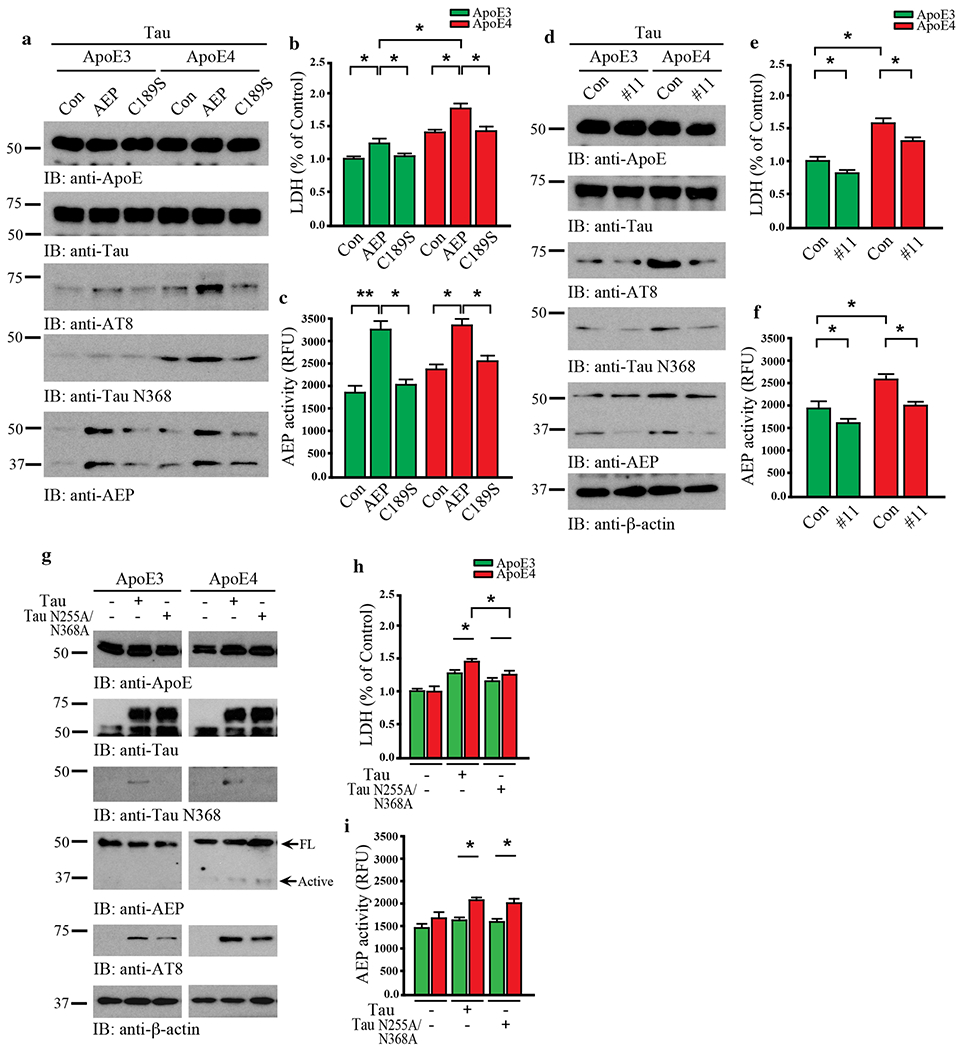
Primary neurons were cultured and infected with AAV-hTau, AAV-ApoE3 or ApoE4, and AAV-AEP or AEP C189S. a. Western blot analysis showed that ApoE4 overexpression induced Tau phosphorylation and Tau N368 cleavage with AEP overexpression but not inactive AEP C189S. b. LDH assay showed that ApoE4 or AEP overexpression in neurons induced cell death. c. The activation of AEP was confirmed by enzymatic assay. Data are shown as mean ± SEM. N=3 per group. * p<0.05. Primary neurons were infected with AAV-hTau and AAV-ApoE3/E4, followed by treatment with vehicle or the AEP inhibitor, compound 11(10 μM). d. Western blot analysis showed that Tau and ApoE4-induced AEP activation, Tau phosphorylation, and Tau N368 cleavage were blocked by compound 11. e. LDH assay showed that cell death by Tau and ApoE4 was inhibited by compound 11. f. The inhibition of AEP was confirmed by enzymatic assay. Data are shown as mean ± SEM. N=3 per group. * p<0.05. Primary neurons were infected with AAV-ApoE3/E4 and AAV-Tau/Tau N255A/N368A. g. Western blot analysis showed that non-cleavable Tau N255A/N368A repressed the effects of Tau and ApoE4 in AEP activation, Tau phosphorylation, and Tau N368 cleavage, h. LDH assay indicating the blocking effect of Tau N225A/N368A in cell death by Tau and ApoE4. i. The activation of AEP was confirmed by enzymatic assay. Data are shown as mean ± SEM. N=3 per group. * p<0.05, ** p<0.01.
To interrogate whether Tau fragmentation by AEP is necessary for these events, we conducted experiments with AEP-resistant Tau N255/368A mutant in the presence of ApoE3 or ApoE4. Immunoblotting showed that ApoE4 neurons contained more N368 and AT8-positivie Tau than ApoE3 cells. Consistent with these findings, AEP was proteolytically truncated into its active form in the presence of ApoE4. By contrast, Tau N368 cleavage was completely blocked in Tau N255/368A expressing neurons, and associated phenotypes activities were also reduced accordingly (Fig. 3g, Supplementary Fig. 2c). LDH assay showed that neuronal cell death tightly correlated with AEP and Tau N368 activities, and was substantially decreased by AEP-resistant Tau N255/368A (Fig. 3h & i). Thus, AEP cleavage of Tau N368 plays a crucial role in ApoE4-elicited neurotoxicity.
To further assess cross-talk between ApoE4 and Tau, we co-transfected GST-Tau and ApoE3 or E4 into SH-SY5Y cells. Tau was pulled down with glutathione beads, and co-precipitated proteins were analyzed by immunoblotting. ApoE3, but not ApoE4, selectively interacted with Tau, and AEP was more activated by ApoE4 than ApoE3. Subcellular fractionation revealed more aggregated Tau in the insoluble fractions in the presence of ApoE4 compared with ApoE3. AT8 immunoreactivity was tightly coupled with insoluble Tau aggregates (Supplementary Fig. 3a). LDH assay demonstrated that ApoE4 significantly enhanced cell death compared to ApoE3 in Tau co-transfected cells. AEP enzymatic activity tightly coupled with LDH (Supplementary Fig. 3b & c). Mapping assay revealed that the repetitive microtubule-binding domain (amino acids 255-368) of Tau was implicated in binding to ApoE3 (Supplementary Fig. 3d). Because Tau is proteolytically cleaved by AEP, we speculated that ApoE3 binding to Tau protects it from fragmentation. In vitro cleavage assay showed that ApoE3 dose-dependently prevented AEP cleavage of recombinant Tau, while ApoE4 had minimal protective effects (Supplementary Fig. 3e). Similar results were observed in Tau transfected intact cells (Supplementary Fig. 3f). Hence, ApoE3 but not E4 preferentially binds to Tau and protects it from AEP cleavage.
AEP is required for ApoE4-induced AD-like pathology and cognitive deficits in MAPT mice
Tau pathology can be generated intrinsically, but can also propagate between interconnected brain regions. Infusion of synthetic Tau preformed fibrils (PFFs) into the hippocampus travel retrogradely and induce Tau pathology in the LC [29]. To determine whether this form of propagated tau pathology in the LC also involves ApoE4, we injected an AAV expressing ApoE3 or E4 into the hippocampus of 3-month old MAPT and MAPT/AEP −/− mice and assessed Tau pathology 3 months later. We first confirmed the role of AEP and Tau on ApoE4-triggered pathology in the hippocampus itself. Although hippocampal neurons do not express VMAT2, we have shown that ApoE4 can also induce Tau pathology via a C/EBPβ/AEP pathway that is independent of monoamine metabolism [50]. We conducted IF co-staining in the hippocampus and found that AEP, Tau N368, and AT8 immunoreactivity were greater in ApoE4 expressing cells compared to ApoE3 (Fig. 4a & b). Aggregated Tau-specific antibody T22 staining revealed more Tau aggregates in ApoE4-infected neurons than ApoE3 or control, and these inclusions were also Thioflavin S positive, indicating that they were fibrillary (Fig. 4c). Quantification of Nissl staining revealed that ApoE4 significantly shrank the hippocampal volume, while lateral ventricle volume was greatly elevated compared to ApoE3 (Fig. 4d). Aβ40 and Aβ42 ELISA assays showed no difference among the groups, indicating that the effects of ApoE4 are independent of Aβ production (Supplementary Fig. 4a). However, pro-inflammatory cytokines including IL-1β, IL-6 and TNFα were all significantly increased in ApoE4-infected MAPT mice as compared to ApoE3 (Supplementary Fig. 4b). Golgi-impregnated dendritic spines in hippocampal neurons were substantially reduced by ApoE4 compared to ApoE3 in MAPT mice (Supplementary Fig. 4c & d). Finally, electronic microscopy (EM) analysis revealed a significant reduction of hippocampal synapses in ApoE4-infected MAPT mice (Supplementary Fig. 4e & f). Remarkably, all effects of ApoE4 were absent in MAPT/AEP −/− mice, demonstrating that AEP is absolutely necessary for ApoE4-induced Tau pathology (Fig. 4, Supplementary Fig. 4).
Fig. 4. AEP is required for ApoE4-stimulated AD pathology and cognitive defects in MAPT mice.
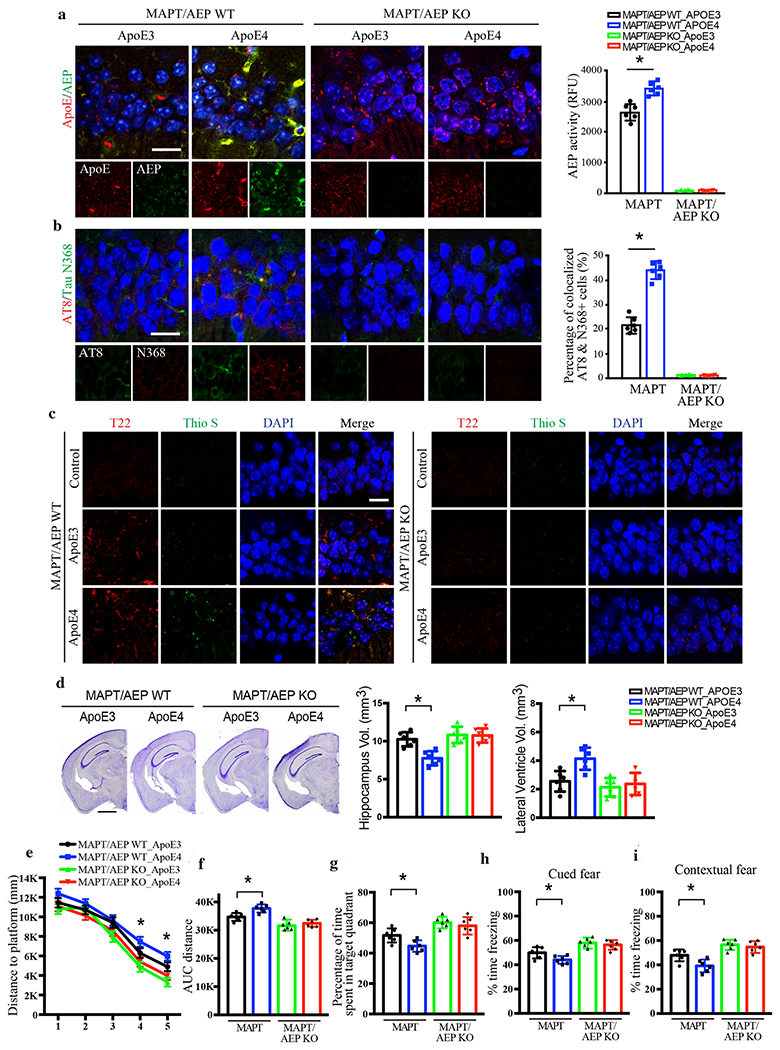
AAV-ApoE3 or AAV-ApoE4 was injected into the hippocampus of MAPT/AEP WT or MAPT/AEP KO mice, and mice were assessed for Tau pathology and memory dysfunctions 3 months later, a. Representative images of ApoE and AEP immunofluorescence co-staining verifying viral infection of AAV-ApoE3 or AAV-ApoE4, and AEP enzymatic assay for MAPT/AEP WT and MAPT/AEP KO mice. Scale bar is 20 μm. Data are shown as mean ± SEM. N=6 per group. * p<0.05. b. Representative images of immunofluorescence staining of AT8 (red), Tau N368 (green), and DAPI (blue) and quantification of co-localized cells to show Tau phosphorylation and cleavage in the hippocampus. Scale bar is 20 μm. Data are shown as mean ± SEM. N=6 per group. * p<0.05. c. Representative images of immunofluorescence staining of Thioflavin S (green), T22 (red), and DAPI (blue) to show Tau aggregation and fibrillization. Scale bar is 20 μm. d. Cresyl violet staining and the quantitative analysis of hippocampus and lateral ventricle volumes demonstrate that ApoE4 mediated the hippocampal atrophy in MAPT mice but not MAPT/AEP KO mice. Scale bar is 1 mm. Data are shown as mean ± SEM. N=6 per group. * p<0.05. Behavioral tests for Morris water maze (e-g) and fear conditioning test (h & i) demonstrating memory dysfunction mediated by ApoE4 in MAPT/AEP WT mice, not MAPT/AEP KO mice. Distance travelled to the platform (e), area under the curve for total distance travelled (f), and percent time spent in the quadrant (g) previously containing the platform during the probe trial in the Morris water maze. Percent time spent freezing during the cued fear (h) and contextual fear (i) tests following fear conditioning. All data were analyzed using one-way ANOVA and shown as mean ± SEM. N = 6 per group. * p < 0.05.
To determine the impact of ApoE4-induced pathology on cognition, we conducted Morris Water Maze (MWM) and fear conditioning tests on ApoE3 and ApoE4-expressing MAPT and MAPT/AEP−/− mice. In general, ApoE4 elicited mild but significant impairment on spatial and associative learning and memory compared to ApoE3. While ApoE4-expressing mice learned both tasks, their swim distance to the hidden platform during training was increased (Fig. 4e & f) and time spent in the target quadrant of the probe trial was decreased (Fig. 4g) in the MWM, while freezing to the cue (Fig. 4h) and context (Fig. 4i) was reduced. Similar to the pathological results, ApoE4-induced cognitive deficits were abrogated by knockout of AEP (Fig. 4e–i). Together, these findings demonstrate that AEP plays a crucial role in ApoE4-mediated AD-like pathology and cognitive dysfunction.
ApoE4 provokes AD-like pathology and cognitive dysfunction through Tau
To determine the contribution of Tau to ApoE-induced pathology, primary neurons from Tau −/− mice or MAPT mice were infected with viruses expressing human ApoE3 or ApoE4. Overexpressing ApoE4 in MAPT neurons triggered AEP activation, associated with Tau N368 cleavage, hyperphosphorylation (AT8+), and aggregation (T22+), as well as cell death (LDH) (Supplementary Fig. 5a–c). By contrast, AEP activation was substantially abrogated in Tau −/− neurons. There was no difference in Aβ levels between MAPT and Tau −/− neurons (Supplementary Fig. 5d). Therefore, Tau is indispensable for ApoE4-stimulated AEP activation and neuronal cell death. To confirm these results in vivo, 3 month-old MAPT transgenic mice and MAPT −/− mice were injected with AAVs expressing ApoE3 or ApoE4 in the hippocampus. Three months after viral infusion, AEP, AT8, Tau N368, T22, and ThS immunoreactivity were substantially increased in ApoE4 expressed neurons as compared to ApoE3 in MAPT mice, but not in MAPT −/− mice (Fig. 5a–c). Aβ40 and Aβ42 were not altered under any condition (Supplementary Fig. 6a), but IL-1β, IL-6 and TNFα were significantly increased in ApoE4-infected MAPT mice compared to ApoE3 (Supplementary Fig. 6b). Knocking out Tau prevented these pro-inflammatory effects. Golgi staining and EM demonstrated that overexpression of ApoE4, but not ApoE3, incurred dendritic spine and synapse reduction in the hippocampus of MAPT mice, while MAPT −/− mice were protected (Supplementary Fig. 6c–f). Quantitative analysis of Nissl staining showed that ApoE4-elicited hippocampal atrophy was inversely related to lateral ventricle volume augmentation in MAPT mice, which were diminished in Tau −/− mice (Fig. 5d).
Fig. 5. Tau is necessary for ApoE4-mediated AD pathology and cognitive deficits.
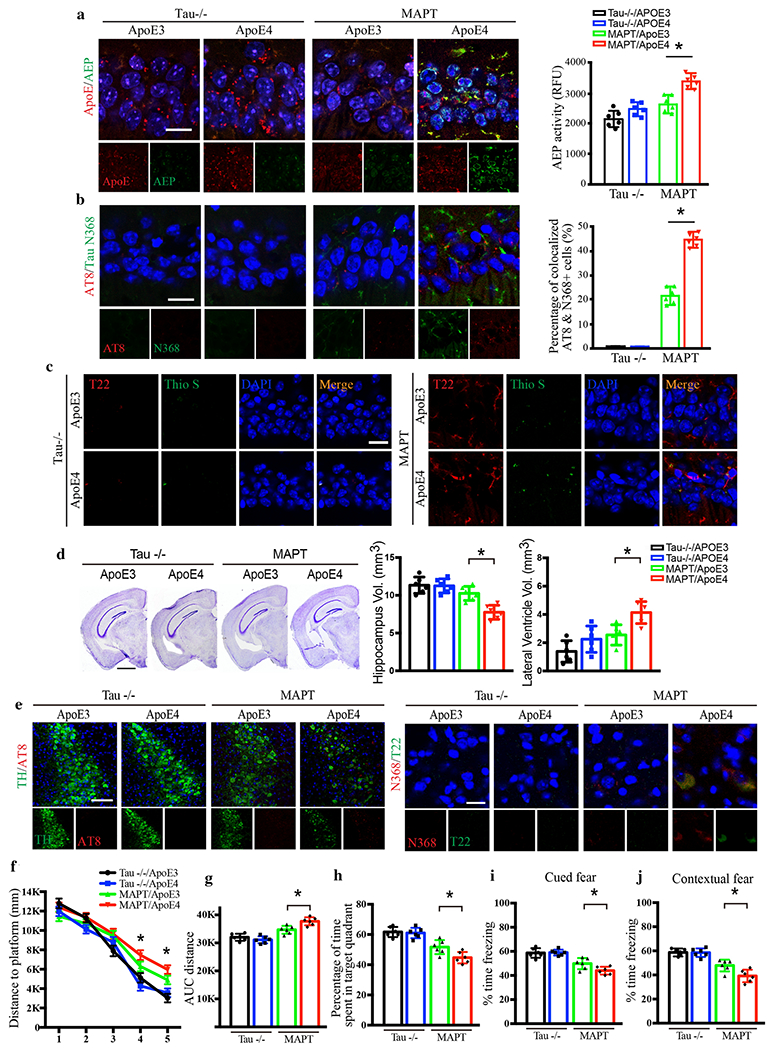
AAV-ApoE3 or AAV-ApoE4 was injected into the hippocampus of MAPT or Tau −/− mice, and then mice were assessed for Tau pathology and memory dysfunctions 3 months later, a. Representative images of ApoE and AEP immunofluorescence co-staining verifying viral infection of AAV-ApoE3 or AAV-ApoE4, and AEP enzymatic assay for MAPT and Tau−/− mice. Scale bar is 20 μm. Data are shown as mean ± SEM. N=6 per group. * p<0.05. b. Representative images of immunofluorescence staining of AT8 (red), Tau N368 (green), and DAPI (blue) and quantification of co-localized cells to show Tau phosphorylation and cleavage in the hippocampus. Scale bar is 20 μm. Data are shown as mean ± SEM. N=6 per group. * p<0.05. c. Representative images of immunofluorescence staining of Thioflavin S (green), T22 (red), and DAPI (blue) to show Tau aggregation and fibrillization. Scale bar is 20 μm. d. Cresyl violet staining and quantitative analysis of hippocampus and lateral ventricle volumes indicate that ApoE4 mediated the hippocampal atrophy in MAPT mice but not Tau −/− mice. Scale bar is 1 mm. Data are shown as mean ± SEM. N=6 per group. * p<0.05. e. Representative images of immunofluorescence staining of TH (green), AT8 (red), and DAPI (blue) in the LC regions of ApoE-injected Tau −/− and MAPT mice (left panels). Scale bar is 100 μm. Right panels are representative images by N368 (red) and T22 (green) immunofluorescence co-staining to show Tau cleavage and aggregation in the LC. Scale bar is 20 μm. Behavioral tests for Morris water maze (f-h) and fear conditioning test (i & j) demonstrating memory dysfunctions mediated by ApoE4 in MAPT mice. Distance travelled to the platform (f), area under the curve for total distance travelled (g), and percent time spent in the quadrant (h) previously containing the platform during the probe trial in the Morris water maze. Percent time spent freezing during the cued fear (i) and contextual fear (j) tests following fear conditioning. All data were analyzed using one-way ANOVA and shown as mean ± SEM. N = 6 per group. * p < 0.05.
ApoE4-induced hippocampal Tau pathology propagates to the LC and causes cognitive impairment
Having established the mechanisms underlying ApoE4-induced pathology in the hippocampus, we next asked whether aberrant Tau was transported retrogradely and affected the LC. We found that hippocampally-expressed ApoE4 triggered Tau N368, AT8, and T22 in the LC, as well as substantial noradrenergic (TH+) neuronal loss compared to ApoE3 in MAPT mice, and these pathologies and degeneration were mitigated by Tau knockout (Fig. 5e, Supplementary Fig. 6g & h). Because no ApoE virus was infused into the LC, these results suggest that hippocampal truncated Tau and its aggregates were retrogradely transported to the LC neurons, where they exerted neurotoxicity.
Because LC-NE transmission is critical for hippocampal-dependent LTP and learning and memory under both normal conditions and in disease states [15, 26, 42], we determined whether dual hippocampal and LC Tau pathology induces cognitive deficits. MWM and fear conditioning cognitive behavioral tests revealed that ApoE4 significantly impaired learning and memory in MAPT mice compared to ApoE3, and these effects were nullified in Tau −/− mice (Fig. 5f–j). Combined, these results indicate that Tau is required for ApoE4-provoked AD-like neuropathology in the hippocampus, its retrograde propagation to the LC, and cognitive dysfunction in vivo.
Deletion of VMAT2 exacerbates ApoE4-provoked AD pathology and cognitive impairment
Our data suggest that ApoE4 facilitates the early onset of AD, and hyperphosphorylated Tau in LC neurons is the earliest detectable AD-like pathology [5–7]. Given our results that ApoE4 interacts with VMAT2, we speculated that ApoE4 may trigger Tau pathology in LC by interfering with VMAT2 function. To test this possibility, we injected AAVs expressing ApoE3 or ApoE4 into the LC of 3-month old MAPT mice. In a separate experiment, we co-administered AAV-ApoE4 or ApoE4 with virus expressing shRNA for VMAT2 into the LC of MAPT and MAPT −/− mice. As validation for the viral expression efficiency, IF co-staining showed that ApoE3 and ApoE4 were robustly expressed in VMAT2-positive LC neurons, while VMAT2 was substantially reduced by shRNA in both MAPT and MAPT −/− mice (Fig. 6a, top panels and Supplementary Fig. 7a). Three and six months later, we found that AT8 and N368 Tau were evident in ApoE4-infected MAPT mice, and were associated with a reduction of TH-positive LC neurons were in a time-dependent manner (Fig. 6a & b). Importantly, knocking down VMAT2 accelerated ApoE4-induced Tau pathology and LC neuron degeneration (Fig. 6a–d), and even conferred some susceptibility to ApoE3 (Fig. 6c & d). These pathological events were completed abolished in MAPT −/− mice regardless of ApoE overexpression or VMAT2 depletion. To further characterize the severity of Tau pathology in LC neurons, we performed T22/ThS co-staining and found that ApoE4 elicited T22- and ThS-positive aggregates in MAPT mice, indicating the presence of bona fide NFTs (Supplementary Fig. 7b & c). Deletion of VMAT2 further elevated, while Tau knockout abolished, these effects. MAO-A and AEP enzymatic activity in the LC lysates were also escalated 6 months after the viral infection (Fig. 6d, Supplementary Fig. 7d). Consistent with increased MAO-A, DOPEGAL was increased and NE was decreased in ApoE4-infected MAPT mice compared to ApoE3. These results suggest that ApoE4 inhibits VMAT2 in the LC neurons, leading to impaired NE uptake and its cytosolic oxidation by MAO-A. In support of this notion, deletion of VMAT2 increased DOPEGAL in both ApoE3- and ApoE4-infected in MAPT transgenic mice and MAPT −/− mice (Supplementary Fig. 7e). MWM and fear conditioning tests showed that LC ApoE4 expression impaired cognition compared to ApoE3 in MAPT mice (Supplementary Fig. 7f–j). Knockdown of VMAT2 in the LC neurons exacerbated learning and memory deficits, while Tau knockout prevented the deleterious effects of ApoE4. Thus, Tau-dependent LC degeneration is mediated by ApoE4 inhibition of VMAT2.
Fig. 6. Deletion of VMAT2 aggravates ApoE4-triggered AD pathology and LC degeneration.
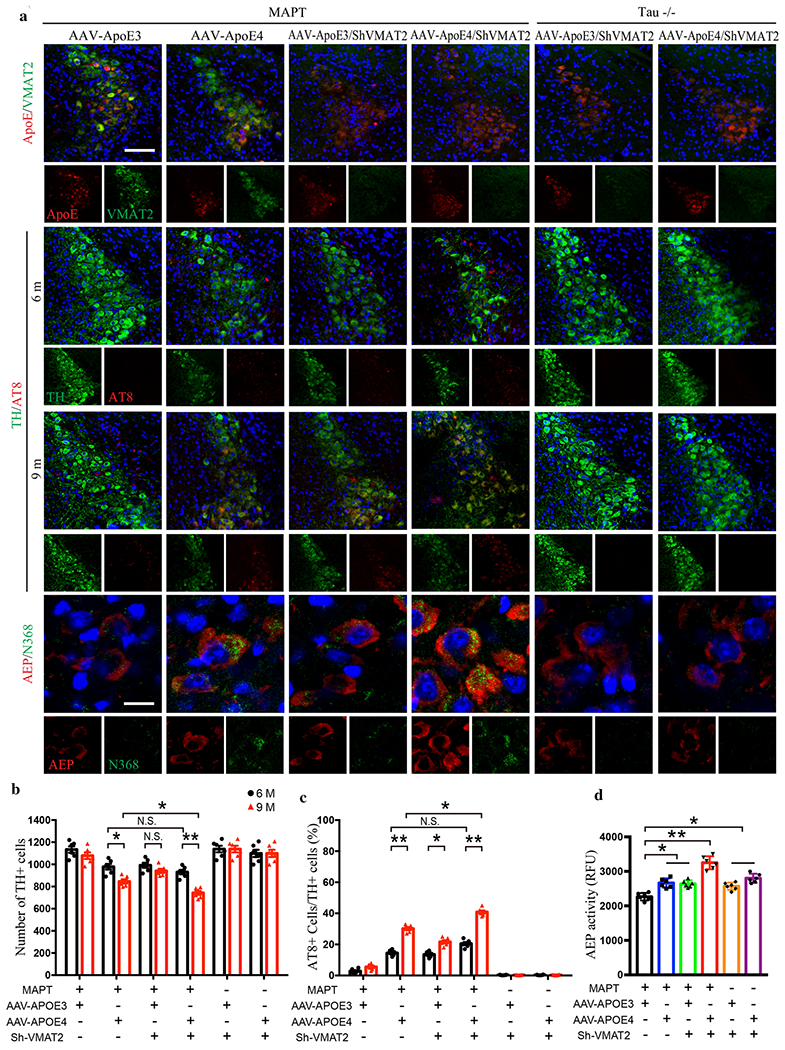
AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2 were injected into the LC of MAPT or Tau −/− mice, and then mice were assessed for Tau pathology and neuronal loss 3 months and 6 months later (6 and 9-month-old group, respectively), a. The upper panels are the representative images of ApoE and VMAT2 immunofluorescence co-staining verifying viral infection of AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2. Representative images of immunofluorescence staining of TH (green), AT8 (red), and DAPI (blue) in 2nd panel (6 month) and 3rd panel (9 month). The last panel’s representative images are AEP (red) and Tau N368 (green) immunofluorescence co-staining to show AEP activation and Tau cleavage in the LC. Scale bar is 100 μm. b. Stereological cell counting of TH+ cells in the LC region showed neuronal loss mediated by ApoE4 overexpression and VMAT2 deficiency, c. Quantification of AT8+ cells in TH+ cells in the LC to show Tau phosphorylation, d. AEP enzymatic assay in the LC for AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2-injected MAPT or Tau −/− mice. All data in b-d were analyzed using two-way ANOVA and shown as mean ± SEM. N=6 per group. * <0.05, ** <0.01.
Previous studies show that VMAT2-deficient mice (VMAT2 LO with 95% gene reduction) display progressive noradrenergic neuronal degeneration in the LC, associated with escalation of deaminated metabolites [49]. To explore whether AEP is activated by DOPEGAL in VMAT2-deleted LC neurons, we conducted IF co-staining and western blot with antibodies against TH and VMAT2 and confirmed that TH-positive LC neurons were lost in an age-dependent manner in VMAT2 LO mice (Supplementary Fig. 8a, b, & f). We further found that AEP and MAO-A were significantly activated in older VMAT2 LO mice (Supplementary Fig. 8c & d). Consistent with increased MAO-A, NE levels were reduced and DOPEGAL was progressively escalated in VMAT2 LO mice (Supplementary Fig. 8e). These results indicate that genetic reduction of VMAT2 elicits DOPEGAL production, AEP activation, and LC neuronal loss in an age-dependent manner.
Discussion
In the current study, we provide evidence that ApoE4 specifically binds and inhibits VMAT2 and leads to cytoplasmic NE oxidation by MAO-A into the metabolite DOPEGAL, resulting in AEP activation in LC neurons. The active AEP subsequently cleaves Tau N368 and promotes its hyperphosphorylation and aggregation, enhancing its neurotoxicity. Conversely, ApoE3 binds Tau and protects it from proteolytic cleavage by active AEP, suppressing its neurotoxicity (Fig. 7). Notably, viral-mediated overexpression of ApoE4 in the hippocampus of triggered Tau neurotoxicity without affecting Aβ production in MAPT mice, leading to hippocampal atrophy and cognitive deficits. These observations are consistent with the recent reports describing aggravation of Tau neurotoxicity by ApoE4 [43]. In addition to inhibition of VMAT2 in LC neurons, our recent study shows that ApoE4 strongly stimulates C/EBPβ/AEP pathway in the hippocampus that triggers Tau upregulation and subsequent Tau N368 fragmentation by elevated AEP, resulting in escalation of Tau pathologies [50]. Conceivably, ApoE4 exacerbates Tau pathologies via multiple mechanisms. Nevertheless, our finding that ApoE4 but not ApoE3 selectively binds and inhibits VMAT2 provides an innovative insight into the molecular mechanism of why Tau pathology is initiated in the LC in AD patient brains. It is possible that an interaction of Tau with prion protein also impacts on the acceleration of Tau pathology spreading [23]. To define the contribution of AEP and Tau to these processes, we employed AEP and MAPT knockout mice, and found that ApoE4 lost its pro-inflammatory and neurodegenerative properties in the absence of these proteins (Fig. 4 & 5, Supplementary Fig. 4 & 6). We and others have reported that aberrant Tau can spread bidirectionally between the LC and forebrain [21, 29, 33] and we found that fibrillary Tau aggregates elicited by ApoE4 expression in the hippocampus propagated to the LC neurons (presumably via retrograde transport), resulting in noradrenergic neuron degeneration (Fig. 5d, Supplementary Fig. 6g & h). Hence, this exciting finding provides an innovative molecular mechanism underlying the development of pretangle Tau pathology in the LC neurons. This discovery provides an explanation for why ApoE4 promotes the early onset of AD and why Tau pathology in the LC is the earliest pathological event in the AD brains. Nonetheless, a few potential limitations remain in this study. For instance, there is no biochemical analysis in human brains for translating the finding, and only immunofluorescent or immunohistochemistry staining was conducted on the human brain sections. In addition, the size of human bio-specimen is quite small. However, the consistent and significant data from human staining analysis support the role of VMAT2 in AD pathogenesis. For Tau LC pathology, mainly MAPT and Tau P301S models were employed. In the future, models that incorporate both Tau and beta-amyloid pathologies (e.g. 3×Tg AD mice) will be valuable for further interrogate the role of ApoE4 on VMAT inhibition in AD. In addition, catecholaminergic SH-SY5Y cells and primary cortical neurons were employed to explore the molecular mechanisms, but do not faithfully recapitulate all features of central noradrenergic neurons. While technically challenging, extension of this research to primary LC neurons will yield additional information.
Fig. 7. Schematic model for the pathological role of ApoE4 through inhibiting VMAT2 in LC neurodegeneration.
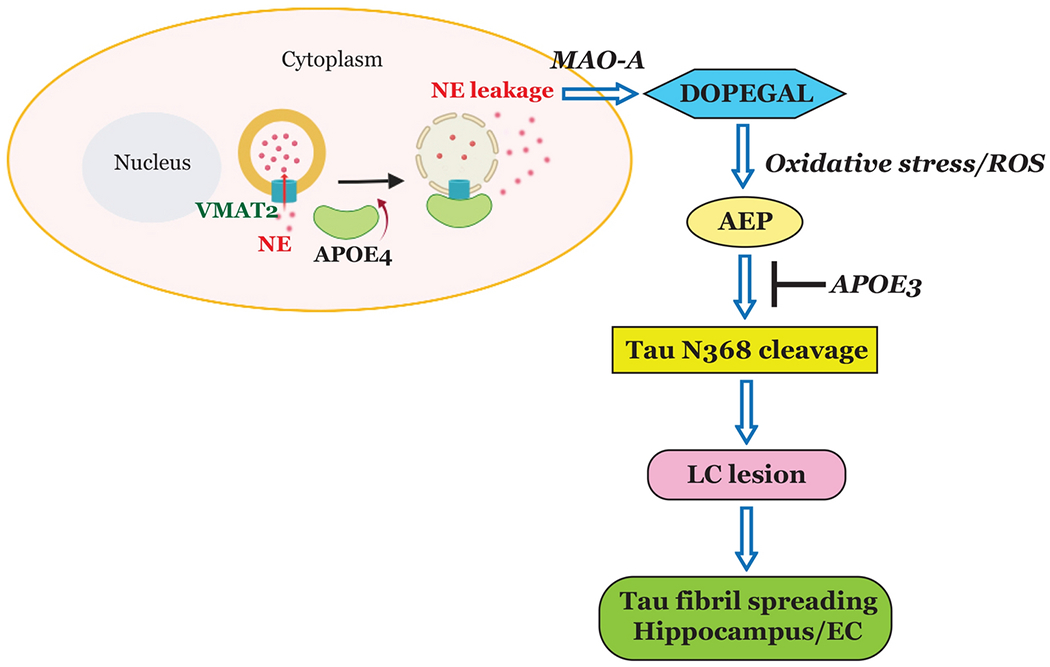
Our data support a model in which ApoE4 blocks VMAT2 and elevates DOPEGAL concentrations in the cytoplasm via NE leakage, which in turn activates AEP, exacerbates Tau toxicity, and prompts LC neurodegeneration. In contrast, ApoE3 binds to Tau and prevents Tau N368 cleavage by AEP.
More than half of AD patients carry at least one copy of ApoE4, and ApoE4 accelerates the onset of cognitive impairment by 7-9 years per allele [34, 41]. ApoE isoform-specific formation of soluble ApoE/Aβ complex modulates levels of neurotoxic Aβ, providing a basis for ApoE4-induced AD risk [28, 48]. Nevertheless, ApoE4 causes age-dependent learning and memory impairment in mice without amyloidopathy [22]. Moreover, Tau aggregates in AD brains and in primary tauopathies correlate more strongly with clinical signs and neurodegeneration than Aβ [2, 32, 51], and recent genome-wide association studies (GWAS) show a strong and significant association of ApoE with cerebrospinal fluid (CSF) Tau and p-Tau after correcting for the effect of APOE on Aβ42 levels [17]. These findings indicate that ApoE4 influences Tau-induced pathology in AD independent of Aβ. Most recently, Holtzman and colleagues showed that P301S/ApoE4 mice develop markedly more brain atrophy and neuroinflammation than P301S/ApoE2 or P301S/ApoE3 mice, whereas P301S/ApoE KO mice are largely protected from these changes [43]. However, it is unclear how ApoE4 enhances Tau neurotoxicity. Our data demonstrate that ApoE4, but not E3, associates with VMAT2 and inhibits its NE uptake in catecholaminergic SH-SY5Y cells. Accordingly, ApoE4 knockin mice show augmented oxidative stress, AEP activation, and Tau N368 cleavage in LC neurons compared to ApoE3 TR mice (Fig. 2). Employing a dominant-negative AEP (C189S) mutant, pharmacologic AEP inhibitor #11, and AEP-resistant Tau N255/368A mutant, we demonstrate that AEP cleavage of Tau N368 is indispensable for ApoE4-driven Tau neurotoxicity. Consequently, viral-mediated expression of ApoE4 but not E3 in the hippocampus of MAPT mice induces hippocampal atrophy, robust AEP activation and Tau N368 cleavage, and cognitive deficits that are substantially diminished in MAPT/AEP −/− mice (Fig. 4, Supplementary Fig. 4). ApoE4 fails to activate AEP and loses its pathogenic properties in MAPT −/− mice (Fig. 5), indicating that Tau is required for ApoE4 to exert its deleterious effects.
We recently reported that DOPEGAL, the NE metabolite produced by MAO-A oxidation, triggers AEP activation via oxidative stress, resulting in Tau N368 cleavage that is prone to hyperphosphorylation, aggregation, and propagation, and triggers noradrenergic neuronal death [33]. In addition, DOPEGAL, but not NE, facilitates the fibrillization of purified recombinant Tau. Tau hyperphosphorylation and N368 cleavage are age-dependently increased in the LC of both MAPT and 3×Tg mice, consistent with our previous finding that AEP is upregulated and activated in the brain during aging. Importantly, we made the same observations in the LC of human AD brains, indicating that active AEP contributes to Tau pathology in the LC [33]. We confirmed and expanded on those findings in the current study by demonstrating that TH and VMAT2 immunoreactivity are significantly reduced in the LC of AD brains and Tau P301S mice, while oxidative stress (as measured by 4-HNE) is elevated. This unstable environment facilitates the production of the toxic DOPEGAL metabolite, indicating that loss of VMAT2 excludes NE from vesicles and promotes its oxidation. In support of this model, VMAT2 LO mice exhibit age-dependent increases in DOPEGAL levels and LC degeneration (Fig. 1 and Supplementary Fig. 8). To interrogate the potential role of VMAT2 in ApoE4-triggered AD pathology, we knocked down VMAT2 in the LC region of MAPT mice in the presence of ApoE3 or E4. Depletion of VMAT2 significantly augmented DOPEGAL levels and AEP activity, leading to extensive LC neuronal degeneration and cognitive impairment in ApoE4 mice, while ApoE3 animals were protected. These adverse effects of ApoE4 were attenuated in MAPT −/− mice, even though DOPEGAL and AEP elevation persisted (Fig. 6 and Supplementary Fig. 7). These data strongly suggest that Tau is required for ApoE4-induced neuropathology and degeneration.
We next turned our attention to understanding the mechanism underlying ApoE3-mediated protection. In patients with frontotemporal dementia, a large percentage of whom have tauopathy, ε4 allele frequency is significantly elevated [38, 44], and ε4 carriers have greater atrophy in affected brain regions [1] and exacerbated behavioral deficits [20]. We reasoned that ApoE may directly influence Tau pathology and neurodegeneration. Interestingly, lipidated-ApoE, which is secreted from astrocytes, as well as recombinant ApoE have been shown to directly bind Tau in vitro [46, 56], and neuronal expression of human ApoE in vivo results in Tau hyperphosphorylation (E4 > E3) [8]. Strikingly, we found that ApoE3, but not E4, selectively binds to the repetitive microtubule binding domains (a.a. 256-368) of Tau. Remarkably, the ApoE3-Tau interaction protected Tau from AEP cleavage and reduced Tau N368 despite strong activation of AEP (Fig. 3, Supplementary Fig. 3), while ApoE4 was ineffective.
ApoE is the major apolipoprotein present in the CNS. ApoE is produced primarily by glia (astrocytes and microglia), although under certain conditions neuronal ApoE expression may occur [52]. In addition to directly facilitating Aβ internalization and aggregation, ApoE4 also alters brain cholesterol homeostasis by modifying lipoprotein-particle formation. In the brain, ApoE primarily directs the mobilization and redistribution of cholesterol during membrane remodeling associated with the plasticity of synapses [39]. Blocking Tau cleavage by AEP using an uncleavable Tau N255/368A mutant, we found that ApoE4-provoked neuronal cell death and AEP activities were attenuated (Fig. 3). It is worth noting that AEP activation by ApoE4 is substantially diminished in Tau −/− neurons as compared to wild-type neurons (Supplementary Fig. 5). This finding indicates that Tau feeds back and somehow mediates AEP activation, although the mechanism is unclear. Hence, these observations indicate that AEP and its proteolytic cleavage of Tau are necessary for mediating ApoE4’s pathological effects. Taken together, these data identify a novel neurochemical/molecular pathway involving NE metabolism and AEP that help explain the selective vulnerability of LC neurons for developing Tau pathology, and provide a potential mechanism underlying exacerbation of Tau toxicity by ApoE4 and the protection conferred by ApoE3. Our findings suggest that aberrant Tau pathology provoked by ApoE4-induced inhibition of VMAT2 in the LC may be a key early trigger in the progression of AD, and highlight the promise of AEP inhibition therapy.
Supplementary Material
Supplementary Fig. 1. ApoE4 selectively interacts with VMAT2. a. The binding between ApoE4 and VMAT2 was investigated in SH-SY5Y cells, which were co-transfected with GFPAPOE3, GFP-APOE4, and VMAT2 (3 more N number of Fig. 2A). Transfected cell lysates were immunoprecipitated with anti-GFP, and the precipitated proteins were analyzed by immunoblotting with anti-VMAT2. b. ApoE TR mouse brain cell lysates were immunoprecipitated with anti-ApoE and the precipitated proteins were analyzed by immunoblotting with anti-VMAT2 (3 more N number of Fig. 2L). c. The interaction between ApoE4 and VMAT2 was confirmed in LC of ApoE TR mice. The interacted proximity signals of anti-ApoE4 and anti-VMAT2 are shown in red, and the nuclei are shown in blue by Proximity ligation assay. Scale bar is 20 μm.
Supplementary Fig. 2. ApoE4 triggers AEP activation and Tau cleavage and phosphorylation. Primary neurons were cultured and infected with AAV-hTau, AAV-ApoE3 or ApoE4, and AAV-AEP or AEP C189S. a. Western blot analysis showed that ApoE4 overexpression induced Tau phosphorylation and Tau N368 cleavage with AEP overexpression, but not inactive AEP C189S (2 more N number of Fig. 3A). b. Western blot analysis showed that Tau and ApoE4-induced AEP activation, Tau phosphorylation, and Tau N368 cleavage were blocked by AEP inhibitor compound 11 (2 more N number of Fig. 3d). c. Western blot analysis showed that non-cleavable Tau N255A/N368A suppressed the effects of Tau and ApoE4 on AEP activation, Tau phosphorylation, and Tau N368 cleavage (2 more N number of Fig. 3g).
Supplementary Fig. 3. ApoE3 but not ApoE4 selectively binds to Tau and protects it from AEP cleavage. SH-SY5Y cells were co-transfected with ApoE3/E4 and GST-Tau. a. GST pull down and western blot analysis showed that ApoE3 but not ApoE4 bound to Tau and prevented AEP activation, Tau phosphorylation and Tau aggregation. b. LDH assay indicating Tau and ApoE4 mediated cell death. c. The activation of AEP was confirmed by the enzymatic assay. Data are shown as mean α SEM. N=3 per group. * p<0.05, ** p<0.01. HEK293 cells were co-transfected with GFP-ApoE3 and GST-Tau Fragments to investigate ApoE3’s binding domain in Tau. d. GST pull down analysis showed that ApoE3 bound to Tau in the repeat motifs (fragment 256-368) including AEP cleavage sites. e. Recombinant Tau (5 μg) was incubated with recombinant AEP (0.05 μg) and escalating doses of recombinant ApoE3/E4 (0.1, 0.5, and 1 μg). Western blot and densitometric quantification of cleaved Tau band showed that rTau cleavage by rAEP was attenuated by rApoE3. f. HEK293 cells were co-transfected with ApoE3/ApoE4 and Tau. Western data and densitometric analysis of cleaved Tau band presented the protective effect of ApoE3 in Tau cleavage by AEP. Data are shown as mean ± SEM. N=3 per group. * p<0.05, ** p<0.01.
Supplementary Fig. 4. AEP is required for mediating ApoE4-provoked AD pathologies in MAPT mice. AAV-ApoE3 or AAV-ApoE4 was injected into the hippocampus of MAPT/AEP WT or MAPT/AEP KO mice, and the pathologic events in the hippocampus for AD were measured by various analysis. a. ELISA analysis showed that Aβ40 and Aμ42 in the hippocampus were not changed by ApoE4 in MAPT mice. b. ELISA assay for IL-6, TNF-α, and IL-1β showed that inflammatory cytokines were increased in the hippocampus by ApoE4 in MAPT/AEP WT mice, but not MAPT/AEP KO mice. Data of ELISA assay in a & b are shown as mean ± SEM. N=6 per group. * p<0.05. c. Representative images of Golgi staining demonstrating the reduction of spines in CA1 region of ApoE4-injected MAPT/AEP WT mice. Scale bar is 5 μm. d. Quantification of dendritic spines in Golgi staining of CA1 region. Data are shown as mean ± SEM. N=6 per group. * p<0.05. e. Representative images by electron microscopy demonstrating the reduction of synapses (red arrows) in CA1 region of ApoE4-injected MAPT/AEP WT mice. Scale bar is 2 ¼m. f. Quantification of synapse number in electron microscopy images of CA1 region. Data are shown as mean ± SEM. N=6 per group. *pμ0.05.
Supplementary Fig. 5. Tau is required for ApoE4-induced AEP activation and neuronal cell death. Primary neurons from MAPT or Tau −/− mice brains were infected with AAV-ApoE3 or AAV-ApoE4. a. Western blot analysis showed that ApoE4 induced AEP activation, Tau aggregation, Tau phosphorylation, and Tau N368 cleavage in MAPT mice primary neurons. b. LDH assay demonstrated that the cell death by ApoE4 was mediated in a Tau-dependent manner. c. The activation of AEP was confirmed by enzymatic assay. d. ELISA assay showed that the change of Aβ40 content by ApoE4 was not affected by Tau. Data of b-d are shown as mean 넼 SEM. N=3 per group. * p<0.05, ** p<0.01.
Supplementary Fig. 6. Tau is required for ApoE4-triggered AD pathologies and LC neuronal loss. AAV-ApoE3 or AAV-ApoE4 was injected into the hippocampus of Tau −/− or MAPT mice, and pathologic events in the hippocampus for AD were assessed. a. ELISA analysis showed that Aβ40 and Aβ42 in the hippocampus were not changed by ApoE4 in MAPT mice. b. ELISA assay for IL-6, TNF-α, and IL-1β showed that inflammatory cytokines were increased in the hippocampus by ApoE4 in MAPT mice but not Tau −/− mice. Data of ELISA assay in a & b are shown as mean ± SEM. N=6 per group. * p<0.05. c. Representative images of Golgi staining demonstrating the reduction of spines in CA1 region of ApoE4-injected MAPT mice. Scale bar is 5 μm. d. Quantification of the dendritic spines in Golgi staining of CA1 region. Data are shown as mean ± SEM. N=6 per group. * p<0.05. e. Representative images by electron microscopy demonstrating the reduction of synapses (red arrows) in CA1 region of ApoE4-injected MAPT mice. Scale bar is 2 μm. f. Quantification of synapse number in electron microscopy images of CA1 region. Data are shown as mean ± SEM. N=6 per group. * p<0.05. g. Stereological cell counting of TH+ cells in the LC region of Fig. 5d showed LC neurodegeneration mediated by ApoE4 injection into the hippocampus of MAPT mice. h. Quantification of Tau N368+ cells and T22+ cells in the LC of Fig. 5d to show the retrograde spread of Tau pathologies. Data are shown as mean ± SEM. N=6 per group. * p<0.05.
Supplementary Fig. 7. ApoE4 inhibits VMAT2 and increases DOPEGAL and triggers cognitive impairments in MAPT mice. AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2 were injected into the LC of MAPT or Tau −/− mice, and then mice were assessed for Tau pathology and memory dysfunction 6 months later. a. Quantification of ApoE+ cells and VMAT2+ cells in the LC of Fig. 6A to verify virus infection. b. Representative images of T22 (red) and Thioflavin S immunofluorescence co-staining verifying Tau aggregation and fibrillization. Scale bar is 100 μm. c. Quantification of Thioflavin S/T22+ cells in the LC shows Tau fibrillization is mediated by ApoE4 overexpression and VMAT2 deficiency. d. MAO-A enzymatic assay in the LC for AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2-injected MAPT or Tau −/− mice. e. Measurement of DOPEGAL in the LC following ApoE4 overexpression and VMAT2 deficiency via HPLC analysis. Behavioral tests for Morris water maze (f-h) and fear conditioning test (i & j) demonstrating memory dysfunctions mediated by ApoE4 overexpression and VMAT2 deficiency in MAPT mice. Distance travelled to the platform (f), area under the curve for total distance travelled (g), and percent time spent in the quadrant (h) previously containing the platform during the probe trial in the Morris water maze. Percent time spent freezing during the cued fear (i) and contextual fear (j) tests following fear conditioning. All data were analyzed using two-way ANOVA and shown as mean ± SEM. N=6 per group. * <0.05, ** <0.01.
Supplementary Fig. 8. VMAT2 knockout elicits DOPEGAL production, AEP activation, and LC neuronal loss in an age-dependent manner. LC neurodegeneration and DOPEGAL concentrations in VMAT2 LO mice were examined by immunofluorescence staining, enzymatic analysis, and HPLC assay. a. Representative images of staining in LC sections of VMAT2 LO mice by TH (red), VMAT2 (green), and DAPI (blue). Scale bar is 100 μm. b. Quantification of TH + cells representing the slight but significant reduction of LC neurons at 12 months of age in VMAT2-LO mice. c & d. Enzymatic assays in LC of age-dependent VMAT2 LO mice for AEP activity (c) and MAO-A activity (d). e. Measurement of age-dependent increase of DOPEGAL in the LC of VMAT2-LO mice via HPLC analysis. Data of b-e were analyzed using two-way ANOVA and shown as mean ± SEM, N = 3 per group. * p<0.05, ** p < 0.01. f. Western blot analysis showed the change of endogenous protein expression for Tau and TH in LC tissue of VMAT2 LO mice.
Supplementary Fig. 9. The full western bolts used in Figures are presented.
Supplementary Fig. 10. The full western bolts used in Supplementary Figures are presented.
Supplementary Fig. 10. The full western bolts used in Supplementary Figures are presented.
Supplementary Table 1. Information of postmortem human patients for LC sections
Acknowledgements
We thank the Emory Goizueta Alzheimer’s Disease Research Center for postmortem human AD and healthy control samples. This study was supported in part by the Rodent Behavioral Core (RBC), Viral Vector Core, and HPLC Bioanalytical Core, which are subsidized by the Emory University School of Medicine and are part of the Emory Integrated Core Facilities.
Funding:
This work was supported by the NIH (R01AG051538; RF1 AG061175 to KY and DW). Additional support was provided by the Emory Neuroscience NINDS Core Facilities (P30NS055077). Further support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of Interest
The authors have declared that no conflict of interests exists.
References
- 1.Agosta F, Vossel KA, Miller BL, Migliaccio R, Bonasera SJ, Filippi M, Boxer AL, Karydas A, Possin KL, Gorno-Tempini ML (2009) Apolipoprotein E epsilon4 is associated with disease-specific effects on brain atrophy in Alzheimer’s disease and frontotemporal dementia. Proc Natl Acad Sci U S A 106: 2018–2022 Doi 10.1073/pnas.0812697106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42: 631–639 Doi 10.1212/wnl.42.3.631 [DOI] [PubMed] [Google Scholar]
- 3.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ et al. (1997) Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17: 263–264 Doi 10.1038/ng1197-263 [DOI] [PubMed] [Google Scholar]
- 4.Bernstein AI, Stout KA, Miller GW (2014) The vesicular monoamine transporter 2: an underexplored pharmacological target. Neurochem Int 73: 89–97 Doi 10.1016/j.neuint.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak H, Del Tredici K (2011) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121: 171–181 Doi 10.1007/s00401-010-0789-4 [DOI] [PubMed] [Google Scholar]
- 6.Braak H, Del Tredici K (2012) Where, when, and in what form does sporadic Alzheimer’s disease begin? Curr Opin Neurol 25: 708–714 Doi 10.1097/WCO.0b013e32835a3432 [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: 960–969 Doi 10.1097/NEN.0b013e318232a379 [DOI] [PubMed] [Google Scholar]
- 8.Brecht WJ, Harris FM, Chang S, Tesseur I, Yu GQ, Xu Q, Dee Fish J, Wyss-Coray T, Buttini M, Mucke L et al. (2004) Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci 24: 2527–2534 Doi 10.1523/JNEUROSCI.4315-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke WJ, Kristal BS, Yu BP, Li SW, Lin TS (1998) Norepinephrine transmitter metabolite generates free radicals and activates mitochondrial permeability transition: a mechanism for DOPEGAL-induced apoptosis. Brain Res 787: 328–332 Doi 10.1016/s0006-8993(97)01488-1 [DOI] [PubMed] [Google Scholar]
- 10.Burke WJ, Li SW, Chung HD, Ruggiero DA, Kristal BS, Johnson EM, Lampe P, Kumar VB, Franko M, Williams EA et al. (2004) Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. Neurotoxicology 25: 101–115 Doi 10.1016/S0161-813X(03)00090-1 [DOI] [PubMed] [Google Scholar]
- 11.Burke WJ, Li SW, Schmitt CA, Xia P, Chung HD, Gillespie KN (1999) Accumulation of 3,4-dihydroxyphenylglycolaldehyde, the neurotoxic monoamine oxidase A metabolite of norepinephrine, in locus ceruleus cell bodies in Alzheimer’s disease: mechanism of neuron death. Brain Res 816: 633–637 Doi 10.1016/s0006-8993(98)01211-6 [DOI] [PubMed] [Google Scholar]
- 12.Burke WJ, Schmitt CA, Gillespie KN, Li SW (1996) Norepinephrine transmitter metabolite is a selective cell death messenger in differentiated rat pheochromocytoma cells. Brain Res 722: 232–235 Doi 10.1016/0006-8993(96)00129-1 [DOI] [PubMed] [Google Scholar]
- 13.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C et al. (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3: 89ra57 Doi 10.1126/scitranslmed.3002156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caudle WM, Richardson JR, Wang MZ, Taylor TN, Guillot TS, McCormack AL, Colebrooke RE, Di Monte DA, Emson PC, Miller GW (2007) Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J Neurosci 27: 8138–8148 Doi 10.1523/JNEUROSCI.0319-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chalermpalanupap T, Schroeder JP, Rorabaugh JM, Liles LC, Lah JJ, Levey AI, Weinshenker D (2018) Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J Neurosci 38: 74–92 Doi 10.1523/JNEUROSCI.1483-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chartier-Harlin MC, Parfitt M, Legrain S, Perez-Tur J, Brousseau T, Evans A, Berr C, Vidal O, Roques P, Gourlet V et al. (1994) Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum Mol Genet 3: 569–574 [DOI] [PubMed] [Google Scholar]
- 17.Deming Y, Li Z, Kapoor M, Harari O, Del-Aguila JL, Black K, Carrell D, Cai Y, Fernandez MV, Budde J et al. (2017) Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol 133: 839–856 Doi 10.1007/s00401-017-1685-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djordjevic J, Roy Chowdhury S, Snow WM, Perez C, Cadonic C, Fernyhough P, Albensi BC (2020) Early Onset of Sex-Dependent Mitochondrial Deficits in the Cortex of 3×Tg Alzheimer’s Mice. Cells 9: Doi 10.3390/cells9061541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrenberg AJ, Nguy AK, Theofilas P, Dunlop S, Suemoto CK, Di Lorenzo Alho AT, Leite RP, Diehl Rodriguez R, Mejia MB, Rub U et al. (2017) Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathol Appl Neurobiol 43: 393–408 Doi 10.1111/nan.12387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Engelborghs S, Dermaut B, Marien P, Symons A, Vloeberghs E, Maertens K, Somers N, Goeman J, Rademakers R, Van den Broeck M et al. (2006) Dose dependent effect of APOE epsilon4 on behavioral symptoms in frontal lobe dementia. Neurobiol Aging 27: 285–292 Doi 10.1016/j.neurobiolaging.2005.02.005 [DOI] [PubMed] [Google Scholar]
- 21.Ghosh A, Torraville SE, Mukherjee B, Walling SG, Martin GM, Harley CW, Yuan Q (2019) An experimental model of Braak’s pretangle proposal for the origin of Alzheimer’s disease: the role of locus coeruleus in early symptom development. Alzheimers Res Ther 11: 59 Doi 10.1186/s13195-019-0511-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM et al. (2016) Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron 90: 740–751 Doi 10.1016/j.neuron.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S et al. (2019) Abeta-induced acceleration of Alzheimer-related tau-pathology spreading and its association with prion protein. Acta Neuropathol 138: 913–941 Doi 10.1007/s00401-019-02053-5 [DOI] [PubMed] [Google Scholar]
- 24.Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM (2007) Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 28: 327–335 Doi 10.1016/j.neurobiolaging.2006.02.007 [DOI] [PubMed] [Google Scholar]
- 25.Guillot TS, Miller GW (2009) Protective actions of the vesicular monoamine transporter 2 (VMAT2) in monoaminergic neurons. Mol Neurobiol 39: 149–170 Doi 10.1007/s12035-009-8059-y [DOI] [PubMed] [Google Scholar]
- 26.Harley CW (2007) Norepinephrine and the dentate gyrus. Prog Brain Res 163: 299–318 Doi 10.1016/S0079-6123(07)63018-0 [DOI] [PubMed] [Google Scholar]
- 27.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A et al. (2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41: 1088–1093 Doi 10.1038/ng.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holtzman DM (2001) Role of apoe/Abeta interactions in the pathogenesis of Alzheimer’s disease and cerebral amyloid angiopathy. J Mol Neurosci 17: 147–155 Doi 10.1385/JMN:17:2:147 [DOI] [PubMed] [Google Scholar]
- 29.Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM (2013) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33: 1024–1037 Doi 10.1523/JNEUROSCI.2642-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iba M, McBride JD, Guo JL, Zhang B, Trojanowski JQ, Lee VM (2015) Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol 130: 349–362 Doi 10.1007/s00401-015-1458-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Irizarry MC, Rebeck GW, Cheung B, Bales K, Paul SM, Holzman D, Hyman BT (2000) Modulation of A beta deposition in APP transgenic mice by an apolipoprotein E null background. Ann N Y Acad Sci 920: 171–178 Doi 10.1111/j.1749-6632.2000.tb06919.x [DOI] [PubMed] [Google Scholar]
- 32.Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Dickson DW et al. (2008) Beta-amyloid burden is not associated with rates of brain atrophy. Ann Neurol 63: 204–212 Doi 10.1002/ana.21223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang SS, Liu X, Ahn EH, Xiang J, Manfredsson FP, Yang X, Luo HR, Liles LC, Weinshenker D, Ye K (2020) Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J Clin Invest 130: 422–437 Doi 10.1172/JCI130513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leoni V (2011) The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer’s disease. Clin Chem Lab Med 49: 375–383 Doi 10.1515/CCLM.2011.088 [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Ye K, Weinshenker D (2015) Norepinephrine Protects against Amyloid-beta Toxicity via TrkB. J Alzheimers Dis 44: 251–260 Doi 10.3233/JAD-141062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahley RW (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240: 622–630 [DOI] [PubMed] [Google Scholar]
- 37.Mahley RW, Rall SC Jr. (2000) Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet 1: 507–537 Doi 10.1146/annurev.genom.1.1.507 [DOI] [PubMed] [Google Scholar]
- 38.Mishra A, Ferrari R, Heutink P, Hardy J, Pijnenburg Y, Posthuma D, International FTDGC (2017) Gene-based association studies report genetic links for clinical subtypes of frontotemporal dementia. Brain 140: 1437–1446 Doi 10.1093/brain/awx066 [DOI] [PubMed] [Google Scholar]
- 39.Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE (1994) Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 264: 850–852 Doi 10.1126/science.8171342 [DOI] [PubMed] [Google Scholar]
- 40.Payami H, Montee K, Grimslid H, Shattuc S, Kaye J (1996) Increased risk of familial late-onset Alzheimer’s disease in women. Neurology 46: 126–129 Doi 10.1212/wnl.46.1.126 [DOI] [PubMed] [Google Scholar]
- 41.Reitz C, Mayeux R (2010) Use of genetic variation as biomarkers for mild cognitive impairment and progression of mild cognitive impairment to dementia. J Alzheimers Dis 19: 229–251 Doi 10.3233/JAD-2010-1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rorabaugh JM, Chalermpalanupap T, Botz-Zapp CA, Fu VM, Lembeck NA, Cohen RM, Weinshenker D (2017) Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 140: 3023–3038 Doi 10.1093/brain/awx232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC et al. (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: 523–527 Doi 10.1038/nature24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevens M, van Duijn CM, de Knijff P, van Broeckhoven C, Heutink P, Oostra BA, Niermeijer MF, van Swieten JC (1997) Apolipoprotein E gene and sporadic frontal lobe dementia. Neurology 48: 1526–1529 Doi 10.1212/wnl.48.6.1526 [DOI] [PubMed] [Google Scholar]
- 45.Stratmann K, Heinsen H, Korf HW, Del Turco D, Ghebremedhin E, Seidel K, Bouzrou M, Grinberg LT, Bohl J, Wharton SB et al. (2016) Precortical Phase of Alzheimer’s Disease (AD)-Related Tau Cytoskeletal Pathology. Brain Pathol 26: 371–386 Doi 10.1111/bpa.12289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD (1994) Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci U S A 91: 11183–11186 Doi 10.1073/pnas.91.23.11183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sudhof TC (2004) The synaptic vesicle cycle. Annu Rev Neurosci 27: 509–547 Doi 10.1146/annurev.neuro.26.041002.131412 [DOI] [PubMed] [Google Scholar]
- 48.Tai LM, Mehra S, Shete V, Estus S, Rebeck GW, Bu G, LaDu MJ (2014) Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk. Mol Neurodegener 9: 2 Doi 10.1186/1750-1326-9-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor TN, Alter SP, Wang M, Goldstein DS, Miller GW (2014) Reduced vesicular storage of catecholamines causes progressive degeneration in the locus ceruleus. Neuropharmacology 76 Pt A: 97–105 Doi 10.1016/j.neuropharm.2013.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang ZH, Xia Y, Liu P, Liu X, Edgington-Mitchell L, Lei K, Yu SP, Wang XC, Ye K (2021) ApoE4 Activates C/EBPbeta/delta-secretase with 27-hydroxycholesterol, Driving the Pathogenesis of Alzheimer’s Disease. Prog Neurobiol: 102032 Doi 10.1016/j.pneurobio.2021.102032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams DR, Holton JL, Strand C, Pittman A, de Silva R, Lees AJ, Revesz T (2007) Pathological tau burden and distribution distinguishes progressive supranuclear palsy-parkinsonism from Richardson’s syndrome. Brain 130: 1566–1576 Doi 10.1093/brain/awm104 [DOI] [PubMed] [Google Scholar]
- 52.Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 26: 4985–4994 Doi 10.1523/JNEUROSCI.5476-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Z, Obianyo O, Dall E, Du Y, Fu H, Liu X, Kang SS, Song M, Yu SP, Cabrele C et al. (2017) Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nat Commun 8: 14740 Doi 10.1038/ncomms14740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Z, Song M, Liu X, Kang SS, Kwon IS, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang JZ et al. (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20: 1254–1262 Doi 10.1038/nm.3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Z, Song M, Liu X, Su Kang S, Duong DM, Seyfried NT, Cao X, Cheng L, Sun YE, Ping Yu S et al. (2015) Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat Commun 6: 8762 Doi 10.1038/ncomms9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, Heckman MG, Diehl NN, Shinohara M, Martens YA et al. (2018) APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat Commun 9: 4388 Doi 10.1038/s41467-018-06783-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. ApoE4 selectively interacts with VMAT2. a. The binding between ApoE4 and VMAT2 was investigated in SH-SY5Y cells, which were co-transfected with GFPAPOE3, GFP-APOE4, and VMAT2 (3 more N number of Fig. 2A). Transfected cell lysates were immunoprecipitated with anti-GFP, and the precipitated proteins were analyzed by immunoblotting with anti-VMAT2. b. ApoE TR mouse brain cell lysates were immunoprecipitated with anti-ApoE and the precipitated proteins were analyzed by immunoblotting with anti-VMAT2 (3 more N number of Fig. 2L). c. The interaction between ApoE4 and VMAT2 was confirmed in LC of ApoE TR mice. The interacted proximity signals of anti-ApoE4 and anti-VMAT2 are shown in red, and the nuclei are shown in blue by Proximity ligation assay. Scale bar is 20 μm.
Supplementary Fig. 2. ApoE4 triggers AEP activation and Tau cleavage and phosphorylation. Primary neurons were cultured and infected with AAV-hTau, AAV-ApoE3 or ApoE4, and AAV-AEP or AEP C189S. a. Western blot analysis showed that ApoE4 overexpression induced Tau phosphorylation and Tau N368 cleavage with AEP overexpression, but not inactive AEP C189S (2 more N number of Fig. 3A). b. Western blot analysis showed that Tau and ApoE4-induced AEP activation, Tau phosphorylation, and Tau N368 cleavage were blocked by AEP inhibitor compound 11 (2 more N number of Fig. 3d). c. Western blot analysis showed that non-cleavable Tau N255A/N368A suppressed the effects of Tau and ApoE4 on AEP activation, Tau phosphorylation, and Tau N368 cleavage (2 more N number of Fig. 3g).
Supplementary Fig. 3. ApoE3 but not ApoE4 selectively binds to Tau and protects it from AEP cleavage. SH-SY5Y cells were co-transfected with ApoE3/E4 and GST-Tau. a. GST pull down and western blot analysis showed that ApoE3 but not ApoE4 bound to Tau and prevented AEP activation, Tau phosphorylation and Tau aggregation. b. LDH assay indicating Tau and ApoE4 mediated cell death. c. The activation of AEP was confirmed by the enzymatic assay. Data are shown as mean α SEM. N=3 per group. * p<0.05, ** p<0.01. HEK293 cells were co-transfected with GFP-ApoE3 and GST-Tau Fragments to investigate ApoE3’s binding domain in Tau. d. GST pull down analysis showed that ApoE3 bound to Tau in the repeat motifs (fragment 256-368) including AEP cleavage sites. e. Recombinant Tau (5 μg) was incubated with recombinant AEP (0.05 μg) and escalating doses of recombinant ApoE3/E4 (0.1, 0.5, and 1 μg). Western blot and densitometric quantification of cleaved Tau band showed that rTau cleavage by rAEP was attenuated by rApoE3. f. HEK293 cells were co-transfected with ApoE3/ApoE4 and Tau. Western data and densitometric analysis of cleaved Tau band presented the protective effect of ApoE3 in Tau cleavage by AEP. Data are shown as mean ± SEM. N=3 per group. * p<0.05, ** p<0.01.
Supplementary Fig. 4. AEP is required for mediating ApoE4-provoked AD pathologies in MAPT mice. AAV-ApoE3 or AAV-ApoE4 was injected into the hippocampus of MAPT/AEP WT or MAPT/AEP KO mice, and the pathologic events in the hippocampus for AD were measured by various analysis. a. ELISA analysis showed that Aβ40 and Aμ42 in the hippocampus were not changed by ApoE4 in MAPT mice. b. ELISA assay for IL-6, TNF-α, and IL-1β showed that inflammatory cytokines were increased in the hippocampus by ApoE4 in MAPT/AEP WT mice, but not MAPT/AEP KO mice. Data of ELISA assay in a & b are shown as mean ± SEM. N=6 per group. * p<0.05. c. Representative images of Golgi staining demonstrating the reduction of spines in CA1 region of ApoE4-injected MAPT/AEP WT mice. Scale bar is 5 μm. d. Quantification of dendritic spines in Golgi staining of CA1 region. Data are shown as mean ± SEM. N=6 per group. * p<0.05. e. Representative images by electron microscopy demonstrating the reduction of synapses (red arrows) in CA1 region of ApoE4-injected MAPT/AEP WT mice. Scale bar is 2 ¼m. f. Quantification of synapse number in electron microscopy images of CA1 region. Data are shown as mean ± SEM. N=6 per group. *pμ0.05.
Supplementary Fig. 5. Tau is required for ApoE4-induced AEP activation and neuronal cell death. Primary neurons from MAPT or Tau −/− mice brains were infected with AAV-ApoE3 or AAV-ApoE4. a. Western blot analysis showed that ApoE4 induced AEP activation, Tau aggregation, Tau phosphorylation, and Tau N368 cleavage in MAPT mice primary neurons. b. LDH assay demonstrated that the cell death by ApoE4 was mediated in a Tau-dependent manner. c. The activation of AEP was confirmed by enzymatic assay. d. ELISA assay showed that the change of Aβ40 content by ApoE4 was not affected by Tau. Data of b-d are shown as mean 넼 SEM. N=3 per group. * p<0.05, ** p<0.01.
Supplementary Fig. 6. Tau is required for ApoE4-triggered AD pathologies and LC neuronal loss. AAV-ApoE3 or AAV-ApoE4 was injected into the hippocampus of Tau −/− or MAPT mice, and pathologic events in the hippocampus for AD were assessed. a. ELISA analysis showed that Aβ40 and Aβ42 in the hippocampus were not changed by ApoE4 in MAPT mice. b. ELISA assay for IL-6, TNF-α, and IL-1β showed that inflammatory cytokines were increased in the hippocampus by ApoE4 in MAPT mice but not Tau −/− mice. Data of ELISA assay in a & b are shown as mean ± SEM. N=6 per group. * p<0.05. c. Representative images of Golgi staining demonstrating the reduction of spines in CA1 region of ApoE4-injected MAPT mice. Scale bar is 5 μm. d. Quantification of the dendritic spines in Golgi staining of CA1 region. Data are shown as mean ± SEM. N=6 per group. * p<0.05. e. Representative images by electron microscopy demonstrating the reduction of synapses (red arrows) in CA1 region of ApoE4-injected MAPT mice. Scale bar is 2 μm. f. Quantification of synapse number in electron microscopy images of CA1 region. Data are shown as mean ± SEM. N=6 per group. * p<0.05. g. Stereological cell counting of TH+ cells in the LC region of Fig. 5d showed LC neurodegeneration mediated by ApoE4 injection into the hippocampus of MAPT mice. h. Quantification of Tau N368+ cells and T22+ cells in the LC of Fig. 5d to show the retrograde spread of Tau pathologies. Data are shown as mean ± SEM. N=6 per group. * p<0.05.
Supplementary Fig. 7. ApoE4 inhibits VMAT2 and increases DOPEGAL and triggers cognitive impairments in MAPT mice. AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2 were injected into the LC of MAPT or Tau −/− mice, and then mice were assessed for Tau pathology and memory dysfunction 6 months later. a. Quantification of ApoE+ cells and VMAT2+ cells in the LC of Fig. 6A to verify virus infection. b. Representative images of T22 (red) and Thioflavin S immunofluorescence co-staining verifying Tau aggregation and fibrillization. Scale bar is 100 μm. c. Quantification of Thioflavin S/T22+ cells in the LC shows Tau fibrillization is mediated by ApoE4 overexpression and VMAT2 deficiency. d. MAO-A enzymatic assay in the LC for AAV-ApoE3/ApoE4 and Lenti-Sh-VMAT2-injected MAPT or Tau −/− mice. e. Measurement of DOPEGAL in the LC following ApoE4 overexpression and VMAT2 deficiency via HPLC analysis. Behavioral tests for Morris water maze (f-h) and fear conditioning test (i & j) demonstrating memory dysfunctions mediated by ApoE4 overexpression and VMAT2 deficiency in MAPT mice. Distance travelled to the platform (f), area under the curve for total distance travelled (g), and percent time spent in the quadrant (h) previously containing the platform during the probe trial in the Morris water maze. Percent time spent freezing during the cued fear (i) and contextual fear (j) tests following fear conditioning. All data were analyzed using two-way ANOVA and shown as mean ± SEM. N=6 per group. * <0.05, ** <0.01.
Supplementary Fig. 8. VMAT2 knockout elicits DOPEGAL production, AEP activation, and LC neuronal loss in an age-dependent manner. LC neurodegeneration and DOPEGAL concentrations in VMAT2 LO mice were examined by immunofluorescence staining, enzymatic analysis, and HPLC assay. a. Representative images of staining in LC sections of VMAT2 LO mice by TH (red), VMAT2 (green), and DAPI (blue). Scale bar is 100 μm. b. Quantification of TH + cells representing the slight but significant reduction of LC neurons at 12 months of age in VMAT2-LO mice. c & d. Enzymatic assays in LC of age-dependent VMAT2 LO mice for AEP activity (c) and MAO-A activity (d). e. Measurement of age-dependent increase of DOPEGAL in the LC of VMAT2-LO mice via HPLC analysis. Data of b-e were analyzed using two-way ANOVA and shown as mean ± SEM, N = 3 per group. * p<0.05, ** p < 0.01. f. Western blot analysis showed the change of endogenous protein expression for Tau and TH in LC tissue of VMAT2 LO mice.
Supplementary Fig. 9. The full western bolts used in Figures are presented.
Supplementary Fig. 10. The full western bolts used in Supplementary Figures are presented.
Supplementary Fig. 10. The full western bolts used in Supplementary Figures are presented.
Supplementary Table 1. Information of postmortem human patients for LC sections