

Abstract
Nociception and opioid antinociception in females are pliable processes, varying qualitatively and quantitatively over the reproductive cycle. Spinal estrogenic signaling via membrane estrogen receptors (mERs), in combination with multiple other signaling molecules [spinal dynorphin, kappa-opioid receptors (KOR), glutamate and metabotropic glutamate receptor 1 (mGluR1)], appears to function as a master coordinator, parsing functionality between pronociception and antinociception. This provides a window into pharmacologically accessing intrinsic opioid analgesic/anti-allodynic systems. In diestrus, membrane estrogen receptor alpha (mERα) signals via mGluR1 to suppress spinal endomorphin 2 (EM2) analgesia. Strikingly, in the absence of exogenous opioids, interfering with this suppression in a chronic pain model elicits opioid anti-allodynia, revealing contributions of endogenous opioid(s). In proestrus, robust spinal EM2 analgesia is manifest but this requires spinal dynorphin/KOR and glutamate-activated mGluR1. Furthermore, spinal mGluR1 blockade in a proestrus chronic pain animal (eliminating spinal EM2 analgesia) exacerbates mechanical allodynia, revealing tempering by endogenous opioid(s). A complex containing mu-opioid receptor, KOR, aromatase, mGluRs, and mERα are foundational to eliciting endogenous opioid anti-allodynia. Aromatase-mERα oligomers are also plentiful, in a central nervous system region-specific fashion. These can be independently regulated and allow estrogens to act intracellularly within the same signaling complex in which they are synthesized, explaining asynchronous relationships between circulating estrogens and central nervous system estrogen functionalities. Observations with EM2 highlight the translational relevance of extensively characterizing exogenous responsiveness to endogenous opioids and the neuronal circuits that mediate them along with the multiplicity of estrogenic systems that concomitantly function in phase and out-of-phase with the reproductive cycle.
INTRODUCTION
Despite the discovery of endorphins dating back to the 1980s, little is known regarding the regulatory parameters governing the role of endogenous opioids in endogenous pain management. Thus, it is not surprising that the pharmacopeias contain virtually no drugs designed to act indirectly on pain control via their activation of endorphins. Until such time as the null hypothesis is proven, ie, that in situ, naturally occurring endorphins have little or no role in endogenous nociception and/or allodynia, the naturally occurring pool of opioids must be viewed as an as of yet untapped reservoir of analgesic/antiallodynic potential.
Effectively tapping into endogenous opioids for pain relief requires a somewhat detailed understanding of parameters that influence the in situ manifestation of the analgesic/antiallodynic functionality of endorphins, enabling them to act within a time frame commensurate with currently available narcotic and alternative pain-relieving drugs. Among the factors that have emerged to be relevant, central nervous system (CNS) estrogens and their receptors hold center stage along with glutamate, metabotropic glutamate receptors (mGluRs), dynorphin and the kappa-opioid receptor (KOR).
Estrogens:
Estrogenic signaling is a major determinant of opioid functionality. Pain and its relief by opioids vary over the reproductive cycle in both laboratory animals as well as humans.1–7 In fact, there is a prominent divergence in the employment of the highly selective endogenous μ-opioid receptor (MOR) ligand endomorphin 2 (EM2) in nociceptive processing in males vs. females, that tracks the estrous cycle, oscillating between analgesically active and inactive states (in proestrous and diestrous females, respectively). This is in stark contrast to stable analgesic responsiveness to intrathecal EM2 in males.7 Adding to this complexity, the predominant estrogen 17-β-estradiol (E2), is pronociceptive,8–13 and antinociceptive (analgesic),14–22 both occurring via multiple mechanisms.
Estrogens can act via classical nuclear estrogen receptors (ERs) that function as estrogen-activated transcription factors,23 or via membrane estrogen receptors (mERs) (that passage to the plasma membrane from the nucleus), eg, estrogen receptor α (ERα)/estrogen receptor β (ERβ)24,25 and, a G protein-coupled ER (GPR30).26–29 These mERs concentrate in caveolae subsequent to being palmitoylated, and activate membrane signaling sequelae.30–33 Armed with this knowledge, one can hypothesize a number of mechanisms that could mediate the fluctuation of the magnitude of intrathecal EM2 analgesia in proestrus and diestrus. These include: (1) augmented facilitation during proestrus, when plasma E2 levels are elevated, (2) alterations in the balance between estrogenic pronociception and antinociception, (3) disengagement of spinal ERs from EM2 analgesic responsiveness, etc., each of which would have differing implied therapeutic implications. Unexpectedly, however, an opposite relationship exists between estrogenic regulation of spinal EM2 analgesic responsiveness and peripheral levels of estrogens. This relationship is associated with stage of estrous cycle-temporally correlated interactions among components of a recently discovered signaling complex that is comprised of an oligomer containing MOR, KOR, mGluRs, ERα, GPR30 and aromatase (aka estrogen synthase).
Paradoxically, in females during proestrus, when circulating estrogens are at their highest, analgesic responsiveness to intrathecal EM2 is indistinguishable from males and are not influenced by blockers of either ERα, ERβ or GPR30.34 However, during diestrus, when peripheral estrogens are relatively low, blockade of spinal mERα or GPR30 restores spinal EM2 analgesia to that manifest in untreated proestrous female or male rats.34 Thus, relatively low levels of circulating estrogens in females are (temporally) associated with maximum estrogenic suppression of spinal EM2 analgesia, the neutralization of which could have substantial translational value in pain control. Additionally, this underscores the dichotomous relationship between spinal and circulating estrogens.
CNS synthesis of estrogens:
The spinal and supraspinal CNS contain a wide distribution of the enzyme aromatase, which enables CNS in situ synthesis of estrogens. Moreover, aromatase is present in many spinal areas involved in nociception/opioid antinociception.36–40 Estrogens intrinsic to the spinal cord are in fact essential to the suppression of intrathecal EM2 analgesia, as evidenced by the ability of the intrathecally applied aromatase inhibitor fadrozole to uncover spinal EM2 analgesia similar in robustness to the EM2 analgesia observed in proestrous female and male rats.34
Estrogens and ERs partner with mGluR1:
In addition to spinal mERs (mERα, GPR30) and aromatase, mGluR1 is also required for diestrous-associated suppression of spinal EM2 analgesia, but even this effect is inextricably linked to estrogenic signaling. During diestrus, the noncompetitive mGluR1 antagonist YM298198 rapidly (within 5 min) uncovers a robust spinal analgesic response to intrathecal EM2 that is neither distinguishable from that manifest in proestrus female and male rats34 nor from the response elicited by intrathecal EM2 during diestrus following mERα blockade or aromatase inhibition. The parallelism between effects of blocking mGluR1 and blockade of mERα/aromatase inhibition led to our hypothesizing that mERα acted to modify mGluR1 signaling by physically interacting with it, as has been described for other signaling proteins.41,42 In this scenario, the presence of mERα in mGluR1 spinal cord immunoprecipitate is significantly greater in immunoprecipitate obtained during diestrus than proestrus, consistent with mGluR1 collaborating with estrogenic-mERα signaling to suppress intrathecal EM2 analgesia during diestrus but not proestrus.
Parallelism between pharmacological and intrinsic regulation of spinal EM2 antinociception:
Notably, MOR, aromatase, mERα, and mGluR1 not only have wide distribution in the dorsal horn, but also are coexpressed and colocalize in or near the plasma membrane of neurons during diestrus.34 Furthermore, EM2-immunoreactive varicosities appose the dendrite of a MOR-immunoreactive neuron (in the lumbar region of the spinal cord) coexpressing mGluR1 and mERα. This provides a cellular context for spinal mERα, mGluR1 and estrogens not only to be synthesized within the same neuron, but also to negatively modulate intrathecal EM2 analgesic responsiveness during diestrus. Thus, estrous cycle stage-correlated targets are emerging (eg, mERs, mGluR1 aromatase) for magnifying opioid analgesic responsiveness to spinally applied EM2, and perhaps endogenously generated EM2 that have substantial potential translational value.
Stage of cycle-correlated emergence of signaling sequelae that facilitate spinal EM2 analgesia:
In addition to signaling molecules that are active in suppressing intrathecal EM2 analgesia, there are also signaling molecules that sustain spinal EM2 analgesia during proestrus; the emergence of robust intrathecal EM2 analgesia during proestrus does not result solely from the loss of aromatase-mERα-mGluR1 suppression (prominent in diestrus) of intrathecal EM2 analgesia, but also requires the emergence of alternative facilitative signaling. These include a switch from mERα to glutamate activation of mGluR1, and a critical requirement for threshold levels of spinal dynorphin release and KOR activation.43
Reciprocal relationship between intrathecal EM2 analgesic responsiveness and spinal.
Release of endogenous EM2:
Release of endogenous spinal EM2 is itself also tightly controlled over the rat estrous cycle. Notably, however, negative regulation of spinal EM2 release is mediated by estrogens and mERs. Additionally, in contrast to intrathecal EM2 analgesic responsiveness, it is robust during proestrus and much less so during diestrus (and absent during estrus.44 This parallels the highs and lows of peripheral estrogens, highest in proestrus (146.8–367 pM), lower in diestrus (up to 135.8 pM), reaching the nadir in estrus (down to 18.40 pM). Furthermore, underscoring the intricacy and complexity of this regulation, not only is the concomitant action of both mERα and GPR30 a prerequisite for suppression of i.t. EM2 analgesia,44 but both peripherally as well as spinally synthesized estrogens are required. This is evidenced by the ability of either ovariectomy or spinal aromatase inhibition to eliminate proestrous-associated estrogenic suppression of spinal EM2 release.44 The ability of estrogens to function as a biological lock on EM2 release is underscored by the inverse association between basal EM2 release and peripheral levels of estrogens. The mechanism(s) mediating combinatorial interactions between centrally and peripherally synthesized estrogens are not currently understood.
Endogenous biased agonism:
The difference in spinal EM2 analgesic responsiveness over the estrous/menstrual cycle is contingent on whether or not mGluR1 is activated by glutamate or mERα, as well as the ebb and flow in spinal dynorphin/KOR signaling. Suppressive vs. facilitative variation of spinal EM2 analgesia by mGluR1 signaling that depends on the endogenous activator of mGluR1 most likely reflects endogenous biased agonism—agonist-induced conformations of receptor that preferentially stimulate certain signaling pathways over others. This is potentially particularly relevant to signaling by mGluR1, since mGluR1 functionally associates with Gq,45 as well as Gs46,47 and Gi/o,48,49 the degree of this association being influenced by mERα vs. glutamate activation of mGluR1. Our present finding that spinal EM2 analgesia is both inhibited and facilitated by spinal mGluR1, depending on its activator, strongly suggests that ligand bias is not only relevant to exogenously applied agonists, as a pharmacological construct, but is, additionally, also likely to be an endogenous controlling mechanism. Interestingly, EM2 itself is reported to be a biased agonist at MOR.50,51
Endogenous spinal estrogenic signaling does not alter intrathecal EM2 analgesia during proestrus,43 indicating the disengagement of estrogens from causally associated underlying processes. However, paradoxically, acute spinal mGluR1 blockade (via intrathecal YM298198) (that reveals intrathecal EM2 analgesia during diestrus) actually abolishes spinal EM2 analgesia during proestrus. This indicates that the conversion from spinal EM2 non-analgesic to analgesic responsivity during diestrus and proestrus, respectively, results from the emergence of mGluR1 facilitative effects during proestrus that was not present during diestrus, in addition to the negation of suppressive mERα-mGluR1 modulation.
Relevance of the ebb and flow of spinal dynorphin/KOR signaling to intrathecal EM2 analgesia:
The ability of intrathecal EM2 to produce analgesia is determined by variability in spinal dynorphin release, repressed in diestrus but facilitated in proestrus. During proestrus, dynorphin release into spinal perfusate is augmented nearly 2-fold relative to that achieved during diestrus.43 This is consistent with our earlier pharmacological demonstration that spinal dynorphin/KOR activity is essential for female, but not male, intrathecal EM2 analgesic responsiveness.7 In fact, spinal dynorphin/KOR activity is a prerequisite for the ability of mERα/mGluR1 blockade to uncover intrathecal EM2 analgesic responsiveness during diestrus.43 Either intrathecal anti-dynorphin antibodies (30 min prior to EM2) or intrathecal norbinaltorphimine (norBNI; KOR-selective antagonist; 18 h prior to EM2 intrathecal treatments) abolished the intrathecal EM2 analgesia that emerged after blocking either spinal mERα or mGluR1.43 These data suggest that during diestrus, unmasking spinal EM2 analgesia by either spinal mERα or mGluR1 blockade results from disinhibiting spinal dynorphin release and KOR signaling, as well as facilitating signaling by glutamate/mGluR1, implying the prerequisite for threshold levels of their endogenous signaling activities for intrathecal EM2 analgesia to be manifest. This formulation is buttressed by the ability of intrathecal EM2 to produce analgesia during diestrus when rats are intrathecally pretreated (30 min prior to intrathecal EM2) with intrathecal dynorphin itself (3 nmol)35,43 and the facts that acute blockade of spinal glutamate release with intrathecal pretreatment with riluzole (glutamate release inhibitor52,53; 43 nmol, 1 hr) eliminated intrathecal proestrous-associated intrathecal EM2 analgesia, reducing it to levels observed in diestrus females,43 while blockade of glutamate transport (reuptake) (which enhances synaptic glutamate) unmasks spinal EM2 analgesia during diestrus (Liu and Gintzler, unpublished observations).
Anatomical organization of spinal EM2, mGluR1, ERα, glutamate and dynorphin; exogenous vs. intrinsic regulation:
Confocal imaging reveals that a thin shell of dynorphin-immunoreactivity envelops neurons, as we have previously described.54 Moreover, such neurons coexpress mGluR1 and ERα in or adjacent to the plasma membrane, as well as within the cell body. Importantly, these glutamate transport vesicles (VGLUT)-expressing (glutamatergic) terminals appose these neurons, affording a cellular basis for cycling among ERα-activated and glutamate-activated mGluR1 signaling over the estrous cycle. This organization permits modulation of spinal dynorphin release by glutamate, thereby coordinating glutamate activation of mGluR1 with dynorphin release, both of which are essential for spinal EM2 analgesia during proestrus.43
Do endogenous opioids mediate endogenous antinociception?
An abundance of evidence indicates that endogenous opioids (β-endorphins,55,56 endomorphins,57 dynorphins,58 and enkephalins)59 mediate placebo-induced analgesia.60–64 Moreover, they do so via the same neural mechanisms that mediate opioid analgesia produced by narcotics.65 This is underscored by reports that the magnitude of analgesia produced by synthetic opioids directly correlates with the magnitude of placebo-induced opioid analgesia.65 This portends that endogenous and exogenous opioids share common mechanisms and, furthermore, that harnessing endogenous opioid analgesia for chronic pain management is likely to have translational utility. Amazingly, spatially-directed expectation of pain relief not only produces endogenous opioid-mediated pain reduction,66 but does so only on the body part targeted by the expectation.67
Additional evidence that endogenous opioids are active as analgesics in situ include the following: (1) placebo-induced elimination of postoperative dental pain is abolished by opioid receptor block,60 which also augments clinical nociception61; (2) anticipation of pain relief itself activates human MOR62; (3) analgesia triggered by a placebo occurs concomitant with amplified endogenous opioid action68; (4) transcranial direct current stimulation-induced analgesia enhances MOR recruitment69; (5) tissue injury constitutively activates MOR, suppressing spinal nociception63; (6) endogenous opioid activity is elicited by transcranial magnetic stimulation resulting in opioid analgesia64; (7) opioid antagonists block analgesic effects of acupuncture and electroacupuncture.70–72 Moreover, responsiveness to opioid analgesics can predict the magnitude of opioid placebo responsiveness, underscoring shared opioid mechanisms and the likely clinical utility of eliciting endogenous analgesia for chronic pain management. These instances validate that harnessing endogenous opioids can be an effective analgesic strategy. However, they do not provide pharmacological targets for effectively doing so under commonly encountered clinical situations, nor the likely success in doing so.
Endogenous opioids and clinical pain control.
A critical question is whether or not endogenous opioid analgesia can be subject to pharmacological activation, within the time frame required for the opioid analgesia elicited by exogenous synthetic narcotics. The unleashing of intrathecal EM2 analgesia during diestrus by either (1) inhibiting spinal aromatase or (2) blocking spinal mERα, or (3) blocking spinal mGluR1, combined with the active facilitation of intrathecal EM2 analgesia during proestrus by spinal glutamate and dynorphin release/KOR activation,34,43 provides a roadmap for pharmacologically turning on CNS endogenous opioid analgesia (particularly that resulting from EM2), and assessing the potential clinical utility of doing so (Fig 1). Both are required if tapping into endogenous opioid analgesia is to fulfill its promise of a viable clinical alternative to synthetic prescription opioids, enabling access to the powerful analgesic properties of opioids while minimizing their abuse potential.
Fig 1.
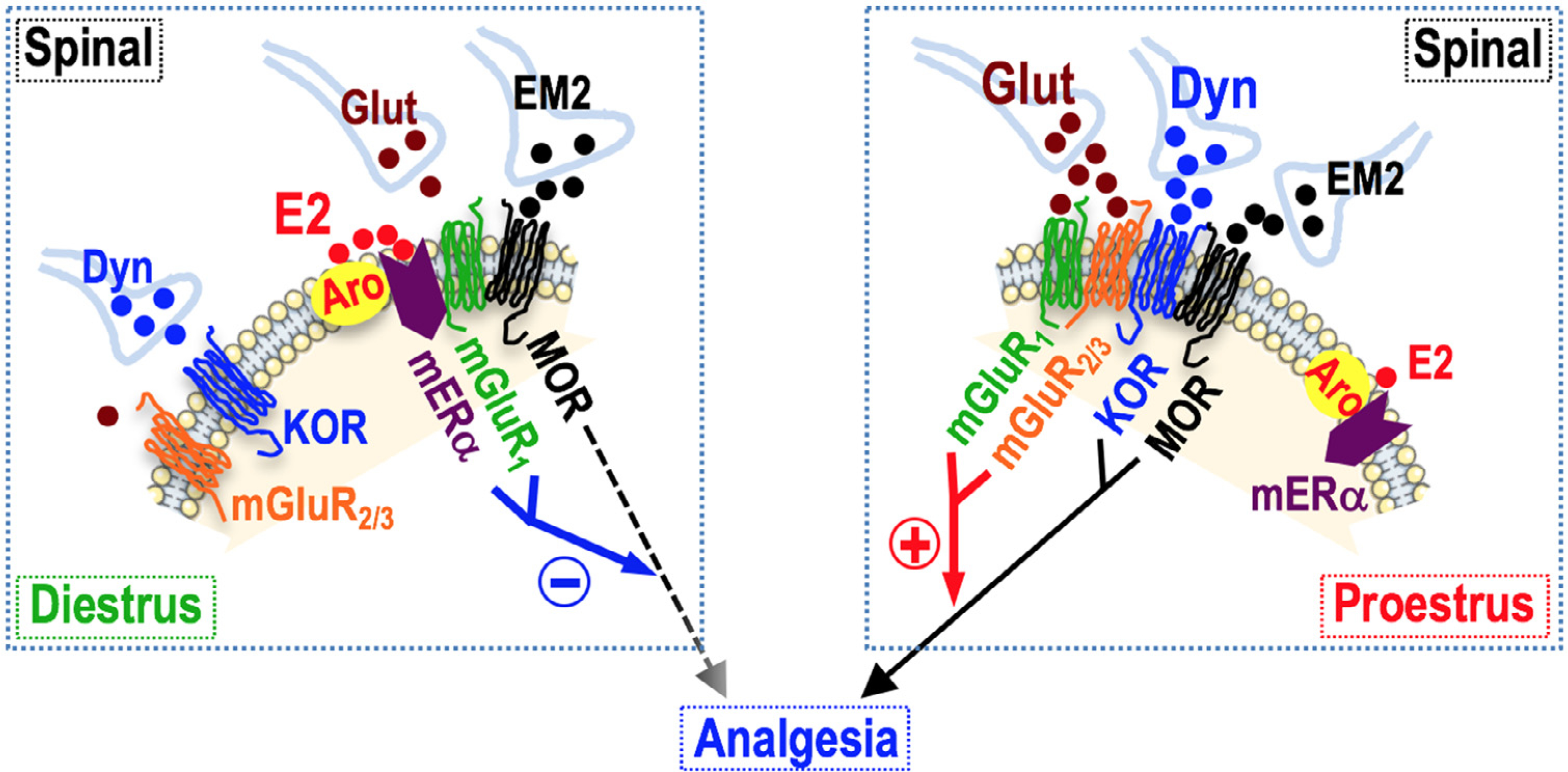
Analgesic responsiveness to spinal EM2 is governed by dynamic, pliable interactions among MOR, KOR, mGluR1, mGluR2/3, mERα and aromatase (Aro) within an oligomer that tracks the estrous cycle. In diestrus, E2 synthesized within the oligomer comprised of Aro-mERα-mGluR1-MOR stimulates mGluR1 via signaling by mERα to suppress analgesic responsiveness to intrathecal EM2 by inhibiting MOR signaling. Blockade of mERα/mGluR1 or inhibition of Aro neutralizes MOR inhibition, unmasking endogenous MOR-mediated (EM2) analgesia. The disconnection of suppressive mERα-mGluR1 signaling, the transition from mERα to glutamate (Glut) activation of mGluR1, which now signals in partnership with mGluR2/3, and augmented spinal Dyn/KOR signaling, which signals in collaboration with MOR in an oligomer of mGluR1-mGluR2/3-KOR-MOR that is different from that of diestrus, triggers the appearance of spinal EM2 analgesia. Inhibition of Glut release and thus a decrease in mGluR1/mGluR2/3 signaling activity eliminates the expression of endogenous spinal opioid analgesia, resulting in the worsening of allodynia in neuropathic pain rats. An organizational framework in which the spinal neurons coexpressing the pertinent signaling proteins (oligomerized therein) are in apposition to EM2-expressing, Dyn/Glut-containing varicosities likely underlies these observations. This organization would permit individual neurons to vary responsiveness to EM2 as a function of the ebb and flow of spinal Dyn and Glut signaling.
Understanding the on/off switch of intrathecal EM2 analgesic mechanisms could point the way for developing pharmacotherapies for manipulating endogenous EM2 activity for medicinal purposes. Plastic interactions within a membrane-bound oligomer that contains ERs, aromatase, mGluR1, mGluR2/3, MOR and KOR43 underlies estrous cycle-associated phasic changes in analgesic responsiveness to spinal EM2. As discussed above and shown in Fig 1, spinal cord contains an anatomical organization that permits endogenous interactions among modulatory components of EM2 analgesia analogous to those pharmacological treatments that ‘turn on and off’ analgesia elicited by the exogenous (intrathecally) applied EM2. Thus, pharmacologic perturbations that unveil analgesic responses to intrathecal EM2 during diestrus (eg, mERα/mGluR1 blockade, aromatase inhibition, inhibition of glutamate transport) would be expected to enhance endogenous spinal opioid analgesia. In analogous fashion, pharmacologic perturbations that sustain analgesic responses to intrathecal EM2 during proestrus (eg, glutamate activation of mGluR1, dynorphin release), would also be expected to undergird endogenous opioid analgesia, both sets of pharmacological perturbations producing effects in an estrous cycle-correlated fashion. Alternatively, whereas spinal mGluR1 blockade in diestrus would be expected to be analgesic/antiallodynic, the same treatment during proestrus would be expected to produce the opposite, be pronociceptive, ie, exacerbate nociception. Accordingly, intrathecal EM2 modulatory dynamics defined thus far establishes guardrails for translational forays into establishing the ‘reasonableness’ of pharmacologically tapping into intrinsic opioid systems for clinical pain relief in women.
Proof of principle that intrinsic opioid analgesic systems can be pharmacologically activated within a requisite time frame for clinical utility:
During physiologically quiescent conditions (ie, in the absence of nociception), endogenous opioid systems are dormant; opioid receptor block fails to alter basal nociceptive thresholds in laboratory animals73,74 and humans.75,76 However, nociceptive stimuli do produce endogenous opioid analgesia,60,77–80 indicating the ability of those stimuli to release endogenous opioids. This suggests the utility of using a chronic pain model to investigate whether or not pharmacological interventions that enhance analgesic responsiveness to exogenous, intrathecal EM2, also augment endogenous opioid analgesia. Accordingly, we utilized spinal nerve ligated diestrous rats, a known pain model to establish that opioid-mediated anti-allodynia could be provoked in the absence of exogenous opioids via the same pharmacological treatments that unveil spinal analgesic responsiveness to intrathecal EM2 in intact diestrous rats. Spinal nerve ligation (SpNL) was selected as the chronic pain model,81–83 since it augments release of endogenous opioids. This is reflected by the ability of spinal opioid receptor blockade to exacerbate mechanical allodynia,84 which is not observed in surgically naïve rats (Liu and Gintzler, Unpublished observations).
As we had predicted, the mechanical allodynia manifest by SpNL in diestrous rats, is markedly attenuated by either spinal aromatase inhibition or mERα/mGluR1 blockade (Table I). This is manifest only on the paw ipsilateral to SpNL and, importly, is eliminated by naloxone. The latter indicates endogenous opioid mediation, notwithstanding that no exogenous opioid had been administered. In other words, the opioid anti-allodynia resulted from harnessing the activity of an intrinsic opioid(s).
Table I.
Endogenous opioid blunting of nociceptive processing is regulated by dynamic interactions among signaling molecules
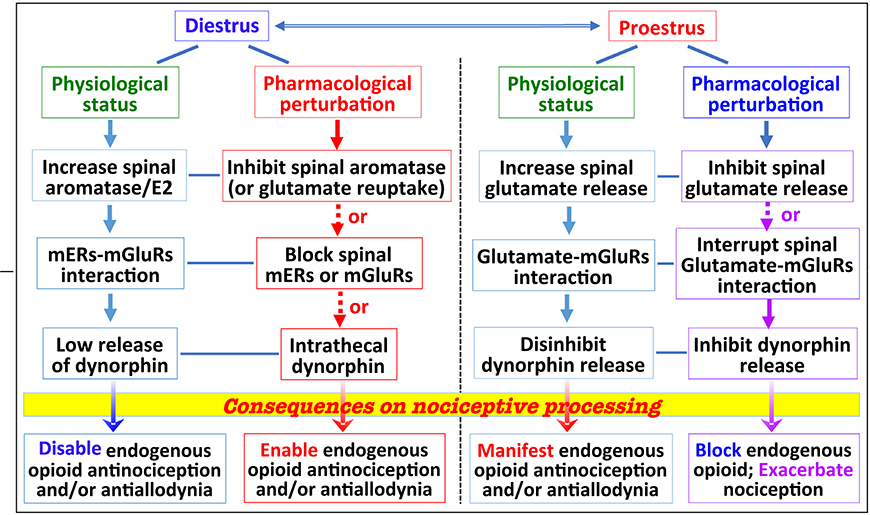 |
In diestrous female rats, interactions between mERs activated by spinally synthesized estrogen and mGluR1, combined with attenuated release of spinal dynorphin, actively suppress intrathecal EM2 analgesia, as well as the ability of endogenous spinal opioids to dampen the allodynia resulting from SpNL. This suppression can be overcome by enhancing spinal synaptic glutamate and/or spinal mGluR1 and KOR activity. The robust endogenous opioid dampening of SpNL-induced allodynia during proestrus requires spinal glutamate (in lieu of estrogen) activation of spinal mGluRs, as well as a threshold activation of spinal KOR by spinal dynorphin, whose release is augmented during proestrus. Linkage of endogenous opioid analgesia/antiallodynia to physiological state, and deciphering responsible molecular underpinning(s), holds out promise of defining pharmacological interventions to harness endogenous opioids for medicinal pain relief, while minimizing opioid abuse potential.
Asynchronous relationship between circulating estrogens and CNS estrogen functionalities:
Estrogenic signaling in the spinal cord is a crucial parameter influencing spinal EM2 analgesia. In diestrus, rapid signaling spinal mERs, activated by spinally synthesized estrogens, suppress spinal EM2/MOR analgesia, consistent with the presence of aromatase in many spinal areas involved in nociception and opioid analgesia.36,37,39 Surprisingly, the adverse effect of spinally synthesized estrogens on intrathecal EM2 analgesia occurs during diestrus,34 when circulating levels of estrogens are relatively low, not during proestrus, when systemic levels of estrogens are at their highest. This enigmatic relationship between spinal and peripheral estrogens indicates that cycle stage does not always dependably forecast the magnitude of estrogenic signaling in the CNS, informing attempts to modulate CNS estrogenic signaling for medicinal purposes. Furthermore, this inverse relationship is likely a basis for many inconsistent findings regarding nociception and opioid antinociception over the estrous and menstrual cycle,85 often a major confound in investigating the male-female dichotomy in pain, as well as pain management in women.
Existing data supports the existence of at least 2 estrogenic systems, one in the CNS and one, ovarian-based, in the periphery. The interrelationships between these estrogenic systems are mostly unexplored but have been the subject of much speculation.86 Peripheral estrogens reach the CNS by penetrating the blood brain barrier and diffusing from cerebrospinal fluid to extracellular fluid87 and sites of action. Such estrogens likely act directly on spinal ERs and might be expected to produce a generalized stimulation of CNS mERs, trivializing the functionality of estrogens synthesized in the CNS. However, systemic estrogens do not have unhampered access to all CNS ERs, the accessibility being influenced by the activity/distribution of estrogen-metabolizing enzymes,88–93 estrogen binding proteins, etc. In keeping with restricted access of systemic estrogens to CNS sites of action, some signaling by estrogens in the CNS is strikingly out-of-phase with peripheral concentrations of estrogens,34 which would not be expected if systemic estrogens had unrestricted access to CNS ERs.
In parallel with systemic estrogens, CNS estrogens are synthesized by spinal aromatase near synaptic structures and also stimulate proximal ERs.36,37,39,40 Spinal cord estrogens act in-phase as well as out-of-phase with peripheral estrogens. Since diffusion of centrally synthesized estrogens is highly spatially restricted,86,94–96 it is not improbable that CNS-and ovarian-derived estrogens not only activate different populations of spinal ERs that are either functionally convergent or parallel and independent, but also have variable temporality. Furthermore, the proclivity of membrane aromatase to physically pair with or exist separately from membrane estrogen receptor a97 creates 2 populations of aromatase, each of which themselves can be in-phase and/or out-of-phase with the ebb and flow of estrogens across the estrous cycle.
Our studies compare two well-characterized endocrinological states, diestrus and proestrous (the longest estrous cycle stages facilitating behavioral analyses) in order to generate two relative homogeneous populations of animals as well as to have markers of estrogenic activity in the periphery. However, direct causality between stage of cycle and endogenous spinal EM2 functionality is not, necessarily to be inferred. Consequently, given the often dichotomous relationship between central and peripheral estrogenic activity, further refinement correlating spinal EM2 analgesic functionality with additional estrous cycle stages (eg, metestrus, estrus) have not been pursued. Similarly, in this review, we do not provide a comparison between stage of estrous and menstrual cycle circulating estrogen levels since points of intersection between central and peripheral estrogens across rodents and primates are not understood and thus stage of cycle comparisons could prove very misleading regarding CNS analgesic functionality.
In rat, continuing the parallel between the ebb and flow of intrathecal EM2 analgesic responsiveness and stage of cycle, in proestrus, mGluR1 antagonism, which abolishes intrathecal EM2 analgesia in untreated proestrous rats, markedly aggravated the SpNL-induced mechanical allodynia of the ipsilateral paw in proestrous rats (Table I).84 Temporal correlation of pain management outcome on stage of reproductive cycle has substantial translational consequences, mandating that stage of menstrual cycle be tracked and considered when employing pharmacotherapies to tap into endogenous opioid analgesia in women of childbearing age. This is particularly important since pharmacotherapies that are antinociceptive in one stage can be pronociceptive in another.
Oligomerization of aromatase and mERα:
As part of investigating mechanisms responsible for the ups and downs of intrathecal EM2 analgesia, we discovered that aromatase and mERα oligomerize to form signaling complexes.34 This enables a novel modality of estrogenic signaling that we termed ‘oligocrine’, the ability of estrogens to perform as intracellular messengers, which are synthesized and act within the same macromolecular signaling complex.34 From a translational perspective, it can be important to note that these complexes can be independently regulated,34 constituting a molecular structure mediating the differential influence of stage of reproductive cycle on nociception and opioid analgesia.
As an exemplar, variable connection of cycle stage with the activity of discrete subpopulations of estrogenic signaling complexes34 could underlie inconstant nociception/opioid antinociception across the reproductive cycle.1–7 Potential translational relevance of discrete subpopulations of mERα-aromatase signaling complexes is bolstered by the fact that the extent of the oligomerization between aromatase and mERα vary in spinal cord (that has predominantly neural functionalities) and hypothalamus that has both endocrine and neural functionalities. Specifically, in spinal cord, regardless of reproductive cycle stage, virtually all membrane aromatase is oligomerized with mERα.97 In contrast, only ≈15% is oligomerized in hypothalamus.97 Furthermore, the prevalence of non-mERα-associated membrane aromatase in hypothalamus, in combination with the fact that numerous hypothalamic aromatase immunoreactive neurons are retrogradely labeled with peripherally injected Fluoro-Gold97 (ie, extend outside of the blood brain barrier), implies that some estrogens are secreted from the hypothalamus, possibly to regulate pituitary function, which could be exploited for medicinal purposes. The occurrence of membrane aromatase and mERα associated and non-associated subpopulations in the CNS holds out the promise that their selective targeting restores impaired estrogen-dependent CNS functionalities while curtailing undesirable effects.
Oligocrine estrogenic signaling also provides a structural context for fluid relationships of cycle stage-associated peripheral estrogens with estrogenic functionalities. For example, in proestrus, spinal aromatase and mERα activity are both essential for MOR-KOR heterodimerization,98 the analgesic form of KOR.99 In stark contrast, whereas spinal aromatase/mERα activity does not modulate spinal EM2 analgesia in proestrus, it does during diestrus.34 These variances infer that separate populations of locally synthesized estrogens independently activate discrete pools of spinal mERα. This realization underscores the imperative to be cautious when associating particular CNS functionalities with circulating ovarian steroid levels, and to consider cycle stage as a covariate in females of childbearing age in all investigations of nociception, as well as opioid analgesia and translational forays therein.
In many ways, the opioid field developed backwards. We extensively characterized narcotic drugs and the pharmacology of the receptors it was theorized they acted upon before we were able to appreciate the endogenous ligands whose effects they were mimicking. As a result, salient functional characteristics of narcotic drugs were thought to apply, often inappropriately, to endogenous opioids. We now know that this was overly simplistic. A poignant example of such differences is the characteristics of the spinal analgesia produced by EM2100-103 versus the intrathecal opioid analgesia resulting from the decidedly selective MOR-selective agonists sufentanil or [D-Ala2,N-Me-Phe4,Gly-ol5]-enkephalin (DAMGO). Intrathecal application of either sufentanil or DAMGO produces strong analgesia in both female (irrespective of the stage of estrous cycle) and male rats that is naloxone-reversible and does not oscillate between analgesically active and inactive states,7 as does intrathecal EM2. Additionally, mechanisms underlying sufentanil or DAMGO spinal opioid analgesia vary from those utilized by EM2, notwithstanding that all 3 MOR ligands are highly selective for their targeted receptor. Sufentanil and DAMGO act entirely via MOR to produce analgesia, irrespective of sex or stage of cycle, whereas EM2 requires spinal dynorphin and KOR signaling, concomitant with MOR activation, to produce analgesia in proestrous female rats.7 These variances are consistent with the fact that spatiotemporal activation patterns of opioid receptors are differentially produced by native opioid peptides and narcotics drugs.104 Such observations profile the translational relevance of extensive characterization of exogenous responsiveness to endogenous opioids and the neuronal circuits that mediate them when attempting to develop pharmacodynamic therapies designed to harness intrinsic opioid analgesic systems for acute and chronic pain management.
Pain management in the era of the epidemic of prescription opioid abuse:
Treating chronic pain is confounded by concerns regarding addiction to and misuse of opioid analgesics. The persisting quandary is how to continue benefiting from the unparalleled pain-mitigating properties of opioids while Eradicating addiction to and misapplication of opioids, ie, balancing the social command to equalize the urgency to allay the unrelenting opioid misuse epidemic with the ethical necessity to heal and effectively manage pain. The pharmacological harnessing of endogenous opioids for targeted pain relief may hold promise for pain management by allowing utilization of opioids while avoiding confounds of addiction and prescription opioid misuse.
Undermanaging pain has substantial deleterious functional consequences:
The present environment encourages the implementation and enforcement of policies that rigidly restrain the medical employment of synthetic opioids for pain management. As well intentioned as that might be, it is critical to understand that inadequately managed pain is itself a health risk. Uncontrolled or poorly managed pain has been reported to alter brain structure and function, modifying the functional connectivity of cortical regions105 and decreasing gray matter in pain-transmitting areas,106 prefrontal cortex and thalamus.107 In addition to these physical deleterious consequences, chronic pain has been associated with poor sleep, depression and anxiety,108 impaired emotional decision-making109 and diminished motivation (via long-term depression in the nucleus acumbens).110
CONCLUSION
The catalogue of negative effects of poorly managed pain emphasizes that just saying no to the medicinal use of synthetic opioids for pain management is not an acceptable way to get a handle on the opioid misuse crisis that is plundering society. A vital problem demanding consideration is how to resolve the medical and ethical necessities to assuage chronic pain with the ongoing rampant opioid crisis. Endogenous opioids represent an untapped reservoir of less easily abused opioids. Utilization of the newfound complexities of CNS estrogenic functionalities and the ability of glutamate/mGluR1 and dynorphin/KOR to sustain endogenous opioid analgesia (during proestrus) could bring drugs that act to harness endogenous opioids into the mainstream pharmacopeia, expanding effective pain management options.
The biological underpinnings for the strikingly severity and frequency of chronic pain in women than men remain an enigma. Notably, while the flexibility of EM2 utilization may contribute to women’s elevated risk of developing chronic pain conditions, it also provides an opportunity for medicinal intervention. CNS mechanisms underlying any cooperative/synergistic actions of central and peripheral estrogens could represent novel pharmacological targets for ameliorating pain in women, facilitating enhanced utilization of endogenous EM2. Given the variable in-phase and out-of-phase nature of estrogenic functionalities with stage of cycle, it appears that generalizations across estrous and menstrual cycling humans would have to be empirically determined.
ACKNOWLEDGMENTS
Funding: Supported by a grant from the National Institute on Drug Abuse, R01DA043774 to A.R.G. and N.-J.L.
Abbreviation:
- Aro
aromatase
- CNS
central nervous system
- DAMGO
D-Ala2,N-Me-Phe4, Gly-ol5]-enkephalin
- E2
17-β-estradiol
- EM2
endomorphin 2
- ERβ
estrogen receptor beta
- ERs
estrogen receptors
- Glut
glutamate
- hr
hour
- KOR
kappa-opioid receptors
- mERα
membrane estrogen receptor alpha
- mERs
membrane estrogen receptors
- mGluRs
metabotropic glutamate receptors
- mGluR1
metabotropic glutamate receptor 1
- MOR
μ-opioid receptor
- norBNI
norbinaltorphimine
- SpNL
spinal nerve ligation
- VGLUT
vesicular glutamate transport vesicles
Footnotes
Conflicts of interest: Authors declare that there are no conflicts of interest or competing financial/nonfinancial interests to disclose. We confirm that this work is original and has not been published as peer reviewed material elsewhere, nor is it currently under consideration for publication elsewhere.
The manuscript was written by the authors without external editorial support. All authors have read the journal’s authorship statement. The manuscript was reviewed and approved by all named authors.
The paper conforms to the relevant ethical guidelines for human and animal research.
All authors have read the journal’s policy on disclosure of potential conflicts of interest, and declare no potential conflicts of interest for this work.
REFERENCES
- 1.Fillingim RB, King CD, Ribeiro-Dasilva MC, et al. Sex, gender, and pain: a review of recent clinical and experimental findings. J Pain 2009;10:447–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fillingim RB, Gear RW. Sex differences in opioid analgesia: clinical and experimental findings. Eur J Pain 2004;8:413–25. [DOI] [PubMed] [Google Scholar]
- 3.Craft RM. Sex differences in opioid analgesia: “from mouse to man”. Clin J Pain 2003;19:175–86. [DOI] [PubMed] [Google Scholar]
- 4.Teepker M, Peters M, Vedder H, et al. Menstrual variation in experimental pain: correlation with gonadal hormones. Neuropsychobiology 2010;61:131–40. [DOI] [PubMed] [Google Scholar]
- 5.Ibironke GF, Aji KE. Pain threshold variations in female rats as a function of the estrus cycle. Niger J Physiol Sci 2011;26:67–70. [PubMed] [Google Scholar]
- 6.Mogil JS, Chesler EJ, Wilson SG, et al. Sex differences in thermal nociception and morphine antinociception in rodents depend on genotype. Neurosci Biobehav Rev 2000;24:375–89. [DOI] [PubMed] [Google Scholar]
- 7.Liu NJ, Gintzler AR. Spinal endomorphin 2 antinociception and the mechanisms that produce it are both sex- and stage of estrus cycle-dependent in rats. J Pain 2013;14(11):1522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu YC, Chen CW, Wang SY, et al. 17Beta-estradiol mediates the sex difference in capsaicin-induced nociception in rats. J Pharmacol Exp Ther 2009;331:1104–10. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Fan X, Warner M, et al. Ablation of estrogen receptor alpha or beta eliminates sex differences in mechanical pain threshold in normal and inflamed mice. Pain 2009;143:37–40. [DOI] [PubMed] [Google Scholar]
- 10.Ji Y, Murphy AZ, Traub RJ. Estrogen modulates the visceromotor reflex and responses of spinal dorsal horn neurons to colorectal stimulation in the rat. J Neurosci 2003;23:3908–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji Y, Tang B, Traub RJ. Spinal estrogen receptor alpha mediates estradiol-induced pronociception in a visceral pain model in the rat. Pain 2011;152:1182–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanoja R, Cervero F. Estrogen-dependent abdominal hyperalgesia induced by ovariectomy in adult mice: a model of functional abdominal pain. Pain 2005;118:243–53. [DOI] [PubMed] [Google Scholar]
- 13.Bradshaw H, Miller J, Ling Q, et al. Sex differences and phases of the estrous cycle alter the response of spinal cord dynorphin neurons to peripheral inflammation and hyperalgesia. Pain 2000;85:93–9. [DOI] [PubMed] [Google Scholar]
- 14.Fischer L, Torres-Chavez KE, Clemente-Napimoga JT, et al. The influence of sex and ovarian hormones on temporomandibular joint nociception in rats. J Pain 2008. [DOI] [PubMed] [Google Scholar]
- 15.Lawson KP, Nag S, Thompson AD, et al. Sex-specificity and estrogen-dependence of kappa opioid receptor-mediated antinociception and antihyperalgesia. Pain 2010;151:806–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sarajari S, Oblinger MM. Estrogen effects on pain sensitivity and neuropeptide expression in rat sensory neurons. Exp Neurol 2010;224:163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kramer PR, Bellinger LL. The effects of cycling levels of 17beta-estradiol and progesterone on the magnitude of temporomandibular joint-induced nociception. Endocrinology 2009;150:3680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aloisi AM, Affaitati G, Ceccarelli I, et al. Estradiol and testosterone differently affect visceral pain-related behavioural responses in male and female rats. Eur J Pain 2010;14:602–7. [DOI] [PubMed] [Google Scholar]
- 19.Riley JL 3rd, Robinson ME, Wise EA, et al. A meta-analytic review of pain perception across the menstrual cycle. Pain 1999;81:225–35. [DOI] [PubMed] [Google Scholar]
- 20.Giamberardino MA, Affaitati G, Valente R, et al. Changes in visceral pain reactivity as a function of estrous cycle in female rats with artificial ureteral calculosis. Brain Res 1997;774:234–8. [DOI] [PubMed] [Google Scholar]
- 21.Mannino CA, South SM, Quinones-Jenab V, et al. Estradiol replacement in ovariectomized rats is antihyperalgesic in the formalin test. J Pain 2007;8:334–42. [DOI] [PubMed] [Google Scholar]
- 22.Cao DY, Ji Y, Tang B, et al. Estrogen receptor beta activation is antinociceptive in a model of visceral pain in the rat. J Pain 2012;13:685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev 1999;20:358–417. [DOI] [PubMed] [Google Scholar]
- 24.Razandi M, Pedram A, Greene GL, et al. Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol 1999;13:307–19. [DOI] [PubMed] [Google Scholar]
- 25.Levin ER. G protein-coupled receptor 30: estrogen receptor or collaborator? Endocrinology 2009;150:1563–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bonini JA, Anderson SM, Steiner DF. Molecular cloning and tissue expression of a novel orphan G protein-coupled receptor from rat lung. Biochem Biophys Res Commun 1997;234:190–3. [DOI] [PubMed] [Google Scholar]
- 27.Carmeci C, Thompson DA, Ring HZ, et al. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997;45:607–17. [DOI] [PubMed] [Google Scholar]
- 28.Feng Y, Gregor P. Cloning of a novel member of the G protein-coupled receptor family related to peptide receptors. Biochem Biophys Res Commun 1997;231:651–4. [DOI] [PubMed] [Google Scholar]
- 29.Takada Y, Kato C, Kondo S, et al. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun 1997;240:737–41. [DOI] [PubMed] [Google Scholar]
- 30.Micevych P, Dominguez R. Membrane estradiol signaling in the brain. Front Neuroendocrinol 2009;30:315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mermelstein PG. Membrane-localised oestrogen receptor alpha and beta influence neuronal activity through activation of metabotropic glutamate receptors. J Neuroendocrinol 2009;21:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasudevan N, Pfaff DW. Non-genomic actions of estrogens and their interaction with genomic actions in the brain. Front Neuroendocrinol 2008;29:238–57. [DOI] [PubMed] [Google Scholar]
- 33.Vasudevan N, Pfaff DW. Membrane-initiated actions of estrogens in neuroendocrinology: emerging principles. Endocr Rev 2007;28:1–19. [DOI] [PubMed] [Google Scholar]
- 34.Liu NJ, Murugaiyan V, Storman EM, et al. Estrogens synthesized and acting within a spinal oligomer suppress spinal endomorphin 2 antinociception: ebb and flow over the rat reproductive cycle. Pain 2017;158:1903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang D, Zeng J, Li Q, et al. Contribution of adrenomedullin to the switch of G protein-coupled mu-opioid receptors from Gi to Gs in the spinal dorsal horn following chronic morphine exposure in rats. Br J Pharmacol 2016;173:1196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evrard HC, Balthazart J. Rapid regulation of pain by estrogens synthesized in spinal dorsal horn neurons. J Neurosci 2004;24:7225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evrard HC. Estrogen synthesis in the spinal dorsal horn: a new central mechanism for the hormonal regulation of pain. Am J Physiol Regul Integr Comp Physiol 2006;291:R291–9. [DOI] [PubMed] [Google Scholar]
- 38.Hojo Y, Hattori TA, Enami T, et al. Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017alpha and P450 aromatase localized in neurons. Proc Natl Acad Sci U S A 2004;101:865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naftolin F, Horvath TL, Jakab RL, et al. Aromatase immunoreactivity in axon terminals of the vertebrate brain. An immunocytochemical study on quail, rat, monkey and human tissues. Neuroendocrinology 1996;63:149–55. [DOI] [PubMed] [Google Scholar]
- 40.Peterson RS, Yarram L, Schlinger BA, et al. Aromatase is presynaptic and sexually dimorphic in the adult zebra finch brain. Proc Biol Sci 2005;272:2089–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kahlert S, Nuedling S, van Eickels M, et al. Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem 2000;275:18447–53. [DOI] [PubMed] [Google Scholar]
- 42.Razandi M, Pedram A, Park ST, et al. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem 2003;278:2701–12. [DOI] [PubMed] [Google Scholar]
- 43.Liu NJ, Murugaiyan V, Storman EM, et al. Plasticity of signaling by spinal estrogen receptor alpha, kappa-opioid receptor and mGluRs over the rat reproductive cycle regulates spinal endomorphin 2 antinociception: relevance of endogenous biased agonism. J Neurosci 2017;37:11181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar A, Storman EM, Liu NJ, et al. Estrogens suppress spinal endomorphin 2 release in female rats in phase with the estrous cycle. Neuroendocrinology 2015;102:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Masu M, Tanabe Y, Tsuchida K, et al. Sequence and expression of a metabotropic glutamate receptor. Nature 1991;349:760–5. [DOI] [PubMed] [Google Scholar]
- 46.Aramori I, Nakanishi S. Signal transduction and pharmacological characteristics of a metabotropic glutamate receptor, mGluR1, in transfected CHO cells. Neuron 1992;8:757–65. [DOI] [PubMed] [Google Scholar]
- 47.Miyashita T, Kubo Y. Extracellular Ca2+ sensitivity of mGluR1alpha induces an increase in the basal cAMP level by direct coupling with Gs protein in transfected CHO cells. Receptors Channels 2000;7:77–91. [PubMed] [Google Scholar]
- 48.Akam EC, Carruthers AM, Nahorski SR, et al. Pharmacological characterization of type 1alpha metabotropic glutamate receptor-stimulated [35S]-GTPgammaS binding. Br J Pharmacol 1997;121:1203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharon D, Vorobiov D, Dascal N. Positive and negative coupling of the metabotropic glutamate receptors to a G proteinactivated K+ channel, GIRK, in Xenopus oocytes. J Gen Physiol 1997;109:477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rivero G, Llorente J, McPherson J, et al. Endomorphin-2: a biased agonist at the mu-opioid receptor. Mol Pharmacol 2012;82:178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McPherson J, Rivero G, Baptist M, et al. mu-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol 2010;78:756–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci 2003;23:2899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mao J, Sung B, Ji RR, et al. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci 2002;22:8312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu NJ, Schnell S, Wessendorf MW, et al. Sex, pain and opioids: inter-dependent influences of sex and pain modality on dynorphin-mediated antinociception in rats. J Pharmacol Exp Ther 2013;344:522–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crine P, Benjannet S, Seidah NG, et al. In vitro biosynthesis of beta-endorphin in pituitary glands. Proc Natl Acad Sci U S A 1977;74:1403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crine P, Benjannet S, Seidah NG, et al. In vitro biosynthesis of beta-endorphin, gamma-lipoprotein, and beta-lipotropin by the pars intermedia of beef pituitary glands. Proc Natl Acad Sci U S A 1977;74:4276–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zadina JE, Hackler L, Ge LJ, et al. A potent and selective endogenous agonist for the mu-opiate receptor [see comments]. Nature 1997;386:499–502. [DOI] [PubMed] [Google Scholar]
- 58.Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 1982;215:413–5. [DOI] [PubMed] [Google Scholar]
- 59.Lord JA, Waterfield AA, Hughes J, et al. Endogenous opioid peptides: multiple agonists and receptors. Nature (Lond) 1977;267:495–9. [DOI] [PubMed] [Google Scholar]
- 60.Levine JD, Gordon NC, Fields HL. The mechanism of placebo analgesia. Lancet 1978;2:654–7. [DOI] [PubMed] [Google Scholar]
- 61.Levine JD, Gordon NC, Jones RT, et al. The narcotic antagonist naloxone enhances clinical pain. Nature 1978;272:826–7. [DOI] [PubMed] [Google Scholar]
- 62.Zubieta JK, Bueller JA, Jackson LR, et al. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J Neurosci 2005;25:7754–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Corder G, Doolen S, Donahue RR, et al. Constitutive mu-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science 2013;341:1394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taylor JJ, Borckardt JJ, George MS. Endogenous opioids mediate left dorsolateral prefrontal cortex rTMS-induced analgesia. Pain 2012;153:1219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrovic P, Kalso E, Petersson KM, et al. Placebo and opioid analgesia—imaging a shared neuronal network. Science 2002;295:1737–40. [DOI] [PubMed] [Google Scholar]
- 66.Amanzio M, Benedetti F. Neuropharmacological dissection of placebo analgesia: expectation-activated opioid systems versus conditioning-activated specific subsystems. J Neurosci 1999;19:484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benedetti F, Arduino C, Amanzio M. Somatotopic activation of opioid systems by target-directed expectations of analgesia. J Neurosci 1999;19:3639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wager TD, Scott DJ, Zubieta JK. Placebo effects on human mu-opioid activity during pain. Proc Natl Acad Sci USA. 2007;104:11056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DosSantos MF, Martikainen IK, Nascimento TD, et al. Building up analgesia in humans via the endogenous mu-opioid system by combining placebo and active tDCS: a preliminary report. PLoS One 2014;9:e102350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ulett GA, Han S, Han JS. Electroacupuncture: mechanisms and clinical application. Biol Psychiatry 1998;44:129–38. [DOI] [PubMed] [Google Scholar]
- 71.Huang C, Wang Y, Chang JK, et al. Endomorphin and mu-opioid receptors in mouse brain mediate the analgesic effect induced by 2 Hz but not 100 Hz electroacupuncture stimulation. Neurosci Lett 2000;294:159–62. [DOI] [PubMed] [Google Scholar]
- 72.Han JS. Acupuncture and endorphins. Neurosci Lett 2004;361: 258–61. [DOI] [PubMed] [Google Scholar]
- 73.Berkowitz BA, Ngai SH, Finck AD. Nitrous oxide “analgesia”: resemblance to opiate action. Science 1976;194:967–8. [DOI] [PubMed] [Google Scholar]
- 74.Goldstein A, Pryor GT, Otis LS, et al. On the role of endogenous opioid peptides: failure of naloxone to influence shock escape threshold in the rat. Life Sci 1976;18:599–604. [DOI] [PubMed] [Google Scholar]
- 75.El-Sobky A, Dostrovsky JO, Wall PD. Lack of effect of naloxone on pain perception in humans. Nature 1976;263:783–4. [DOI] [PubMed] [Google Scholar]
- 76.Grevert P, Goldstein A. Effects of naloxone on experimentally induced ischemic pain and on mood in human subjects. Proc Natl Acad Sci U S A 1977;74:1291–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kajii TS, Okamoto T, Yura S, et al. Elevated levels of beta-endorphin in temporomandibular joint synovial lavage fluid of patients with closed lock. J Orofac Pain 2005;19:41–6. [PubMed] [Google Scholar]
- 78.Petraschka M, Li S, Gilbert TL, et al. The absence of endogenous beta-endorphin selectively blocks phosphorylation and desensitization of mu opioid receptors following partial sciatic nerve ligation. Neuroscience 2007;146:1795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Niikura K, Narita M, Butelman ER, et al. Neuropathic and chronic pain stimuli downregulate central mu-opioid and dopaminergic transmission. Trends Pharmacol Sci 2010;31:299–305. [DOI] [PubMed] [Google Scholar]
- 80.Clemente JT, Parada CA, Veiga MC, et al. Sexual dimorphism in the antinociception mediated by kappa opioid receptors in the rat temporomandibular joint. Neurosci Lett 2004;372:250–5. [DOI] [PubMed] [Google Scholar]
- 81.Blenk KH, Habler HJ, Janig W. Neomycin and gadolinium applied to an L5 spinal nerve lesion prevent mechanical allodynia-like behaviour in rats. Pain 1997;70:155–65. [DOI] [PubMed] [Google Scholar]
- 82.Ringkamp M, Eschenfelder S, Grethel EJ, et al. Lumbar sympathectomy failed to reverse mechanical allodynia- and hyperalgesia-like behavior in rats with L5 spinal nerve injury. Pain 1999;79:143–53. [DOI] [PubMed] [Google Scholar]
- 83.Abbadie C, Gultekin SH, Pasternak GW. Immunohistochemical localization of the carboxy terminus of the novel mu opioid receptor splice variant MOR-1C within the human spinal cord. Neuroreport 2000;11:1953–7. [DOI] [PubMed] [Google Scholar]
- 84.Liu NJ, Storman EM, Gintzler AR. Pharmacological modulation of endogenous opioid activity to attenuate neuropathic pain in rats. J Pain 2019;20:235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Craft RM, Mogil JS, Aloisi AM. Sex differences in pain and analgesia: the role of gonadal hormones. Eur J Pain 2004;8:397–411. [DOI] [PubMed] [Google Scholar]
- 86.Schlinger BA, Remage-Healey L, Rensel M. Establishing regional specificity of neuroestrogen action. Gen Comp Endocrinol 2014;205:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Guyton A, Hall JE. Guyton and Hall Textbook of Medical Physiology. 12th ed. Philadelphia, PA: Saunders; 2010. [Google Scholar]
- 88.Balthazart J, Stoop R, Foidart A, et al. Distribution and regulation of estrogen-2-hydroxylase in the quail brain. Brain Res Bull 1994;35:339–45. [DOI] [PubMed] [Google Scholar]
- 89.Timmers RJ, Granneman JC, Lambert JG, et al. Estrogen-2-hydroxylase in the brain of the male African catfish, Clarias gariepinus. Gen Comp Endocrinol 1988;72:190–203. [DOI] [PubMed] [Google Scholar]
- 90.Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis 1998;19:1–27. [DOI] [PubMed] [Google Scholar]
- 91.Purinton SC, Wood CE. Ovine fetal estrogen sulfotransferase in brain regions important for hypothalamus-pituitary-adrenal axis control. Neuroendocrinology 2000;71:237–42. [DOI] [PubMed] [Google Scholar]
- 92.Miki Y, Nakata T, Suzuki T, et al. Systemic distribution of steroid sulfatase and estrogen sulfotransferase in human adult and fetal tissues. J Clin Endocrinol Metab 2002;87:5760–8. [DOI] [PubMed] [Google Scholar]
- 93.Albert C, Barbier O, Vallee M, et al. Distribution of uridine diphosphate-glucuronosyltransferase (UGT) expression and activity in cynomolgus monkey tissues: evidence for differential expression of steroid-conjugating UGT enzymes in steroid target tissues. Endocrinology 2000;141:2472–80. [DOI] [PubMed] [Google Scholar]
- 94.Remage-Healey L, Maidment NT, Schlinger BA. Forebrain steroid levels fluctuate rapidly during social interactions. Nat Neurosci 2008;11:1327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Charlier TD, Newman AE, Heimovics SA, et al. Rapid effects of aggressive interactions on aromatase activity and oestradiol in discrete brain regions of wild male white-crowned sparrows. J Neuroendocrinol 2011;23:742–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chao A, Schlinger BA, Remage-Healey L. Combined liquid and solid-phase extraction improves quantification of brain estrogen content. Front Neuroanat 2011;5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Storman EM, Liu NJ, Wessendorf MW, et al. Physical linkage of estrogen receptor alpha and aromatase in rat: oligocrine and endocrine actions of CNS-produced estrogens. Endocrinology 2018;159:2683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu NJ, Chakrabarti S, Schnell S, et al. Spinal synthesis of estrogen and concomitant signaling by membrane estrogen receptors regulate spinal {kappa}- and {micro}-opioid receptor heterodimerization and female-specific spinal morphine antinociception. J Neurosci 2011;31:11836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chakrabarti S, Liu NJ, Gintzler AR. Formation of mu-/kappa-opioid receptor heterodimer is sex-dependent and mediates female-specific opioid analgesia. Proc Natl Acad Sci U S A 2010;107:20115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martin-Schild S, Zadina JE, Gerall AA, et al. Localization of endomorphin-2-like immunoreactivity in the rat medulla and spinal cord. Peptides 1997;18:1641–9. [DOI] [PubMed] [Google Scholar]
- 101.Martin-Schild S, Gerall AA, Kastin AJ, et al. Endomorphin-2 is an endogenous opioid in primary sensory afferent fibers. Peptides 1998;19:1783–9. [DOI] [PubMed] [Google Scholar]
- 102.Martin-Schild S, Gerall AA, Kastin AJ, et al. Differential distribution of endomorphin 1- and endomorphin 2-like immunoreactivities in the CNS of the rodent. J Comp Neurol 1999;405:450–71. [PubMed] [Google Scholar]
- 103.Zadina JE, Hackler L, Ge LJ, et al. A potent and selective endogenous agonist for the mu-opiate receptor. Nature 1997;386:499–502. [DOI] [PubMed] [Google Scholar]
- 104.Stoeber M, Jullie D, Lobingier BT, et al. A genetically encoded biosensor reveals location bias of opioid drug action. Neuron 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Baliki MN, Geha PY, Apkarian AV, et al. Beyond feeling: chronic pain hurts the brain, disrupting the default-mode network dynamics. J Neurosci 2008;28:1398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rodriguez-Raecke R, Niemeier A, Ihle K, et al. Brain gray matter decrease in chronic pain is the consequence and not the cause of pain. J Neurosci 2009;29:13746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Apkarian AV, Sosa Y, Sonty S, et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 2004;24:10410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nicholson B, Verma S. Comorbidities in chronic neuropathic pain. Pain Med 2004;5(suppl 1):S9–S27. [DOI] [PubMed] [Google Scholar]
- 109.Apkarian AV, Sosa Y, Krauss BR, et al. Chronic pain patients are impaired on an emotional decision-making task. Pain 2004;108:129–36. [DOI] [PubMed] [Google Scholar]
- 110.Schwartz N, Temkin P, Jurado S, et al. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 2014;345:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]