

Abstract
Background:
Soil salinity is a major abiotic stress that limits plant growth and yield worldwide.
Objective:
To better understand the mechanism of salt stress adaptation in maize (Zea may), proteomic analysis of maize responses to salt stress were analyzed in seedling.
Materials and Methods:
Taking maize seedlings untreated and treated with NaCl for 24 h as material, isobaric tags for relative and absolute quantitation (iTRAQ) were used to analyze the protein expression profile of maize seedlings after salt stress.
Results:
The result showed that 270 differentially expression proteins (DEPs) were identified in maize seedlings after salt stress. The majority proteins had functions related to translation, ribosomal structure and biogenesis (15%), posttranslational modification, protein turnover, chaperones (14%) and others metabolism. Quantitative real-time PCR analysis showed that the EF-Tu, peroxiredoxin, FoF1-type ATP synthase, glutamate dehydrogenase, glyceraldehyde-3-phosphate dehydrogenase, Acetyl-CoA acetyltransferase and nucleoside diphosphate kinase genes were up-regulated in the adaptation of maize to salt stress.
Conclusions:
The coped with salt stress of maize seedlings might be included nitrogen and glutamate (Glu) metabolism and energy homeostasis, nucleotide transport and metabolism, soluble sugar, fatty acid and nucleoside triphosphates synthesis. Moreover, the enhancement of plant to scavenge ROS, such as peroxiredoxin, might play significant roles in the adaptation of maize to salt stress.Taken together, these proteins might have important roles in defense mechanisms against salt stress in maize.We hope that this study provides valuable information for the further utilization and study on the molecular mechanisms of defense mechanisms in maize.
Keywords: iTRAQ, Maize seedlings, Proteomic analysis, Quantitative Real-Time PCR, Salt stress
1. Background
Salinity affect plant growth and yield, and it is one of the most important environmental factors throughout the world. Soil salinity are becoming more severe every year and have been predicted to cause a loss of up to 50% of the cultivatable land by 2050 ( 1). It is estimated that 19.5% of irrigated lands are salt-affected and 40% of the food production worldwide was lossed. High NaCl concentration in the soil can trigger various effects, including photosynthesis and protein synthesis depression, excess reactive oxygen species (ROS) generation, oxidative stress, ionic toxicity, nutritional disorders, and stunted plant growth ( 2, 3). Plants have evolved a series of regulatory mechanisms to cope with the deleterious effects of salt stress. Salt response of plants involves express regulating of specific proteins for the re-establishment of proper cellular ion, osmotic homeostasis with other concomitant processes of repair, detoxification and so on ( 4). Investigating the molecular and biochemical basis of plant stress tolerance is very helpful to improve crop yeild.
Maize is the third most important food crop in the world. It is grown in a variety of soil and climatic conditions. Soil salinity is a major constraint on maize growth because it affects seed germination, the uptake of essential nutrients, grain weight and its productivity ( 5). Understanding biochemical and molecular basis of maize response to salinity stress will help devise strategies for maize improvement in salinity environments.
Proteomics, the large-scale analysis of proteins, have been receiving an increased attention in studying cellular functions. The isobaric tag for relative and absolute quantitation(iTRAQ) is a mass spectrometry-based proteomics technique and this technique overcomes some of the limitations of 2-DE ( 6, 7) . It has been used to analyze abiotic stress induced alterations in different plant species, such as cotton, Arabidopsis, rice, wheat and Brassica napus ( 8, 9). In these species, the identified salt-responsive proteins had various cellular functions such as regulation of carbohydrate, nitrogen and energy metabolism, ROS scavenging, detoxification, signal transduction and so on.
These proteins are mainly play role in the signal transduction, water conservation, protein synthesis, biotic cross-tolerance and so on ( 10). However, proteomics studies of maize response to abiotic stress was few. Zrb et al (2010) studied proteomic changes in maize roots after a short-term adjustment to saline growth conditions ( 11). A set of phosphoproteins such as fructokinase, UDP-glucosyl transferase BX9, 2-Cys-peroxyredoxine and 40-S-ribosomal protein in maize was detected.The expression of these proteins were different after adjustment to saline conditions. Cui D et al (2015) taking salt-tolerant genotype F63 and the salt-sensitive genotype F35 as materials, comparative proteomic analysis were done in seedling roots after 160 mM NaCl treatment for 2 days. Twenty-eight proteins that showed more than 2.0- fold changes in abundance were regarded as salt-responsive proteins. These proteins were mainly in volved in signal processing, water conservation, protein synthesis and biotic cross-tolerance.
2. Objectives
In our study, an iTRAQ-based proteomic technique was used to identify the differentially expressed proteins (DEPs) in maize seedlings treated with NaCl for 24 h.The resluts of this study may lay the foundation for further elucidating salt tolerance mechanisms of maize.
3. Materials and Methods
3.1. Plant Materials,NaCl Treatment and Experimental Design
Maize Huangzao 4 line is an adaptable variety which obtained from Zhangjiakou Academy of Agricultural Sciences, Hebei province, China. Maize seeds were surface-sterilized with 2% hypochlorite solution for 10 min.Then, seeds was germinated on moistened filter paper after washed with distilled water. The seedlings were grown hydroponically in an incubator.The condition of plant was 25 ℃/ 20 ℃(light/dark) amd 14h/10h (light/dark). Maize seedlings were filled with Hoagland’s full-strength nutrient solution.
When the sixth leaves of each plant were fully expanded, the seedlings were cultured in nutrient solution containing 100 mM NaCl. The remaining (control) seedlings were grown in nutrient solution lacking NaCl. The control and treated samples were harvested and washed with distilled water three times before being immersed into liquid nitrogen after 24 h of NaCl treatment.
The samples were stored at -80 °C for proteomic analysis. Three independent biological replicates were conducted. Figure 1 showed the experimental design in this study.
Figure 1.
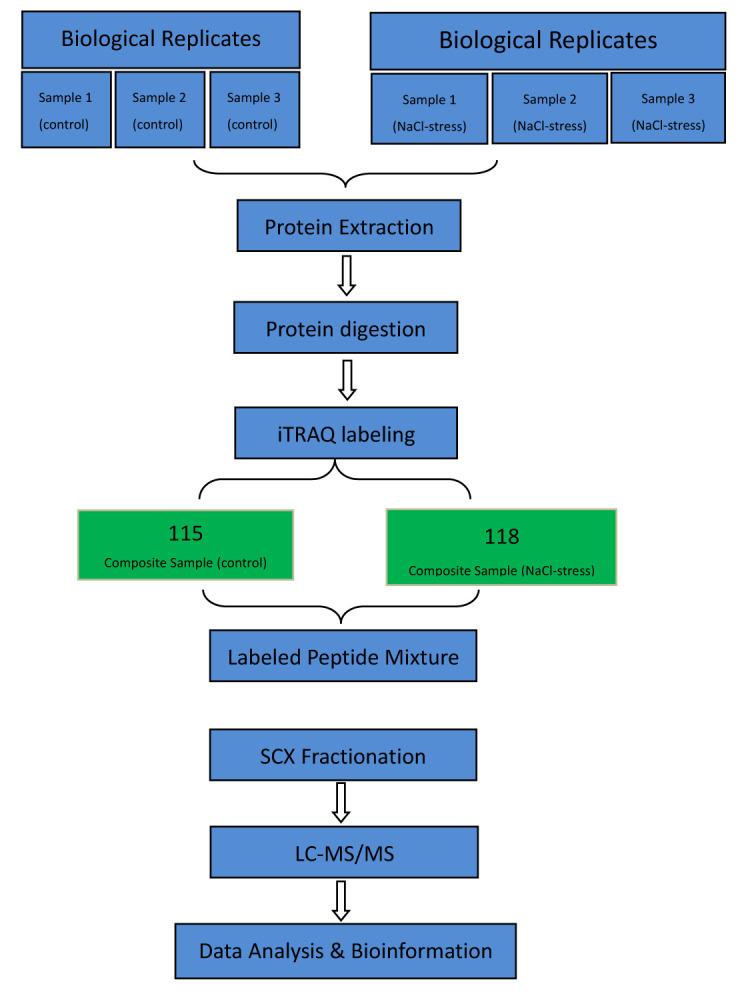
Flow chart of experimental design.
3.2. Protein Extraction
Total protein was extracted from maize seedlings according to the following procedure ( 12). Approximately 1.0 g maize seedling powder was precipitated with trichloroacetic acid (TCA)/acetone precipitation method. The samples were incubated at −20 °C for 2 h and centrifuged at 20,000 g for 30 min at 4 °C. The precipitating step was repeated until the pellets were white. The dried pellets were lysed with lysis buffer [8 M urea, 30 mM 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid (HEPES), 1 mM phenylmethanesulfonyl fluoride (PMSF), 2 mM ethylene diamine tetraacetic acid (EDTA) and 10 mM DL-dithiothreitol (DTT)]. After 5 min, the samples was centrifuged at 20,000 g for 30 min at 4 °C.The supernatant were added 10 mM DTT and incubated at 56 °C for 1 h.Then, 55 mM iodoacetamide (IAM) was added and resting at room temperature for 1 h in the dark. The supernatant was mixed with 4x volumes of chilled acetone and incubated at -20 ℃overnight. After centrifugation at 20,000 g 30 min, the supernatant were discarded. The pellets were dissolved by 50% borane-triethylamine complex (TEAB) buffer with 0.1% sodium dodecyl sulfate (SDS). Finally,the samples were centrifuged at 20,000 g for 30 min at 4 °C.The quantify the protein was used the method of Bradford.
3.3. Protein Digestion, iTRAQ Labeling
A total of 100 μg protein was collected from sample and the protein was digested with 3.3 μg of trypsin (1 μg.μL-1) (Promega, Madison, WI, USA) at 37 °C for 24 h. Samples were labeled using the iTRAQ reagents 8-plexkit according to the manufacturer’s instructions (ABSciexInc., MA, USA). The salt-treated samples’ replicates were labeled with the tags 118 and the control labeled with tags 115, respectively.
3.4. High Performance Liquid Chromatography (HPLC)
The labelled samples were fractionated using an HPLC system. The peptides mixtures were eluted with Buffer A (10 mmoL.L-1 KH2PO4 in 25% acetonitrile, pH 3.0) and Buffer B (10 mmoL.L-1 KH2PO4 and 2M KCl in 25% acetonitrile, pH 3.0). The gradient elution of the chromatographic column was conducted as follows: 0–35 min 100% Buffer A; 36 min 5% Buffer B; 56 min 30% Buffer B; 61 min 50% Buffer B, 66 min 50% Buffer B; 71 min 100% Buffer B. The flow rate was set at 1 mL.min-1, and fractions were collected every minute after sample injection for 31 min. Based on the chromatogram, collected elution ingredients were merged, which were desalted using C18 solid-phase extraction cartridge and vacuum dried. C18 column was activated by 1 mL methanol at a speed of 2–3 drops/s, and then was balanced by 5% ACN (acetonitrile) at a speed of 1 drop/s.
3.5. Reversed-Phase Nano Liquid Chromatography Tandem MS
Peptides were loaded onto a C18 analytical reverse phase column (100 mm × 75 μm, 300 Å, 5 μm). LC-MS/MS was performed on a Q-exact mass spectrometer (Thermo Fisher Scientific,Waltham, MA,USA) combined with a Proxeon Easy Nano-LC system. A 10 μL sample from each fraction was injected to the Q-exact mass spectrometer. The MS/MS scans from 50–2000 m·z-1 were recorded. Nitrogen was used as the collision gas. The ionization tip voltage and interface temperature were set at 1 250 V and 150 °C, respectively.
3.6. Data Analysis and Protein Identification
Protein identification was performed by searching the Maize database (87 406 sequences) with following parameters:MS/MS ion search; enzyme: trypsin with one missed cleavage; monoisotopic mass; peptide mass tolerance: 15ppm; MS/MS tolerance:20mmu; oxidation of methionine and tyrosine labeled by iTRAQ 8-plex as variable modifications, while carbamidomethylation on cysteine, iTRAQ 8-plex labeled N-term and lysine as fixed modifications.To reduce the probability of false peptide identification, only peptides with significance scores(≥20 ) at the 95% confidence interval by a Paragon probability analysis greater than “identity” were counted as identified. Each confidently identified protein involved at least one unique peptide.Only data with a false discovery rate (FDR) <5% were used for sequence data analysis identified by the MASCOT software 2.3.0.
3.7. Bioinformatics
The functional annotations of DEPs were conducted using Gene Ontology (http://www.geneontology.org/). The metabolic pathways and biochemical signals transduction pathways that involved the DEPs were analysis through the Kyoto Encyclopedia of Gene and Genomens (KEGG) (http://www.genomen.jp/kegg) and Clusters of Orthologous Groups (COG) databases (http://www.ncbi.nlm.nih.gov/COG/).
3.8. Verification of Differentially Expression Proteins Using Quantitative Real-time PCR (qRT-PCR)
To investigate whether gene expression is correlated between transcript and protein level, quantitative real-time PCR was performed for 10 genes selected based on proteomic data. Total RNA was extracted from seedlings of maize with Trizol reagent (Invitrogen) accordancing the manufacturer’s instructions. Genomic DNA contamination was removed with DNaseI (Qiagen, Inc) treatment. The SuperScript™ III First-Strand Synthesis SuperMix (Invitrogen) was used for total RNA to reverse transcribe into first strand cDNA. The qRT-PCR were conducted using the SYBR Green JumpStar™ Taq ReadyMix™ (Sigma-Aldrich). The qRT-PCR was conducted in the Bio-Rad CFX 96 sequence detection system according to the manufacturer’s instructions. The reaction system consisted of 2 μL primers (25pmol·uL-1, sense /antisenese primer sequences seen in Supplementary Table), 0.5 μL Sybr green I, 2 μL cDNA template, and diethy pyrocarbonate (DEPC) water was used to complement to 25 μL. The amplification procedure was as follows: pre-denaturation at 94 °C for 4 min; 35 cycles of amplification at 94 °C for 20 s, 60 °C for 30 s and 72 °C for 30 s; and 10 min of extension at 72 °C. Each experiment was repeated three times. The actin 2 gene was used as the internal reference for normalization.
4. Results
4.1. Primary Data Analysis and Protein Identification
A total of 309,857 spectra were generated from the iTRAQ experiment between control and NaCl treatment maize seedlings. Mascot identified a total of 42,771 spectra matched to known spectra, 15,418 peptides, 9,503 unique peptides and 2873 proteins ( Fig. 2).
Figure 2.
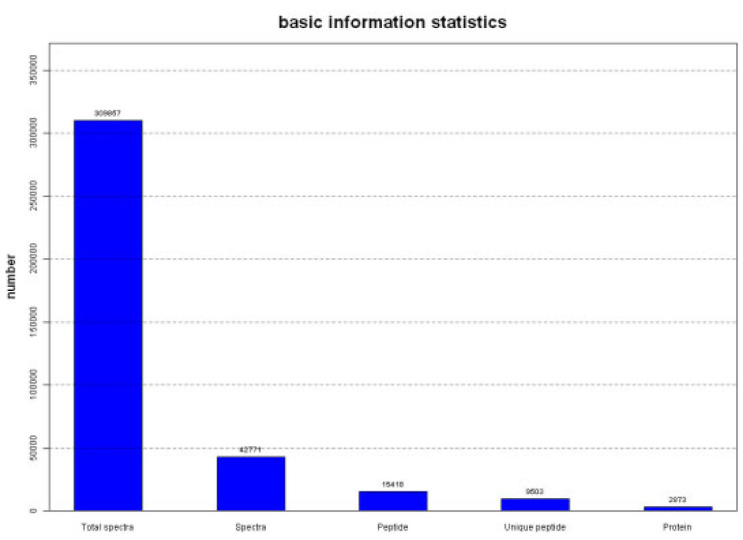
Spectrum peptides and proteins identified from iTRAQ proteomics by searching against NCBI Zea mays database.
Figure 3B showed that more than 79.6% of the proteins included at least two peptides. Protein sequences coverage with 40–100%, 30–40%, 20–30%, 10–20%, and under 10% variation accounted for 12.4%, 9.95%, 17.4%, 26.8%, 33.4%, respectively (Fig. 3A, Fig. 3D). 77 low molecular weight protein (Mr<10kDa) and 115 high molecular weight proteins (Mr>100kDa) were identified (Fig. 3C)
Figure 3.

The distribution of length and number of peptides(A and B), sequence coverage and mass of proteins(C and D) identified by iTRAQ proteomics.
4.2. Identification of DEPs by iTRAQ
When the expression level of one protein exhibited a difference (a > 2-fold or <0.5-fold) compared to the control line, it was considered to be differentially expressed.Using these criteria, 270 DEPs were identified, of which 166 were up-accumulated and 104 were down-accumulated between NaCl treated and control seedlings.
4.3. Bioinformatic Analysis of DEPs
GO functional classification results showed that 101 DEPs (37.4%) were classified into 746 functional groups ( Fig. 4), of which biological processes accounted for 406 GO terms (the most representative were “biological process”), cellular components accounted for 48 GO terms (the most representative were “cellular component”), and molecular functions accounted for 278 GO terms (the most representative was “molecular function”). Those annotated protein species were strongly enriched in GO categories“porphyrin-containing compound biosynthetic process” (P=1.97002E-05), “tetrapyrrole metabolic process” (P=1.97002E-05), “porphyrin-containing compound metabolic process” (P = 1.97002E-05), “tetrapyrrole biosynthetic process” (P=1.97002E-05), “cofactor biosynthetic process” (P = 8.82921E-05).
Figure 4.

Gene Ontology (GO) annotation of the differentially expressed proteins (DEPs) in seedlings of salt stress and control conditions.
KEGG pathway analysis result showed that 139 different metabolic pathways were summarized. The most represented pathway was the “Metabolic pathways”, followed by the pathway of “biosynthesis of secondary metabolites” and “biosynthesis of amino acids”. Those annotated protein species were significantly enriched in pathways of “metabolic pathways” (P = 0.35058E-11), “biosynthesis of secondary metabolites” (P = 2.09419E-05), “photosynthesis” (P=0.000698791), “porphyrin and chlorophyll metabolism” (P = 0.000740518).
A total of 56 DEPs (20.7%) were classified into 14 COG categories.The main biological functions were follow: translation, ribosomal structure and biogenesis (15%), amino acid transport and metabolism (13%), energy production and conversion (12%), posttranslational modification, protein turnover, chaperones (12%), coenzyme transport and metabolism (10%), lipid transport and metabolism (8%), carbohydrate transport and metabolism (6%), cell wall/membrane/envelope biogenesis (6%), nucleotide transport and metabolism (4%), secondary metabolites biosynthesis, transport and catabolism (4%), inorganic ion transport and metabolism (4%), other metabolism(4%), signal transduction mechanism (2%), general function prediction only (2%). Detailed information were showed in Figure 5 and Table1.
Figure 5.

Functional classification of the identified proteins based on COG analysis.
Table 1.
Differentially expressed proteins in maize seedlings to salt stress (100mM NaCl).
| Number | Gene | Annotation | Percent coverage | No.of unique peptides | Mean ratioa | Up/downb |
|---|---|---|---|---|---|---|
| Translation, ribosomal structure and biogenesis | ||||||
| 1 | sp|P08530|RR8_MAIZE | Ribosomal protein S8 | 33.09% | 4 | 0.813 | ↓ |
| 2 | sp|P17788|RK2_MAIZE | Ribosomal protein L2 | 22.34% | 4 | 0.833 | ↓ |
| 3 | tr|B6U1J2|B6U1J2_MAIZE | Ribosomal protein L11 | 19.82% | 3 | 1.372 | ↑ |
| 4 | tr|B4FVB2|B4FVB2_MAIZE | Translation elongation factor EF-Tu, a GTPase | 28.32% | 2 | 1.391 | ↑ |
| 5 | tr|A0A096U6X9|A0A096U6X9_MAIZE | Translation elongation factor EF-G, a GTPase | 29.39% | 2 | 0.706 | ↓ |
| 6 | tr|K7TKZ6|K7TKZ6_MAIZE | Ribosome-binding ATPase YchF, GTP1/OBG family | 20.87% | 6 | 1.202 | ↑ |
| 7 | tr|O82108|O82108_MAIZE | Seryl-tRNA synthetase | 6.35% | 3 | 1.457 | |
| 8 | tr|K7TUG6|K7TUG6_MAIZE | Protein chain release factor A | 14.56% | 4 | 0.764 | ↓ |
| Post-translational modification, protein turnover, chaperones | ||||||
| 9 | tr|B6UFB3|B6UFB3_MAIZE | Molecular chaperone DnaK (HSP70) | 33.71% | 3 | 0.77 | ↓ |
| 10 | tr|A0A096RH43|A0A096RH43_MAIZE | Chaperonin GroEL (HSP60 family) | 58.43% | 1 | 0.699 | ↓ |
| 11 | tr|A0A096PXR7|A0A096PXR7_MAIZE | Peroxiredoxin | 46.86% | 8 | 1.378 | ↑ |
| 12 | tr|B4FSF1|B4FSF1_MAIZE | ATP-dependent Zn proteases | 18.48% | 5 | 1.425 | ↑ |
| 13 | tr|B4FTV9|B4FTV9_MAIZE | ATP-dependent Zn proteases | 9.75% | 2 | 1.212 | ↑ |
| 14 | tr|B4FZZ2|B4FZZ2_MAIZE | Peptidyl-prolyl cis-trans isomerase (rotamase) - cyclophilin family | 40.70% | 5 | 1.311 | ↑ |
| Energy production and conversion;Inorganic ion transport and metabolism | ||||||
| 15 | tr|K7URG3|K7URG3_MAIZE | Citrate synthase | 32.97% | 3 | 1.356 | ↑ |
| 16 | tr|A0A096S8W2|A0A096S8W2_MAIZE | Citrate synthase | 9.94% | 4 | 1.219 | ↑ |
| 17 | tr|Q49HD8|Q49HD8_MAIZE | 2,4-dienoyl-CoA reductase | 13.78% | 4 | 1.297 | ↑ |
| 18 | tr|B6TS21|B6TS21_MAIZE | Succinyl-CoA synthetase, beta subunit | 52.84% | 3 | 1.252 | ↑ |
| 19 | sp|P19023|ATPBM_MAIZE | FoF1-type ATP synthase, beta subunit | 48.64% | 2 | 1.627 | ↑ |
| 20 | tr|A0A096QV77|A0A096QV77_MAIZE | Inorganic pyrophosphatase | 20.90% | 1 | 0.736 | ↓ |
| Amino acid transport and metabolism | ||||||
| 21 | sp|Q43260|DHE3_MAIZE | Glutamate dehydrogenase/leucine dehydrogenase | 37.23% | 9 | 1.253 | ↑ |
| 22 | tr|A0A096TF85|A0A096TF85_MAIZE | 3-deoxy-D-arabino-heptuloso-nate 7-phosphate (DAHP) synthase, class II | 13.83% | 3 | 0.688 | ↓ |
| 23 | tr|B4F7V1|B4F7V1_MAIZE | 3-deoxy-D-arabino-heptuloso-nate 7-phosphate (DAHP) synthase | 13.06% | 3 | 1.235 | ↑ |
| 24 | tr|B4G1Z7|B4G1Z7_MAIZE | 3-dehydroquinate synthetase | 20.57% | 2 | 0.773 | ↓ |
| 25 | tr|A0A096TVA7|A0A096TVA7_MAIZE | O-acetylserine sulfhydrylase, pyridoxal phosphate- dependent | 12.68% | 5 | 2.253 | ↑ |
| 26 | tr|B6T7Q7|B6T7Q7_MAIZE | glycine/serine hydroxymethyltransferase | 18.13% | 8 | 1.242 | ↑ |
| 27 | tr|B4FAI1|B4FAI1_MAIZE | N-acetyl-gamma-glutamylphosphate reductase | 12.26% | 4 | 1.376 | ↑ |
| Carbohydrate transport and metabolism | ||||||
| 28 | sp|P08735|G3PC1_MAIZE | glyceraldehyde-3-phosphate dehydrogenase/Erythrose-4-phosphate ehydrogenase | 67.95% | 8 | 1.484 | ↑ |
| 29 | tr|B4FQW6|B4FQW6_MAIZE | glyceraldehyde-3-phosphate dehydrogenase /erythrose-4-phosphate dehydrogenase | 77.63% | 7 | 1.549 | ↑ |
| 30 | tr|A0A096R9A2|A0A096R9A2_MAIZE | Pentose-5-phosphate-3-epimer-ase | 7.38% | 1 | 1.205 | ↑ |
| General function prediction only | ||||||
| 31 | tr|B6T5H6|B6T5H6_MAIZE | Predicted oxidoreductase (related to aryl-alcohol dehydrogenase) | 34.80% | 10 | 1.339 | ↑ |
| Lipid transport and metabolism | ||||||
| 32 | tr|A0A096Q2N4|A0A096Q2N4_MAIZE | Biotin carboxylase | 4.69% | 2 | 1.27 | ↑ |
| 33 | tr|A0A096UC49|A0A096UC49_MAIZE | Acetyl-CoA acetyltransferase | 47.88% | 12 | 1.33 | ↑ |
| 34 | tr|H2BJB6|H2BJB6_MAIZE | Acyl-CoA synthetase (AMP-forming) | 19.54% | 8 | 1.202 | ↑ |
| 35 | tr|A0A096QRK2|A0A096QRK2_MAIZE | 1-deoxy-D-xylulose 5-phosphate reductoisomerase | 25.21% | 8 | 0.814 | ↓ |
| Nucleotide transport and metabolism | ||||||
| 36 | tr|A0A096S8D2|A0A096S8D2_MAIZE | Nucleoside diphosphate kinase | 42.61% | 4 | 1.723 | ↑ |
| 37 | tr|A0A096UA28|A0A096UA28_MAIZE | Nucleoside diphosphate kinase | 34.44% | 6 | 1.393 | ↑ |
| Secondary metabolites biosynthesis, transport and catabolism | ||||||
| 38 | tr|A2T1W7|A2T1W7_MAIZE | Aldo/keto reductase, related to diketogulonate reduct-ase | 23.55% | 7 | 1.515 | ↑ |
| Inorganic ion transport and metabolism | ||||||
| 39 | tr|B6SKA7|B6SKA7_MAIZE | Sulfate adenylyltransferase subunit 1 | 38.03% | 2 | 1.211 | ↑ |
| Coenzyme transport and metabolism | ||||||
| 40 | tr|B4FAD1|B4FAD1_MAIZE | S-adenosylmethionine synthetase | 57.58% | 7 | 0.709 | ↓ |
| 41 | tr|B4FSE1|B4FSE1_MAIZE | Archaeal ribulose 1,5-bisphosphate synthetase | 28.01% | 1 | 0.587 | ↓ |
| 42 | sp|Q41739|THI42_MAIZE | Archaeal ribulose 1,5-bisphosphate synthetase | 28.81% | 3 | 0.805 | ↓ |
| 43 | tr|B6SJR7|B6SJR7_MAIZE | 5,10-methylene-tetrahydrof-olate dehydrogenase | 11.65% | 3 | 1.226 | ↑ |
| 44 | tr|A0A096TKH4|A0A096TKH4_MAIZE | Delta-aminolevulinic acid dehydratase, porphobilinogen synthase | 42.25% | 14 | 0.79 | ↓ |
| Cell wall/membrane/envelope biogenesis | ||||||
| 45 | tr|B4FAG0|B4FAG0_MAIZE | Nucleoside-diphosphate-sugar epimerase | 29.71% | 3 | 1.216 | ↑ |
| 46 | tr|B4FF24|B4FF24_MAIZE | dTDP-D-glucose 4,6- dehydratase | 22.51% | 7 | 0.797 | ↓ |
| Cell cycle control, cell division, chromosome partitioning;Cell wall/membrane/envelope biogenesis;Signal transduction mechanisms | ||||||
| 47 | tr|A0A096S4K3|A0A096S4K3_MAIZE | Chromosome partitioning ATPase, Mrp family, contains Fe-S cluster | 22.51% | 7 | 0.797 | ↓ |
| 48 | tr|C0P5X1|C0P5X1_MAIZE | AAA+-type ATPase, SpoVK/Ycf46/Vps4 family | 61.36% | 15 | 0.798 | ↓ |
| Other metabolism | ||||||
| 49 | tr|A0A096QFD4|A0A096QFD4_MAIZE | 14.53% | 5 | 1.215 | ↑ | |
| 50 | tr|A0A059Q6U2|A0A059Q6U2_MAIZE | 16.76% | 12 | 0.786 | ↓ | |
| 51 | tr|A0A059Q6W8|A0A059Q6W8_MAIZE | 13.60% | 11 | 0.734 | ↓ | |
| 52 | tr|A0A096PU70|A0A096PU70_MAIZE | 14.75% | 5 | 0.824 | ↓ | |
| 53 | tr|A0A096SA34|A0A096SA34_MAIZE | 3.04% | 2 | 0.787 | ↓ | |
| 54 | tr|K7USR3|K7USR3_MAIZE | 18.63% | 6 | 0.667 | ↓ | |
| 55 | tr|C0HIU5|C0HIU5_MAIZE | 18.56% | 4 | 1.434 | ↑ | |
| 56 | tr|B6T681|B6T681_MAIZE | 56.38% | 2 | 0.634 | ↓ | |
aMean ratio corresponds to the protein reporter ion intensity originating from salt-treated protein samples(113 and 114) relative to fully control samples (115 and 116)with a 1.2 fold-changes and p<0.05.
bProteins increased in abundance(↑) or decreased in abundance(↓)
A. 2873 proteins.
B. Differentially expressed proteins in salt stress maize seedlings as compared to the control. The percentage for each class is shown and represented in the pie-chart.
4.4. Verification of Differentially Expression Proteins Using Quantitative Real-time PCR (qRT-PCR)
To further confirm the iTRAQ data, transcriptional analysis of 10 protein species was performed by qRT-PCR ( Fig. 6). Maize actin2 gene taken as reference genes. The transcript levels of ten genes displayed the same trend with the abundance of the corresponding protein species. The transcript levels of EF-Tu, peroxiredoxin, FoF1-type ATP synthase, glutamate dehydrogenase, glyceraldehyde-3-phosphate dehydrogenase, Acetyl-CoA acetyltransferase and nucleoside diphosphate kinase genes were increased in the adaptation of maize to salt stress. While, the transcript levels of molecular chaperone DnaK, 1-deoxy-D-xylulose 5-phosphate reductoisomerase and ribosomal protein were decreased.
Figure 6.
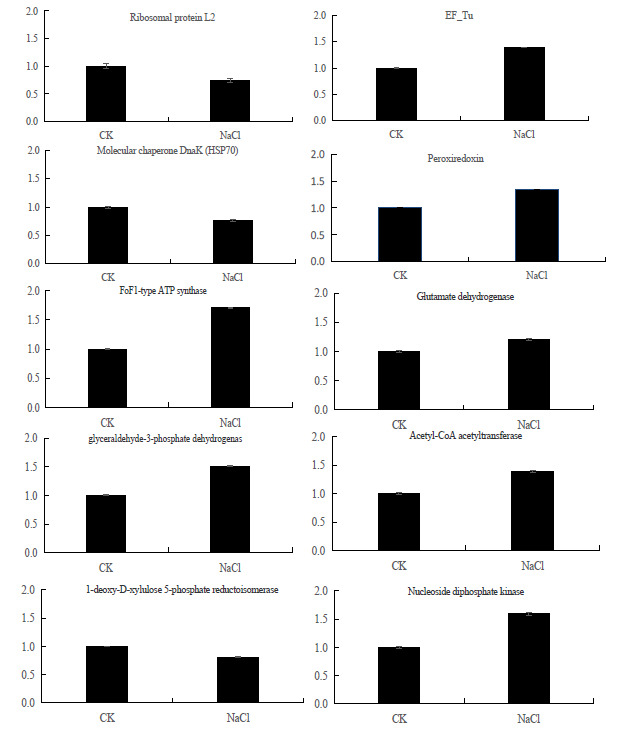
Verification result of differentially expressed proteins (DEPs) using quantitative real-time PCR (qRT-PCR).CK:control;NaCl: salt-stressed maize seedlings
5. Discussion
5.1. DEPs Involved Proteins Involved in Translation, Ribosomal Structure and Biogenesis
Gene expression is regulated transcriptionally and post-transcriptionally. The proteins species, involved in protein translation, ribosomal structure and bio-synthesis, were identified in maize seedlings after salt stress (Table 1). Consistent with the level change of ribosomal proteins in Upland cotton ( 13), the abundance of ribosomal protein (tr|B6U1J2) was up-accumulated. The up-regulation of ribosomal protein may indicate that an overall increased translation machinery in salt-stressed maize seedlings.
The elongation factor Tu play an important role in protein biosynthesis and chaperones. In Brassica napus, soybean seedlings and cucumber species, the expression of elongation factor Tu was up-accumulated in response to salt stresses ( 14). In our study,elongation factor EF-Tu (tr|B4FVB2) was also observed and the abundance of it was also increased in maize after salt stress(Table 1). The cumulative abundance of elongation factor Tu may enhance protein biosynthesis and repair salt stress damage of chloroplast photosynthetic proteins. Interestingly, the abundance of another translation elongation factor EF-G (tr|A0A096U6X9) was decreased under salt stress conditions (Table 1). The function of it in salt stress is unknown now.
5.2. DEPs Involved Posttranslational Modification, Protein Turnover, Chaperones
Hsp70, a member of HSPs, assists in proper folding of newly synthesized proteins, protein transport, translocation and control of regulatory protein functions in various environmental conditions ( 15) (Scarpeci, Zanor et al. 2008). In Arabidopsis and cotton , the HSPs contributed to the enhancement to salt tolerance ( 16) (Ali, Uliaie et al. 2017) . In this study, the abundance of hsp70 proteins (tr|B6UFB3) was decreased after salt stress (Table 1). Moreover, the abundance of chaperonin GroEL (Hsp60 family) (tr|A0A096RH43) was also decreased under salt stress (Table 1). These results showed that members of plant HSPs play various role in response to salt stress of plant.
Reactive oxygen species (ROS) could induced by salt stress and cause oxidative damage to the plant cell, metabolic processes. Peroxiredoxins (Prxs), a group of antioxidant enzymes, were identified as an important component of the oxidative defense system. Jianwen X et al. (2015) showed that the expression of rice putative peroxiredoxin Q (PrxQ) protein was increased by1.688-fold after salt stress treatment. Consist with this result, the abundance of peroxiredoxin protein (tr|A0A096PXR7) was increased in maize seedlings under salt stress (Table 1). The up-accumulated abun-dance of the peroxiredoxin protein could confer salt-stress tolerance in maize seedlings.
5.3. DEPs Involved Energy Production and Conversion
Tricarboxylic acid (TCA) cycle is a key metabolic pathway that unifies carbohydrate, fat, and protein metabolism. In cotton roots, the abundance of proteins that involved in the process of TCA was altered after salt stress. In this study, three proteins related to TCA were identified. The abundance of two citrate synthase (tr|K7URG3 and tr|A0A096S8W2) was increased under salt stress (Table 1). The proteins 2,4-dienoyl-CoA reductase (tr|Q49HD8) and Succinyl-CoA synthetase beta subunit (tr|B6TS21), involved in the process of the carboxylic acid cycle, were also found and the abundance of them was also increased in salt stress (Table 1).
ATP synthase is one of salt-responsive enzyme which plays a significant role in plant response to salt stress ( 17, 18). The ATP synthase expression in plant species was different in response to salt stress. In Brassica napus seedlings, the abundance of ATP synthase beta subunit was changed. It showed significantly decreased at 24 h and then increased at 48h. In rice and black locust, ATP synthase was induced upon salinity. In a salt tolerant cowpea cultivar, ATP synthase was significantly down-regulated. While, it was up-regulated in salt-sensitive cowpea cultivar under salt treatment. The abundance of ATP synthase decreased in cotton seedling, soybean leaves, roots of Arabidopsis and cucumber under salt stress. Gao et al.(2011)described that H+-ATPases was up-regulated in the wheat (Triticum aestivum L.) cultivar Zhengmai 9023 under salt stress ( 19). In our study, the abundance of FoF1-type ATP synthase beta subunit (sp|P19023) was increased after salt treatment (Table 1). Our result consisted with the expression profile of ATP synthase protein in the wheat. These results showed that energy synthesis might be promoted in salt-stressed maize seedlings.
5.4. DEPs Involved Amino Acid Transport and Metabolism
Glutamate dehydrogenase involves and plays a key role in the process of nitrogen and glutamate (Glu) metabolism and energy homeostasis. In rice roots, glutamate dehydrogenase was up-regulated in response to salt-stress ( 20). However, in Arabidopsis, the expression of glutamate dehydrogenase 2 (GDH2, At5g07440) was decreased at the 6 h time point, but increased at the 48 h time point after salt treatment. In our experiment, the abundance of one glutamate dehydrogenase (sp|Q43260) was increased at 24h after salt stress treatment in maize seedlings (Table 1).
Notably, three new proteins, including 3-Deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) synthase (sp|Q43260 and tr|A0A096TF85) and 3-dehydroquinate synthetase (tr|B4G1Z7), associated with amino acid metabolism were also found in maize seedlings after salt stress (Table 1). The abundance of 3-dehydroquinate synthetase and 3-Deoxy-D-arabino-
heptulosonate 7-phosphate (DAHP) synthase was decreased, while the other DAHP synthase was increased in maize seedlings after treated with NaCL.
5.5. DEPs Involved Carbohydrate Transport and Metabolism
Glyceraldehyde-3-phosphate dehydrogenase is one of dehydrogenases and catalyzes the oxidation of glyceraldehyde 3-phosphate to 1,3-biphosphoglycerate in the glycolytic pathway.
In sugarcane, halophyte grass Aeluropus lagopoides and cucumber, the abundance of glyceraldehyde 3-phosphate dehydrogenase was increased after salt treatment ( 21, 22). Consistent with these results, two glyceraldehyde-3-phosphate dehydrogenases (tr|B4FQW6 and sp|P08735) were obtained in our study and the abundance of them was increased in response to salt-stress of maize seedlings (Table 1). The increased abundance of these protein may reflect the pattern of carbon flux in response to a reduction in photosynthesis.
5.6. DEPs Involved General Function Prediction only
Oxidoreductases are a superfamily of proteins and play role in secondary metabolism in cells ( 23). One pepper oxidoreductase CaOXR1 which interacts with the CaRAV1 transcription factor was described in the nucleus ( 24). In response to high salinity and drought stress, the expression of oxidoreductase CaOXR1 was attenuated in the CaOXR1-silenced pepper plants. However, the functions of CaOXR1 protein has not been investigated. In our study, one predicted oxidoreductase related to aryl-alcohol dehydrogenase (tr|B6T5H6) enzyme (a oxidoreductase family) was obtained (Table 1).This enzyme belongs to the family of oxidoreductases, specifically those acting on the CH-OH group of donor with NAD+ or NADP+ as acceptor. The aryl-alcohol dehydrogenase, specifically those acting on the CH-OH group of donor with NAD+ or NAD+ as acceptor. The abundance of predicted oxidoreductase related to aryl-alcohol dehydrogenase showed increased under salt stress. The function of predicted oxidoreductase related to aryl-alcohol dehydrogenase in maize response to salt stress will need further investigation.
5.7. DEPs Involved Lipid Transport and Metabolism
Reports showed that the lipid compositions were changed in broccoli roots and alfalfa cultivars after treated with salt (Rahman, Alam et al. 2015).In our study, acetyl-CoA acetyltransferase (tr|A0A096UC49), biotin carboxylase (tr|A0A096Q2N4), and acyl-CoA synthetase (tr|H2BJB6) which participated in fatty acid synthesis were obtained. The abundance of three proteins was increased in salt treated maize seedlings (Table 1). The increased abundance of these proteins indicated that fatty acid synthesis may be participate in the defense response of maize to salt stress.
1-Deoxy-D-xylulose reductoisomerase (DXR) plays role in the first committed step of plastidial isoprenoid-precursor biosynthesis. In the gray poplar leaves, the transcript levels of 1-deoxy-d-xylulose 5-phosphate reductoisomerase (PcDXR) responded slightly to the salt stress. In our study, one 1-Deoxy-D-xylulose reductoisomerase (tr|A0A096QRK2) was obtained and the abundance was decreased in maize seedlings after NaCL treatment (Table 1).
5.8. DEPs Involved Nucleotide Transport and Metabolism
Nucleoside-diphosphate kinases (NDPKs, also NDP Kinase) involved in the process of nucleotide triphosphates production. It catalyzed the exchange of terminal phosphate between different nucleoside diphosphates (NDP) and triphosphates (NTP) in a reversible manner. In Arabidopsis, NDPK-2 regulated the cellular redox state through interacts with two oxidative stress-activated mitogen-activated protein kinases (MAPKs) and enhanced multiple stress tolerance. Over-expression of AtNDPK2 could also enhance plant tolerance in response to various environmental stresses such as cold, salt, and H2O2. In rice, NDPK gene was upregulated against bacterial pathogens infection ( 25, 26). In our study, two nucleoside diphosphate kinases (tr|A0A096S8D2 and tr|A0A096UA28) were obtained and the abundance of them was increased in response to salt-stress of maize (Table 1). The increased abundance of nucleoside diphosphate kinases may enhance salt adaption or resistance of maize seedlings by promote the synthesis of nucleotide triphosphates.
5.9. Verification of DEPs Using Quantitative Real-Time PCR (qRT-PCR)
The qRT-PCR results show that EF-Tu, peroxiredoxin, FoF1-type ATP synthase, glutamate dehydrogenase, glyceraldehyde-3-phosphate dehydrogenase, Acetyl-CoA acetyltransferase and nucleoside diphosphate kinase genes were upregulated in the adaptation of maize to salt stress.
In this study, the abundance of elongation factor Tu was increased. It indicated that protein biosynthesis and repair the damage of photosynthetic proteins in chloroplasts was enhanced under salt stress.Salinity could stimulate plants to produce excessive ROS and cause oxidative damage.Therefore, ROS scavening and keep ROS homeostasis is very important to improve plant tolerance to salt stress.In this study, peroxiredoxin protein invovlved in ROS scavenging was accumulated. The increased activities of peroxiredoxin showed that ROS homeostasis was also necessary for maize seedlings to enhance salt tolerance. Glutamate dehydrogenase plays a key role in nitrogen and glutamate (Glu) metabolism and energy homeostasis. Consist with rice, maize glutamate dehydrogenase detected in our study was also up-regulated in response to salt-stress. GAPDH plays important role in process of gene transcription, DNA replication and repair, etc. In P. cathayana, over-expression of GAPDH does not only enhance the soluble sugar accumulation, but also provides more energy after salt treatment. Consist with it, expression of GAPDH was also accumulated and invovled in enhancement of maize seedlings salt tolerance.
Meantime, the activities of the Acetyl-CoA acetyltransferase and nucleoside diphosphate kinases which participate in fatty acid and nucleoside triphosphates synthesis were all increased.
6. Conclusion
The iTRAQ results showed that 270 DEPs were detected between control and salt stress seedlings. 166 were up-accumulated and 104 were down-accumulated after salt stress compared to control. The function analysis of GO term and KEGG pathway showed that DEPs were significantly enriched in the translation, ribosomal structure and biogenesis, posttranslational modification, protein turnover, chaperones and others metabolism which was hypothesized to participate in salt stress response.While seven of them were identified as salt stress-responsive proteins.These proteins are involved in regulation of protein biosynthesis, reactive oxygen species scavenging,nitrogen and glutamate (Glu) metabolism, energy homeostasis, nucleic acid synthesis and so on. Our study provieds valuable insights to mechanisms underlying salt response of maize seedlings.
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding
This research was funded by Major Projects Foundation of Hebei North University (ZD201407), Major Projects Foundation of Hebei North University (ZD201305), Major national science and technology projects (2014ZX0800909B), Selection and training of talents of higher discipline plan of Hebei Province (BR2-234) and Hebei Province Natural Science Fund(C2018405042),The national Key Research and Development Program of China (2017YFD0300305).
Footnotes
Conflict of Interest: None .
References
- 1.Mahajan S, Tuteja N. “Cold, Salinity and Drought Stresses: An Overview.”. Arcs of Biochem Biophys. 2005;444(2):139–158. doi: 10.1016/j.abb.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Yadav S, Wholey HM, Kuntzer “Causes of Salinity and Plant Manifestations to Salt Stress: A Review.” . J Environ Biol. 2011;32(5):667–685. doi: 10.1007/s11852-010-0106-3. [DOI] [PubMed] [Google Scholar]
- 3.Barkla BJ, Castellanos-Cervantes T, Diaz dLJL, Matros A, Mock HP HP, Perez-Alfocea F, et al. “Elucidation of Salt Stress Defense and Tolerance Mechanisms of Crop Plants Using Proteomics-Current Achievements and Perspectives.”. J Proteomics. 2013;13(12-13):1885–1900. doi: 10.1002/pmic.201200-399. [DOI] [PubMed] [Google Scholar]
- 4.Chinnusamy V, Jagendorf A, Zhu J. “Understanding and Improving Salt Tolerance in Plants.”. Crop Sci. 2005;45(2):437–448. doi: 10.2135/cropsci2005.0437. [DOI] [Google Scholar]
- 5.Farooq M, Hussain M, Siddique KHM. “Salt Stress in Maize: Effects, Resistance Mechanisms, and Management. A Review.” . Agron Sustain Dev. 2015;35(2):461–481. doi: 10.1007/s13593-015-0287-0. [DOI] [Google Scholar]
- 6.Karp NA, Huber W, Sadowski PG, Charles PD, Hester PD, Lilley KS. “Addressing Accuracy and Precision Issues in iTRAQ Quantitation.”. Mol Cell Proteomics. 2010;9(9):1885–1897. doi: 10.1074/mcp.m900628-mcp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulze WX, Usadel BR. “Quantitation in Mass-Spectrometry-Based Proteomics.”. Annu Rev Plant Biol. 2010;61(1):491–516. doi: 10.1146/annurev-arplant-042809-112132. [DOI] [PubMed] [Google Scholar]
- 8.Jia H, Shao M, He Y, Guan R, Chu P, Jiang H, et al. “Proteome Dynamics and Physiological Responses to Short-Term Salt Stress in Brassica napus Leaves.” . Plos One. 2015;10(12):e0144808. doi: 10.1371/journal.pone.0144808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qilin M, Wenying Z, Yunbo Z, Haoliang L, Matsui T, et al. “iTRAQ-Based Quantitative Proteomics Analysis on Rice Anther Responding to High Temperature.” . Intl J Mol Sci. 2017;18(9):1811. doi: 10.3390/ijms18091811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui D, Wu D, Liu J, Li D, Xu C, Li S, et al. “Proteomic Analysis of Seedling Roots of Two Maize Inbred Lines That Differ Significantly in The Salt Stress Response.”. Plos One. 2015;10(2):e0116697. doi: 10.1371/journal.pone.0116697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christian Z, Zrb C, Schmitt S, Mühling KH. “Proteomic Changes in Maize Roots After Short-Term Adjustment to Saline Growth Conditions.” . J Proteomics. 2010;10(24):4441–4449. doi: 10.1002/pmic.201000231. [DOI] [PubMed] [Google Scholar]
- 12.Yin S, Xue J, Sun H, Wen B, Wang Q, Perkins G, et al. “Quantitative Evaluation of The Mitochondrial Proteomes of Drosophila Melanogaster Adapted to Extreme Oxygen Conditions.” . Plos One. 2013;8(9):e74011. doi: 10.1371/journal.pone.0074011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Uribe L, Higbie SM, Stewart JM, Wilkins T, Lindemann W, Sengupta-Gopalan C, et al. “Identification of Salt Responsive Genes Using Comparative Microarray Analysis in Upland Cotton (Gossypium hirsutum L.).” . Plant Sci. 2011;180(3):461–469. doi: 10.1016/j.plantsci.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 14.Ma H, Song L, Shu Y, Shuang W, Niu J, Shu Y, Wang Z, et al. “Comparative Proteomic Analysis of Seedling Leaves of Different Salt Tolerant Soybean Genotypes.” . J Proteomics. 2012;75(5):1529–1546. doi: 10.1016/j.jprot.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 15.Scarpeci TE, Zanor MI, Carrillo N, Mueller-Roeber B, Valle EM. “Generation of Superoxide Anion in Chloroplasts of Arabidopsis thaliana During Active Photosynthesis: A Focus on Rapidly Induced Genes.” . Plant Mol Bio. 2008;66(4):361–378. doi: 10.1007/s11103-007-9274-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ali B, Uliaie ED, Salekdeh GH. “Proteomic Analysis of Rapeseed (Brassica napus L.) Seedling Roots Under Salt Stress.” . Ann Biol Res. 2017 [Google Scholar]
- 17.Liu CW, Chang TS, Hsu YK, Wang AZ, Yen HC, Wu YP, et al. “Comparative Proteomic Analysis of Early Salt Stress Responsive Proteins in Roots and Leaves of Rice.”. J Proteomics. 2014;14(15):1759–1775. doi: 10.1002/pmic.201300276. [DOI] [PubMed] [Google Scholar]
- 18.Yi X, Yang Q, Guo AP, Chang LL, Wang D, Tong Z, et al. “Quantitative Proteomics of Sesuvium Portulacastrum Leaves Revealed That Ion Transportation by V-ATPase and Sugar Accumulation in Chloroplast Played Crucial Roles in Halophyte Salt Tolerance.” . J Proteomics. 2014;99:84–100. doi: 10.1016/j.jprot.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Gao L, Yan X, Guo G, Hu Y, Ma W, Yan Y. “Proteome Analysis of Wheat Leaf Under Salt Stress by Two-Dimensional Difference Gel Electrophoresis (2D-DIGE).” . Phytochemistry. 2011;72(10):180–1191. doi: 10.1016/j.phytochem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Nam MH, Sun MH, Kim KM, Park WJ, Seo JB, Cho K, et al. “Proteome Analysis of Wheat Leaf Under Salt Stress by Two-Dimensional Difference Gel Electrophoresis (2D-DIGE).” . Proteome Sci. 2012;10(1):25. doi: 10.1186/1477-5956-10-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang F, Xiao X, Sheng Z, Korpelainen H , Li C. “Salt Stress Responses in Populus Cathayana Rehder.” . Plant Sci. 2009;176(5):669–677. doi: 10.1016/j.plantsci.2009.02.008. [DOI] [Google Scholar]
- 22.Sobhanian H, Motamed N, Jazii FR, Nakamura T, Komatsu S. “Salt Stress Induced Differential Proteome and Metabolome Response in The Shoots of Aeluropus Lagopoides (Poaceae), A Halophyte C-4 Plant.” . J Proteome Res. 2010;9(6):2882–2897. doi: 10.1021/pr900974k. [DOI] [PubMed] [Google Scholar]
- 23.Reddy AM, Reddy VS, Scheffler BE, Wienand U, Reddy AR. “Salt Stress Induced Differential Proteome and Metabolome Response in The Shoots of Aeluropus Lagopoides (Poaceae), A Halophyte C-4 Plant.” . Metab Eng. 2007;9(1):95–111. doi: 10.1016/j.ymben.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Lee SC, Du SC, Hwang IS, Hwang BK. “The Pepper Oxidoreductase CaOXR1 Interacts With The Transcription Factor CaRAV1 and Is Required For Salt and Osmotic Stress Tolerance.” . Plant Mol Bio. 2010;73(s4-5):409–424. doi: 10.1007/s11103-010-9629-0. [DOI] [PubMed] [Google Scholar]
- 25.Harris N, Taylor JE, Roberts JA. “Isolation of A mRNA Encoding A Nucleoside Diphosphate Kinase From Tomato That Is Up-Regulated by Wounding.” . Plant Mol Bio. 1994;25(4):739–742. doi: 10.1007/bf00029611. [DOI] [PubMed] [Google Scholar]
- 26.Song MC, Shin SH, Kim KS, Kim YC, Cho BH. “Enhanced Expression of A gene Encoding A nucleoside Diphosphate Kinase 1 (OsNDPK1) in Rice Plants Upon Infection With Bacterial Pathogens.” . Mol Cell. 2004;18(3):390–395. doi: 10.1074/jbc.M411910200. [DOI] [PubMed] [Google Scholar]