

Abstract
Background:
Ankylosing spondylitis (AS) is a type of arthritis which can cause inflammation in the vertebrae and joints between the spine and pelvis. However, our understanding of the exact genetic mechanisms of AS is still far from being clear.
Objective:
To study and find the mechanisms and possible biomarkers related to AS by surveying inter-gene correlations of networks.
Materials and Methods:
A weighted gene co-expression network was constructed among genes identified by microarray analysis, gene co-expression network analysis, and network clustering. Then receiver operating characteristic (ROC) curves were conducted to identify a significant module with the genes implicated in the AS pathogenesis. Real-time PCR was performed to validate the results of microarray analysis.
Results:
In the significant module obtained from the network analysis there were eight AS related genes (LSM3, MRPS11, NSMCE2, PSMA4, UBL5, RPL17, MRPL22 and RPS17) which have been reported in previous studies as hub genes. Further, in this module, eight significant enriched pathways were found with adjusted p-values < 0.001 consisting of oxidative phosphorylation, ribosome, nonalcoholic fatty liver disease, Alzheimer's, Huntington's, and Parkinson's diseases, spliceosome, and cardiac muscle contraction pathways which have been linked to AS. Furthermore, we identified nine AS related genes (UQCRB, UQCRH, UQCRHL, UQCRQ, COX7B, COX5B, COX6C, COX6A1 and COX7C) in these pathways which can play essential roles in controlling mitochondrial activity and pathogenesis of autoimmune diseases. Real-time PCR results showed that three genes including UQCRH, MRPS11, and NSMCE2 in AS patients were significantly differentially expressed compared with normal controls.
Conclusions:
The results of the present study may contribute to understanding of AS molecular pathogenesis, thereby aiding the early prognosis, diagnosis, and effective therapies of the disease.
Keywords: Ankylosing spondylitis, Autoimmune, Gene co-expression network, Microarray, Real-Time PCR
1. Background
Ankylosing spondylitis (AS), a type of persistent inflammatory arthritis, is a complex genetic disease which mainly affects the sacroiliac joints (SI joints) and axial skeleton. It can cause dramatic resorption and formation of bone, which eventually results in the occurrence of ankylosis ( 1). Until now, several factors including genetic susceptibility loci such as human leukocyte antigen HLA-B27 ( 2) and endoplasmic reticulum aminopeptidase 1 (ERAP1) ( 3), as well as infections and environmental factors have been proposed which are involved in the onset and development of the AS ( 4). However, the pathogenesis mechanism, progression, and diagnosis of AS have not been well identified ( 5). Therefore, exploration and finding the genes that may have an important role in the AS pathogenesis would be a necessity.
Genome-wide association studies (GWAS) unveiled the involvement of numerous susceptible genes in the AS pathogenesis ( 6). On the other hand, genome-wide transcriptome analysis has become the popular method to understand the pathogenesis mechanism of a disease, molecular classification, and identification of biomarkers. Recently network-based approaches have also been recommended to identify genetic determinants of human diseases.
While the description of differentially expressed genes (DEGs) can offer clues for ensuing functional analysis, finding patterns in the gene expression data may disclose the overlooked perspectives of one-dimensional differential gene expression method. Various studies show that gene interaction networks can help to identify new biomarkers of complex diseases ( 7). The co-expression network is the most common gene network that is based on the correlations between gene expression profiles. In this network, enrichment for gene ontology (GO) or multivariate statistical methods are utilized to group highly co-expressed (correlated) genes into co-expression modules (gene sets).
2. Objective
The main objective of this research is to find the possible biomarkers of AS disease by surveying inter-gene correlations of networks and identifying the AS related mechanisms. For this purpose, a co-expression network was constructed and various modules were identified. The module with the highest area under curve (AUC) values in ROC curve was selected for further analysis. Moreover, Real time-PCR analysis was conducted to validate the hub genes which were found from the network analysis.
3. Materials and Methods
3.1. Microarray Dataset and Preprocessing
The microarray dataset was downloaded from the Gene Expression Omnibus (GEO) database (GEO accession GSE25101) ( 8). Sample sequencing was done with the Illumina platform (GPL6947-Illumina HumanHT-12 V3.0 expression beadchip). The dataset comprises the gene expression profiles of 16 AS patients and 16 age, sex-matched normal subjects ( 8). Lumi package ( 9) was applied to quantile normalization. The probes which annotated to several genes were excluded and among the probes which annotated to a gene, the one with the max variance expression was chosen. After applying the preprocessing steps, we gained an expression matrix, where rows represent genes and columns represent samples.
3.2. Construction of Gene Co-Expression Network
In this research, weighted gene co-expression analysis (WGCNA) was applied to predict significant genes in the development of AS ( 10). By such analysis, the strongly correlated gene modules and the gene membership in a module are identified. Firstly, hierarchical clustering was used to detect sample outlier ( 11). Then, Pearson’s correlation cor(i, j)|, between every pair of genes was calculated. This correlation matrix was transformed into a matrix of connection strengths (i.e. an adjacency matrix) by a power function , which resulted in a weighted co-expression network. The various power β value was used to estimate the scale-free topology criterion, as described in the original publication( 12.
Signed and unsigned networks are two of the more frequently used co-expression networks which are constructed based on the correlation coefficient values and absolute values, respectively. In this study, the signed network was constructed ( 13). Subsequently, the topological overlap measure (TOM) ( 14) was employed to combine the adjacency of each two genes and also their connection strengths with other neighbor genes. The genes inside a module can be summarized with the module eigengene (ME), which is defined as the first principal component of the expression profiles ( 15). Next, the dissimilarity matrix 1-TOM was calculated as the input for hierarchical clustering by flashClust package. Afterward, the modules were identified by the cutreeDynamic function ( 16) with parameters deepSplit = 2 and minClusterSize = 30, and default values were used for other parameters. Flow diagram of the underlying protocol is presented in Figure 1.
Figure 1.
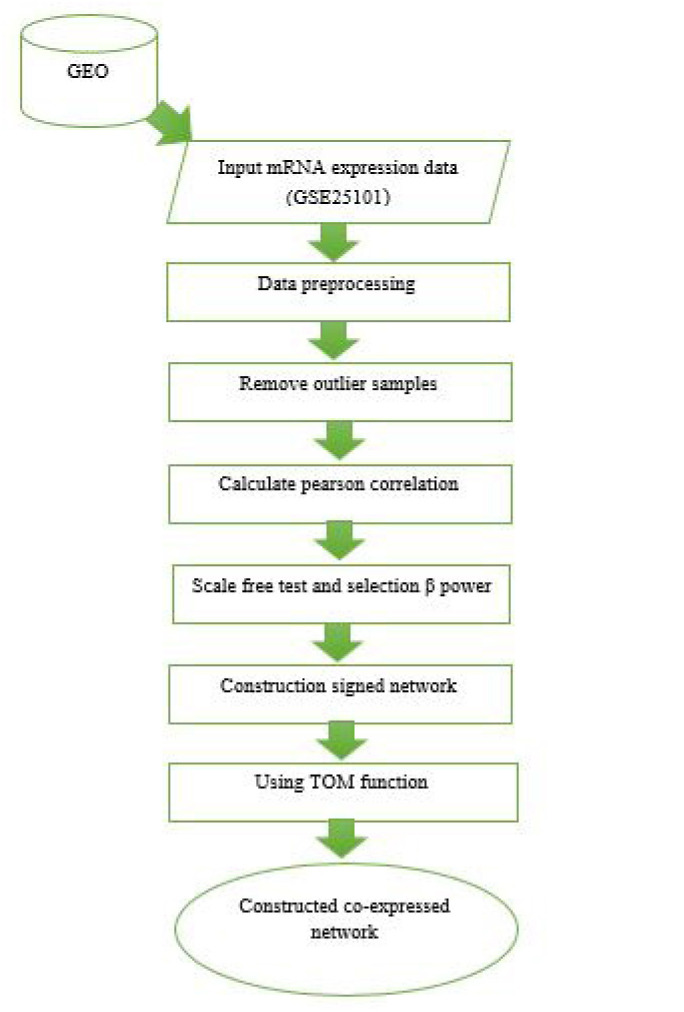
Flow diagram of Construction of Gene Co-expression Network.
In order to select the module with the highest correlation with AS disease, the support vector machine (SVM) classification procedure was utilized. SVM classifier explores the optimal hyperplane that can fulfill the classification requirement. The distance between two support vectors is considered as the criteria for the optimal margin evaluation ( 17). The samples were classified based on the genes of each module and class label (normal or patient). The SVM was operated for each module, then the results were evaluated using the receiver operating characteristic (ROC) curves. The module with the highest AUC values was selected for further analysis. To find genes with higher membership value in the selected module, the module membership value was calculated. The module membership determines the gene significance within the module, in which higher absolute value of MM(i) specifies the more important gene i in the module ( 10). The genes in the candidate module were chosen when their membership to the candidate module was more than 0.8 and their adjusted p-values were significant (adjusted p-values < 0.01). The module co-expression network was visualized in Cytoscape_v3.6.0 ( 18).
3.3. Functional Enrichment Analysis
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used to carry out functional enrichment analysis ( 19). Pathways with adjusted p-values < 0.001 were selected as significant. The genes which enriched in the significant pathways were chosen for future studies.
3.4. Candidate Genes Validation Using Quantitative Real-Time Polymerase Chain Reaction
Real-time PCR was performed to validate six representative genes which randomly selected from
AS related genes in significant module. In the current study, 53 AS patients attending Shariati Hospital, Tehran, Iran were recruited along with 49 healthy controls. The healthy group had no clinical evidence or family history of any type of autoimmune disease. Healthy controls were matched to the patients in terms of gender and age. Diagnosis of AS was performed by a rheumatologist based on the modified New York criteria ( 20). Since neither two AS patients nor two controls were selected from the same family, the intrafamily correlation was excluded. All AS patients and healthy subjects declared their agreement to enter the study by signing the consent form. This study was approved by The Human Research Ethics Committee, Tehran University of Medical Sciences. Disease severity and functional disabilities of patients were assessed through Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), ( 21) Bath Ankylosing Spondylitis Functional Index (BASFI) ( 22), and Bath Ankylosing Spondylitis Metrology Index (BASMI) ( 23). HLA-B27 screening for each AS patient was performed. Baseline characteristics and clinical manifestations of AS patients are present in Table S1 (Supplementary Materials).
The experiment was performed by obtaining 10 mL of peripheral blood from all the subjects. Then, peripheral blood mononuclear cells (PBMCs) were isolated using Ficoll-Hypaque (inno-Train Diagnostik GmbH, Taunus, Germany) density gradient centrifugation approach. High Pure RNA Isolation Kit (Roche, Mannheim, Germany) was used to extract total cellular RNA, according to the manufacturer’s instruction. Next, DNA and RNA contents of samples were quantified spectrophotometrically by recording absorbance at 260 and 280 nm (NanoDrop 2000c Spectrophotometer, Thermo Fisher Scientific, Wilmington, DE, USA).
cDNA was synthesized from 1 μg of total RNA by Transcriptor First Strand cDNA Synthesis Kit (Roche, Germany) according to the manufacturer’s instructions. StepOnePlus Real-time PCR system (Applied Biosystems, Foster City, CA, USA) and SYBR Green (Ampliqon, Odense, Denmark) master mix were employed. Primer sequences are listed in Table S2 (Supplementary Materials). The relative amount of target mRNA in each test sample was calculated and normalized to the corresponding β2-microglobulin (β2M) mRNA transcript level as a housekeeping gene. Comparative CT method was used to evaluate the quantitative mRNA expression, as formerly described by Schmittgen and Livak ( 24). Then, the relative expression for each sample was calculated according to the following equation: relative mRNA expression = (2− ΔCt) ×103.
3.5. Statistical Analysis
The data were analyzed using SPSS software version 22 (SPSS, Chicago, IL, USA). The normality of data distribution was evaluated using the Kolmogorov–Smirnov test. In the cases of normally distributed data, the independent sample t-test was used to compare differences between two groups but if data were not normal, the Man-Whitney test was used. The GraphPad Prism version 6 (GraphPad Software, La Jolla, CA USA, www.graphpad.com) was used to draw the graphs. The correlations between the expression of genes and clinicopathological characteristics were analyzed employing Spearman’s Rank-Order. Data were expressed as mean ± standard deviation (SD) with statistical significance set (P = 0.05).
4. Results
4.1. Data Preprocessing and Sample Selection
Gene expression dataset GSE25101 was firstly quantile normalized and log 2 transformed. The samples were assessed in terms of missing entries. Finally, 11484 annotated genes were used to construct the co-expression network. No sample was detected as the outlier.
4.2. Construction of Weighted Gene Co-Expression Network
To specify the required criterion for construction of WGCN, the scale-free topology fit index was calculated for different soft-thresholding power. It reached above 0.8 for a power of 8 while a relatively high mean connectivity remained. Next, the signed co-expression network was constructed based on the correlation between gene expression levels. After clustering the co-correlated genes, 25 modules with corresponding color assignments were identified. Each color represents a module in the constructed gene co-expression network by WGCNA (Fig. 2).
Figure 2.

Dendrogram plot displaying the co-expression modules determined by the WGCNA, labeled by color. Each color represents a certain gene module. 25 modules with corresponding color are illustrated.
4.3. Module-Trait Relationships
To find the most AS associated module, accuracy and ROC diagram for SVM classification has been calculated and depicted in Figure 3. Among these 25 modules, green-yellow and black modules demonstrated statistically significant relation to the traits (AUC (green-yellow) = 0.89, AUC (black) = 0.67). The number of genes in green-yellow and black modules was 350 and 421, respectively. Since the AUC of black module was calculated as 0.67, the green-yellow module was selected for further enrichment analysis.
Figure 3.

Module-Trait Relationships ROC (MTR_ROC). Among 25 modules, green-yellow and black modules demonstrated statistically significant related to the traits (AUC (green-yellow) = 0.89, AUC (black) = 0.67).
The genes co-expression network in module green-yellow is visualized in Figure S1 (Supplementary Materials). It consists of 350 nodes and 756 edges.
4.4. Modules Preservation Across Independent Dataset
A second dataset (i.e., the samples of the GSE73754 dataset) was assessed to examine whether the modules resulting from the first dataset (GSE25101) can be replicated. To this purpose, modulePreservation function in the WGCNA package was applied ( 10). Generally, the modules with Zsummary <2 considered as no preservation, 2 < Zsummary < 10 implies moderate preservation, while a Z-statistic > 10 implies strong module preservation. Altogether, a Zsummary ≥ 2 indicates that the modules are preserved (or reproducible) ( 25). The green-yellow module showed moderate preservation (2 ≤ Z-statistic ≤ 10) between the two datasets (Fig. S2, Supplementary Materials).
4.5. Pathway Enrichment Analysis
Pathway enrichment analysis of green-yellow module was performed using the DAVID online tools. It showed the enrichment of significant genes in eight remarkable pathways under the condition of adjusted p-values < 0.001 (Table 1). These pathways were Ribosome, Oxidative phosphorylation, Parkinson’s disease, nonalcoholic fatty liver disease, Alzheimer’s disease, Huntington’s disease, spliceosome, and cardiac muscle contraction pathways. Nine genes were involved in most of the mentioned pathways, which may have potential as biomarkers and treatment purposes.
Table1.
Significant pathways with adjusted p-value < 0.001.
| Terms | Count | adjusted p-value | Genes |
|---|---|---|---|
| Ribosome | 36 | 7.4E-27 | RPL4, RPL31, MRPS11, RPL11, MRPL36, MRPL15, RPL9, MRPL35, RPL6, MRPL32, RPL7, MRPL11, MRPL20, MRPL3, RPS17, MRPL1, RPL36AL, RPL35, RPL17, RPL39, RPL41, RPS7, RPL23, MRPL27, MRPS21, RPL35A, RPS3A, MRPL22, MRPL30, RPS27, RPL37A, FAU, RPL26, RPS21, RPL26L1, RPS24 |
| Oxidative phosphorylation | 29 | 8.3E-19 | COX7B, NDUFB8, NDUFB7, NDUFB10, UQCRB, NDUFA11, NDUFA12, COX17, NDUFB3, NDUFB2, ATP5C1, ATP5J, COX7A2, ATP5I, ATP5H, COX6A1, ATP5O, COX5B, COX7C, UQCRH, NDUFV2, NDUFA4, NDUFA1, COX6C, UQCRHL, ATP5J2, UQCRQ, NDUFS5, NDUFS4 |
| Parkinson's disease | 26 | 5.18E-15 | COX7B, NDUFB8, NDUFB7, NDUFB10, UQCRB, NDUFA11, NDUFA12, NDUFB3, NDUFB2, ATP5C1, ATP5J,COX7A2, ATP5H, COX6A1, ATP5O, COX5B, COX7C, UQCRH, NDUFV2, NDUFA4, NDUFA1, COX6C, UQCRHL, UQCRQ, NDUFS5, NDUFS4 |
| Huntington's disease | 28 | 1.1E-13 | COX7B, NDUFB8, NDUFB7, NDUFA11, NDUFB10, UQCRB, NDUFA12, NDUFB3, NDUFB2, ATP5C1, CLTA, ATP5J, COX7A2, ATP5H, COX5B, COX6A1, ATP5O, COX7C, UQCRH, NDUFV2, NDUFA4, NDUFA1, COX6C, UQCRHL, UQCRQ, NDUFS5, NDUFS4, TAF4B |
| Alzheimer's disease | 26 | 2.8E-13 | COX7B, NDUFB8, NDUFB7, NDUFB10, UQCRB, NDUFA11, NDUFA12, NDUFB3, NDUFB2, ATP5C1, ATP5J, COX7A2, ATP5H, COX6A1, ATP5O, COX5B, COX7C, UQCRH, NDUFV2, NDUFA4, NDUFA1, COX6C, UQCRHL, UQCRQ, NDUFS5, NDUFS4 |
| Non-alcoholic fatty liver disease (NAFLD) | 22 | 1.5E-10 | COX7B, NDUFB8, NDUFB7, NDUFB10, UQCRB, NDUFA11, NDUFA4, NDUFA12, NDUFB3, NDUFB2, NDUFA1, COX7A2, COX6A1, COX6C, COX5B, COX7C, UQCRH, UQCRHL, UQCRQ, NDUFS5, NDUFS4, NDUFV2 |
| Spliceosome | 13 | 5.4 E-4 | SF3B5, SF3B6, BUD31, LSM5, LSM3, PQBP1, LSM7, PRPF18, SNRPD1, TRA2B, SNRPG, SNRNP27, SNRPF |
| Cardiac muscle contraction | 10 | 5.2E-04 | COX7B, UQCRB, UQCRQ, COX7A2, COX6C, COX6A1, COX5B, COX7C, UQCRHL, UQCRH |
4.6. Demographic Data
The mean age of patient and healthy groups were 40 ± 10.9 and 38 ± 9.9 years, respectively. Any significant difference was found between the ages of two groups. Moreover, the patient group composed of 13 females and 40 males whereas the healthy group included 11 females and 38 males.
4. 7. Quantitative Real-time PCR Validation
The expression levels of six genes (COX7C, UQCRH, LSM3, PSMA4, NSMCE2, and MRPS11) in the green-yellow module were evaluated to validate the results of the network-based analysis. These genes were downregulated in the AS patients rather than normal subjects in the microarray dataset. The expression levels of MRPS11 and NSMCE2 were downregulated (both 0.8-fold, with P = 0.044 and P = 0.029, respectively) in the patient validation samples, however, the UQCRH gene was upregulated (1.2-fold, P = 0.007; Fig. 4). The difference between the expression levels of COX7C, LSM3, and PSMA4 in normal and AS patients was not significant. Further, the relative UQCRH gene expression level was significantly higher in HLA-B27 positive than in the HLA-B27 negative patients’ group (1.3-fold, P = 0.041; Fig. S3, Supplementary Materials).
Figure 4.

Relative expression levels of three selected genes (MRPS11, NSMCE2 and UQCRH) according to the network and sub-networks findings. The expression of genes in AS patients (black bars) compared to normal controls (white bars) are shown (*P <0.05, ** P <0.01, *** P < 0. 0.001). MRPS11 and NSMCE2 were downregulated (both 0.8-fold, with P = 0.044 and P = 0.029, respectively) and UQCRH gene was upregulated (1.2-fold, P = 0.007).
4.8. Correlations between Gene Expression and Clinical Indices
The correlations between the expression of UQCRH, NSMCE2, and MRPS11 genes and clinical indices of AS patients are demonstrated in Table 2. Although the significant correlation was found between the expression of MRPS11 gene and BASDAI scores (r = 0.287, P = 0.041) there were no significant correlations between the expression of three significant genes and clinical manifestations of the patients.
Table2.
Correlation between differentially expressed genes validated by Real-time PCR and AS clinical indices.
| Gene | Clinical Index | Correlation Coefficient | P |
|---|---|---|---|
| UQCRH Gene Expression | BASDAI | 0.21 | 0.14 |
| BASMI | 0.17 | 0.23 | |
| BASFI | 0.16 | 0.25 | |
| ASQoL | 0.26 | 0.06 | |
| ESR | 0.12 | 0.42 | |
| MRPS11 Gene Expression | BASDAI | 0.29 | 0.04 |
| BASMI | 0.01 | 0.96 | |
| BASFI | 0.13 | 0.34 | |
| ASQoL | 0.24 | 0.08 | |
| ESR | -0.02 | 0.89 | |
| NSMCE2 Gene Expression | BASDAI | 0.18 | 0.19 |
| BASMI | -0.8 | 0.59 | |
| BASFI | -0.13 | 0.92 | |
| ASQoL | 0.08 | 0.55 | |
| ESR | -0.11 | 0.46 |
Bold item demonstrates statistically significant value.
5. Discussion
In the present study, WGCNA was employed to identify the gene signatures involved in the pathogenesis of AS disease. The co-expression analysis revealed the green-yellow module as the most related module to AS. The remarkable genes in this module were determined through pathway analysis since the common genes in statistically significant pathways were considered as the AS biomarkers.
The presence of genes such as LSM3 homolog, U6 small nuclear RNA and mRNA degradation associated (LSM3), Mitochondrial ribosomal protein S11 (MRPS11), E3 SUMO-protein ligase NSE2 (NSMCE2), Proteasome subunit, alpha type 4 (PSMA4), Ubiquitin-like 5 (UBL5), ribosomal protein L17 (RPL17), Mitochondrial ribosomal protein L22 (MRPL22), and ribosomal protein S17 (RPS17) in the significant module (green-yellow) that have been reported in previous AS studies, supported the validity of module.
LSM3 gene is involved in pre-mRNA splicing. Herein, we identified that LSM3 gene was downregulated in the AS cases. Perturbations in processing and splicing of pre-mRNAs might affect the expression quality and quantity of other genes which might be involved in the AS pathogenesis. Also, the expression of spliceosome members in the various leukocyte subsets is associated with the inflammatory, oxidative, and proatherogenic profile of AS patients ( 26).
MRPS11 gene was downregulated in the AS subjects with the largest effect size. NSMCE2 gene is one of the tops downregulated genes in AS patients as well. These findings are the results of the analysis that have led to the identification of genes with unknown transcriptional changes in AS( 27). PSMA4 gene is the 20S proteasome core which is required for proteasome function ( 28). The proteasome complex is a proteolytic system which has key role in the function of the adaptive immune system through the major histocompatibility complex (MHC) class I ( 29).
Previous studies revealed the upregulation of proteins in the ubiquitin-proteasome pathway for AS patients. Ubiquitin-like 5 gene encodes UBL5 protein, which is similar to ubiquitin. UBL5 may not directly bind to target proteins but impresses the ubiquitin-proteasome pathway (UPP) through conjugation with the protein by an isopeptide bond.
The ubiquitin-proteasome pathway of protein degradation involves the conjugation of ubiquitin molecules to the targeted protein that is recognized by proteasome subunits. It is an ATP-dependent process with the involvement of three enzymes ( 30). The ribosomal protein and proteasome domains may be disturbed through the abnormal expression of DEGs. This event leads to the incidence of the AS disease. Wright et al. demonstrated a significant differential UPP protein expression in the monocytes from patients with AS and emphasized the important role of UPP in AS ( 31). Moreover, ribosomal proteins such as RPL17, MRPL22 ( 32), and RPS17 ( 33) might have a close correlation with AS.
The pathway analysis demonstrated the signaling pathways like oxidative phosphorylation ( 34- 36), ribosome ( 31) and Parkinson’s ( 37), Alzheimer’s ( 38), Huntington’s ( 39) diseases, as well as spliceosome ( 40), cardiac muscle contraction ( 41), and nonalcoholic fatty liver disease (NAFLD) ( 42) that all of them are associated with AS. Moreover, according to pathway enrichment, nine genes were involved in various pathways. Ubiquinol-cytochrome c reductase binding protein (UQCRB), ubiquinol-cytochrome c reductase hinge protein (UQCRH), ubiquinol-cytochrome c reductase hinge protein-like (UQCRHL), ubiquinol-cytochrome c reductase complex III subunit VII (UQCRQ), cytochrome c oxidase subunit 7B (COX7B), cytochrome c oxidase subunit 5B (COX5B), cytochrome c oxidase subunit 6C (COX6C), cytochrome c oxidase subunit 6A1 (COX6A1), and cytochrome c oxidase subunit 7C (COX7C) are implicated in six remarkable pathways and seems to have a higher significance versus other genes. UQCRB, UQCRH, UQCRHL, and UQCRQ encode proteins that are parts of the complex III or cytochrome b-c1 complex, and a component of the mitochondrial respiratory chain (https://www.ncbi.nlm.nih.gov/protein). Furthermore, COX7B, COX5B, COX6C, COX6A1, and COX7C encode proteins that are the nuclear-coded polypeptide chains of cytochrome c oxidase (https://www.ncbi.nlm.nih.gov/protein). The association of cytochrome c and cytochrome bc1 complex (complex III, ubiquinol: cytochrome c oxidoreductase) with the autoimmune diseases through reactive oxygen species (ROS) production has been proposed. Cytochrome c can amplify ROS production in mitochondria ( 43). Moreover, cytochrome bc1 complex and complex I (NADH: ubiquinone oxidoreductase) are known as the major sources of superoxide anion radicals ( 44). The critical role of ROS in the pathophysiology of autoimmune diseases has been suggested. ROS can cause the “neo-antigen” generation by changing the structure of cellular antigens, which leads to increase the autoimmune response against the original antigen. ROS are also implicated in the activation of antigen-presenting cells, apoptosis, and amplification of diverse immunologic reactions ( 45) as well as in the pathophysiology of several autoimmune diseases including Parkinson’s, Huntington’s, Alzheimer’s diseases, and systemic lupus erythematosus (SLE) disease ( 46, 47).
Moreover, we validated the expression of UQCRH, MRPS11, and NSMCE2 by Real-time PCR. Expression of UQCRH gene was increased in our samples while it was downregulated in the co-expression network. The difference in samples can be regarded as a possible cause to justify such inconsistency. Moreover, several confounding factors, such as disease stage, sample formation, cell-specific frequently, and experimental factors that can severely influence the outcome of a transcriptome analysis could be the underlying reasons for such discrepancy ( 48).
The UQCRH gene encodes a hinge protein consisting of 91 amino acids that are distributed in the nucleus and mitochondria. It is also primarily involved in mitochondrial oxidative phosphorylation. As the main subunit of ubiquinol-cytochrome c reductase complex, UQCRH is responsible for electron transfer between cytochrome c and cytochrome c1 during oxidative phosphorylation. The abnormal high expression level of this gene can lead to cellular ROS production ( 49). In addition to upregulation of UQCRH in the AS patients compared with controls, it also overexpressed in the HLA-B27 positive AS subjects in comparison to HLA-B27 negative AS patients (1.3 times; P = 0.041). These data indicate that UQCRH gene may intensify AS pathogenesis in HLA-B27 positive subjects, since HLA-B27 molecule was already reported to be involved in TNF production ( 50) and, hence an inflammation in AS.
MRPS11 is a component of the ribosomal small subunit that binds to 12S rRNA ( 51). All of the Mitochondrial Ribosomal Proteins (MRPs) are encoded by nuclear genes and involved in protein synthesis within the mitochondrion. The mitoribosomes of mammals are 55-60S particles and consist of a small 28S and a large 39S subunits ( 52). Mutations or deficiencies of ribosome assembly proteins implicated in mitochondrial translation are potential candidates for mitochondrial diseases since the mitochondrial ribosome translates mRNAs for the 13 vital components of the oxidative phosphorylation system ( 53). Several studies have shown that mitochondrial dysfunction is evidence of neurodegenerative diseases, some autoimmune disorders, and cancers ( 54, 55). Analysis based on the Spearman’s rho test indicated a positive correlation between disease activity (measured by BASDAI) and MRPS11 gene expression. Some researchers have affirmed that the disease severity of AS might be related to the HLA-B27 status. Indeed, Freeston et al. reported that HLA-B27 positive patients with AS have significantly longer disease duration and present worse BASDAI, BASFI, and ankylosing spondylitis quality of life (ASQoL) compared with HLA-B27 negative AS ( 56). However, there is no study on the correlation between expression of MRPS11 gene and disease activity indices in the course of AS disease, and more studies are needed.
NSMCE2 gene is a component of the structural maintenance of chromosomes (Smc) 5/6 complex, which is involved in DNA repair ( 57). To prevent DNA damage from compromising the genetic integrity of an organism, cells employ surveillance mechanisms to sense damaged DNA and to elicit concordant cellular responses such as DNA repair, cell cycle arrest, and apoptosis. Mutations of genes involved in the DNA damage response pathways can also lead to diseases such as cancer, immune deficiencies ( 58), and some connective tissue diseases such as rheumatoid arthritis. Furthermore, defects in DNA damage and DNA repair mechanisms may contribute to the AS process ( 59) as we detected differentially expression pattern of NSMCE2 in the AS subjects.
6. Conclusion
In conclusion, we successfully identified eight pathways in Ankylosing spondylitis based on co-expression network analysis. Moreover, we predicted 17 genes as potential biomarkers for AS and confirmed UQCRH, MRPS11, and NSMCE2 as the differently expressed genes in Ankylosing spondylitis patients using RT-PCR assay. These genes are proposed as the biomarkers for early detection and therapy for Ankylosing spondylitis.
Declarations
Ethics, consent and permissions: The ethical committee of Tehran University of Medical Sciences approved this study. All study subjects signed the consent forms before being included in the study.
Consent for publication: Not applicable.
Availability of data and material
The datasets used for the current study are available in the NCBI GEO repository, (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE25101)
Competing interests
Leila Najafzadeh, Mahdi Mahmoudi, Mostafa Ebadi, Marzieh Dehghan Shasaltaneh and Ali Masoudi-Nejad, declare that they have no conflict of interest.
Funding: NA
Acknowledgement
We would like to thank Saber Ghanbari Ara and Saeed Aslani without whom we could not complete this study. We also appreciate great help of Dr. Mohadese Zaree for critical reading the manuscript.
Conflict of Interest: None.
References
- 1.Assassi S, Reveille JD, Arnett FC, Weisman MH, Ward MM, Agarwal SK, et al. Whole-Blood Gene Expression Profiling in Ankylosing Spondylitis Shows Upregulation of Toll-Like Receptor 4 and 5. J Rheumatol. 2011;38(1):87–98. doi: 10.3899/jrheum.100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colbert RA, Tran TM, Layh-Schmitt G. Hla-B27 Misfolding and Ankylosing Spondylitis. Mol Immunol. 2014;57(1):44–51. doi: 10.1016/j.molimm.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keidel S, Chen L, Pointon J, Wordsworth P. Erap1 and Ankylosing Spondylitis. Curr Opin Immunol. 2013;25(1):97–102. doi: 10.1016/j.coi.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 4.Zambrano-Zaragoza JF, Agraz-Cibrian JM, González-Reyes C, Durán-Avelar M, Vibanco-Pérez N. Ankylosing Spondylitis: From Cells to Genes. Int J Inflam. 2013;2013 doi doi: 10.1155/2013/501653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Machado P, Landewe R, Lie E, Kvien TK, Braun J, Baker D, et al. Ankylosing Spondylitis Disease Activity Score (Asdas): Defining Cut-Off Values for Disease Activity States and Improvement Scores. Ann Rheum Dis. 2011;70(1):47–53. doi: 10.1136/ard.2010.138594. [DOI] [PubMed] [Google Scholar]
- 6.Brionez TF, Reveille JD. The Contribution of Genes Outside the Major Histocompatibility Complex to Susceptibility to Ankylosing Spondylitis. Curr Opin Rheumatol. 2008;20(4):384–391. doi: 10.1097/bor.0b013e32830460fe. [DOI] [PubMed] [Google Scholar]
- 7.Browne BC, Hochgräfe F, Wu J, Millar EK, Barraclough J, Stone A, et al. Global Characterization of Signalling Networks Associated with Tamoxifen Resistance in Breast Cancer. FEBS J. 2013;280(21):5237–5257. doi: 10.1111/febs.12441. [DOI] [PubMed] [Google Scholar]
- 8.Pimentel-Santos FM, Ligeiro D, Matos M, Mourão AF, Costa J, Santos H, et al. Whole Blood Transcriptional Profiling in Ankylosing Spondylitis Identifies Novel Candidate Genes That Might Contribute to the Inflammatory and Tissue-Destructive Disease Aspects. Arthritis Res Ther. 2011;13(2):R57. doi: 10.1186/ar3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du P, Kibbe WA, Lin SM. Lumi: A Pipeline for Processing Illumina Microarray. Bioinformatics. 2008;24(13):1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 10.Langfelder P, Horvath S. Wgcna: An R Package for Weighted Correlation Network Analysis. BMC bioinformatics. 2008;9(1):559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu M, Li W, James GM, Mehan MR, Zhou XJ. Automated Multidimensional Phenotypic Profiling Using Large Public Microarray Repositories. Proc Natl Acad. 2009;106(30):12323–12328. doi: 10.1073/pnas.0900883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Horvath S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat Appl Genet Mol Biol. 2005;4(1) doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 13.Mousavian Z, Nowzari-Dalini A, Stam RW, Rahmatallah Y, Masoudi-Nejad A. Network-Based Expression Analysis Reveals Key Genes Related to Glucocorticoid Resistance in Infant Acute Lymphoblastic Leukemia. Cell Oncol. 2017;40(1):33–45. doi: 10.1007/s13402-016-0303-7. [DOI] [PubMed] [Google Scholar]
- 14.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási A-L. Hierarchical Organization of Modularity in Metabolic Networks. Science. 2002;297(5586):1551–1555. doi: 10.1126/science.1073374. [DOI] [PubMed] [Google Scholar]
- 15.Yip AM, Horvath S. Gene Network Interconnectedness and the Generalized Topological Overlap Measure. BMC bioinformatics. 2007;8(1):22. doi: 10.1186/1471-2105-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langfelder P, Zhang B, Horvath S. Defining Clusters from a Hierarchical Cluster Tree: The Dynamic Tree Cut Package for R. Bioinformatics. 2007;24(5):719–720. doi: 10.1093/bioinformatics/btm563. [DOI] [PubMed] [Google Scholar]
- 17.Suykens JA, Vandewalle J. Least Squares Support Vector Machine Classifiers. Neural Process Lett. 1999;9(3):293–300. [Google Scholar]
- 18.Kohl M, Wiese S, Warscheid B. Data Mining in Proteomics: Springer . 2011. Cytoscape: Software for Visualization and Analysis of Biological Networks; pp. 291–303. [DOI] [PubMed] [Google Scholar]
- 19.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. David: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(9):R60. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 20.Van Der Linden S, Valkenburg HA, Cats A. Evaluation of Diagnostic Criteria for Ankylosing Spondylitis. Arthritis Rheum. 1984;27(4):361–368. doi: 10.1002/art.1780270401. [DOI] [PubMed] [Google Scholar]
- 21.Garrett S, Jenkinson T, Kennedy LG, Whitelock H, Gaisford P, Calin A. A New Approach to Defining Disease Status in Ankylosing Spondylitis: The Bath Ankylosing Spondylitis Disease Activity Index. J Rheumatol. 1994;21(12):2286–2291. doi: 10.1093/rheumatology/34.8.793. [DOI] [PubMed] [Google Scholar]
- 22.Calin A, Garrett S, Whitelock H, Kennedy L, O’hea J, Mallorie P, et al. A New Approach to Defining Functional Ability in Ankylosing Spondylitis: The Development of the Bath Ankylosing Spondylitis Functional Index. J Rheumatol. 994;21(12):2281–2285. doi: 10.1093/rheumatology/34.8.793. [DOI] [PubMed] [Google Scholar]
- 23.Bidad K, Fallahi S, Mahmoudi M, Jamshidi A, Farhadi E, Meysamie A, et al. Evaluation of the Iranian Versions of the Bath Ankylosing Spondylitis Disease Activity Index (Basdai), the Bath Ankylosing Spondylitis Functional Index (Basfi) and the Patient Acceptable Symptom State (Pass) in Patients with Ankylosing Spondylitis. Rheumatol Int. 2012;32(11):3613–3618. doi: 10.1007/s00296-011-2186-2. [DOI] [PubMed] [Google Scholar]
- 24.Schmittgen TD, Livak KJ. Analyzing Real-Time Pcr Data by the Comparative C T Method. Nat Protoc. 2008;3(6):1101. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 25.Langfelder P, Luo R, Oldham MC, Horvath S. Is My Network Module Preserved and Reproducible? PLoS Comput Biol. 2011;7(1) doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jimenez-Gomez Y, Lopez-Pedrera C, Pedraza-Arévalo S, Άbalos-Aguilera M, Ruiz-Limon P, Perez-Sanchez C, et al. ri0429 Dysregulation of the Splicing Machinery in Leukocytes from Ankylosing Spondylitis Patients Is Associated to Disease Pathogenesis. BMJ Open. 2017 doi: 10.1136/annrheumdis-2017-eular.4845. [DOI] [Google Scholar]
- 27.Lee Y, Song G. Meta-Analysis of Differentially Expressed Genes in Ankylosing Spondylitis. Genet Mol Res. 2015;14(2):5161–5170. doi: 10.4238/2015.may.18.6. [DOI] [PubMed] [Google Scholar]
- 28.Davoli R, Fontanesi L, Russo V, Čepica S, Musilová P, Stratil A, et al. The Porcine Proteasome Subunit A4 (Psma4) Gene: Isolation of a Partial Cdna, Linkage and Physical Mapping. Anim Genet. 1998;29(5):385–388. doi: 10.1046/j.1365-2052.1998.295354.x. [DOI] [PubMed] [Google Scholar]
- 29.Saiki T, Kawai T, Morita K, Ohta M, Saito T, Rokutan K, et al. Identification of Marker Genes for Differential Diagnosis of Chronic Fatigue Syndrome. Mol Med. 2008;14(9-10):599–607. doi: 10.2119/2007-00059.saiki. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lecker SH, Goldberg AL, Mitch WE. Protein Degradation by the Ubiquitin–Proteasome Pathway in Normal and Disease States. J Am Soc Nephrol. 2006;17(7):1807–1819. doi: 10.1681/asn.2006010083. [DOI] [PubMed] [Google Scholar]
- 31.Wright C, Edelmann M, Kollnberger S, Kramer H, McGowan S, McHugh K, et al. Ankylosing Spondylitis Monocytes Show Upregulation of Proteins Involved in Inflammation and the Ubiquitin Proteasome Pathway. Ann Rheum Dis. 2009;68(10):1626–1632. doi: 10.1136/ard.2008.097204. [DOI] [PubMed] [Google Scholar]
- 32.Aldea G, Výprachtický D, Cimrová V, editors. Modification of Poly (Styrene‐Alt‐Maleic Anhydride) with 1, 3, 4‐Oxadiazole Units for Electroluminescent Devices. Macromolecular Symposia. Wiley Online Library. 2004 doi: 10.1002/masy.200450868. [DOI] [Google Scholar]
- 33.Zhao H, Wang D, Fu D, Xue L. Predicting the Potential Ankylosing Spondylitis-Related Genes Utilizing Bioinformatics Approaches. Rheumatol Int. 2015;35(6):973–979. doi: 10.1007/s00296-014-3178-9. [DOI] [PubMed] [Google Scholar]
- 34.DiMauro S, Schon EA. Mitochondrial Disorders in the Nervous System. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- 35.Lian Z, Chai W, Shi LL, Chen C, Liu J, Wang Y. Analysis of Ppargc1b, Runx3 and Tbkbp1 Polymorphisms in Chinese Han Patients with Ankylosing Spondylitis: A Case-Control Study. PloS One. 2013;8(4):e61527. doi: 10.1371/journal.pone.0061527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ranganathan V, Gracey E, Brown MA, Inman RD, Haroon N. Pathogenesis of Ankylosing Spondylitis—Recent Advances and Future Directions. Nat Rev Rheumatol. 2017;13(6):359. doi: 10.1038/nrrheum.2017.56. [DOI] [PubMed] [Google Scholar]
- 37.Barker RA, Cahn AP. Parkinson’s Disease: An Autoimmune Process. Int. J. Neurosci. 1988;43(1-2):1–7. doi: 10.3109/00207458808985773. [DOI] [PubMed] [Google Scholar]
- 38.Dinkins MB, Dasgupta S, Wang G, Zhu G, He Q, Kong JN, et al. The 5xfad Mouse Model of Alzheimer’s Disease Exhibits an Age-Dependent Increase in Anti-Ceramide Igg and Exogenous Administration of Ceramide Further Increases Anti-Ceramide Titers and Amyloid Plaque Burden. J Alzheimers Dis. 2015;46(1):55–61. doi: 10.3233/jad-150088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, et al. A Novel Pathogenic Pathway of Immune Activation Detectable before Clinical Onset in Huntington’s Disease. J Exp Med. 2008;205(8):1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tabrizi Z, Mansouri R, Aslani S, Jamshidi A, Mahmoudi M. Expression Levels of the Microrna Maturing Microprocessor Complex Components. Drosha, Dicer, and Dgcr8 in Pbmcs from Ankylosing Spondylitis Patients. 2017 doi: 10.31138/mjr.28.2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ryall NH, Helliwell P. A Critical Review of Ankylosing Spondylitis. Crit Rev Phys Rehabil Med. 1998;10(3) doi: 10.1615/critrevphysrehabilmed.v10.i3.40. [DOI] [Google Scholar]
- 42.Zhu J-Z, Zhu H-T, Dai Y-N, Li C-X, Fang Z-Y, Zhao D-J, et al. Serum Periostin Is a Potential Biomarker for Non-Alcoholic Fatty Liver Disease: A Case–Control Study. Endocr. 2016;51(1):91–100. doi: 10.1007/s12020-015-0735-2. [DOI] [PubMed] [Google Scholar]
- 43.Akopova O, Kolchinskaya L, Nosar V, Bouryi V, Mankovska I, Sagach V. Cytochrome C as an Amplifier of Ros Release in Mitochondria. Fiziologichnyi Zhurnal. 2012;58(1):3–12. doi: 10.1615/intjphyspathophys.v3.i3.70. [DOI] [PubMed] [Google Scholar]
- 44.Kussmaul L, Hirst J. The Mechanism of Superoxide Production by Nadh: Ubiquinone Oxidoreductase (Complex I) from Bovine Heart Mitochondria. Proc Natl Acad Sci. 2006;103(20):7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Namazi M. Cytochrome-P450 Enzymes and Autoimmunity: Expansion of the Relationship and Introduction of Free Radicals as the Link. J Autoimmune Dis. 2009;6(1):4. doi: 10.1186/1740-2557-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benzi G, Moretti A. Are Reactive Oxygen Species Involved in Alzheimer’s Disease? Neurobiol Aging. 1995;16(4):661–674. doi: 10.1016/0197-4580(95)00066-n. [DOI] [PubMed] [Google Scholar]
- 47.Leishangthem B, Sharma A, Bhatnagar A. Role of Altered Mitochondria Functions in the Pathogenesis of Systemic Lupus Erythematosus. Lupus. 2016;25(3):272–281. doi: 10.1177/0961203315605370. [DOI] [PubMed] [Google Scholar]
- 48.Greene JG. Current Status and Future Directions of Gene Expression Profiling in Parkinson’s Disease. Neurobiol Dis. 2012;45(1):76–82. doi: 10.1177/0961203315605370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okazaki M, Ishibashi Y, Asoh S, Ohta S. Overexpressed Mitochondrial Hinge Protein, a Cytochromec-Binding Protein, Accelerates Apoptosis by Enhancing the Release of Cytochromecfrom Mitochondria. Biochem Biophys Res Commun. 1998;243(1):131–136. doi: 10.1006/bbrc.1997.7979. [DOI] [PubMed] [Google Scholar]
- 50.Poddubnyy DA, Märker-Hermann E, Kaluza-Schilling W, Zeidler H, Braun J, Listing J, et al. Relation of Hla-B27, Tumor Necrosis Factor-Α Promoter Gene Polymorphisms, and T Cell Cytokine Production in Ankylosing Spondylitis—a Comprehensive Genotype-Phenotype Analysis from an Observational Cohort. J Rheumatol. 2011:110130. doi: 10.3899/jrheum.110130. [DOI] [PubMed] [Google Scholar]
- 51.Koc EC, Burkhart W, Blackburn K, Moseley A, Spremulli LL. The Small Subunit of the Mammalian Mitochondrial Ribosome Identification of the Full Complement of Ribosomal Proteins Present. J. Biol. Chem. 2001;276(22):19363–19374. doi: 10.1074/jbc.m100727200. [DOI] [PubMed] [Google Scholar]
- 52.Pietromonaco S, Denslow N, O’Brien T. Proteins of Mammalian Mitochondrial Ribosomes. Biochimie. 1991;73(6):827–835. doi: 10.1016/0300-9084(91)90062-6. [DOI] [PubMed] [Google Scholar]
- 53.Lightowlers RN, Rozanska A, Chrzanowska-Lightowlers ZM. Mitochondrial Protein Synthesis: Figuring the Fundamentals, Complexities and Complications, of Mammalian Mitochondrial Translation. FEBS lett. 2014;588(15):2496–2503. doi: 10.1016/j.febslet.2014.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corrado M, Scorrano L, Campello S. Mitochondrial Dynamics in Cancer and Neurodegenerative and Neuroinflammatory Diseases. Int J Cell Biol. 2012;2012 doi: 10.1155/2012/729290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrera G, Gentile F, Pizzimenti S, Canuto RA, Daga M, Arcaro A, et al. Mitochondrial Dysfunction in Cancer and Neurodegenerative Diseases: Spotlight on Fatty Acid Oxidation and Lipoperoxidation Products. Antioxidants. 2016;5(1):7. doi: 10.3390/antiox5010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freeston J, Barkham N, Hensor E, Emery P, Fraser A. Ankylosing Spondylitis, Hla-B27 Positivity and the Need for Biologic Therapies. Joint Bone Spine. 2007;74(2):140–143. doi: 10.1016/j.jbspin.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Park HJ, Park HC, Choi J, Choi W, Chung WS, Kim S, et al. Identification of Sumo-Modified Proteins by Affinity Purification and Tandem Mass Spectrometry in Arabidopsis Thaliana. J. Plant Bio. 2013;56(3):176–185. doi: 10.1007/s12374-013-0127-1. [DOI] [Google Scholar]
- 58.Sancar A, Lindsey-Boltz LA, Ünsal-Kaçmaz K, Linn S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu Rev Biochem. 2004;73(1):39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 59.Frank O, Klein W, Kocsis F, Altmann H. DNA--” Rejoining” in Lymphycytes of Patients with a Minifestation of Rheumatoid Arthritis in Older Age (Author’s Transl) Aktuelle Gerontol. 1977;7(12):629–631. [PubMed] [Google Scholar]