

Abstract
High throughput and high-resolution lipid analyses are important for many biological model systems and research questions. This comprises both monitoring at the individual lipid species level and broad lipid classes. Here, we present a nontarget semiquantitative lipidomics workflow based on ultrahigh performance supercritical fluid chromatography (UHPSFC)-mass spectrometry (MS). The optimized chromatographic conditions enable the base-line separation of both nonpolar and polar classes in a single 7-minute run. Ionization efficiencies of lipid classes vary 10folds in magnitude and great care must be taken in a direct interpretation of raw data. Therefore, the inclusion of internal standards or experimentally determined Response factors (RF) are highly recommended for the conversion of raw abundances into (semi) quantitative data. We have deliberately developed an algorithm for automatic semiquantification of lipid classes by RF. The workflow was tested and validated using a bovine liver extract with satisfactory results. The RF corrected data provide a more representative relative lipid class determination, but also the interpretation of individual lipid species should be performed on RF corrected data. In addition, semiquantification can be improved by using internal or also external standards when more accurate quantitative data are of interest but this requires validation for all new sample types. The workflow established greatly extends the potential of nontarget UHPSFC–MS/MS based analysis.
Introduction
Lipids have multiple roles and functions across species (e.g. energy storage, cell protection, cell division and signaling) and their analysis has had growing interest in recent decades. That ranges from applied focus on biofuels and lipid accumulating microorganisms to more basic studies, e.g. changes in lipidome are associated with many metabolic and neurodegenerative disorders and even with cancer (1).
Lipids represent a broad group of compounds with high variation in their structure and polarity, which is demanding for analytical techniques available. In Table I we summarize the strength and shortcomings of the main analytical technologies used in lipid analysis. This leads to the conclusion that only liquid based separation combined with mass spectrometry (MS) offers the versatility needed for high throughput, high selectivity, high resolution and high sensitivity lipid analysis (2–4). In addition to chromatographic methods, direct infusion-MS(/MS) also became popular in lipidomic analysis since it is very fast and can be quite easily established in any lab with suitable equipment, but it suffers from ion suppression effect, and thus analysis of low abundant lipids might be compromised. Reversed-phase liquid (RP) chromatography is a great choice for separation of individual lipid compounds, which is driven by the carbon chain length and the number of double bonds, however lipids belonging to different classes frequently coelute (5). Hydrophilic-interaction liquid chromatography (HILIC) provides good separation of complex lipids according to their polarity, but nonpolar lipids elute in the void volume and remain nonresolved (6). Complete separation of lipids by class has been shown using normal phase liquid chromatography (7) (NP), however NP suffers from long elution and re-equilibration times and proves challenging for ionization and introduction intoMS.
Table I.
Overview of Most Popular Technologies for Lipidome Analysis
| Analytical technology | Throughput | LC coverage and selectivity | LS coverage and selectivity | Sensitivity | Quantification |
|---|---|---|---|---|---|
| RP-MS | + | + | +++ | +++ | ++ |
| HILIC-MS | + | ++ | ++ | +++ | ++ |
| NP-MS | + | ++ | ++ | +++ | ++ |
| SFC-MS | +++ | +++ | +++ | +++ | ++ |
| NMR | ++ | ++ | + | + | ++ |
| TLC | − | ++ | − | + | + |
Abbreviations: LC, lipid class; LS, lipid species; NMR, nuclear magnetic resonance; TLC, thin-layer chromatography.
1Classification: +++ superior properties, − not useful.
Few years ago a reliable commercial system for supercritical fluid chromatography (SFC) employing columns packed with sub-2 μm particles has been introduced (known as UHPSFC). SFC is essentially a form of a NP as it usually utilizes nonpolar mobile phase (with a polar modifier) and polar stationary phase. However, SFC offers several significant benefits in comparison with NP. One of the main advantages is given by the increased diffusivity of solutes, allowing better separation efficiency at higher eluent linear velocities and thus higher sample throughput (2). Moreover, SFC does not request a long time for re-equilibration of the column and is compatible with MS detection. We have adapted a previously developed and published method for nontarget lipid characterization of lipids using UHPSFC-ESI-MS (8) and utilized its potential to suggest and establish a strategy for lipid class quantification and a more precise interpretation at individual lipid species level.
Reliable quantification of multiple lipid classes remains challenging. Although we have observed a high demand for such methods and data, there are not many options available now and most of the strategies published suffer from various shortcomings. The conventional quantification method used in MS analyses is typically based on multiple-point internal standardization, which involves calibration curve and internal standards (ISTD) for each compound quantified (9). This approach is popular and widely used when single reaction monitoring (SRM) or multiple reaction monitoring (MRM) is employed but has limited application in nontarget lipidomics because of the wide variety of the lipidome. Relative quantification is often sufficient when relative changes are of interest, it is a simple method and provides a comprehensive comparison between biological samples (for example between diseased and control populations). Relative quantification, which does not require a calibration curve, and relies on the addition of a set of ISTDs representative of the classes of lipids analyzed, is a widely used and accepted quantification strategy in nontargeted lipidomics (10).
The UHPSFC method separates lipids based on their polarity, each class is thereby eluted in a discrete zone characterized by retention time, which allows to find total abundances of individual lipid classes and estimate their concentration. In this paper we introduce fast and simple strategy for lipid class quantification and compare different quantification strategies such as single-point ISTD method, multiple-point ISTD method and application of response factors (RF) adjusting variations in the abundances. Our overall Lipidomics strategy based on UHPSFC–MS is visualized in Figure 1. We have developed an algorithm (Lipid Class Algorithm) for efficient workflow. The algorithm allows an automatic filtering of lipid compounds, based on retention time and m/z thresholds (for more details, see Section 3.3), and collection of total lipid class abundances as well as correction of compound and lipid class response by experimentally determined RF. Raw or RF-corrected data can be extracted, depending on what type of question is raised. Also, raw data can be further used for quantification of compounds or/and lipid classes by applying a single-point or multiple-point ISTD method, but internal or also external standards are needed in such cases. The presented workflow greatly extends the potential of the nontargetdata.
Figure 1.
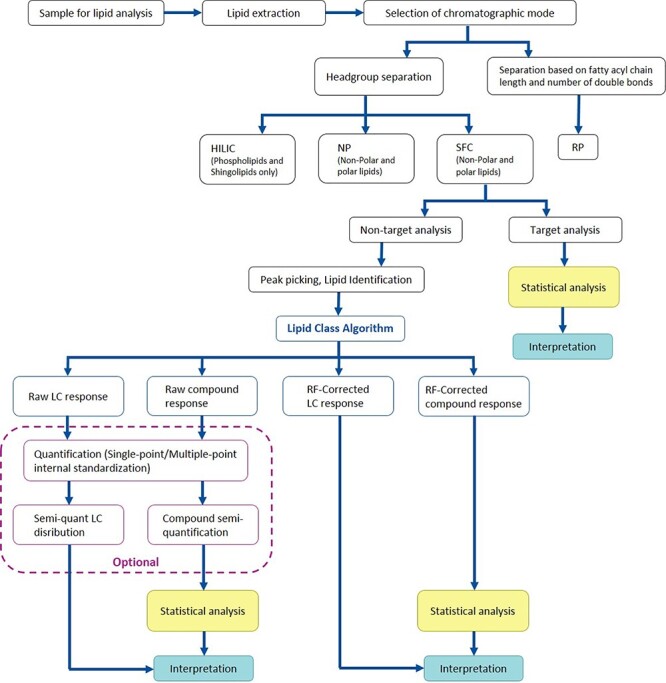
UHPSFC-MS lipidomics workflow for improved quantitative analysis of lipids. The workflow established enables characterization of lipid profile as well as automatic semiquantification of lipid classes or lipid compounds using experimentally determined response factors.
Materials and methods
Chemicals
MS grade water, 2-propanol (LC–MS grade), dichloromethane (GC grade, stabilized with 0.0020% of 2-methyl-2-butene) and chloroform (reagent Ph. Eur., stabilized with 0.6% of ethanol) were purchased from VWR International, Radnor, PA, USA. Methanol (LC–MS grade) were obtained from Merck KGaA (Darmstadt, Germany). Ammonium acetate (for MS, 98%) were purchased from Honeywell International Inc. (Morris Plains, NJ,USA).
ISTDs 1,2-di-tridecanoyl-sn-glycero-3-phosphocholine, PC 13:0/13:0; 1-docosanoyl-sn-glycero-3-phosphocholine, LPC 22:0/0:0; 1-tridecanoyl-sn-glycero-3-phosphoethanol-amine, LPE 13:0/0:0; N-lauroyl-D-erythro-sphingosylphosphorylcholine, SM d18:1/12:0; N-(2′-(R)-hydroxyheptadecanoyl)-D-erythro-sphingosine, Cer d18:1/17:0(OH) and cholesterol-d7 were purchased from Avanti Polar Lipids (Alabaster, AL). ISTDs 1-Monoheptadecanoin, MG 17:0/0:0/0:0; (1-hydroxy-3-pentadecanoyloxypropan-2-yl) pentadecanoate DG 15:0/15:0/0:0; 1,2-dimyristoyl-sn-glycero-3-phosphoethanolamine, PE 14:0/14:0; 1,2-dimyristoyl-sn-glycero-3-phosphatidyl-rac-glycerol (Na-salt), PG 14:0/14:0 and cholesteryl heptadecanoate, CE 17:0 were purchased from Larodan AB (Solna, Sweden). ISTD 1,2,3-tripentadecanoyl-sn-glycerol, TG(15:0/15:0/15:0) were obtained from Sigma–Aldrich (St Louis, MO,USA).
Lipid standards Cholesterol, FC; 1,2-distearoyl-sn-glycero-3-phosphocholine, PC 18:0/18:0; 1,2-dilinolenoyl-sn-glycero-3-phosphocholine, PC 18:3/18:3; 1-(1Z-octadecenyl)-2-oleoyl-sn-glycero-3-phosphocholine, PC P-18:0/18:1; 1,2-distearoyl-sn-glycero-3-phosphoethanolamine, PE 18:0/18:0; 1-heptadecenoyl-2-hydroxy-sn-glycero-3-phosphoethanolamine, LPE 17:1/0:0; N-heptadecanoyl-D-erythro-sphingosine, Cer d18:1/17:0; D-glucosyl-β-1,1′-N-heptadecanoyl-D-erythro-sphingosine, GlcCer d18:1/17:0; D-galactosyl-β-1,1′-N-stearoyl-D-erythro-sphingosine, GalCer d18:1/18:0 and N-palmitoyl-D-erythro-sphingosylphosphorylcholine, SM d18:1/16:0 were purchased from Avanti Polar Lipids (Alabaster, AL). Lipid standards glyceryl tristearate, TG 18:0/18:0/18:0; glyceryl trioleate, TG 18:1/18:1/18:1; glyceryl trilinoleate, TG 18:2/18:2/18:2; 1,2-di (cis-9-octadecenoyl)-rac-glycerol, DG 18:1/18:1/0:0; 1,3-di (cis-9-octadecenoyl) glycerol, DG 18:1/0:0/18:1; 1-(cis-9-Octadecenoyl)-rac-glycerol, MG 18:1/0:0/0:0; 1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-(1-glycerol) ammonium salt, PG 16:0/18:1 and cholesteryl oleate CE 18:1 were purchased from Sigma–Aldrich (St Louis, MO, USA). Lipid standard 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine, LPC 16:0/0:0 was purchased from Larodan AB (Solna, Sweden).
Egg Sphingomyelin extract (chicken) and liver total lipid extract (bovine) were obtained from Avanti Polar Lipids (Alabaster,AL).
Lipid extraction
Lipids from salmon liver were extracted using the solvent system based on the Folch method (11). Moreover, a Precellys®24 bead homogenizer equipped with a Cryolys temperature controller (all Bertin Technologies SAS), have been employed to ensure disruption and homogenization of the tissue. Snap frozen salmon tissue or cell pellet (20–50 mg) was homogenized with zirconium oxide beads (0.5 ± 0.01 g, Ø 1.4 mm) in 500 μL of a cold mixture of chloroform:methanol (2:1, v/v). The tissue was kept frozen during cutting and weighing. 2–3 cycles of 30 s with an intermediate 15 s pause of bead-beating at 6,500 rpm, were needed to ensure a homogeneous sample. After that, another portion of 500 μL of a cold mixture of chloroform:methanol (2:1, v/v) was added to the sample and the tube was shaken at 800 rpm for 10 min at 16°C using a thermoshaker (Thermal shake lite, VWR). Phase separation was induced by adding 200 μL of water. After 10 min of shaking the tube was centrifuged for 4–5 min at maximum speed (13,400 rpm) using a small centrifuge (MiniSpin, Eppendorf). About 500 μL of chloroform layer (lower) were collected and the resulting extract was filtrated through a syringe filter with GHP membrane, 0.2 μm, Ø 13 mm (Acrodisc®, Pall Laboratory) and kept in a dark glass vial with a PTFE lined lid. Lipid extracts were stored at −20°C prior further analysis with SFC tandem MS (UPC2–MS/MS). Dichloromethane was used as a diluent for lipid extracts.
Chromatographic analysis of lipids
A lipid profile analysis was performed using a UHPSFC separation system coupled to a hybrid quadrupole orthogonal time-of-flight mass spectrometer SYNAPT G2-S HDMS (both Waters, Milford, MA, USA). The previously described analytical method (8) was adopted and modified. The separation was performed on an Acquity BEH UPC2 (100 mm × 3 mm, 1.7 μm) column protected with a VanGuard precolumn (BEH 2.1 × 5 mm, both Waters). The column temperature was 50°C, flow rate 1.9 mL/min and automated back-pressure regulator (ABPR) was set to 1800 psi. Mixture of methanol:water (99:1,v/v) containing 30 mM of ammonium acetate was used as modifier. The gradient of the modifier was set as follows: 0 min, 1%; 4.0 min, 30% (6); 4.4 min, 50% (2); 6.25 min 50% (1); 7.25 min, 50% (6); 7.35 min, 1% (6), 8.50 min, 1%. Mixture of methanol:isopropanol:water (50:49:1, v/v/v) was used as make-up liquid and the flow rate was set to 0.2 mL/min.
Mass spectrometer operated in MSE mode and the collision energy ramped from 20 to 30 eV. Data were acquired over the mass range of 50–1200 Da and resolution of mass spectrometer was 20,000. Positive ion electrospray ionization mode was applied and the MS tuning parameters were set as follows: capillary voltage 3.0 kV, the source temperature 150°C, the sampling cone 40 V, the source offset 60 V, the desolvation temperature 500°C, the cone gas flow 50 L/h, the desolvation gas flow 900 L/h, and the nebulizer gas pressure 4 bar. Leucine enkephalin was used as the lockmass.
Data processing
Data were collected using the MassLynx 4.1 (Waters Corporation) software program. Raw data were processed using a Progenesis QI software (Nonlinear Dynamics, Waters) with an in-built LipidBlast database (12) and Lipid Maps Structure Database (http://lipidmaps.org/) for lipid identification. Further, the data were filtered using an in-house developed script collecting total abundances for each individual lipid class (details are described in Section 3.3).
Lipid identification and nomenclature
Identification of a lipid compound is based on the following main characteristics: retention time of the appropriate lipid class (Table IV.), accurate mass (ppm error <5), isotope pattern similarity (>80%) and fragmentation pattern. The lipid nomenclature and shorthand notation described by Lipid Maps (13,14) and Liebisch et al. (15) were followed throughout this paper.
Table IV.
Absolute Quantification of Lipid Classes in Bovine Liver; Comparison of Single-Point Internal Standard Method and Multiple-Point Internal Standard Method
| Lipid class | BLE 50 ug mL−1 | BLE 100 ug mL−1 | Declared lipid content wt/wt% | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Single-point | Multiple-point | Single-point | Multiple-point | ||||||
| Mean | RSD | Mean | RSD | Mean | RSD | Mean | RSD | ||
| MG | 0.21 | 5.88 | 0.16 | 8.45 | 0.21 | 0.74 | 0.16 | 1.07 | |
| DG | 0.54 | 1.36 | 0.49 | 1.46 | 1.07 | 5.35 | 1.01 | 5.54 | |
| TG | 12.60 | 15.72 | 14.41 | 15.79 | 18.24 | 7.02 | 20.88 | 7.04 | 120 |
| CE | 1.19 | 1.14 | 0.16 | 3.50 | 1.75 | 1.85 | 0.40 | 3.42 | |
| PC | 14.61 | 3.02 | 9.27 | 2.99 | 26.94 | 1.97 | 40.93 | 3.57 | 42 |
| Cer | 0.94 | 5.22 | 0.62 | 5.78 | 1.86 | 1.10 | 1.29 | 1.15 | |
| SM | 0.74 | 2.65 | 0.77 | 1.61 | 1.43 | 2.11 | 1.22 | 1.59 | |
| LPC | 0.16 | 7.72 | 1.06 | 1.01 | 0.29 | 1.80 | 1.17 | 0.38 | |
| PE | 8.73 | 1.18 | 11.88 | 1.18 | 17.79 | 0.78 | 24.21 | 0.78 | 22 |
| 2PI | NA | NA | NA | NA | 8 | ||||
| 2LPI | NA | NA | NA | NA | 1 | ||||
| FC | NA | NA | NA | NA | 7 | ||||
| Total | 39.72 | 38.82 | 69.59 | 91.26 | 100 | ||||
n = 3.
1Others, incs. neutral lipids.
2Not detectable in POSmode.
Results
Chromatographic performance and optimization
SFC provides fast and good separation of individual lipid classes which is crucial for the development of a nontargeted quantification methodology. We adopted previously published UHPSFC-based lipidomic method by Lisa et al. (8) with modifications in gradient profile and composition of make-up solvent (for details, see Section 2.3). The gradient elution needed some optimization since we observed coelution of LPCs and SMs. Also, isopropanol was added into the make-up solvent to improve stability of the electrospray. The baseline was stable and retention times of lipid compounds were reproducible. The method enables separation of 13 individual classes in 6 min (Figure 2).
Figure 2.
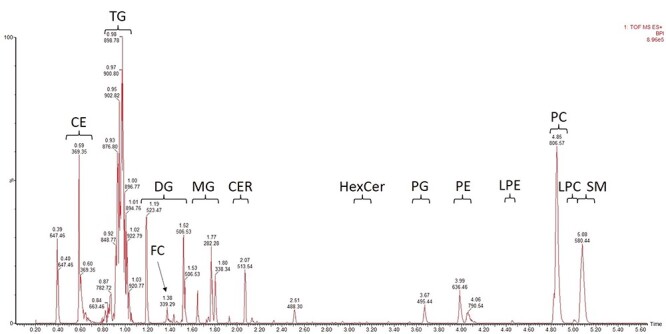
Chromatogram of salmon liver lipid extract with ISTDs added. Peak annotation: CE, cholesteryl esters; TG, triacylglycerols; DG, diacylglycerols; MG, monoacylglycerols; FC, cholesterol; CER, ceramides; HexCer, hexosylceramides (glucosyl- and galactosylceramides); PG, phosphatidylglycerols; PE, phosphatidylethanolamines; LPE, lysophosphatidylethanolamines; PC, phosphatidylcholines; LPC, lysophosphatidylcholines; SM, sphingomyelins.
Selection of injection solvent is crucial in SFC analysis since the sample diluent strongly affects peak shape (16). Peak deterioration is particularly manifested for early eluting compounds when solvents with high elution strength are used as is shown in Figure 3. Five different solvents (acetonitrile, isopropanol, modifier, chloroform and hexane) were tested on a model mixture of lipid standard containing TG, DG, MG, PC, PE and SM. Hexane provided great peak shape for compounds with lower retention but response of polar lipids dropped. Peaks of nonpolar lipids showed distortion when acetonitrile, isopropanol or mobile phase were used as sample solvent. Chloroform and dichloromethane displayed similar behavior and provided good peak shape for both nonpolar and polar lipids. We have also tested mixture of chloroform and isopropanol at various ratios, but without any improvement (data not shown).
Figure 3.
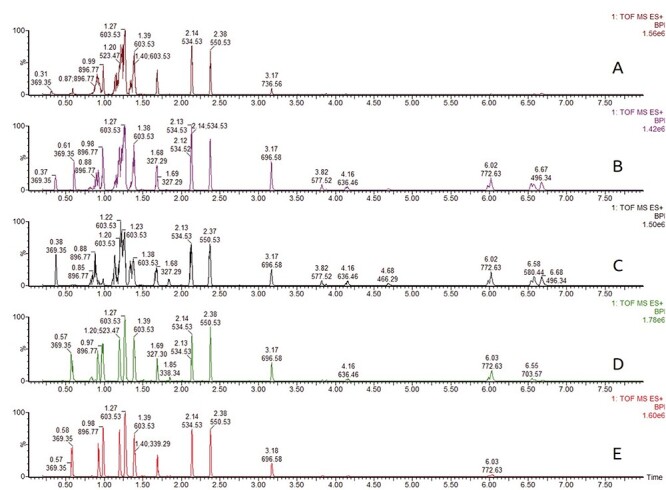
Effect of sample diluent on peak shape and response has been tested on a model mixture of lipid standards. Legend: A—acetonitrile, B—isopropanol, C—modifier (CH3OH:H2O, 99:1, containing 30 mM CH3COONH4), D—chloroform, E—hexane.
Even though the chromatographic system was washed thoroughly after each use with methanol and CO2 (50:50, v/v) followed by pure CO2 as it is common practice, high ion background, particularly m/z 406, was occasionally observed. The source of the ion remains unclear, however, short washing with higher isopropanol content (typically CO2: isopropanol, 55:45, v:v), followed by brief wash with pure CO2 is advisable to eliminate eventual impurities. A cleaning with 100% isopropanol showed to be effective as well, but it should not be used regularly only as a last resort.
Development of the quantitative method
Nontargeted MS analysis is very beneficial as it provides comprehensive information about the general composition of a sample. However, interpretation in a biological context is highly benefitted from both knowing the identity of individual analytes as well as the concentration levels. Lipid samples contain hundred/thousands of lipid species and their abundance vary greatly among sample species and type. It is not feasible to use hundreds of isotopically labeled ISTDs, even if they were available. But as an approximation one can use that lipid species with identical polar head group show similar RFs since ionization efficiency is given by physicochemical properties of polar head group (17). Here we aimed to develop a simple and reliable quantification strategy and for this reason, different quantification methods such as a RF correction, single-point ISTD or multiple-point ISTD were tested and compared.
Since availability of 13C-labeled lipid standards is very limited, lipids with short or very long fatty acyls are frequently used instead and compounds with odd number of carbons in fatty acyls are preferred since the probability of their natural occurrence is lower (18). Lipid compounds with lauroyl, tridecanoyl, myristoyl, pentadecanoyl, heptadecanoyl and docosanoyl fatty acyls and deuterated cholesterol were used as ISTDs.
Single-point ISTD method requires analysis of a sample containing analytes of unknown concentrations and known amount of ISTD. Amount of the unknown analyte is calculated using equation: 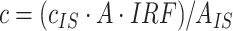 , where c is concentration, A is peak area, IRF is internal RF and IS stands for ISTD (19). Since we use set of ISTDs, one for each individual lipid class, IRF value was considered to be equal to 1, assuming that all lipid species in the same lipid class shows identical ionization efficiency.
, where c is concentration, A is peak area, IRF is internal RF and IS stands for ISTD (19). Since we use set of ISTDs, one for each individual lipid class, IRF value was considered to be equal to 1, assuming that all lipid species in the same lipid class shows identical ionization efficiency.
Multiple-point ISTD calibration curve method is based on series of standards with different concentrations of analyte and constant concentration of ISTD. Individual calibration curves were constructed by plotting the ratio (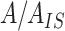 ) versus concentration of analytes
) versus concentration of analytes 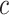 . Lipid species with palmitoyl, heptadecanoyl, heptadecenoyl, stearoyl or oleoyl fatty acyls were selected as representatives for construction of multiple-point ISTD calibration curves. Parameters of individual calibration curves (
. Lipid species with palmitoyl, heptadecanoyl, heptadecenoyl, stearoyl or oleoyl fatty acyls were selected as representatives for construction of multiple-point ISTD calibration curves. Parameters of individual calibration curves (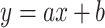 ) for the multiple-point ISTD method are presented in Table II.
) for the multiple-point ISTD method are presented in Table II.
Table II.
Parameters of Calibration Curves for Individual Lipid Classes and Lipid Species Added into the Egg Lipid Extract; Experimentally Determined Response Factors (RF) Used for Relative Quantification of Lipid Classes
| Compound | Lipid class | Linear range ug/mL | Slope, a | Intercept, b | R2 | RF |
|---|---|---|---|---|---|---|
| 1Cholesterol | FC | 0.05-10 | 0.58 | 0.02 | 0.994 | 72.64 |
| 1PE 18:0/18:0 | PE | 0.2–40 | 0.17 | 0.18 | 0.992 | 3.59 |
| 1LPE 17:1/0:0 | LPE | 2.5–10 | 0.61 | -0.50 | 0.997 | 23.69 |
| 1PC 18:0/18:0 | PC | 0.2–10 | 0.66 | -0.07 | 0.998 | 1.00 |
| PC P-18:0/18:1 | PC(P) | 0.2–10 | 0.57 | -0.06 | 0.997 | 1.16 |
| 1SM d16:0/18:1 | SM | 0.2–10 | 0.39 | -0.12 | 0.998 | 1.98 |
| 1LPC 16:0/0:0 | LPC | 0.8–10 | 0.30 | -0.27 | 0.993 | 4.40 |
| 1PG 16:0/18:1 | PG | 0.8–38.7 | 0.31 | 0.33 | 0.992 | 41.77 |
| 1TG 18:0/18:0/18:0 | TG | 0.05–2.5 | 1.19 | -0.03 | 0.992 | 0.29 |
| TG 18:2/18:2/18:2 | TG | 0.1–5 | 0.87 | 0.06 | 0.994 | 0.30 |
| 1DG 18:1/18:1/0:0 | DG | 0.05–2.5 | 1.02 | 0.04 | 0.995 | 0.35 |
| DG 18:1/0:0/18:1 | DG | 0.05–2.5 | 1.10 | 0.16 | 0.983 | 0.39 |
| 1MG 18:1/0:0/0:0 | MG | 0.05–2.5 | 0.93 | 0.06 | 0.997 | 1.25 |
| 1Cer d18:1/17:0 | Cer | 0.05–1.25 | 1.38 | 0.09 | 0.982 | 0.32 |
| GlcCer d18:1/17:0 | HexCer | 0.05–2.5 | 0.53 | -0.02 | 0.999 | 0.68 |
| 1GalCer d18:1/18:0 | HexCer | 0.05–10 | 0.56 | 0.05 | 0.998 | 0.56 |
| 1CE 18:1 | CE | 0.2–5 | 0.60 | 0.20 | 0.989 | 1.70 |
1Calibration curves used for quantification of lipid classes.
It was observed in most of the calibration curves measured that linearity was lost at high concentrations. Therefore, samples with high levels of lipids require to be diluted prior to analysis. This problem can also be overcome by building an additional linear calibration curve for higher concentrations or by using a quadratic or cubic calibration curve. However, loss of the linear response might be caused by an ability of lipids to form aggregates at higher concentrations, which affects ionization efficiencies (9).
Evaluation of single-point versus multiple-point quantification in egg and bovine liver extracts
An accuracy of the single-point and multiple-point ISTD quantification methods was evaluated by addition of known amount of selected lipid representatives into a commercially available egg extract of sphingomyelins (SM) to mimic matrix effect. Further, measured and expected concentrations were compared, and deviation was expressed as percentages. Precision was assessed as the relative standard deviation (RSD) of three or four replicates. Three different concentration levels were tested for each lipid individual and results are summarized in the Table III. Response of individual lipid compound is affected by length of fatty acyls and level of saturation, therefore quantification of lipid class using a single reference compound is not truly correct and might introduce a systematic error. Taking that into account, we considered accuracy of 25% as acceptable. Good precision, <5%, was obtained for most of the lipids tested (Table III). DG, Cer, GalCer, LPE show precision >10% at low concentration levels. In general, accuracy was better when multiple-point calibration was applied for quantification, e.g. particularly evident for the cholesteryl ester CE 18:1 where single-point quantification had >100% deviation for all three levels whereas multiple-point provided much better accuracy (Table III). Lower accuracy was also frequently found at low concentration levels tested, indicating that absolute quantification of low abundant lipid species is not reliable when nontarget approach is employed. Lower accuracy can be caused by matrix induced effects or ion suppression. Moreover, inappropriately selected concentration of ISTDs can also lead to strong ion suppression effect and compromise analysis. Thus, we recommend optimizing the composition of the ISTD mixture to individual lipid extracts, since the distribution varies from source to source, e.g. from high to lowTGs.
Table III.
Egg Lipid Extract Spiked with a Known Amount of Standards; Comparison of Single-Point Internal Standard Method and Multiple-Point Internal Standard Method
| Lipid compound | tR (min) | Amount added (ug/ml) | Single-Point | Multiple-Point | Accuracy (dev %) | |||
|---|---|---|---|---|---|---|---|---|
| Mean | RSD | Mean | RSD | Single point | Multiple point | |||
| CE 18:1 | 0.61 | 0.53 | 1.51 | 0.8 | 0.29 | 1.8 | 183.1 | 44.8 |
| 2.13 | 5.36 | 3.6 | 1.90 | 4.2 | 151.1 | 10.8 | ||
| 4.27 | 9.27 | 1.2 | 3.54 | 1.3 | 117.4 | 17.1 | ||
| TG 18:0/18:0/18:0 | 0.93 | 0.13 | 0.12 | 1.6 | 0.13 | 1.3 | 8.5 | 4.9 |
| 0.53 | 0.47 | 1.5 | 0.42 | 1.5 | 12.0 | 21.7 | ||
| 1.07 | 0.93 | 12.9 | 0.80 | 12.5 | 13.3 | 25.1 | ||
| TG 18:1/18:1/18:1 | 0.95 | 0.13 | 0.11 | 2.1 | 0.06 | 4.6 | 16.5 | 56.1 |
| 0.53 | 0.46 | 2.1 | 0.46 | 2.4 | 14.2 | 14.4 | ||
| 1.07 | 0.91 | 14.1 | 0.98 | 15.1 | 14.6 | 13.4 | ||
| TG 18:2/18:2/18:2 | 0.99 | 0.10 | 0.09 | 2.3 | 0.04 | 6.7 | 7.8 | 63.5 |
| 0.40 | 0.36 | 1.4 | 0.34 | 1.7 | 10.5 | 14.6 | ||
| 0.80 | 0.72 | 11.0 | 0.76 | 12.0 | 11.0 | 10.5 | ||
| DG 18:1/18:1/0:0 | 1.36 | 0.15 | 0.12 | 2.1 | 0.08 | 3.0 | 15.2 | 42.2 |
| 0.59 | 0.52 | 1.3 | 0.47 | 1.4 | 11.8 | 20.2 | ||
| 1.17 | 0.96 | 1.0 | 0.90 | 1.0 | 17.9 | 22.9 | ||
| DG 18:1/0:0/18:1 | 1.25 | 0.10 | 0.14 | 13.7 | NA | NA | 43.7 | NA |
| 0.40 | 0.57 | 7.6 | 0.37 | 10.7 | 42.0 | 8.3 | ||
| 0.80 | 1.08 | 3.0 | 0.84 | 3.6 | 35.6 | 4.6 | ||
| MG 18:1/0:0/0:0 | 1.63 | 0.19 | 0.19 | 2.6 | 0.13 | 4.0 | 1.9 | 29.6 |
| 0.75 | 0.70 | 8.8 | 0.68 | 9.7 | 6.8 | 9.2 | ||
| 1.49 | 1.18 | 3.2 | 1.20 | 3.4 | 20.9 | 19.8 | ||
| Cer d18:1/17:0 | 2.03 | 0.10 | 0.13 | 8.8 | 0.03 | 28.6 | 31.8 | 70.5 |
| 0.40 | 0.54 | 0.4 | 0.33 | 0.4 | 35.8 | 17.9 | ||
| 0.80 | 0.91 | 3.0 | 0.59 | 3.4 | 13.1 | 26.1 | ||
| GlcCer d18:1/17:0 | 3.03 | 0.13 | 0.06 | 3.1 | 0.15 | 2.4 | 53.8 | 10.1 |
| 0.53 | 0.23 | 0.1 | 0.46 | 0.1 | 57.7 | 14.4 | ||
| 1.07 | 0.43 | 3.1 | 0.85 | 2.9 | 59.6 | 20.8 | ||
| GalCer d18:1/18:0 | 3.11 | 0.12 | 0.05 | 3.5 | 0.01 | 37.6 | 56.5 | 92.7 |
| 0.48 | 0.22 | 0.3 | 0.31 | 0.4 | 54.6 | 36.0 | ||
| 0.96 | 0.43 | 2.2 | 0.69 | 2.5 | 55.0 | 27.8 | ||
| PG 16:0/18:1 | 3.60 | 0.72 | 0.72 | 3.1 | NA | NA | 2.2 | NA |
| 2.89 | 4.04 | 3.6 | 2.20 | 5.3 | 39.7 | 23.9 | ||
| 5.78 | 7.26 | 1.0 | 4.82 | 1.2 | 25.7 | 16.5 | ||
| PE 18:0/18:0 | 3.89 | 0.51 | 0.37 | 0.7 | 0.50 | 0.7 | 27.2 | 1.0 |
| 2.03 | 1.64 | 0.9 | 2.23 | 0.9 | 19.2 | 10.0 | ||
| 4.05 | 3.12 | 0.4 | 4.24 | 0.4 | 23.1 | 4.6 | ||
| LPE 17:1/0:0 | 4.40 | 0.48 | 0.96 | 11.7 | 1.21 | 3.8 | 99.0 | 152.6 |
| 1.92 | 4.08 | 1.7 | 2.49 | 1.1 | 112.3 | 29.5 | ||
| 3.84 | 7.25 | 1.4 | 3.78 | 1.1 | 88.8 | 1.5 | ||
| PC 18:0/18:0 | 4.76 | 0.40 | 0.37 | 1.8 | 0.33 | 1.3 | 8.2 | 16.3 |
| 1.60 | 1.56 | 0.2 | 1.08 | 0.1 | 2.3 | 32.2 | ||
| 3.20 | 2.99 | 0.7 | 1.98 | 0.7 | 6.5 | 38.1 | ||
| PC P-18:0/18:1 | 4.78 | 0.19 | 0.18 | 1.5 | 0.23 | 0.8 | 4.9 | 21.8 |
| 0.75 | 0.72 | 0.4 | 0.63 | 0.4 | 3.0 | 16.0 | ||
| 1.49 | 1.38 | 0.4 | 1.11 | 0.4 | 7.3 | 25.8 | ||
| PC 18:3/18:3 | 4.82 | 0.59 | 0.91 | 0.4 | 0.68 | 0.3 | 55.7 | 15.5 |
| 2.35 | 4.07 | 0.9 | 2.65 | 0.9 | 73.2 | 13.1 | ||
| 4.69 | 7.68 | 0.6 | 4.92 | 0.6 | 63.7 | 4.9 | ||
| LPC 16:0/0:0 | 5.06 | 0.43 | 0.20 | 0.3 | 1.10 | 0.0 | 52.1 | 157.0 |
| 1.71 | 1.04 | 0.7 | 1.80 | 0.4 | 39.0 | 5.7 | ||
| 3.41 | 2.11 | 1.4 | 2.71 | 0.9 | 38.1 | 20.7 | ||
| Cholesterol | 1.36 | 1.07 | 1.17 | 2.7 | 0.23 | 3.0 | 9.8 | 78.8 |
| 4.27 | 4.40 | 0.8 | 0.92 | 0.8 | 3.1 | 78.3 | ||
| 8.53 | 8.40 | 2.0 | 1.79 | 2.1 | 2.1 | 79.0 | ||
n = 3.
The single-point and multiple-point internal standardization methods for lipid class quantification were also evaluated using a commercial bovine liver extract (BLE) and results were compared with the composition declared by the manufacturer (Table IV). Two concentration levels (50 μg/mL−1 and 100 μg/mL) of bovine liver total lipid extract were tested. Declared lipid content is expressed as weight percentage and data are directly comparable with sample of BLE with concentration of 100 μg/mL. Reproducibility of the results is <6% for most of lipid classes, except for TGs which permanently show a higher variation in measured data. High levels of PCs are present in BLE, therefore an additional calibration curve for higher concentration was used. The content of PC, as the most abundant lipid class, was underestimated when single-point ISTD method was utilized. A good agreement with declared lipid composition was achieved by the application of multiple-point internal standardization.
In conclusion, multiple-point internal standardization provides better overall accuracy, whereas the less laborious single-point method can be used for dedicated analysis of most lipid classes, of which particularly MG, DG, TG show good accuracy, but not for cholesteryl esters.
Semiquantitative lipid class determination using RFs
During the trials to establish a quantitative lipid profiling method, both single and multiple points, it was realized that a simpler strategy was needed that require less resources (both labor and consumables) and that can be used on all model systems without comprehensive adjustments. In such cases, experimentally determined correction factors (also known as RFs) can be applied to adjust raw data and reflect real content of compounds. This approach does not request utilization of any expensive internal and external standards and still provide reliable semiquantitative picture of lipid class distribution in samples. RF were determined by comparing of abundances of lipid standards at the same concentration level. The large variation in ionization efficiencies of different lipid species is easily observable when inspecting the range of RF in Table II.
Processing of nontarget data with e.g. Progenesis QI software involves several steps: alignment of chromatograms, peak extraction, compound identification and statistical analysis. For lipid class quantification we have deliberately developed a script which collects and summarizes a total peak area for individual lipid classes. Each class is characterized by a retention window and low mass to charge (m/z) threshold for lipid individual with very short fatty acyl/acyls (typically lauroyl) and high m/z threshold for lipid species with very long fatty acyl/acyls (typically hexacosanoyl). The comprehensive list of real lipid species available on the website www.lipidmaps.org was used as a base. Lower and higher m/z thresholds were further corrected based on ion adducts observed and expected. The script simply utilizes filtering based on retention time and m/z thresholds (Table V), thus only compounds that meet the conditions are retained. The total lipid class and compound abundances are further corrected based on the relative RFs being experimentally determined for each lipid class. Five data files, containing both original raw and RF-corrected measurements, are automatically generated: (i) a total peak area of each individual lipid class, (ii) RF-corrected lipid class abundances, (iii) list of compounds with abundances and lipid classification, (iv) RF-corrected compound abundances and (v) number of lipid species in an individual lipid class. The filtering parameters are summarized in an editable table and can therefore be easily adjusted if necessary. The script is written in Python and is freely available along with a data template from the following link: https://github.com/andrevo/Lipid-class.
Table V.
Criteria Used for Data Filtering and Response Correction—Retention Time tR, m/z Thresholds and Response Factors for Each IndividualLC
| Lipid class | Start tR (min) | End tR (min) | Lower m/z threshold | Upper m/z threshold | RF |
|---|---|---|---|---|---|
| CE | 0.56 | 0.82 | 550 | 700 | 1.7 |
| TG | 0.82 | 1.18 | 710 | 1081 | 0.29 |
| DG | 1.18 | 1.55 | 450 | 741 | 0.35 |
| FC | 1.36 | 1.39 | 369.35 | 369.4 | 72.64 |
| MG | 1.6 | 1.86 | 280 | 430 | 1.25 |
| CER | 2.02 | 2.13 | 460 | 764 | 0.32 |
| HexCer | 3.02 | 3.13 | 623 | 864 | 0.56 |
| HexCer(OH) | 3.26 | 3.3 | 665 | 894 | 10.56 |
| PG | 3.60 | 3.75 | 667 | 914 | 41.77 |
| PE | 3.89 | 4.03 | 580 | 900 | 3.59 |
| LPE | 4.39 | 4.45 | 397 | 590 | 23.69 |
| PC | 4.76 | 4.83 | 620 | 980 | 1 |
| SM | 4.91 | 5.05 | 625 | 870 | 1.98 |
| LPC | 5.00 | 5.25 | 410 | 610 | 4.4 |
1The same RF as for HexCer.
The power of this simple strategy is illustrated in the Figure 4., where we have processed data in three different ways and expressed lipid class content of BLE as fraction of total abundance or amount. Lipid class concentrations of BLE (100 μg/mL) determined by application of the multiple-point ISTD method were used as a control (data are presented in Table IV). Quantity of each individual lipid class was related to the total lipid content (91.26 μg/mL) and expressed as a percentage form (Figure 4C). The pie chart in Figure 4A shows lipid class content determined based on raw data, whereas Figure 4B demonstrates the impact of using RFs to convert raw data to semi-quant state. The chart in the Figure 4A clearly misrepresents the real lipid class distribution in the BLE since TGs seem to be the most abundant lipid class (64.5%) and content of PEs is <6%. On the contrary, application of RFs (Figure 4B) reflects better the real content of TGs (down to 28.6%), PCs, PEs, DGs, CERs, SMs and LPCs and proved to be useful. RFs are stable under the same experimental conditions but need to be verified regularly.
Figure 4.
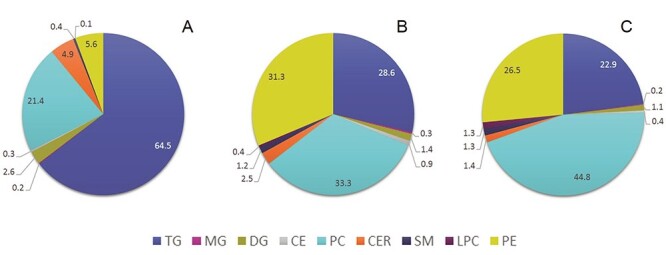
Lipid class distribution (%) of bovine liver extract. Legend: A—raw data, B—raw data corrected with RFs, C—based on lipid class concentration found in the BLE (see Section 3.4 for more details).
The matrix enhancement/suppression effect (ME) was investigated using the standard addition method. Calibration series of selected lipid class representatives of both neutral and polar lipids (MG, DG, TG, PC, PE, SM and CE) were prepared in diluent (dichloromethane) and BLE and obtained slopes were compared: 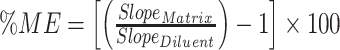 (20) Most of the standards selected do not show strong suppression/enhancement effect (data not shown). We expected stronger suppression effect for TGs because of their high abundancy, short retention time and narrow retention window, which was confirmed in case of TG 18:0/18:0/18:0 (ME: 35%) but not for TG 18:2/18:2/18:2 (ME: 11%). Phospholipids did not show higher ME than 15% with exception of PE 18:0/18:0 where ME found was 39%. This shows that matrix effects are very unpredictable and it should be considered for accurate quantification, however it is not feasible to determine matrix enhancement/suppression effect for each individual lipid species in a nontarget profiling like this presented method. We believe the semiquantitative approach as it is suggested here is still valuable even though the matrix effect is not considered. Moreover, if needed can matrix effects to a certain extent be equalized by means of ISTDs.
(20) Most of the standards selected do not show strong suppression/enhancement effect (data not shown). We expected stronger suppression effect for TGs because of their high abundancy, short retention time and narrow retention window, which was confirmed in case of TG 18:0/18:0/18:0 (ME: 35%) but not for TG 18:2/18:2/18:2 (ME: 11%). Phospholipids did not show higher ME than 15% with exception of PE 18:0/18:0 where ME found was 39%. This shows that matrix effects are very unpredictable and it should be considered for accurate quantification, however it is not feasible to determine matrix enhancement/suppression effect for each individual lipid species in a nontarget profiling like this presented method. We believe the semiquantitative approach as it is suggested here is still valuable even though the matrix effect is not considered. Moreover, if needed can matrix effects to a certain extent be equalized by means of ISTDs.
Discussion
A robust SFC system was implemented for laboratory practice quite recently and although it is not as widespread as LC, SFC offers high-throughput separations and it is also compatible with MS detection systems and has been proven useful for analysis of hydrophobic compounds and metabolites, such as vitamins, steroids, fatty acids or lipids (21–23). SFC based methods showed to be an effective tool for fast lipid screening in samples of various origin (24–26). However, nontargeted data allow not only to reveal changes in lipidome but also can be used for quantification of lipid classes or lipid species. Positive ionization allows detection of most common lipid classes, such as MG, DG, TG, CE, SM, CER, HexCer, PG, LPE, PE, LPC, PC and cholesterols. Negative ionization allows detection of free fatty acids, PA, PS, PI and sulfatides, but do not enable detection of TGs, which are usually very abundant in lipid extracts of various origin. Moreover, we have observed that PS and PA show poor peak shape including peak broadening and tailing which has negative effect on signal/noise ratio. However, we recommend to run pooled samples in negative ionization mode to inspect whether studied lipid extracts contain higher levels of above mentioned anionic lipids or their relative changes are expected, e.g. in analysis of cell membranes.
We also find it necessary to stress the importance of manual inspection of the chromatogram processing including peak picking results from both commercial providers, e.g. Waters Progenesis QI as well as free ware from academic labs. One main concern is if the software performs correct deisotoping, e.g. is not reporting M + 2, 4, 6 peaks, etc as one lipid if these are unique closely coeluting lipid species. This is illustrated for the TG52:0, TG52:1, TG52:2 series with <1 s retention time differences, but Progenesis QI is able to correctly report them as three individual lipid species (Supplementary Figure S1). Rapid chromatography requires mass spectrometers to be operated in fast scanning mode (>5 scans per second), otherwise must the elution conditions be changed to broader peaks if it is realized during manual inspection that the software processes the raw chromatograms incorrectly.
In this study we present a high-throughput nontarget semiquantitative lipidomics workflow, and different quantification strategies has been tested and compared. We have developed an automatic data processing script allowing semiquantification of lipid compounds or lipid classes by relative RFs. The RF corrected data provide a more correct picture of the relative lipid class distribution and could also be used for evaluation and interpretation of individual lipid species. The methodology developed was applied to study changes in lipid composition of nerves in goats with genetic mutation and results were published in The Faseb Journal (27). The evaluation and interpretation of the lipid profiling data herein were greatly enhanced with the semiquantitative data compared to the noncorrected raw data. In addition, semiquantification can be improved by using internal or also external standards, but this is more laborious and costly, and is probably not needed for many studies, including the above mentioned. This method has also been applied to a number of samples types (dog, cod, salmon, chicken, neural tissue, liver and muscle) and quite clear separations between sample origins and types were obtained (data not shown). Thus, we demonstrate that nontarget data can easily, and we recommend should be, upgraded to semiquantitative state since the ionization efficiencies varies largely. This provides a more correct internal picture of the individual lipid species within a sample, but also for inter sample comparisons.
Supplementary Material
{kind=link}
Acknowledgments
The authors would acknowledge the administrative and technical support from host Department and the NTNU Natural Science faculty Mass Spectrometry laboratory and the Centre for Digital Life Norway (www.ntnu.edu/dln/centre-for-digital-life-norway). This research was supported by the Research Council of Norway, [grant numbers 248792 DigiSal, 269432 AurOmega].
Contributor Information
Zdenka Bartosova, Department of Biotechnology and Food Science, NTNU Norwegian University of Science and Technology, Sem Sælands vei 6/8, N-7491 Trondheim Norway.
Susana Villa Gonzalez, Department of Chemistry, NTNU Norwegian University of Science and Technology, Høgskoleringen 5, N-7491 Trondheim, Norway.
André Voigt, Department of Biotechnology and Food Science, NTNU Norwegian University of Science and Technology, Sem Sælands vei 6/8, N-7491 Trondheim Norway.
Per Bruheim, Department of Biotechnology and Food Science, NTNU Norwegian University of Science and Technology, Sem Sælands vei 6/8, N-7491 Trondheim Norway.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1. Wang, J.N., Wang, C.Y., Han, X.L.; Tutorial on lipidomics; Analytica Chimica Acta, (2019); 1061: 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cajka, T., Fiehn, O.; Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry; TrAC Trends in Analytical Chemistry, (2014); 61: 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lange, M., Ni, Z., Criscuolo, A., Fedorova, M.; Liquid chromatography techniques in lipidomics research; Chromatographia, (2019); 82: 77–100. [Google Scholar]
- 4. Schwudke, D., Schuhmann, K., Herzog, R., Bornstein, S.R., Shevchenko, A.; Shotgun lipidomics on high resolution mass spectrometers; Cold Spring Harbor Perspectives in Biology, (2011); 3: a004614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ovcacikova, M., Lisa, M., Cifkova, E., Holcapek, M.; Retention behavior of lipids in reversed-phase ultrahigh-performance liquid chromatography-electrospray ionization mass spectrometry; Journal of Chromatography A, (2016); 1450: 76–85. [DOI] [PubMed] [Google Scholar]
- 6. Cífková, E., Holčapek, M., Lísa, M., Ovčačíková, M., Lyčka, A., Lynen, F., et al. ; Nontargeted quantitation of lipid classes using hydrophilic interaction liquid chromatography–electrospray ionization mass spectrometry with single internal standard and response factor approach; Analytical Chemistry, (2012); 84: 10064–10070. [DOI] [PubMed] [Google Scholar]
- 7. Christie, W.W.; Rapid separation and quantification of lipid classes by high-performance liquid-chromatography and mass (light-scattering) detection; Journal of Lipid Research, (1985); 26: 507–512. [PubMed] [Google Scholar]
- 8. Lisa, M., Holcapek, M.; High-throughput and comprehensive lipidomic analysis using ultrahigh-performance supercritical fluid chromatography-mass spectrometry; Anal Chem, (2015); 87: 7187–7195. [DOI] [PubMed] [Google Scholar]
- 9. Wang, M., Wang, C.Y., Han, X.L.; Selection of internal standards for accurate quantification of complex lipid species in biological extracts by electrospray ionization mass spectrometry-what, how and why? Mass spectrometry reviews, (2017); 36: 693–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koelmel, J.P., Cochran, J.A., Ulmer, C.Z., Levy, A.J., Patterson, R.E., Olsen, B.C., et al. ; Software tool for internal standard based normalization of lipids, and effect of data-processing strategies on resulting values; BMC Bioinformatics, (2019); 20: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Folch, J., Lees, M., Sloane Stanley, G.H.; A simple method for the isolation and purification of total lipides from animal tissues; The Journal of biological chemistry, (1957); 226: 497–509. [PubMed] [Google Scholar]
- 12. Kind, T., Liu, K.-H., Yup Lee, D., DeFelice, B., Meissen, J.K., Fiehn, O.; Lipidblast - in-silico tandem mass spectrometry database for lipid identification; Nature methods, (2013); 10: 755–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fahy, E., Subramaniam, S., Brown, H.A., Glass, C.K., Merrill, A.H., Murphy, R.C., et al. ; A comprehensive classification system for lipids; Journal of Lipid Research, (2005); 46: 839–862. [DOI] [PubMed] [Google Scholar]
- 14. Fahy, E., Subramaniam, S., Murphy, R.C., Nishijima, M., Raetz, C.R.H., Shimizu, T., et al. ; Update of the lipid maps comprehensive classification system for lipids; Journal of Lipid Research, (2009); 50: S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liebisch, G., Vizcaino, J.A., Kofeler, H., Trotzmuller, M., Griffiths, W.J., Schmitz, G., et al. ; Shorthand notation for lipid structures derived from mass spectrometry; J Lipid Res, (2013); 54: 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desfontaine, V., Tarafder, A., Hill, J., Fairchild, J., Grand-Guillaume Perrenoud, A., Veuthey, J.-L., et al. ; A systematic investigation of sample diluents in modern supercritical fluid chromatography; Journal of Chromatography A, (2017); 1511: 122–131. [DOI] [PubMed] [Google Scholar]
- 17. Han, X.; Lipidomics : Comprehensive mass spectrometry of lipids. John Wiley & Sons, Inc, (2016), ISBN 978-1-118-89312-8, 496.
- 18. Holcapek, M., Liebisch, G., Elcroos, K.; Lipidomic analysis; Analytical Chemistry, (2018); 90: 4249–4257. [DOI] [PubMed] [Google Scholar]
- 19. Cuadros-Rodríguez, L., Bagur-González, M.G., Sánchez-Viñas, M., González-Casado, A., Gómez-Sáez, A.M.; Principles of analytical calibration/quantification for the separation sciences; Journal of Chromatography A, (2007); 1158: 33–46. [DOI] [PubMed] [Google Scholar]
- 20. Røst, L.M., Shafaei, A., Fuchino, K., Bruheim, P.; Zwitterionic hilic tandem mass spectrometry with isotope dilution for rapid, sensitive and robust quantification of pyridine nucleotides in biological extracts; Journal of Chromatography B, (2020); 1144: 122078. [DOI] [PubMed] [Google Scholar]
- 21. Jumaah, F., Larsson, S., Essén, S., Cunico, L.P., Holm, C., Turner, C., et al. ; A rapid method for the separation of vitamin d and its metabolites by ultra-high performance supercritical fluid chromatography-mass spectrometry; Journal of chromatography. A, (2016); 1440: 191–200. [DOI] [PubMed] [Google Scholar]
- 22. Sen, A., Knappy, C., Lewis, M.R., Plumb, R.S., Wilson, I.D., Nicholson, J.K., et al. ; Analysis of polar urinary metabolites for metabolic phenotyping using supercritical fluid chromatography and mass spectrometry; Journal of chromatography. A, (2016); 1449: 141–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Señoráns, F.J., Ibañez, E.; Analysis of fatty acids in foods by supercritical fluid chromatography; Analytica Chimica Acta, (2002); 465: 131–144. [Google Scholar]
- 24. Lisa, M., Cifkova, E., Khalikova, M., Ovcacikova, M., Holcapek, M.; Lipidomic analysis of biological samples: Comparison of liquid chromatography, supercritical fluid chromatography and direct infusion mass spectrometry methods; Journal of chromatography. A, (2017); 1525: 96–108. [DOI] [PubMed] [Google Scholar]
- 25. Shi, X., Yang, W., Qiu, S., Hou, J., Wu, W., Guo, D.; Systematic profiling and comparison of the lipidomes from panax ginseng, p. Quinquefolius, and p. Notoginseng by ultrahigh performance supercritical fluid chromatography/high-resolution mass spectrometry and ion mobility-derived collision cross section measurement; Journal of chromatography. A, (2018); 1548: 64–75. [DOI] [PubMed] [Google Scholar]
- 26. Gil-Ramirez, A., Al-Hamimi, S., Rosmark, O., Hallgren, O., Larsson-Callerfelt, A.-K., Rodríguez-Meizoso, I.; Efficient methodology for the extraction and analysis of lipids from porcine pulmonary artery by supercritical fluid chromatography coupled to mass spectrometry; Journal of Chromatography A, (2019); 1592: 173–182. [DOI] [PubMed] [Google Scholar]
- 27. Skedsmo, F.S., Malachin, G., Våge, D.I., Hammervold, M.M., Salvesen, Ø., Ersdal, C., et al. ; Demyelinating polyneuropathy in goats lacking prion protein; The FASEB Journal, (2020); 34: 2359–2375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.