


Abstract
ReFOLD3 is unique in its application of gradual restraints, calculated from local model quality estimates and contact predictions, which are used to guide the refinement of theoretical 3D protein models towards the native structures. ReFOLD3 achieves improved performance by using an iterative refinement protocol to fix incorrect residue contacts and local errors, including unusual bonds and angles, which are identified in the submitted models by our leading ModFOLD8 model quality assessment method. Following refinement, the likely resulting improvements to the submitted models are recognized by ModFOLD8, which produces both global and local quality estimates. During the CASP14 prediction season (May–Aug 2020), we used the ReFOLD3 protocol to refine hundreds of 3D models, for both the refinement and the main tertiary structure prediction categories. Our group improved the global and local quality scores for numerous starting models in the refinement category, where we ranked in the top 10 according to the official assessment. The ReFOLD3 protocol was also used for the refinement of the SARS-CoV-2 targets as a part of the CASP Commons COVID-19 initiative, and we provided a significant number of the top 10 models. The ReFOLD3 web server is freely available at https://www.reading.ac.uk/bioinf/ReFOLD/.
Graphical Abstract
Graphical abstract.
Refinement of a 3D model using ReFOLD3.
INTRODUCTION
In silico modelling of protein structures provides a potential solution for bridging the protein sequence-structure gap. Although protein structures can now be predicted with high accuracy, using either advanced template based or deep learning-based methods, the resulting 3D models may still include significant local errors including unfavourable contacts, irregular hydrogen bonds, geometrical clashes and unusual angles. These errors may limit the usage of the modelled structures in cases where high accuracy is required, such as in drug discovery and protein engineering (1–3). The refinement of predicted protein structures refers to the correction of the local errors and improvement of the overall quality of theoretical 3D models, which is often considered as the ‘last mile’ for structure prediction (4).
Methods for the refinement of 3D protein models aim to increase model accuracy by fixing the local errors with the adjustment of secondary structure and modification of the sidechain interactions (3,5). Ironically, until recent years, the refinement of 3D models has more often than not resulted in decreases in average model accuracy and it remains a challenge for refinement groups to consistently improve all 3D models across all categories of target difficulty (3,6).
The employment of physics-based molecular dynamics (MD) simulations has been the main stay for the non-server-based refinement methods. Since the CASP9 experiment, leading MD methods have generated refined 3D models by taking advantage of new force fields, parallel computing and restraint strategies (3). Although the MD-based protocols have been used by many top-performing groups in recent CASP experiments, they have suffered from a high computational time cost and the inadequacy of force fields for directing the generation of the 3D models towards the native basin (2–4,7,8). Both the automated server-based and non-server-based approaches for refinement sample dozens of 3D models in many alternative conformations (9). The most native like conformation among all 3D models are subsequently identified in the scoring stage (3). However, it is often problematic to recognise the improved models using either energy functions or model quality assessment programs (MQAPs), as the similarity among the 3D models generated by the sampling approaches (3).
The original ReFOLD server (10) was developed by our group to refine 3D models using a hybrid approach, which included both rapid and MD-based sampling and leading model quality estimates, to produce the predicted local and global quality scores for each sampled 3D model. The first protocol included the refinement of the initial structure using i3Drefine in 20 refinement cycles (1,11). The second protocol accommodated an MD-based protocol inspired by that of Feig and Mirjalili (7,12,13), but using more modest computational resources compared to the supercomputer scale resources, which were originally used. All 3D models generated by these sampling approaches were ranked using ModFOLD6 (14) at the last stage. The original ReFOLD method (10) was used for the refinement of CASP12 targets, and it showed promising performance, boosting the accuracy of our prediction pipeline (10). Nevertheless, structural drifts from the native state often occurred, especially for the templated based modelling (TBM) targets (10).
The ModFOLD server (14–17) has been a leading approach for predicting the local and global quality of theoretical 3D models for more than a decade, according to both the CASP and CAMEO experiments (2,14–17). The local quality estimates provide significant information about the predicted accuracy score for each residue in each 3D model. In CASP13, ReFOLD2 was developed, which utilized a fixed restraint threshold, based on the per-residue accuracy scores produced by ModFOLD7 (the predicted distances from the native structure for each C-alpha atom), in order to guide the MD-based protocol. The CASP13 targets were relatively larger compared to the previous CASP experiments (3,10,18), and we observed that fixed restraint thresholds were not always appropriate for larger targets containing domains with varying accuracy. Therefore, with ReFOLD3, we developed a more targeted, novel gradual restraint strategy based on the local quality estimates for each starting model, which considered the required level of refinement for each individual residue.
In CASP13, contact prediction methods showed significant progress and reached up to 70% accuracy following the application of advanced deep learning methods (19–21). Residue–residue contact prediction methods have been used for the prediction of 3D models, protein–ligand interactions (22) and 3D model quality estimation (23–26), and more recently, the performance of the tertiary structure prediction has been boosted by contact prediction methods (19–21). Thus for ReFOLD3, we also utilized contact predictions for the refinement of the predicted 3D models with the application of an additional gradual restraint strategy based on our contact distance agreement (CDA) score (14). The CDA score measures the agreement between the contacts, which were predicted by the DeepMetaPSICOV method (21,27,28), and the contacts between residues in the predicted 3D model, measured according to their Euclidean distance (14). The CDA score has been a local scoring component of the ModFOLD server since CASP11, and it will become increasingly important as contact prediction accuracy improves (14,21,27–29).
In CASP14, ReFOLD3 gave us a performance boost in the main tertiary structure prediction category, where it enabled us to further improve the quality of some of the very best initial server models. Our group ranked in the top 10 in the refinement category itself, according to the official assessment. This was significant in the context of the very high-quality models of tertiary structures produced by many modelling groups. In CASP14, the models were much harder to refine as there was less room for improvement. Despite this major progress in modelling, most server models still contained significant local errors, which were both detectable by our ModFOLD8 method and fixable using ReFOLD3. It is notable that ReFOLD3 performed better at refining the most highly accurate starting models and it ranked within the top 5 refinement approaches, for models with GDT-TS scores >70. As 3D models of proteins become more widely used, it is important that biologists have access to freely available tools, such as ReFOLD3, in order to confidently identify and fix local errors in their theoretical models and bring them even closer to experimental quality.
MATERIALS AND METHODS
ReFOLD3, consisted of four protocols, which included improvements on the original version (10). The major improvement for ReFOLD3 was the accommodation of the two new comprehensive MD-based strategies (Supplementary Figure S1). The first and final protocol used i3Drefine (1,11,30) to refine the input model with 20 rapid iterations, the second and third protocols both employed more CPU/GPU intensive molecular dynamic simulation strategies, and models were ranked and selected at each stage using our ModFOLD8 server (14,16).
The second protocol included the introduction of molecular dynamics simulations that were guided by the per-residue accuracy scores obtained from ModFOLD8 (14,16,31). For the gradual restraint strategy, it was assumed that the regions identified as highly accurate should be restrained by applying a stronger harmonic positional restraint to prevent deviations from the native basin (3,7,13). A weaker restraint was also applied to the poorly predicted regions to allow for increases in the quality of these regions towards the native state (Supplementary Figure S2A). Thus, the gradual restraints ranged from weak (0.05 kCal/mol/Å2) to strong (1 kCal/mol/Å2) harmonic positional restraints, which were applied to all atoms, including C-alphas, according to the distribution of the per-residue accuracy scores produced by ModFOLD8 (Supplementary Table S1) (3,7,10,18,31).
For the third protocol, a similar gradual restraint strategy was used to guide the MD simulation, but this time it was based on the residue-residue contact predictions. We utilized the CDA score method (14,31), which is based on the agreement between the residue contacts predicted by DeepMetaPSICOV (21) and the contacts in the model. If the CDA score was high, a stronger restraint was applied to preserve those residue contacts in the predicted 3D model. Lower CDA scores indicated where residues were likely to be further away from the native structure, and so weaker restraints were applied (Supplementary Table S2 and Supplementary Figure S2B).
The MD-based protocols were carried out at normal cellular conditions at 298 K with 1 bar of pressure in explicit solvent using nanoscale molecular dynamics (NAMD) (32) version 2.10 in Graphics processing unit (GPU) mode. The CHARMM22/27 force field (33) and the TIP3P water model (34) were also used for the simulation of the protein system. The system was also neutralized by inserting Na+ or Cl- ions to balance the net charge using Particle Mesh Ewald (PME) (35). The non-bonded interactions (mostly van der Waal’s) were cut off by 12 Å to the exclusion of bonded interactions by using the CHARMM27 default parameter file with the switching distance of 10 Å (10). Using the pairlistdist function with 14 Å distance between atom pairs for inclusion in pair lists made the switching function more efficient (10). The rigidBonds functions were also used to rigidify hydrogen bonds with a 2 fs timestep (10). The system’s electrostatics and the temperature were calculated by PME with the temperature control using Langevin dynamics under the NTP conditions (constant number of particles, temperature and pressure) (10,36). The correction of clashes and the minimization of the system were carried out by 1000 steps in the first step of the MD-based protocol (10). The minimization step was followed by the implementation of the defined MD simulation to refine each target (10). Four parallel simulations were run for 2 ns, making 8 ns in total for a target as in the original MD-based protocol of ReFOLD (10). After the completion of the simulation run, 164 refined models were generated per target by taking a snapshot every 50ps, for each MD-based protocol (10).
Refined models generated from the MD-based protocols were then assessed and ranked using ModFOLD8_rank (10,14,31). The fourth protocol was a combination of the MD-based approaches, where the top-ranked model from the second and third protocol was then further refined using i3Drefine (1,10,11,14,31). Finally, all of the refined models generated by each of these protocols and the input model were pooled and re-ranked again according to the ModFOLD8_rank global scores (1,10,11,14,31).
RESULTS AND DISCUSSION
Server inputs and outputs
The required inputs for ReFOLD3 are the protein sequence and a 3D model (in PDB format) for refinement. Optionally, users may provide a name for their protein sequence and their email address. The ReFOLD3 server results page (Figure 1) includes an accurate estimate of the likely percentage improvement in model quality score for each refined 3D model, and it is unique in providing a series of individual per-residue error plots (Figure 1B), which show the local quality estimates for the refined models compared to the uploaded protein structure. Therefore, users may easily visualize the specific local improvements in every refined 3D model at a glance. Users may also click buttons to compare the refined and original 3D models interactively directly within the browser. The superposition of the refined models with the input models can be visualized using the JSmol/HTML5 framework. Therefore, models can be viewed in 3D directly within in the browser, including on mobile devices, without the requirement of any plugins (Figure 1C). The ReFOLD3 server will typically provide the results within ∼21 h for a target with 100 residues, with the MD simulation stages lasting ∼4 h (with Intel Xeon Platinum 8268 CPUs and Nvidia Tesla T4 GPUs). The server should complete the majority of refinement jobs within 48 h, once they are running.
Figure 1.

The ReFOLD3 server results page for an FM CASP14 target (T1031). In CASP14, the ReFOLD3 protocol was used to refine the top selected server models. (A) The results page provides the superposition of the top model and the input 3D model, a summary of the predicted scores and improvements in model quality estimation, along with the confidence intervals and P-value for each generated 3D model. After clicking on images or plots, users can download and view the data in detail (B) The plot of the predicted per-residue accuracy scores produced by ModFOLD8 for the top refined 3D model (green bars) compared with the original 3D model (red bars). Plots can also be downloaded as PDFs. (C) Interactive superposition of the top 3D model and the original 3D model, which are displayed in 3D using JSmol. Models can also be downloaded which including the per-residue error data in the B-factor column.
Independent benchmarking and cross validation
CASP14 and CASP Commons 2020: The ReFOLD3 protocol was used to provide a significant proportion of the top ten identified 3D models for the ten SARS-2-CoV targets in CASP Commons COVID-19 initiative (Supplementary Tables S3–S10) (1,10,11,14,31). ReFOLD3 was also subsequently blind tested by independent assessors in the CASP14 experiment (May–Aug 2020) (2,10,14,18,31). Our refinement pipeline was employed to refine the best server 3D models for each target, which were selected by ModFOLD8 in the regular prediction category, as well as the starting models provided by the CASP assessors for the refinement category itself (1,10,11,14,31). ReFOLD3 generated numerous improved models compared to the initial structures, and this enhanced the performance of our prediction pipeline in the CASP14 experiment (Table 1 and Figure 2) (1,10,11,14,31). ReFOLD3 was ranked as the ninth best approach according to the assessors’ formula (Supplementary Table S11) and tenth, according to the cumulative GDT-TS score in the CASP14 refinement category (Supplementary Table S12).
Table 1.
The performance of the McGuffin group ReFOLD3 refinement pipeline for all regular CASP14 targets according to the GDT-TS and lDDT scores versus the starting model. The top ModFOLD8 selected server models were taken as the starting models that were refined using ReFOLD3 and submitted during the CASP14 experiment. Data are from https://www.predictioncenter.org/casp14/
| CASP model ID | GDT-TS | LDDT | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Target ID | Prediction category | Starting model | Submitted model | Starting model | Refined model | Diff | Starting model | Refined model | Diff |
| T1024 | TBM-easy | T1024TS326_2 | T1024TS220_1 | 60.68 | 60.93 | 0.25 | 0.67 | 0.68 | 0.01 |
| T1026 | TBM-hard | T1026TS487_4-D1 | T1026TS220_1-D1 | 68.49 | 68.15 | -0.34 | 0.54 | 0.58 | 0.04 |
| T1027 | FM | T1027TS487_1-D1 | T1027TS220_1-D1 | 36.87 | 37.12 | 0.25 | 0.4 | 0.4 | 0 |
| T1029 | FM | T1029TS487_1-D1 | T1029TS220_1-D1 | 40.8 | 40.8 | 0 | 0.47 | 0.47 | 0 |
| T1030 | TBM-hard | T1030TS487_1 | T1030TS220_1 | 39.84 | 43.77 | 3.93 | 0.6 | 0.62 | 0.02 |
| T1031 | FM | T1031TS209_5-D1 | T1031TS220_1-D1 | 24.47 | 24.74 | 0.27 | 0.32 | 0.32 | 0 |
| T1033 | FM | T1033TS209_5-D1 | T1033TS220_1-D1 | 46.25 | 45.25 | -1 | 0.51 | 0.5 | -0.01 |
| T1034 | TBM-easy | T1034TS487_1-D1 | T1034TS220_1-D1 | 82.37 | 82.53 | 0.16 | 0.7 | 0.73 | 0.03 |
| T1035 | FM/TBM | T1035TS351_3-D1 | T1035TS220_1-D1 | 48.28 | 50 | 1.72 | 0.5 | 0.5 | 0 |
| T1037 | FM | T1037TS337_1-D1 | T1037TS220_1-D1 | 51.3 | 51.3 | 0 | 0.52 | 0.53 | 0.01 |
| T1038 | FM/TBM | T1038TS487_4 | T1038TS220_1 | 26.45 | 26.58 | 0.13 | 0.37 | 0.36 | -0.01 |
| T1039 | FM | T1039TS487_4-D1 | T1039TS220_1-D1 | 34.63 | 34.78 | 0.15 | 0.35 | 0.38 | 0.03 |
| T1040 | FM | T1040TS351_3-D1 | T1040TS220_1-D1 | 24.42 | 23.65 | -0.77 | 0.38 | 0.38 | 0 |
| T1041 | FM | T1041TS377_3-D1 | T1041TS220_1-D1 | 52.58 | 53 | 0.42 | 0.53 | 0.53 | 0 |
| T1042 | FM | T1042TS226_2-D1 | T1042TS220_1-D1 | 54.98 | 54.62 | -0.36 | 0.55 | 0.53 | -0.02 |
| T1043 | FM | T1043TS487_3-D1 | T1043TS220_1-D1 | 17.06 | 17.23 | 0.17 | 0.22 | 0.24 | 0.02 |
| T1045s2 | TBM-easy | T1045s2TS487_4-D1 | T1045s2TS220_1-D1 | 69.28 | 70.03 | 0.75 | 0.63 | 0.65 | 0.02 |
| T1046s1 | FM/TBM | T1046s1TS487_3-D1 | T1046s1TS220_1-D1 | 74.31 | 75 | 0.69 | 0.6 | 0.6 | 0 |
| T1046s2 | TBM-hard | T1046s2TS487_4-D1 | T1046s2TS220_1-D1 | 75.53 | 76.06 | 0.53 | 0.57 | 0.63 | 0.06 |
| T1047s1 | FM | T1047s1TS075_1-D1 | T1047s1TS220_1-D1 | 32.58 | 32.94 | 0.36 | 0.58 | 0.56 | -0.02 |
| T1047s2 | FM/TBM | T1047s2TS487_5 | T1047s2TS220_1 | 34.05 | 34.21 | 0.16 | 0.62 | 0.66 | 0.04 |
| T1049 | FM | T1049TS326_3-D1 | T1049TS220_1-D1 | 63.25 | 63.43 | 0.18 | 0.57 | 0.58 | 0.01 |
| T1050 | TBM-easy | T1050TS487_1 | T1050TS220_1 | 55.85 | 55.88 | 0.03 | 0.63 | 0.67 | 0.04 |
| T1052 | FM/TBM | T1052TS487_4 | T1052TS220_1 | 54.48 | 54.15 | -0.33 | 0.67 | 0.66 | -0.01 |
| T1053 | FM/TBM | T1053TS238_4 | T1053TS220_1 | 39.95 | 39.18 | -0.77 | 0.54 | 0.53 | -0.01 |
| T1054 | TBM-hard | T1054TS326_4-D1 | T1054TS220_1-D1 | 67.48 | 68.71 | 1.23 | 0.66 | 0.68 | 0.02 |
| T1055 | FM/TBM | T1055TS238_5-D1 | T1055TS220_1-D1 | 70.9 | 70.29 | -0.61 | 0.57 | 0.55 | -0.02 |
| T1056 | TBM-hard | T1056TS209_3-D1 | T1056TS220_1-D1 | 54.44 | 53.25 | -1.19 | 0.48 | 0.46 | -0.02 |
| T1057 | TBM-easy | T1057TS351_5-D1 | T1057TS220_1-D1 | 79.57 | 79.47 | -0.1 | 0.68 | 0.68 | 0 |
| T1060s3 | TBM-hard | T1060s3TS487_4-D1 | T1060s3TS220_1-D1 | 67.89 | 67.28 | -0.61 | 0.61 | 0.64 | 0.03 |
| T1061 | FM/TBM | T1061TS326_4 | T1061TS220_1 | 30.34 | 30.5 | 0.16 | 0.45 | 0.47 | 0.02 |
| T1064 | FM | T1064TS140_1-D1 | T1064TS220_1-D1 | 20.65 | 20.65 | 0 | 0.22 | 0.23 | 0.01 |
| T1065s1 | TBM-hard | T1065s1TS487_1-D1 | T1065s1TS220_1-D1 | 88.44 | 88.87 | 0.43 | 0.75 | 0.8 | 0.05 |
| T1065s2 | TBM-hard | T1065s2TS277_4-D1 | T1065s2TS220_1-D1 | 90.31 | 91.07 | 0.76 | 0.78 | 0.78 | 0 |
| T1067 | TBM-hard | T1067TS351_3-D1 | T1067TS220_1-D1 | 52.83 | 52.49 | -0.34 | 0.51 | 0.49 | -0.02 |
| T1068 | TBM-hard | T1068TS183_2-D1 | T1068TS220_1-D1 | 57.26 | 55.17 | -2.09 | 0.53 | 0.51 | -0.02 |
| T1073 | TBM-easy | T1073TS140_5-D1 | T1073TS220_1-D1 | 83.47 | 83.47 | 0 | 0.73 | 0.73 | 0 |
| T1074 | FM | T1074TS487_5-D1 | T1074TS220_1-D1 | 35.8 | 35.98 | 0.18 | 0.38 | 0.39 | 0.01 |
| T1076 | TBM-easy | T1076TS487_4-D1 | T1076TS220_1-D1 | 87.41 | 87.41 | 0 | 0.72 | 0.78 | 0.06 |
| T1078 | TBM-easy | T1078TS351_3-D1 | T1078TS220_1-D1 | 76.74 | 76.55 | -0.19 | 0.7 | 0.69 | -0.01 |
| T1079 | TBM-easy | T1079TS487_2-D1 | T1079TS220_1-D1 | 62.03 | 62.36 | 0.33 | 0.64 | 0.68 | 0.04 |
| T1080 | FM/TBM | T1080TS183_1-D1 | T1080TS220_1-D1 | 24.81 | 25.19 | 0.38 | 0.41 | 0.41 | 0 |
| T1082 | FM/TBM | T1082TS487_5-D1 | T1082TS220_1-D1 | 58.67 | 59.33 | 0.66 | 0.43 | 0.42 | -0.01 |
| T1083 | TBM-hard | T1083TS364_4-D1 | T1083TS220_1-D1 | 83.7 | 83.15 | -0.55 | 0.71 | 0.71 | 0 |
| T1084 | TBM-hard | T1084TS252_2-D1 | T1084TS220_1-D1 | 88.73 | 88.03 | -0.7 | 0.76 | 0.75 | -0.01 |
| T1085 | FM/TBM | T1085TS183_3 | T1085TS220_1 | 35.97 | 35.78 | -0.19 | 0.61 | 0.61 | 0 |
| T1086 | FM/TBM | T1086TS238_1 | T1086TS220_1 | 41.8 | 42.06 | 0.26 | 0.73 | 0.71 | -0.02 |
| T1089 | TBM-easy | T1089TS238_4-D1 | T1089TS220_1-D1 | 65.45 | 65.92 | 0.47 | 0.59 | 0.59 | 0 |
| T1090 | FM | T1090TS487_1-D1 | T1090TS220_1-D1 | 53.17 | 53.17 | 0 | 0.51 | 0.53 | 0.02 |
| T1091 | TBM-easy | T1091TS351_4 | T1091TS220_1 | 26.62 | 26.67 | 0.05 | 0.62 | 0.62 | 0 |
| T1092 | TBM-easy | T1092TS319_3 | T1092TS220_1 | 23.24 | 23.47 | 0.23 | 0.45 | 0.49 | 0.04 |
| T1093 | FM | T1093TS487_3 | T1093TS220_1 | 25.44 | 25.4 | -0.04 | 0.36 | 0.39 | 0.03 |
| T1095 | TBM-hard | T1095TS487_2-D1 | T1095TS220_1-D1 | 41.86 | 42.09 | 0.23 | 0.56 | 0.61 | 0.05 |
| T1096 | FM | T1096TS252_1 | T1096TS220_1 | 26.14 | 26.52 | 0.38 | 0.52 | 0.56 | 0.04 |
| T1099 | TBM-hard | T1099TS487_4-D1 | T1099TS220_1-D1 | 57.3 | 55.76 | -1.54 | 0.59 | 0.53 | -0.06 |
| T1101 | TBM-easy | T1101TS487_4 | T1101TS220_1 | 56.65 | 57.16 | 0.51 | 0.65 | 0.69 | 0.04 |
| Total | 2943.86 | 2948.55 | 4.69 | 30.95 | 31.47 | 0.52 | |||
Figure 2.

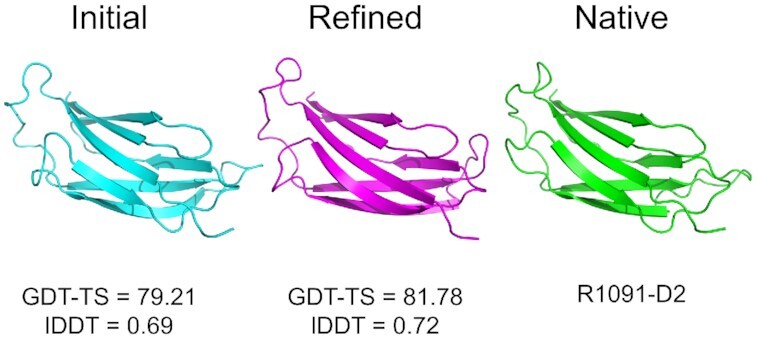
The refinement of four CASP14 targets using the ReFOLD3 protocol. In the left panels, the initial structures were coloured by the per-residue accuracy score produced by ModFOLD8. In the middle panels, superposition of the best predicted 3D server model selected by ModFOLD8 or the starting model provided by CASP in the refinement category (cyan), the top 3D model generated by ReFOLD3 (magenta) and native structure (green). In the right panels, GDT_TS plots for the comparison of the original 3D model (cyan) with the top 3D model generated by ReFOLD3 (magenta). (A) CASP14 regular TBM-hard target T1030: RaptorX_TS1 versus McGuffin_TS1, a GDT_TS improvement from 39.84 to 43.77. (B) CASP14 regular FM/TBM target T1035 domain 1: tFOLD-lDT_TS3 versus McGuffin_TS1, a GDT_TS improvement from 48.28 to 50.00. (C) CASP14 refinement FM target R10909: starting model versus McGuffin_TS1, a GDT_TS improvement from 65.61 to 67.06 (D) CASP14 refinement TBM-easy target R1091-D1: starting model versus McGuffin TS1, a GDT_TS improvement from 79.21 to 81.78. Images are created using PyMOL (http://www.pymol.org). GDT_TS plots are from https://www.predictioncenter.org/casp14/.
For the regular CASP14 prediction category, in our prediction pipeline, the top 3D model was selected by ModFOLD8 and then refined by the ReFOLD3 protocol in an attempt to further increase the accuracy of the predicted structure (1,10,11,14,31). From the results in Table 1, it is clear that ReFOLD3 managed to improve the quality for 68% of the top server 3D models selected by ModFOLD8, according to GDT-TS and lDDT scores (∑ΔGDT TS = 4.69, ∑ΔlDDT = 0.52). The GDT-TS score (37) is based on the superposition of C-alpha atoms between the predicted and native structure, while the lDDT score (38) calculates scores based on the atom–atom distance between the predicted or refined 3D model and the observed structure, considering all atoms (38). Thus, the results show that ReFOLD3 increased the accuracy of both the backbone and local regions for 68% of the structures. Furthermore, whereas the original version of ReFOLD was not successful in increasing the accuracy of the models for TBM targets in CASP12 (10), ReFOLD3 performed well across all target definitions in CASP14, including TBM (Figure 2, Supplementary Tables S13–S15).
The refinement of the best server 3D model in the regular prediction category is arguably closer to a real-world scenario, compared to refinement of the starting models provided by assessors in the refinement category. The starting models chosen by the CASP assessors for the refinement category, may have already been pre-refined in their modelling pipelines and they often represent the very highest quality starting models, which makes their further refinement much more difficult. Nevertheless, ReFOLD3 managed to improve the quality for 38% of the starting models in the refinement category according to the observed scores (Supplementary Table S16). Using ReFOLD3, our group ranked ninth in the refinement category for the regular time-frame targets, according to the official assessors’ formula (Supplementary Table S11). The ReFOLD3 method has also shown similar performance for each of the CASP14 target definitions, for the refinement category as well as the regular prediction category (Supplementary Table S17-S19). The datasets used for benchmarking are freely available for all to download via https://predictioncenter.org/download_area/CASP14/.
The performance of the ReFOLD3 was also analysed according to the size (number of amino acids) and the GDT-TS score intervals of the starting models by the CASP assessors in the refinement category (Supplementary Tables S20–S25). It is clear that the CASP14 refinement targets were much harder to refine overall and there was less room for improvement, as high-quality models of tertiary structures were produced by many modelling groups, which were then chosen by the assessors (20,39). However, it is important to note that many models still contained significant local errors, which were both detected and fixed using our ReFOLD3 pipeline (10,14,31). In that context, while many groups did not perform particularly well for targets with GDT-TS scores >70 (Supplementary Table S25), ReFOLD3 performed relatively better at refining the most highly accurate starting models and was ranked within the top 5 refinement approaches for targets with GDT-TS scores >60, according to the cumulative Z-score (Supplementary Tables S24 and S25).
CONCLUSIONS
ReFOLD3 is one of the leading freely available fully automated servers for the refinement of theoretical 3D models of proteins. The method is unique in utilizing a gradual restraint strategy, based on both contact predictions and local quality estimation, to guide the refinement of protein models. Our application of gradual, rather than fixed, restraints has proved to be more successful for guiding our MD-simulations. As well as existing as an independent server, the ReFOLD3 method is integrated with the latest ModFOLD8 server (14,31). This integration enables users to identify the local errors in their 3D models and then correct them more conveniently, through the targeted improvement of specific regions. Using the ReFOLD3 server, non-expert users can easily visualize the likely improvements to their models, via intuitive per-residue error plots and 3D model superpositions.
DATA AVAILABILITY
The ReFOLD3 server is freely available at: https://www.reading.ac.uk/bioinf/ReFOLD/
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Prof Dan Rigden who was the CASP assessor in CASP14 refinement category and Adam Simpkin (PhD) for sharing their Z-score evaluation data, which was based on the refinement target lengths and accuracy.
Contributor Information
Recep Adiyaman, School of Biological Sciences, University of Reading, Whiteknights, Reading RG6 6AS, UK.
Liam J McGuffin, School of Biological Sciences, University of Reading, Whiteknights, Reading RG6 6AS, UK.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Biotechnology and Biological Sciences Research Council (BBSRC) [BB/T018496/1 to L.J.M., R.A.]. Funding for open access charge: Biotechnology and Biological Sciences Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bhattacharya D., Cheng J.. i3Drefine software for protein 3D structure refinement and its assessment in CASP10. PLoS One. 2013; 8:e69648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hovan L., Oleinikovas V., Yalinca H., Kryshtafovych A., Saladino G., Gervasio F.L.. Assessment of the model refinement category in CASP12. Proteins. 2018; 86:152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adiyaman R., McGuffin L.J.. Methods for the refinement of protein structure 3D models. Int. J. Mol. Sci. 2019; 20:2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feig M. Computational protein structure refinement: almost there, yet still so far to go. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017; 7:e1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heo L., Park H., Seok C.. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013; 41:384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. MacCallum J.L., Pérez A., Schnieders M.J., Hua L., Jacobson M.P., Dill K.A.. Assessment of protein structure refinement in CASP9. Proteins. 2011; 79:74–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mirjalili V., Feig M.. Protein structure refinement through structure selection and averaging from molecular dynamics ensembles. J. Chem. Theory Comput. 2013; 9:1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E.. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010; 78:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D.. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017; 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shuid A.N., Kempster R., McGuffin L.J.. ReFOLD: a server for the refinement of 3D protein models guided by accurate quality estimates. Nucleic Acids Res. 2017; 45:422–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhattacharya D., Nowotny J., Cao R., Cheng J.. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016; 44:W406–W409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feig M., Mirjalili V.. Protein structure refinement via molecular-dynamics simulations: what works and what does not. Proteins. 2016; 84:282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mirjalili V., Noyes K., Feig M.. Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins. 2014; 82:196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maghrabi A.H.A., McGuffin L.J.. ModFOLD6: an accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res. 2017; 45:W416–W421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McGuffin L.J. The ModFOLD server for the quality assessment of protein structural models. Bioinformatics. 2008; 24:586–587. [DOI] [PubMed] [Google Scholar]
- 16. McGuffin J.L., Buenavista M.T., Roche D.B.. The ModFOLD4 server for the quality assessment of 3D protein models. Nucleic Acids Res. 2013; 41:W368–W372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Haas J., Barbato A., Behringer D., Studer G., Roth S., Bertoni M., Mostaguir K., Gumienny R., Schwede T.. Continuous automated model evaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins. 2018; 86:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Read R.J., Sammito M.D., Kryshtafovych A., Croll T.I.. Evaluation of model refinement in CASP13. Proteins. 2019; 87:1249–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shrestha R., Fajardo E., Gil N., Fidelis K., Kryshtafovych A., Monastyrskyy B., Fiser A.. Assessing the accuracy of contact predictions in CASP13. Proteins. 2019; 87:1058–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A.et al.. Improved protein structure prediction using potentials from deep learning. Nature. 2020; 577:706–710. [DOI] [PubMed] [Google Scholar]
- 21. Kandathil S.M., Greener J.G., Jones D.T.. Prediction of interresidue contacts with DeepMetaPSICOV in CASP13. Proteins. 2019; 87:1092–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kliger Y., Levy O., Oren A., Ashkenazy H., Tiran Z., Novik A., Rosenberg A., Amir A., Wool A., Toporik A.et al.. Peptides modulating conformational changes in secreted chaperones: from in silico design to preclinical proof of concept. Proc. Natl. Acad. Sci. USA. 2009; 106:13797–13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miller C.S., Eisenberg D.. Using inferred residue contacts to distinguish between correct and incorrect protein models. Bioinformatics. 2008; 24:1575–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z., Eickholt J., Cheng J.. APOLLO: a quality assessment service for single and multiple protein models. Bioinformatics. 2011; 27:1715–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adhikari B., Bhattacharya D., Cao R., Cheng J.. CONFOLD: residue-residue contact-guided ab initio protein folding. Proteins. 2015; 83:1436–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adhikari B., Cheng J.. Protein residue contacts and prediction methods. Methods Mol Biol. 2016; 1415:463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jones D.T., Singh T., Kosciolek T., Tetchner S.. MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics. 2015; 31:999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kosciolek T., Jones D.T.. Accurate contact predictions using covariation techniques and machine learning. Proteins. 2016; 84:145–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buchan D.W.A., Jones D.T.. Improved protein contact predictions with the MetaPSICOV2 server in CASP12. Proteins. 2018; 86:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhattacharya D., Cheng J.. 3Drefine: consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins. 2013; 81:119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Maghrabi A.H.A., McGuffin L.J.. Estimating the quality of 3D protein models using the ModFOLD7 server. Methods Mol. Biol. 2019; 2165:69–81. [DOI] [PubMed] [Google Scholar]
- 32. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Best R.B., Zhu X., Shim J., Lopes P.E.M., Mittal J., Feig M., MacKerell A.D.. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ 1 and χ 2 dihedral angles. J. Chem. Theory Comput. 2012; 8:3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926. [Google Scholar]
- 35. Götz A.W., Williamson M.J., Xu D., Poole D., Le Grand S., Walker R.C.. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J. Chem. Theory Comput. 2012; 8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Loncharich R.J., Brooks B.R., Pastor R.W.. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N′-methylamide. Biopolymers. 1992; 32:523–535. [DOI] [PubMed] [Google Scholar]
- 37. Zhang Y., Skolnick J.. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005; 33:2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mariani V., Biasini M., Barbato A., Schwede T.. lDDT: a local superposition-free score for comparing protein structures and models using distance difference tests. Bioinformatics. 2013; 29:2722–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A.et al.. Protein structure prediction using multiple deep neural networks in the 13th critical assessment of protein structure prediction (CASP13). Protein Struct. Funct. Bioinform. 2019; 87:1141–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ReFOLD3 server is freely available at: https://www.reading.ac.uk/bioinf/ReFOLD/