

Abstract
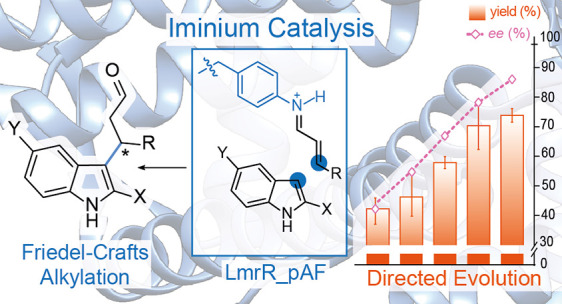
The construction and engineering of artificial enzymes consisting of abiological catalytic moieties incorporated into protein scaffolds is a promising strategy to realize non-natural mechanisms in biocatalysis. Here, we show that incorporation of the noncanonical amino acid para-aminophenylalanine (pAF) into the nonenzymatic protein scaffold LmrR creates a proficient and stereoselective artificial enzyme (LmrR_pAF) for the vinylogous Friedel–Crafts alkylation between α,β-unsaturated aldehydes and indoles. pAF acts as a catalytic residue, activating enal substrates toward conjugate addition via the formation of intermediate iminium ion species, while the protein scaffold provides rate acceleration and stereoinduction. Improved LmrR_pAF variants were identified by low-throughput directed evolution advised by alanine-scanning to obtain a triple mutant that provided higher yields and enantioselectivities for a range of aliphatic enals and substituted indoles. Analysis of Michaelis–Menten kinetics of LmrR_pAF and evolved mutants reveals that different activities emerge via evolutionary pathways that diverge from one another and specialize catalytic reactivity. Translating this iminium-based catalytic mechanism into an enzymatic context will enable many more biocatalytic transformations inspired by organocatalysis.
Keywords: artificial enzymes, Friedel−Crafts alkylation, iminium ion, noncanonical amino acids, stop-codon suppression, directed evolution, catalytic promiscuity
Introduction
The enantioselective Friedel–Crafts alkylation of arenes is an important reaction in both synthetic chemistry and biosynthesis.1−4 The use of enzymes as catalysts for this transformation is of great appeal because of their mild, efficient, benign, and highly selective nature; however, relatively few enzymes are known to perform this chemistry.4−7 Notable examples are tryptophan synthase (TrpB),8 a promiscuous enzyme involved cylindrocyclophane biosynthesis (CylK),9,10 as well as enzymes which catalyze Friedel–Crafts acylation (PpATase) (Figure 1a).11,12 Catalytic machinery in these enzymes includes nucleophilic cysteine residues as well as pyridoxal phosphate, each powerful chemical motifs which have inspired biomimetic catalysis strategies.13,14 Artificial enzymes have also been employed for Friedel–Crafts alkylation reactions by CuII Lewis acid activation of α,β-unsaturated imidazole ketones, employing proteins or DNA as a scaffold to afford rate acceleration and enantioselectivity.15,16 An effective chemocatalytic strategy for Friedel–Crafts alkylation of arenes employs secondary-amine organocatalysts (e.g., MacMillan imidazolidinone catalysts) to activate α,β-unsaturated aldehydes as iminium ions (Figure 1b).1,17,18 The iminium ion species is a highly versatile reaction intermediate19 which has previously been exploited in designer enzymes featuring catalytic lysine or N-terminal proline residues and in artificial enzymes with secondary-amine-containing unnatural cofactors.20−25 However, while Friedel–Crafts alkylation via an iminium ion species has been demonstrated with catalytic peptides with N-terminal proline residues,26 it is a distinct reaction pathway from those employed by natural enzymes for this reaction. We saw great potential in unlocking iminium catalysis to create an artificial Friedel–Crafts alkylase by using expanded genetic code technology to provide an appropriate noncanonical catalytic residue.27
Figure 1.
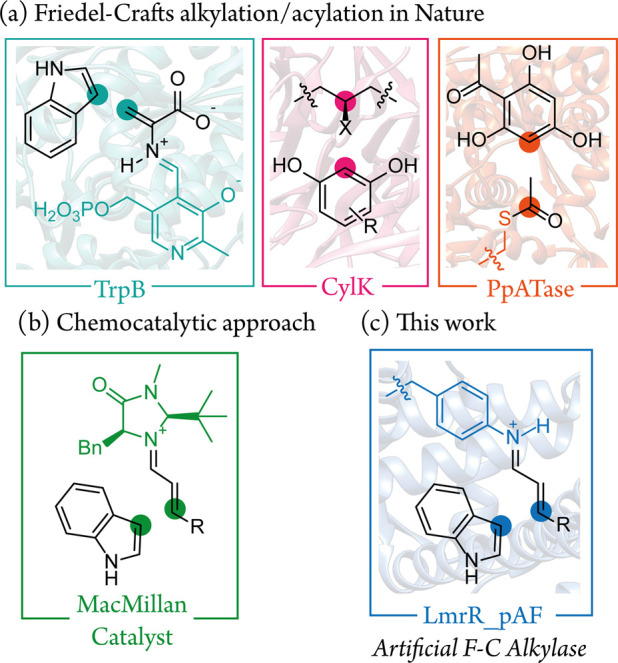
(a) Biocatalytic strategies of natural enzymes which catalyze aromatic alkylation/acylation; (b) organocatalytic approach to vinylogous Friedel–Crafts alkylation; (c) approach followed in this work: an artificial enzyme using expanded genetic code technology to incorporate a noncanonical catalytic residue which operates via a non-natural mechanism.
Artificial enzymes consist of an abiological catalyst incorporated into a protein scaffold which provides stereoselectivity and rate acceleration, as well as the opportunity for genetic optimization.28−30 Strategies to assemble these two components include covalent protein modification, supramolecular, and “trojan-horse” strategies.31,32 Our group has recently pioneered the use of noncanonical amino acids (ncAAs) with inherent catalytic properties for artificial enzyme construction, where the catalytic component is incorporated directly into the protein’s peptide-chain during biosynthesis using expanded genetic code technology.27,33,34 The judicious choice of a protein scaffold is also essential for success; in this study, as in our previous work, we employ the Lactococcal multidrug resistance regulatory (LmrR) protein. This homodimeric protein (15 kDa per monomer) has a unique structural feature, a large hydrophobic pocket situated at the dimer interface, which binds planar organic molecules in its physiological function and affords rate-acceleration and enantio-induction of catalysis conducted inside it.35−37 We incorporated the aniline containing ncAA para-aminophenylalanine (pAF) at position 15 in the hydrophobic pocket of LmrR, creating LmrR_pAF, an artificial enzyme for the hydrazone formation reaction which acts via a trans-amination mechanism employing pAF as the catalytic residue.35,36 A similar strategy has been employed by Green and co-workers, who showed that incorporation of N-methyl histidine into a protein scaffold could endow ester-hydrolase activity.38 More recently, we also employed the pAF catalytic residue in a synergistic system, where α,β-unsaturated aldehydes were activated to iminium ion species at the pAF residue for conjugate addition by enolate nucleophiles activated by a CuII cofactor.39 In this work, we show LmrR_pAF as a means to enable Friedel–Crafts alkylation of indoles via iminium catalysis in a designer enzyme (Figure 1c). Unlike our previous work, where two catalytic components were required, the indole nucleophiles do not require activation by a separate cofactor.39 Situating the pAF residue within the LmrR microenvironment significantly enhances its inherent catalytic properties and paves a path toward further new-to-nature reactions.
Results and Discussion
Intitial Catalysis Experiments
We initially explored the vinylogous Friedel–Crafts alkylation of 2-methyl-indole (2a) with trans-2-hexenal (1a) catalyzed by LmrR_pAF to form product 3a after in situ reduction with sodium borohydride, which makes the product amenable to HPLC analysis.40 We found that with just 2 mol % loading, LmrR_pAF could catalyze the transformation of 1a and 2a to give 3a with a moderate yield and enantioselectivity (Table 1, entry 1). Without pAF incorporated, LmrR itself gives negligible yield and enantioselectivity under the same conditions (Table 1, entry 2). Lysine, which is used as a catalytic residue in nature (e.g., aldolase A),41 when incorporated at position V15 in LmrR fails to endow the protein with significant catalytic activity for this transformation (Table 1, entry 3). Likewise, mutation of the same position to tyrosine—in effect, single-functional group mutagenesis of the amino moiety of pAF to the canonical hydroxy moiety—also abrogates catalytic activity, demonstrating the necessity of the aniline functional group for efficient catalysis (Table 1, entry 4). These control reactions (Table 1, entries 2–4) implicate the pAF side chain as a catalytic residue in this system, where the amino moiety condenses with the enal substrate to form an iminium intermediate (Figure 1c). Such intermediates have been trapped by reductive amination in our previous work.33,39 Even at equimolar catalyst loadings, aniline itself affords only 8% yield of 3a, demonstrating the importance of the chemical environment provided by the hydrophobic pocket of LmrR for rate acceleration (Table 1, entry 5). Finally, we ruled out the efficacy of an artificial enzyme based on the supramolecular assembly between LmrR and aniline by combining these two catalysts in a ratio typically employed in supramolecular catalysis (Table 1, entry 6).15 Nevertheless, we observe that the LmrR protein scaffold itself (Table 1, entries 2–4 and 6) has some modest catalytic properties for this reaction giving small yields above the background reaction in buffer (Table 1, entry 7), presumably because of hydrophobic substrate binding giving enhanced effective molarity, but without significant enantioselectivity.
Table 1. Initial Results of LmrR_pAF Catalyzed Formation of 3a from 1a and 2a and Control Reactionsa.

| entry | catalyst | yield (%)b | ee (%)c |
|---|---|---|---|
| 1 | LmrR_pAF (20 μM) | 42 ± 4 | 45 ± 0 |
| 2 | LmrR (20 μM) | 2 ± 0 | –7 ± 0 |
| 3 | LmrR_V15K (20 μM) | 3 ± 0 | –15 ± 0 |
| 4 | LmrR_V15Y (20 μM) | 2 ± 0 | –11 ± 5 |
| 5 | aniline (1 mM) | 8 ± 0 | - |
| 6 | LmrR (20 μM) + aniline (16 μM) | 4 ± 0 | 1 ± 1 |
| 7 | none | <1 | - |
Reaction conditions: LmrR variants (20 μM dimer concentration) in phosphate buffer (50 mM, pH 6.5) containing NaCl (150 mM) and 8 vol % DMF, [2a] = 1 mM, [1a] = 5 mM; reaction time 16 h at 4 °C with mixing by continuous inversion in 300 μL of total volume. Reduction with NaBH4 (60 μL, 20 mg mL–1 in 0.5 w/v% NaOH) afforded the alcohol product 3a. For each entry, two independent experiments were conducted, each in duplicate, except entries 5 and 7 which were conducted in triplicate. Errors are the standard deviation of the results.
Analytical yields determined by normal-phase HPLC (Chiralcel OJ-H) with 3-(3-hydroxypropyl)indole as internal standard.
Enantiomeric excess determined by chiral normal-phase HPLC (Chiralcel OJ-H), sign of the ee value refers to elution order.
Alanine Scanning
With a good starting activity achieved by LmrR_pAF for the formation of 3a, we sought to probe residues near the active site for their importance in catalysis by alanine scanning. Guided by the structure of LmrR_pAF we selected a total of 12 positions – E7, A11, L18, N19, K22, N88, M89, A92, F93, S95, S97, and D100 (Figure 2), choosing residues which we considered important for catalysis by LmrR-based artificial enzymes based on our previous studies, as well as several others which are close to the catalytic residue but are less well investigated.42−44 We mutated these positions to alanine, except in the cases of A11 and A92 which are already alanine in the wild-type LmrR sequence, where we chose mutations which have previously proved beneficial for catalysis for LmrR-based artificial enzymes, namely, A11L and A92E.42−44 We purified these 12 variants and assessed them in the production of 3a, comparing the enantioselectivity and yield obtained with that obtained with LmrR_pAF (Figure 2, Table S1).
Figure 2.

Analysis of the effects of mutations of residues close to the catalytic residue of LmrR_pAF in catalysis of the reaction of 1a and 2a to produce 3a. The alpha-carbon atoms of residues mutated are shown as spheres in the crystal structure of LmrR_pAF (PDB: 6I8N). Reactions were conducted under the standard reaction conditions outlined in Table 1. Enantioselectivity was visualized as the effect on relative Gibbs’ free energies of the diastereomeric transition states leading to formation of the two enantiomers of 3a which was calculated from the enantiomeric ratio according to the equation ΔΔG‡ = −RTln(e.r.). The results are shown on a scale from green (increased e.r.) to pink (decreased e.r.). Δyield values are the differences in analytical yields obtained when employing different mutants in catalysis for 3a product under the standard conditions, relative to LmrR_pAF, and are shown on a scale from blue (increased yield) to orange (decreased yield).
Of the 12 mutants tested, five (namely, E7A, A11L, N19A, N88A, and M89A) showed no significant effect on the catalytic results. One mutation, D100A, abrogated catalytic activity. Some of our previous work with LmrR-based enzymes also found this position to be immutable for catalysis, suggesting that it may be crucial for proper protein folding.44,45 Mutations at the six remaining residues produced larger, positive or negative, effects on the yield and enantioselectivity obtained in catalysis, indicating a greater importance for catalytic activity. We also tested the 12 mutants in the cell-free extract (CFE), and observed a similar trend in the catalysis results to those obtained with purified proteins (Table S2). Together, these results enabled a focused directed evolution campaign by prioritizing mutations at positions whose importance was highlighted by our alanine scanning as demonstrated in our previous work.43
Directed Evolution
We created randomized codon libraries at the six positions shown to be important for catalysis with alanine scanning (L18, K22, A92, F93, S95, S97).46 We opted for the NDT codon, which encodes 12 amino acids with a good representation of the functional diversity of all 20 proteogenic amino acids.47 The degenerate codon libraries were produced by QuikChange PCR and their qualities were confirmed with pooled plasmid sequencing.48 Cell-free extracts obtained from the resultant libraries were screened by chiral-HPLC and mutants that had beneficial effects on enantioselectivity and yield were verified with purified protein (representative screening results presented in Figure S1, summary of hits identified in Table S3). After one round of screening, we found a few improved mutants, including L18R and S95G, which afford higher yield and enantioselectivity in production of 3a relative to LmrR_pAF (Figure 3, Table S4). Gratifyingly, these particular mutants produced a synergistic effect when combined, giving yet further improvements in the double mutant LmrR_pAF_L18R_S95G (Figure 3, Table S4). In a similar manner, we continued with a second round of library screening, selecting five positions close to the catalytic residue for randomization. This second round of screening identified one further improved mutant, LmrR_pAF_L18R_S95G_M89N (hereafter, LmrR_pAF_RGN), which affords the product 3a with a yield of 74% and an enantiomeric excess of 87% under our standard reaction conditions (Figure 3, Table S4). As with LmrR_pAF, mutation of the catalytic residue in LmrR_pAF_RGN to either lysine or tyrosine abrogates activity (Table S4).
Figure 3.
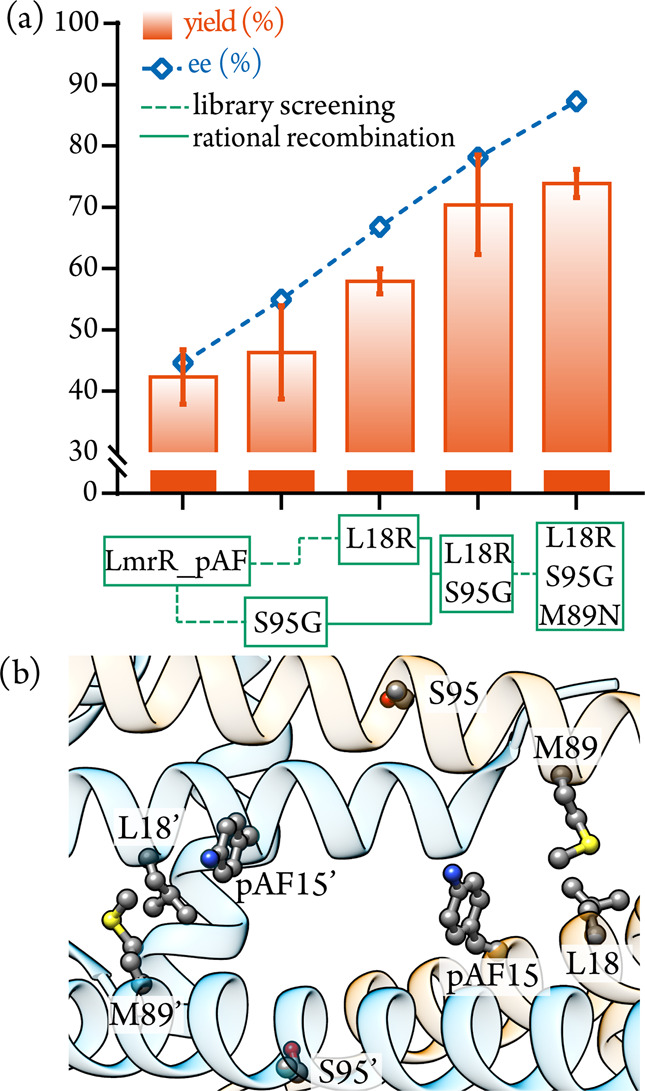
(a) Positions of residues mutated in LmrR_pAF_RGN are shown in the crystal structure of LmrR_pAF (PDB: 6I8N). (b) Analytical yields and enantioselectivities of mutants obtained throughout directed evolution of LmrR_pAF for the formation of 3a; the results with purified protein under standard conditions are outlined in Table 1. Mutations were obtained by screening of degenerate codon libraries (dashed lines) or by rational recombination (solid lines).
While the nature of the contributions of these mutations toward efficient and selective catalysis is hard to rationalize, it is notable that both this directed evolution campaign and our previous campaign with LrmR_pAF found mutants with an arginine residue close to the catalytic residue (previously A92R was identified).42 Positively charged residues in enzymes with catalytic lysine or proline residues are known to promote catalysis by perturbing pKas in the active site or stabilizing negatively charged intermediates.49−52 It is possible that R18 facilitates stereoselective iminium formation, shown to be the stereoselectivity determining step in the Friedel–Crafts reaction,53 acting as a primitive oxyanion hole to promote hemiaminal formation or dehydration. Since the asparagine mutation at position M89 also introduces a new side chain with hydrogen-bond-accepting properties this residue plausibly cooperates with R18 during iminium ion formation. Mutation of the S95 residue to glycine involves a significant shift in helical propensity54 and thus may perturb the secondary structure in the particularly dynamic α4 helix,37,55 thus shifting the conformational ensemble toward more catalytically productive states.56,57
Substrate Scope
We assessed the performance of LmrR_pAF and LmrR_pAF_RGN with a variety of indole and enal substrates, finding that the evolved mutant outperformed the parent in both yields and enantioselectivities for almost all tested substrate combinations. Isopropyl and methyl groups in the beta-position of the enal afforded products 3b and 3c with moderate and excellent yields, respectively. In the case of 3b, only a small loss in enantioselectivity was observed when LmrR_pAF_RGN was used as catalyst, compared with product 3a. Notably, cinnamaldehyde (product 3d) was not tolerated well, giving very low yields with both LmrR_pAF and LmrR_pAF_RGN. However, using the same loading of LmrR (entry 9) did not afford product 3d, suggesting that the pAF residue is involved in catalysis.
When different indole substrates were employed together with trans-2-hexenal 1a the corresponding products were obtained with moderate to good yields, with LmrR_pAF_RGN showing improved results in each case. Using this evolved artificial enzyme, unsubstituted indole, 5-methyl-indole, and 5-methoxy-indole were converted to products 3e–3g without any loss in enantioselectivity compared to the 2-methyl-indole substrate employed in our directed evolution. With the doubly substituted 5-methoxy-2-methyl-indole product 3i was obtained with a good yield and with little loss in enantioselectivity compared with 3a. The electron-poor 5-bromo-indole proved to be unreactive under standard conditions (product 3h), affording no product. Comparison of the order of elution of the enantiomers of the known product 3j obtained with LmrR_pAF and LmrR_pAF_RGN with an enantioenriched reference compound indicates the (S)-isomer as the major product (Table 2, entries 20 and 21). In general, LmrR_pAF, but especially the evolved LmrR_pAF_RGN, maintain enantioselectivity with changes in the steric/electron properties of the substrate. This tolerance toward structurally diverse substrates highlights our evolved mutant as a promising lead for the generation of a highly selective and generally applicable biocatalyst for this Friedel–Crafts alkylation.
Table 2. Substrate Scope of Indoles and Enals Converted to the Corresponding Friedel–Crafts Products 3a–3i by LmrR_pAF and LmrR_pAF_RGNa.

| entry | product | catalyst | yield (%)b | ee (%)c |
|---|---|---|---|---|
| 1 | 3a | A | 42 ± 4 | 45 ± 0 |
| 2 | 3a | B | 74 ± 2 | 87 ± 0 |
| 3d | 3b | A | 24 ± 1 | 58 ± 0 |
| 4d | 3b | B | 34 ± 6 | 83 ± 2 |
| 5 | 3c | A | 76 ± 7 | 33 ± 1 |
| 6 | 3c | B | 95 ± 7 | 58 ± 1 |
| 7 | 3d | A | 3 ± 0 | 20 ± 1 |
| 8 | 3d | B | 4 ± 1 | 13 ± 9 |
| 9e,f | 3d | LmrR | <1 | - |
| 10g | 3e | A | 10 ± 1 | 50 ± 0 |
| 11g | 3e | B | 27 ± 2 | 89 ± 1 |
| 12g | 3f | A | 16 ± 1 | 41 ± 0 |
| 13g | 3f | B | 31 ± 3 | 89 ± 2 |
| 14g | 3g | A | 24 ± 2 | 55 ± 1 |
| 15g | 3g | B | 40 ± 3 | 89 ± 1 |
| 16f | 3h | A | <1 | - |
| 17f | 3h | B | <1 | - |
| 18 | 3i | A | 62 ± 11 | 58 ± 0 |
| 19 | 3i | B | 75 ± 9 | 82 ± 1 |
| 20g | 3j | A | 21 ± 1 | 66 ± 0 (S)h |
| 21g | 3j | B | 18 ± 2 | 73 ± 1 (S)h |
Reaction conditions: LmrR variants (20 μM dimer concentration) in pH 6.5 buffer containing NaCl (150 mM) and NaH2PO4 (50 mM), 8 vol % DMF, [indole] = 1 mM, [enal] = 5 mM, reaction time 16 h, reactions conducted at 4 °C with mixing by continuous inversion in 300 μL total volume. Reduction with NaBH4 (60 μL, 20 mg mL–1 in 0.5 w/v% NaOH) afforded the alcohol products 3a–3i. For each entry, two independent experiments were conducted, each in duplicate. Errors are the standard deviation of the results thus obtained.
Analytical yields determined using normal phase HPLC using 3-(3-hydroxypropyl)indole as internal standard.
Enantiomeric excess determined using chiral normal-phase HPLC (Chiracel OJ-H (3a), AS-H (3b, 3c, 3f), OD-H (3d, 3e, 3h, 3i, 3j) or OB-H (3g)).
Reaction time 40 h.
LmrR was used in place of LmrR_pAF.
No product could be detected.
50 μM artificial enzyme (concentration of dimer) was used, and reaction time was 64 h.
Absolute configuration asigned by comparison of order of elution on chiral normal-phase HPLC with enantioenriched reference compound and the literature;18 see Supporting Information for details.
Kinetic Analysis
Having genetically optimized LmrR_pAF for both Friedel–Crafts alkylation and hydrazone formation,42 we wanted to assess if the mutations in each evolved variant promote catalysis by the pAF residue in a general, or reaction specific, manner.58,59 In order to assess this, we selected three mutants for comparison, LmrR_pAF, LmrR_pAF_RGN, as well as the mutant with the highest kcat for the hydrazone formation reaction studied in our previous work, LmrR_pAF_RMH.42 We then determined the kinetic parameters for one substrate according to the Michaelis–Menten equation for both the transformation of 1a and 2a into 3a (Table 3, Figure S2) as well as the hydrazone formation reaction between 4 and 5 to give 6 (Table 4, Figures S3 and S4).
Table 3. Michaelis–Menten Kinetic Parameters Calculated for a Single Substrate, 1a, for the Friedel–Crafts Reaction between 1a and 2a to Give 3aa.
| mutant | KM-1a, app (mM) | kcat, app (s–1) × 102 | (kcat/KM)app (M–1s–1) | vs LmrR_pAF |
|---|---|---|---|---|
| LmrR_pAF | 18.2 ± 2.8 | 1.45 ± 0.11 | 0.80 ± 0.06 | 1 |
| LmrR_pAF_RGN | 5.2 ± 0.9 | 0.64 ± 0.03 | 1.25 ± 0.17 | 1.56 |
| LmrR_pAF_RMH | 25.7 ± 6.6 | 0.27 ± 0.04 | 0.11 ± 0.02 | 0.14 |
Determined at 25 °C in phosphate buffer (50 mM) containing NaCl (150 mM) and 5% (v/v) DMF at pH 6.5. The estimated errors reflect the standard deviation from three independent experiments. 2a was kept at a concentration of 1 mM, and 1a was varied between 2 mM and 30 mM.
Table 4. Michaelis–Menten Kinetic Parameters Calculated for a Single Substrate, 5, for the Hydrazone Formation Reaction between 4 and 5 to Give 6a.

| mutant | KM-5, app (μM) | kcat, app (s–1) × 103 | (kcat/KM)app (M–1s–1) | vs LmrR_pAF |
|---|---|---|---|---|
| LmrR_pAFb | 122 ± 10 | 0.23 ± 0.01 | 1.85 ± 0.08 | 1 |
| LmrR_pAF_RGN | 203 ± 87 | 0.11 ± 0.03 | 0.56 ± 0.17 | 0.30 |
| LmrR_pAF_RMHb | 38 ± 4 | 3.98 ± 0.16 | 105 ± 7 | 56.7 |
Determined at 25 °C in phosphate buffer (50 mM) containing NaCl (150 mM) and 5% (v/v) DMF at pH 7.4. The estimated errors reflect the standard deviation from three independent experiments. 4 was kept at a constant concentration of 5 mM, and the concentration of 5 was varied from 20 to 200 μM.
Data from ref (42).
Focusing on the Friedel–Crafts alkylation, we saw that the evolved mutant LmrR_pAF_RGN has a decreased KM for the enal 1a, indicating increased affinity toward this substrate (Table 3). The kcat for this mutant is also somewhat decreased relative to LmrR_pAF, which may be an expense paid for selecting based on enhanced stereoselectivity and not on enhanced kinetic properties. An overall increase in catalytic efficiency (kcat/KM) of around 60% is obtained over the course of the evolution. Relative to LmrR_pAF, however, LmrR_pAF_RMH has a similar KM for the enal 1a but a ∼5 fold decrease in kcat and an overall ∼8-fold loss in catalytic efficiency, demonstrating that increased activity for hydrazone formation emerges at the expense of a promiscuous activity (Table 3). Indeed, the same characteristic is observed with LmrR_pAF_RGN in the hydrazone forming reaction, it shows an increased KM for the hydrazine 5 and a ∼2-fold loss in kcat relative to LmrR_pAF, resulting in a ∼3-fold drop in catalytic efficiency (Table 4). Directed evolution of this enzyme for the Friedel–Crafts reaction gave a more modest increase in catalytic efficiency compared with our previous directed evolution for hydrazone formation,42 but a smaller loss in the promiscuous activity not under selection is also observed.
Together these results show that in both this study and our previous work new activities in LmrR_pAF evolve at the expense of others, that is, divergent evolution of function or “evolutionary trade-offs” (Figure 4). Such specialization is often observed in natural and laboratory evolution58,60 and may play an important role in the evolution of new enzyme function in nature after gene duplication.61 Specialisation through mutation has been employed to tune product distributions obtained from promiscuous natural enzymes,62 and to unlock new-to-nature reactivities in engineered enzymes.63 Here we have shown that we can exploit this effect in promiscuous enzymes that operate with noncanonical catalytic machinery in order to specialize them toward selective and useful transformations (Figure 4).
Figure 4.

(a) Promiscuous transformations catalyzed by LmrR_pAF. (b) Directed evolution has specialized LmrR_pAF, increasing catalytic efficiency for reaction selected for, while the promiscuous activity is concomitantly diminished, giving specialized variants LmrR_pAF_RMH and LmrR_pAF_RGN.
Discussion and Implications
Aniline and its derivatives are rather limited in their catalytic capabilities64−66 which include hydrazone formation previously demonstrated with LmrR_pAF33,42 but not the full breadth of iminium-based transformations catalyzed by other amine catalysts.19 Here we have demonstrated that the microenvironment provided by the LmrR scaffold greatly improves the catalytic properties of the pAF aniline side chain, facilitating catalysis that is normally only achieved efficiently with secondary amine catalysts.19,64 However, the lack of steric bulk about the amino moiety in pAF may also permit conversion of substrates that are challenging for secondary amine catalytic groups seen in other promiscuous and artificial enzymes.23,67,68
Compared with the earlier reported Cu(II)-based artificial enzymes for Friedel–Crafts alkylation reactions,70 the reaction developed herein is not only metal-free, but the need for a chelating group in the substrate is also obviated. Instead, both the enal substrate and the product contain aldehyde moieties, whose set of functional-group interconversions is extremely diverse, including the in situ reduction to alcohol shown herein.71 While some natural and engineered enzymes are now known to catalyze Friedel–Crafts alkylation and acylation, the substrate scope of the arene component is limited to (poly)-hydroxylated resorcinol derivatives.9−11 Secondary amine-based organocatalysts catalyze the Friedel–Crafts alkylation of activated arenes with α,β-unsaturated aldehydes.1,17,18 However, often, high catalyst loadings, organic solvents, and cryogenic temperatures are required.72 Here we demonstrate how this reaction can be conducted with reduced catalyst loadings, in aqueous media without significant change from ambient temperature with a catalyst that can be genetically optimized using the Darwinian algorithm.
Conclusions
Here we have shown how iminium-based mechanisms from organocatalysis can be translated into a biocatalytic context by creating artificial enzymes with noncanonical catalytic residues incorporated by expanded genetic code technology. The artificial enzyme described here stereoselectively constructs an important chemical linkage with a different approach to those employed by natural enzymes. While small molecule catalysts often require empirical structural optimization, this artificial enzyme could be optimized using the Darwinian algorithm through directed evolution. We envision that this new approach will enable many more enzymatic reactions via iminium ion reaction intermediates, thus expanding the biocatalytic repertoire to enable further reactions for green synthesis.19,33,73
Acknowledgments
This work was supported by The Netherlands Organisation for Scientific Research (NWO, project 724.013.003), The Netherlands Ministry of Education, Culture and Science (Gravitation program no. 024.001.035) and the European Research Council (ERC starting grant 280010 and ERC advanced grant 885396). We would like to thank Dr. Clemens Mayer for useful discussions and advice throughout this research.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c00996.
Complete experimental procedures and methods, Figures S1–S4, Tables S1–S4, primer sequences, protein mass spectra, HPLC chromatograms, and NMR spectra (PDF)
Author Present Address
† I.D.: Department of Chemistry and Pharmaceutical Sciences, VU Amsterdam, De Boelelaan 1108, 1081 HZ Amsterdam, The Netherlands
The authors declare no competing financial interest.
Supplementary Material
References
- Paras N. A.; MacMillan D. W. C. The Enantioselective Organocatalytic 1,4-Addition of Electron-Rich Benzenes to α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 2002, 124, 7894–7895. 10.1021/ja025981p. [DOI] [PubMed] [Google Scholar]
- Bartoli G.; Bencivenni G.; Dalpozzo R. Organocatalytic Strategies for the Asymmetric Functionalization of Indoles. Chem. Soc. Rev. 2010, 39, 4449. 10.1039/b923063g. [DOI] [PubMed] [Google Scholar]
- Rueping M.; Nachtsheim B. J. A Review of New Developments in the Friedel-Crafts Alkylation - From Green Chemistry to Asymmetric Catalysis. Beilstein J. Org. Chem. 2010, 6, 1–24. 10.3762/bjoc.6.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetzsche L. E.; Narayan A. R. H. Broadening the Scope of Biocatalytic C–C Bond Formation. Nat. Rev. Chem. 2020, 4, 334–346. 10.1038/s41570-020-0191-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon R. A.; Woodley J. M. Role of Biocatalysis in Sustainable Chemistry. Chem. Rev. 2018, 118, 801–838. 10.1021/acs.chemrev.7b00203. [DOI] [PubMed] [Google Scholar]
- Sheldon R. A.; Brady D.; Bode M. L. The Hitchhiker’s Guide to Biocatalysis: Recent Advances in the Use of Enzymes in Organic Synthesis. Chem. Sci. 2020, 11, 2587–2605. 10.1039/C9SC05746C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman M. A.; Fryszkowska A.; Alvizo O.; Borra-Garske M.; Campos K. R.; Canada K. A.; Devine P. N.; Duan D.; Forstater J. H.; Grosser S. T.; Halsey H. M.; Hughes G. J.; Jo J.; Joyce L. A.; Kolev J. N.; Liang J.; Maloney K. M.; Mann B. F.; Marshall N. M.; McLaughlin M.; Moore J. C.; Murphy G. S.; Nawrat C. C.; Nazor J.; Novick S.; Patel N. R.; Rodriguez-Granillo A.; Robaire S. A.; Sherer E. C.; Truppo M. D.; Whittaker A. M.; Verma D.; Xiao L.; Xu Y.; Yang H. Design of an in Vitro Biocatalytic Cascade for the Manufacture of Islatravir. Science 2019, 366, 1255–1259. 10.1126/science.aay8484. [DOI] [PubMed] [Google Scholar]
- Watkins-Dulaney E.; Straathof S.; Arnold F. Tryptophan Synthase: Biocatalyst Extraordinaire. ChemBioChem 2021, 22, 5–16. 10.1002/cbic.202000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H.; Schultz E. E.; Balskus E. P. A New Strategy for Aromatic Ring Alkylation in Cylindrocyclophane Biosynthesis. Nat. Chem. Biol. 2017, 13, 916–921. 10.1038/nchembio.2421. [DOI] [PubMed] [Google Scholar]
- Schultz E. E.; Braffman N. R.; Luescher M. U.; Hager H. H.; Balskus E. P. Biocatalytic Friedel–Crafts Alkylation Using a Promiscuous Biosynthetic Enzyme. Angew. Chem., Int. Ed. 2019, 58, 3151–3155. 10.1002/anie.201814016. [DOI] [PubMed] [Google Scholar]
- Schmidt N. G.; Pavkov-Keller T.; Richter N.; Wiltschi B.; Gruber K.; Kroutil W. Biocatalytic Friedel–Crafts Acylation and Fries Reaction. Angew. Chem., Int. Ed. 2017, 56, 7615–7619. 10.1002/anie.201703270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X.; Kazemi M.; Żądło-Dobrowolska A.; Kroutil W.; Himo F. Mechanism of Biocatalytic Friedel–Crafts Acylation by Acyltransferase from Pseudomonas Protegens. ACS Catal. 2020, 10, 570–577. 10.1021/acscatal.9b04208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko; Satishkumar S.; Panigrahi N.; Arora P. S. Rational Design of an Organocatalyst for Peptide Bond Formation. J. Am. Chem. Soc. 2019, 141, 15977–15985. 10.1021/jacs.9b07742. [DOI] [PubMed] [Google Scholar]
- Chen J.; Gong X.; Li J.; Li Y.; Ma J.; Hou C.; Zhao G.; Yuan W.; Zhao B. Carbonyl Catalysis Enables a Biomimetic Asymmetric Mannich Reaction. Science 2018, 360, 1438–1442. 10.1126/science.aat4210. [DOI] [PubMed] [Google Scholar]
- Bos J.; Browne W. R.; Driessen A. J. M.; Roelfes G. Supramolecular Assembly of Artificial Metalloenzymes Based on the Dimeric Protein LmrR as Promiscuous Scaffold. J. Am. Chem. Soc. 2015, 137, 9796–9799. 10.1021/jacs.5b05790. [DOI] [PubMed] [Google Scholar]
- Boersma A. J.; Feringa B. L.; Roelfes G. Enantioselective Friedel-Crafts Reactions in Water Using a DNA-Based Catalyst. Angew. Chem., Int. Ed. 2009, 48, 3346–3348. 10.1002/anie.200900371. [DOI] [PubMed] [Google Scholar]
- Paras N. A.; MacMillan D. W. C. New Strategies in Organic Catalysis: The First Enantioselective Organocatalytic Friedel–Crafts Alkylation. J. Am. Chem. Soc. 2001, 123, 4370–4371. 10.1021/ja015717g. [DOI] [PubMed] [Google Scholar]
- Austin J. F.; MacMillan D. W. C. Enantioselective Organocatalytic Indole Alkylations. Design of a New and Highly Effective Chiral Amine for Iminium Catalysis. J. Am. Chem. Soc. 2002, 124, 1172–1173. 10.1021/ja017255c. [DOI] [PubMed] [Google Scholar]
- Erkkilä A.; Majander I.; Pihko P. M. Iminium Catalysis. Chem. Rev. 2007, 107, 5416–5470. 10.1021/cr068388p. [DOI] [PubMed] [Google Scholar]
- Garrabou X.; Wicky B. I. M.; Hilvert D. Fast Knoevenagel Condensations Catalyzed by an Artificial Schiff-Base-Forming Enzyme. J. Am. Chem. Soc. 2016, 138, 6972–6974. 10.1021/jacs.6b00816. [DOI] [PubMed] [Google Scholar]
- Obexer R.; Godina A.; Garrabou X.; Mittl P. R. E. E.; Baker D.; Griffiths A. D.; Hilvert D. Emergence of a Catalytic Tetrad during Evolution of a Highly Active Artificial Aldolase. Nat. Chem. 2017, 9, 50–56. 10.1038/nchem.2596. [DOI] [PubMed] [Google Scholar]
- Garrabou X.; Macdonald D. S.; Wicky B. I. M. M.; Hilvert D. Stereodivergent Evolution of Artificial Enzymes for the Michael Reaction. Angew. Chem., Int. Ed. 2018, 57, 5288–5291. 10.1002/anie.201712554. [DOI] [PubMed] [Google Scholar]
- Nödling A. R.; Świderek K.; Castillo R.; Hall J. W.; Angelastro A.; Morrill L. C.; Jin Y.; Tsai Y.-H.; Moliner V.; Luk L. Y. P. Reactivity and Selectivity of Iminium Organocatalysis Improved by a Protein Host. Angew. Chem., Int. Ed. 2018, 57, 12478–12482. 10.1002/anie.201806850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C.; Saifuddin M.; Saravanan T.; Sharifi M.; Poelarends G. J. Biocatalytic Asymmetric Michael Additions of Nitromethane to α,β-Unsaturated Aldehydes via Enzyme-Bound Iminium Ion Intermediates. ACS Catal. 2019, 9, 4369–4373. 10.1021/acscatal.9b00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G.; Crotti M.; Saravanan T.; Kataja K. M.; Poelarends G. J. Enantiocomplementary Epoxidation Reactions Catalyzed by an Engineered Cofactor-Independent Non-natural Peroxygenase. Angew. Chem., Int. Ed. 2020, 59, 10374–10378. 10.1002/anie.202001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagawa K.; Kudo K. Development of Selective Peptide Catalysts with Secondary Structural Frameworks. Acc. Chem. Res. 2017, 50, 2429–2439. 10.1021/acs.accounts.7b00211. [DOI] [PubMed] [Google Scholar]
- Drienovská I.; Roelfes G. Expanding the Enzyme Universe with Genetically Encoded Unnatural Amino Acids. Nat. Catal. 2020, 3, 193–202. 10.1038/s41929-019-0410-8. [DOI] [Google Scholar]
- Yu Y.; Hu C.; Xia L.; Wang J. Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal. 2018, 8, 1851–1863. 10.1021/acscatal.7b03754. [DOI] [Google Scholar]
- Schwizer F.; Okamoto Y.; Heinisch T.; Gu Y.; Pellizzoni M. M.; Lebrun V.; Reuter R.; Köhler V.; Lewis J. C.; Ward T. R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. 10.1021/acs.chemrev.7b00014. [DOI] [PubMed] [Google Scholar]
- Markel U.; Sauer D. F.; Schiffels J.; Okuda J.; Schwaneberg U. Towards the Evolution of Artificial Metalloenzymes—A Protein Engineer’s Perspective. Angew. Chem., Int. Ed. 2019, 58, 4454–4464. 10.1002/anie.201811042. [DOI] [PubMed] [Google Scholar]
- Rosati F.; Roelfes G. Artificial Metalloenzymes. ChemCatChem 2010, 2, 916–927. 10.1002/cctc.201000011. [DOI] [Google Scholar]
- Raines D. J.; Clarke J. E.; Blagova E. V.; Dodson E. J.; Wilson K. S.; Duhme-Klair A.-K. Redox-Switchable Siderophore Anchor Enables Reversible Artificial Metalloenzyme Assembly. Nat. Catal. 2018, 1, 680–688. 10.1038/s41929-018-0124-3. [DOI] [Google Scholar]
- Drienovská I.; Mayer C.; Dulson C.; Roelfes G. A Designer Enzyme for Hydrazone and Oxime Formation Featuring an Unnatural Catalytic Aniline Residue. Nat. Chem. 2018, 10, 946–952. 10.1038/s41557-018-0082-z. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Burke A. J.; Green A. P. Enzymes with Noncanonical Amino Acids. Curr. Opin. Chem. Biol. 2020, 55, 136–144. 10.1016/j.cbpa.2020.01.006. [DOI] [PubMed] [Google Scholar]
- Agustiandari H.; Lubelski J.; van den Berg van Saparoea H. B.; Kuipers O. P.; Driessen A. J. M. LmrR Is a Transcriptional Repressor of Expression of the Multidrug ABC Transporter LmrCD in Lactococcus Lactis. J. Bacteriol. 2008, 190, 759–763. 10.1128/JB.01151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfes G. LmrR: A Privileged Scaffold for Artificial Metalloenzymes. Acc. Chem. Res. 2019, 52, 545–556. 10.1021/acs.accounts.9b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino L.; Splan K. E.; Reddem E.; Alonso-Cotchico L.; Lledós A.; Gutiérrez de Souza C.; Thunnissen A.-M. W. H.; Maréchal J.-D.; Roelfes G. An Artificial Heme Enzyme for Cyclopropanation Reactions. Angew. Chem., Int. Ed. 2018, 57, 7785–7789. 10.1002/anie.201802946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke A. J.; Lovelock S. L.; Frese A.; Crawshaw R.; Ortmayer M.; Dunstan M.; Levy C.; Green A. P. Design and Evolution of an Enzyme with a Non-Canonical Organocatalytic Mechanism. Nature 2019, 570, 219–223. 10.1038/s41586-019-1262-8. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Roelfes G. Synergistic Catalysis in an Artificial Enzyme by Simultaneous Action of Two Abiological Catalytic Sites. Nat. Catal. 2020, 3, 289–294. 10.1038/s41929-019-0420-6. [DOI] [Google Scholar]
- Jin S.; Li C.; Ma Y.; Kan Y.; Zhang Y. J.; Zhang W. Highly Efficient Asymmetric Organocatalytic Friedel-Crafts Alkylation of Indoles with α,β-Unsaturated Aldehydes. Org. Biomol. Chem. 2010, 8, 4011–4015. 10.1039/c0ob00016g. [DOI] [PubMed] [Google Scholar]
- Lorentzen E.; Siebers B.; Hensel R.; Pohl E. Mechanism of the Schiff Base Forming Fructose-1,6-Bisphosphate Aldolase: Structural Analysis of Reaction Intermediates. Biochemistry 2005, 44, 4222–4229. 10.1021/bi048192o. [DOI] [PubMed] [Google Scholar]
- Mayer C.; Dulson C.; Reddem E.; Thunnissen A.-M. W. H.; Roelfes G. Directed Evolution of a Designer Enzyme Featuring an Unnatural Catalytic Amino Acid. Angew. Chem., Int. Ed. 2019, 58, 2083–2087. 10.1002/anie.201813499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chordia S.; Narasimhan S.; Lucini Paioni A.; Baldus M.; Roelfes G. In Vivo Assembly of Artificial Metalloenzymes and Application in Whole-Cell Biocatalysis. Angew. Chem., Int. Ed. 2021, 60, 5913–5920. 10.1002/anie.202014771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino L.; Chordia S.; Alonso-Cotchico L.; Reddem E.; Zhou Z.; Thunnissen A. M. W. H.; Maréchal J.-D.; Roelfes G. Cofactor Binding Dynamics Influence the Catalytic Activity and Selectivity of an Artificial Metalloenzyme. ACS Catal. 2020, 10, 11783–11790. 10.1021/acscatal.0c01619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drienovská I.; Alonso-Cotchico L.; Vidossich P.; Lledós A.; Maréchal J.-D.; Roelfes G. Design of an Enantioselective Artificial Metallo-Hydratase Enzyme Containing an Unnatural Metal-Binding Amino Acid. Chem. Sci. 2017, 8, 7228–7235. 10.1039/C7SC03477F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reetz M. T.; Carballeira J. D. Iterative Saturation Mutagenesis (ISM) for Rapid Directed Evolution of Functional Enzymes. Nat. Protoc. 2007, 2, 891–903. 10.1038/nprot.2007.72. [DOI] [PubMed] [Google Scholar]
- Kille S.; Acevedo-Rocha C. G.; Parra L. P.; Zhang Z.-G.; Opperman D. J.; Reetz M. T.; Acevedo J. P. Reducing Codon Redundancy and Screening Effort of Combinatorial Protein Libraries Created by Saturation Mutagenesis. ACS Synth. Biol. 2013, 2, 83–92. 10.1021/sb300037w. [DOI] [PubMed] [Google Scholar]
- Sullivan B.; Walton A. Z.; Stewart J. D. Library Construction and Evaluation for Site Saturation Mutagenesis. Enzyme Microb. Technol. 2013, 53, 70–77. 10.1016/j.enzmictec.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Westheimer F. H. Coincidences, Decarboxylation, and Electrostatic Effects. Tetrahedron 1995, 51, 3–20. 10.1016/0040-4020(94)00865-R. [DOI] [Google Scholar]
- Highbarger L. A.; Gerlt J. A.; Kenyon G. L. Mechanism of the Reaction Catalyzed by Acetoacetate Decarboxylase. Importance of Lysine 116 in Determining the p K a of Active-Site Lysine 115. Biochemistry 1996, 35, 41–46. 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- Poelarends G. J.; Almrud J. J.; Serrano H.; Darty J. E.; Johnson W. H.; Hackert M. L.; Whitman C. P. Evolution of Enzymatic Activity in the Tautomerase Superfamily: Mechanistic and Structural Consequences of the L8R Mutation in 4-Oxalocrotonate Tautomerase. Biochemistry 2006, 45, 7700–7708. 10.1021/bi0600603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zühlsdorf M.; Werten S.; Klupp B. G.; Palm G. J.; Mettenleiter T. C.; Hinrichs W. Dimerization-Induced Allosteric Changes of the Oxyanion-Hole Loop Activate the Pseudorabies Virus Assemblin PUL26N, a Herpesvirus Serine Protease. PLoS Pathog. 2015, 11, e1005045. 10.1371/journal.ppat.1005045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordillo R.; Carter J.; Houk K. N. Theoretical Explorations of Enantioselective Alkylation Reactions of Pyrroles and Indoles Organocatalyzed by Chiral Imidazolidinones. Adv. Synth. Catal. 2004, 346, 1175–1185. 10.1002/adsc.200404107. [DOI] [Google Scholar]
- Pace C. N.; Scholtz J. M. A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins. Biophys. J. 1998, 75, 422–427. 10.1016/S0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Cotchico L.; Rodríguez-Guerra Pedregal J.; Lledós A.; Maréchal J.-D. The Effect of Cofactor Binding on the Conformational Plasticity of the Biological Receptors in Artificial Metalloenzymes: The Case Study of LmrR. Front. Chem. 2019, 7, 211. 10.3389/fchem.2019.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten R.; Pádua R. A. P.; Bunzel H. A.; Nguyen V.; Pitsawong W.; Patterson M.; Sui S.; Perry S. L.; Cohen A. E.; Hilvert D.; Kern D. How Directed Evolution Reshapes the Energy Landscape in an Enzyme to Boost Catalysis. Science 2020, 370, 1442–1446. 10.1126/science.abd3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom A.; Rakotoharisoa R. V.; Thompson M. C.; Zarifi N.; Nguyen E.; Mukhametzhanov N.; Liu L.; Fraser J. S.; Chica R. A. Ensemble-Based Enzyme Design Can Recapitulate the Effects of Laboratory Directed Evolution in Silico. Nat. Commun. 2020, 11, 4808. 10.1038/s41467-020-18619-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soskine M.; Tawfik D. S. Mutational Effects and the Evolution of New Protein Functions. Nat. Rev. Genet. 2010, 11, 572–582. 10.1038/nrg2808. [DOI] [PubMed] [Google Scholar]
- Aharoni A.; Gaidukov L.; Khersonsky O.; Gould S. M.; Roodveldt C.; Tawfik D. S. The “evolvability” of Promiscuous Protein Functions. Nat. Genet. 2005, 37, 73–76. 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Rueffler C.; Hermisson J.; Wagner G. P. Evolution of Functional Specialization and Division of Labor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E326–E335. 10.1073/pnas.1110521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikuni Y.; Ferrin T. E.; Keasling J. D. Designed Divergent Evolution of Enzyme Function. Nature 2006, 440, 1078–1082. 10.1038/nature04607. [DOI] [PubMed] [Google Scholar]
- Coelho P. S.; Wang Z. J.; Ener M. E.; Baril S. A.; Kannan A.; Arnold F. H.; Brustad E. M. A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in Vivo. Nat. Chem. Biol. 2013, 9, 485–487. 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J.; Zhang Q.; Cai M.; Han Y.; Luo S. Aromatic Aminocatalysis. Chem. - Asian J. 2018, 13, 740–753. 10.1002/asia.201701773. [DOI] [PubMed] [Google Scholar]
- Cordes E. H.; Jencks W. P. Nucleophilic Catalysis of Semicarbazone Formation by Anilines. J. Am. Chem. Soc. 1962, 84, 826–831. 10.1021/ja00864a030. [DOI] [Google Scholar]
- Crisalli P.; Kool E. T. Water-Soluble Organocatalysts for Hydrazone and Oxime Formation. J. Org. Chem. 2013, 78, 1184–1189. 10.1021/jo302746p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L.-W.; Luo J.; Lu Y. Asymmetric Catalysis with Chiral Primary Amine-Based Organocatalysts. Chem. Commun. 2009, 1807. 10.1039/b821070e. [DOI] [PubMed] [Google Scholar]
- Zandvoort E.; Geertsema E. M.; Baas B.-J.; Quax W. J.; Poelarends G. J. Bridging between Organocatalysis and Biocatalysis: Asymmetric Addition of Acetaldehyde to β-Nitrostyrenes Catalyzed by a Promiscuous Proline-Based Tautomerase. Angew. Chem., Int. Ed. 2012, 51, 1240–1243. 10.1002/anie.201107404. [DOI] [PubMed] [Google Scholar]
- Bos J.; Fusetti F.; Driessen A. J. M.; Roelfes G. Enantioselective Artificial Metalloenzymes by Creation of a Novel Active Site at the Protein Dimer Interface. Angew. Chem., Int. Ed. 2012, 51, 7472–7475. 10.1002/anie.201202070. [DOI] [PubMed] [Google Scholar]
- Hammer S. C.; Kubik G.; Watkins E.; Huang S.; Minges H.; Arnold F. H. Anti-Markovnikov Alkene Oxidation by Metal-Oxo–Mediated Enzyme Catalysis. Science 2017, 358, 215–218. 10.1126/science.aao1482. [DOI] [PubMed] [Google Scholar]
- van der Helm M. P.; Klemm B.; Eelkema R. Organocatalysis in Aqueous Media. Nat. Rev. Chem. 2019, 3, 491–508. 10.1038/s41570-019-0116-0. [DOI] [Google Scholar]
- Leveson-Gower R. B.; Mayer C.; Roelfes G. The Importance of Catalytic Promiscuity for Enzyme Design and Evolution. Nat. Rev. Chem. 2019, 3, 687–705. 10.1038/s41570-019-0143-x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.