

Abstract

The current existing methods for the amide bond synthesis via acceptorless dehydrogenative coupling of amines and alcohols all require high reaction temperatures for effective catalysis, typically involving reflux in toluene, limiting their potential practical applications. Herein, we report a system for this reaction that proceeds under mild conditions (reflux in diethyl ether, boiling point 34.6 °C) using ruthenium PNNH complexes. The low-temperature activity stems from the ability of Ru–PNNH complexes to activate alcohol and hemiaminals at near-ambient temperatures through the assistance of the terminal N–H proton. Mechanistic studies reveal the presence of an unexpected aldehyde-bound ruthenium species during the reaction, which is also the catalytic resting state. We further utilize the low-temperature activity to synthesize several simple amide bond-containing commercially available pharmaceutical drugs from the corresponding amines and alcohols via the dehydrogenative coupling method.
Keywords: amides, dehydrogenation, ruthenium pincer, pharmaceuticals, alcohol, amine
Introduction
The amide group is one of the most ubiquitous functional groups found in Nature. The formation of the amide bond is a fundamental reaction with important utility in the synthesis of natural compounds, biologically active pharmaceutical drugs, short-chain peptides, industrial chemicals, and polymers such as nylons as well as in devising liquid organic hydrogen carrier systems.1 Roughly 25% of the currently approved pharmaceutical drugs contain amide groups.2 The traditional method of facile amide bond synthesis has been the coupling of reactive acid derivatives, such as acid chlorides or anhydrides, with amines, but these methods are poorly tolerated by other nucleophilic functional groups and generate waste. Other methods involve direct coupling of carboxylic acids and amines under milder conditions in the presence of coupling promoters, which also generate significant amounts of waste.3 In 2005, ACS GCI Pharmaceutical Roundtable reported the “amide bond formation avoiding poor atom economy” as one of the most preferred reactions to develop.4
In 2007, our group reported a new, environmentally benign synthesis of the amide bond by acceptorless dehydrogenative coupling of alcohols and amines with H2 being the sole byproduct of the reaction (Figure 1).5 The reaction proceeds upon refluxing the alcohol and amine in toluene in the presence of a Ru–PNN catalyst. Since then, many research groups including the groups of Madsen, Crabtree, Hong, Hazari and Bernskoetter, Prakash, and others have reported efficient catalysts that can catalyze the acceptorless dehydrogenative coupling of alcohols and amines under various conditions.6 Importantly, first-row transition-metal-based complexes have also been reported for this transformation by the groups of Bernskoetter6a (iron) and ours7 (manganese). Some heterogeneous catalysts are also known to catalyze this transformation.8
Figure 1.

Selected examples of amide synthesis via dehydrogenative coupling of amines and alcohols.
All the systems mentioned above require a reaction temperature that is generally above 100 °C (typically reflux in toluene), which decreases their practical utility. One of the primary advantages of homogeneous catalysis by well-defined molecular complexes over heterogeneous catalysts is that the activity of the homogeneous complexes can be rationally improved via judicious tuning of the ligand framework. The high reaction temperature required for dehydrogenative amide bond formation is presumably due to the difficulty in the hydride abstraction from the reactant and the hemiaminal intermediate by the employed catalysts, en route to the dihydride intermediate. Wang and co-workers had computationally investigated the mechanism of this amide formation in detail with the traditional Ru–PNNEt Milstein complex, and according to their calculations, hydride abstraction from the alcohol and hemiaminal steps have activation barriers of around 25 and 31 kcal/mol, respectively.9 The high energy requirement, especially for the hemiaminal dehydrogenation, is in line with the elevated temperature required for the amide synthesis. Moreover, the overall formation of amide and H2 from alcohol and amine is generally thermodynamically uphill, also contributing toward the high required temperature for amide synthesis. In this context, development of lower-temperature dehydrogenative amide synthesis protocols by rational catalyst design is desirable in order to improve the applicability of this atom-economical dehydrogenative coupling process.
Results and Discussion
These considerations led us to explore the possibility of a low-temperature amide synthesis pathway via the acceptorless dehydrogenative coupling.10 We started our investigation with the synthesis of N-heptylhexanamide by coupling of 1-hexanol and 1-heptylamine (Table 1). Several ruthenium and manganese pincer complexes were screened for the amide synthesis under Et2O reflux (boiling point 34.6 °C) in the presence of catalytic amounts of t-BuOK. Interestingly, among these complexes, Ru–PNNH complexes111 and 2, featuring a terminal N–H moiety, displayed catalytic activities toward amide formation even at this low temperature (entries 1–2). Ru–PNNH complexes 1–3 are capable of two distinct modes of metal–ligand cooperation (MLC), amido–amine and aromatization–dearomatization,11 unlike complexes 4–6, where only aromatization–dearomatization is possible. Ru–PPhNNH complex 3, with electron-withdrawing Ph substituents on the P donor atom, did not show catalytic activity at this temperature (entry 3). Comparing 1 and 2, the N-benzyl-substituted PNNH complex 2 showed higher activity than the tert-Bu-substituted complex 1. The reason behind the higher activity of complex 2 is investigated in more detail during mechanistic investigations (vide infra).
Table 1. Conditions Screening for Low-Temperature Synthesis of N-Heptylhexanamidea.

| entry | [Ru] | solvent (b.p./°C) | time (h) | amide (%)b |
|---|---|---|---|---|
| 1 | 1 | Et2O (34.6) | 12 | 11 |
| 2 | 2 | Et2O | 12 | 66 |
| 3 | 3 | Et2O | 12 | 0 |
| 4 | 4 | Et2O | 12 | 0 |
| 5 | 5 | Et2O | 12 | 1 |
| 6 | 6 | Et2O | 12 | 0 |
| 7 | 7 | Et2O | 12 | 0 |
| 8 | 8 | Et2O | 12 | 0 |
| 9d | 2 | Et2O | 22 | 92 |
| 10 | 2 | MTBE (55.2) | 12 | 97 (95)c |
Reaction conditions: 1-hexanol (1 mmol), 1-heptylamine (1.1 mmol), [Ru] (0.01 mmol), t-BuOK (0.02 mmol), solvent (2 mL), reflux with bath temperatures of 50 and 70 °C for Et2O and MTBE, respectively, time as specified.
Yields are calculated based on 1H NMR spectra with mesitylene as an internal standard.
Isolated yield.
Reaction scaled down by a factor of 2 (solvent 2 mL).
Traditional PNN complexes, 4–6 (entries 4–6), with only one mode of MLC (aromatization–dearomatization) were not active at low temperature, although they can catalyze the reaction at the higher temperature of 110 °C (entries 4–8).5,6b Thus, the terminal N–H moiety of the Ru–PNNH complexes likely plays an important role in their low-temperature catalytic activity. Mn-based PNN and PNNH complexes were also not active at this low temperature (entries 7–8). High conversion of 1-hexanol and 1-heptylamine to the corresponding amide was obtained with 2 as a pre-catalyst and diethyl ether as a solvent after 22 h of reflux (entry 9). A faster reaction was observed at a higher temperature such as reflux in the methyl tert-butyl ether (MTBE) solvent (b.p.: 55.2 °C) (entry 10). The generation of H2 gas was confirmed by carrying out the reaction in a closed vessel and analyzing the headspace gas by gas chromatography after the reaction (Figure S5).
The substrate scope of this low-temperature amide synthesis system was subsequently explored (Table 2). Simple amides such as N-heptylhexanamide, N-benzylhexanamide, N-heptylbenzamide, N-benzylbenzamide, and N-(furfuryl)hexanamide were synthesized in high yields under refluxing conditions in diethyl ether (Table 2, entries 1–6). Besides primary amines, the secondary amine morpholine and 1-hexanol can also couple at low temperature in the presence of 2, producing the tertiary amide N-hexanoylmorpholine in excellent yield (entry 7). Different halogen substituents such as −F and −Br are also tolerated under the reaction conditions (entries 8–9). For the synthesis of some other amides, such as N-heptyl-2-methoxyacetamide and N-heptylisovaleramide, a slightly higher temperature was required for complete conversions (reflux in MTBE) (entries 10–13).5 Highly reducible groups, such as a C–C double bond, are also tolerated under the conditions despite the reaction being associated with the evolution of H2 gas (entry 14). It is to be noted that the nucleophilicity of the amine plays an important role in the rate of the reactions. For example, in the case of the synthesis of N-phenylhexanamide, the low nucleophilicity of aniline necessitates either a higher reaction temperature (reflux in toluene) or an increased base concentration (50 mol % KOtBu) for effective amidation (entries 15–17). Notably, ethylenediamine and ethanol can also dehydrogenatively couple at low temperatures (reflux in MTBE) in the presence of complex 2 to provide the diamide in 95% yield (entry 18). We also investigated the possibility of the synthesis of chiral amides via this method using a β-chiral alcohol and an α-chiral amine. The chiral centers of the substrates were largely retained in the product amide molecules in both cases, as determined from optical rotation analysis of the products in comparison with literature data, demonstrating the potential utility of this method in the synthesis of biologically active amide molecules (entries 19–20, Figures S41 and S42).
Table 2. Dehydrogenative Synthesis of Various Amides at Low Temperaturea.

Reaction conditions: alcohol (0.5 mmol), amine (0.55 mmol), 2 (0.005 mmol), t-BuOK (0.01 mmol), solvent (2 mL), reflux under Ar in an open system [the bath temperature was 50 °C (Et2O) or 70 °C (MTBE) or 130 °C (toluene)].
b.p. under 1 atmospheric pressure.
Rest of the alcohol unchanged.
Ester 10% observed, rest 50% of alcohol unchanged.
50 mol % KOtBu.
Ethylenediamine (0.5 mmol), EtOH (1.5 mmol), 1 (0.005 mmol), and t-BuOK (0.01 mmol).
Cat (2 mol %), in a 100 mL closed flask.
Cat (2 mol %), in a 25 mL closed flask; the generated gas inside released intermittently after cooling down (24th and 48th h).
In some of the reactions of Table 2 (entries 4, 5, 8, and 14), esters in moderate amounts were detected (∼0–10%). To understand whether the generated ester is converted to the amide at this low temperature or is exclusively a competing reaction pathway, we set up a reaction of hexyl hexanoate with 1-heptylamine. After 24 h of refluxing in Et2O under similar reaction conditions, the formation of the corresponding amide, N-heptylhexanamide, in 76% yield was observed (Scheme 1), signifying that complex 2 can also catalyze the synthesis of amides from esters at low temperatures, presumably via initial nucleophilic substitution of the ester with the amine to form an amide and an alcohol, followed by dehydrogenative coupling between the released alcohol and amine to form amide.12 In the absence of the ruthenium complex, no conversion of ester to amide was observed, signifying that the ruthenium complex catalyzes this reaction, likely acting as a Lewis acid to activate the ester during the initial amine nucleophilic attack on the ester.
Scheme 1. Amide Formation from Ester Catalyzed by 2.

Based on these observations, the reaction pathway for the amide formation is shown in Scheme 2. The initial dehydrogenation of alcohol forms the aldehyde, which further converts to hemiaminal or hemiacetal via nucleophilic attack of the amine or alcohol, respectively. Subsequent dehydrogenation of the hemiaminal and hemiacetal intermediates produces the amide and ester, respectively. The ester is further converted to amide via nucleophilic substitution by the amine, assisted by the ruthenium complex under the reaction conditions.
Scheme 2. Pathway of Amide Formation.

Mechanistic studies were carried out to further understand the mechanism of the low-temperature catalytic activities of the PNNH complexes (Scheme 3). Complex 1, upon addition of 2 eq of t-BuOK, forms the anionic complex 1a which is intensely violet in diethyl ether solution (Scheme 3a).11 Addition of 1-hexanol (4 equiv) to this complex results in the formation of the aromatic alkoxy complex 1b (see Figure S20 for reaction progress). Noteworthily, the alkoxy ligand of 1b exchanges quickly with the free alcohol in solution, and as the excess alcohol is removed from the solution, peak broadening in 31P{1H} and 1H NMR is observed.13 Similar to the formation of the alkoxy complex, addition of benzylamine(4 equiv) to a solution of 1a resulted in the formation of the amido complex 1c. Further addition of alcohol to the amido complex replaced the amido ligand to form the alkoxy complex along with the generation of amine (Scheme 3a; Figure S21). On a similar note, addition of a 1/1 alcohol and amine solution to complex 1a resulted in the selective formation of the alkoxy complex in solution.
Scheme 3. Reactivity of Ru Complexes with the Base, Alcohol, and Amine.

When the resulting solution containing complex 1b, amine, and alcohol was heated at 45 °C in a J. Young NMR tube, the formation of a new complex was observed after 30 min with almost quantitative conversion of 1b (Scheme 3a; Figure S21). This complex exhibits a characteristic peak in the 31P NMR at 119.4 ppm (major isomer) (Figure S14).14 In the proton NMR, a hydride peak corresponding to this complex was observed at −15.2 ppm as a doublet (J = 28.5 Hz) (Figure S8). In the IR spectrum, a strong absorption band at 1900 cm–1 was observed, corresponding to a CO ligand. Interestingly, the 13C NMR spectrum indicated the activation of the N-arm as the secondary picolylic CH2 unit of the N-arm was converted to a tertiary CH unit along with another CH unit, presumably from an alcohol derivative (Figure S11). Based on 1D and 2D NMR analysis, the structure of 1d was assigned to a new complex where an in situ generated aldehyde binds to the N-arm of the ligand through MLC. Single crystals suitable for X-ray diffraction (XRD) analysis were grown by slow evaporation from a THF/pentane solution of 1d, and the XRD analysis confirmed the assigned structure of 1d (Scheme 3a, Figure 2). Interestingly, in the unit cell of the crystal, a product amide molecule of N-benzylhexanamide was found attached to 1d, forming 1d.amide via hydrogen bonding (Figure 2). The aldehyde addition across the metal and ligand arm has been documented before by us with a Ru–PNP system at much lower temperatures (−78 °C)13 and also by Sanford and co-workers with the traditional Ru–PNNEt2 system at room temperature following the addition of benzaldehyde to the dearomatized complexes.15 However, it is quite interesting that this aldehyde complex can also be readily and quantitatively accessed form the alkoxy complex upon brief, mild heating, signifying the possible generation of dearomatized complexes during the reaction.
Figure 2.
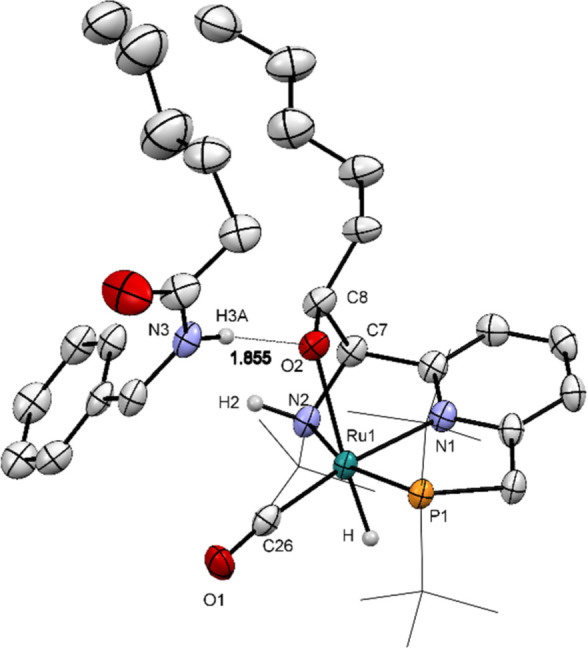
ORTEP diagram of complex 1d.amide. Atoms are drawn with a probability level of 50%. Selected hydrogen atoms omitted for clarity. Tert-butyl groups displayed as a wireframe for clarity. Selected bond lengths (Å) and angles (o): Ru(1)–P(1) 2.2576(5), Ru(1)–O(2) 2.2380(15), Ru(1)–N(1) 2.0850(19), Ru(1)–N(2) 2.2010(17), Ru(1)–C(26) 1.818(2), O(2)–C(8) 1.394(3), C(7)–C(8) 1.570(3); P(1)–Ru(1)–H 81.5(11), O(2)–Ru(1)–P(1) 105.13(4), O(2)–Ru(1)–H 168.8(11), N(1)–Ru(1)–P(1) 81.81(5), N(1)–Ru(1)–O(2) 81.74(7), N(2)–Ru(1)–O(2) 72.92(6), C(8)–O(2)–Ru(1) 113.36(13), O(2)–C(8)–C(7) 109.38(18).
Complex 1d can also be accessed upon heating the alkoxy complex 1b in the presence of alcohol but without amine, forming an alcohol adduct 1d.C6H13OH (Scheme 3a), as verified by NMR analysis (Figures S8 and S9). The formation of 1d from 1b presumably happens via the formation of the dihydride 1e, followed by the formation of amido complex 1f with the evolution of H2 (Scheme 3b). The amido complex 1f can further convert to the N-arm dearomatized complex 1f′, to which the addition of aldehyde affords complex 1d (Scheme 3b). However, the dihydride complex was not observed by NMR, signifying that the H2 elimination from the dihydride complex 1e is facile. A similar aldehyde adduct complex also forms with the traditional Ru–PNNEt2 complex 4 at 45 °C in the presence of a base and alcohol, although, in this case, the dihydride complex is also observed in the NMR (Scheme 3c, Figure S22). This signifies that (i) even in the case of Ru–PNNEt2, the first alcohol dehydrogenation step can proceed at low temperature, at least, stoichiometrically, and (ii) H2 generation from 1e is more facile than from the dihydride complex 4e, presumably due to the new mode of MLC via the involvement of the terminal N–H moiety.
The aldehyde binding to the side arm in complex 1d is reversible and rapidly exchanges with free benzaldehyde in solution as verified by an aldehyde-exchange experiment (Figure S23).16 Complex 1d, upon addition of n-propylamine (5 equiv), followed by mild heating (45 °C), gradually formed a new complex (Scheme 3d) in which the C–C bond was broken as concluded from 1H and 31P{1H} NMR. The transformation is also associated with the formation of H2 gas, as expected. Based on the NMR analysis, the new complex is assigned the structure of the amidate complex 1h, which is formed via addition of the in situ generated amide product to the amido complex 1f (Scheme 4). We have previously observed the reversible formation of similar amidate complexes upon addition of amide to the dearomatized PNNH complexes.17 The formation of complex 1c′ was also observed in a minor amount resulting from the addition of free amine to complex 1f (Scheme 3d, Figure S26). The amidate and amido ligands from 1h and 1c′ are gradually replaced in the presence of 1-hexanol (5 equiv) at 45 °C, regenerating complex 1d (Scheme 3d, Figure S26). These results clearly demonstrate the competing binding routes of different substrates, intermediates, and products to the PNNH amido complex that are operational during active catalysis (Scheme S1).
Scheme 4. Plausible Mechanistic Cycle.

Based on the mechanistic experiments and on prior investigation by our group and others on the dehydrogenative amide bond formation, we propose a catalytic cycle as depicted in Scheme 4. The pre-catalyst, in the presence of the base and alcohol, forms the alkoxy complex 1b. Hydride elimination from the alkoxy ligand through an outer-sphere mechanism (Scheme 5a, vide infra) forms the dihydride complex 1e, along with the aldehyde. Subsequently, H2 elimination from the dihydride forms the amido complex 1f. This H2 evolution from the dihydride complex can happen by itself or through the assistance of an alcohol molecule. The amido complex then binds to the aldehyde to form the likely off-cycle intermediate complex 1d (via a pathway as depicted in Scheme 3b) which is also the catalytic resting state of the reaction. 1d, in the presence of an amine, generates the hemiaminoxy complex 1g, presumably via the formation of 1f. In the subsequent reaction step, hydride elimination from the hemiaminoxy complex generates the amide and the dihydride complex again. Further H2 elimination from the dihydride complex 1e, followed by the addition of alcohol to the resulting amido complex, regenerates the alkoxy complex 1b, closing the catalytic cycle. It is to be noted that based on our experimental evidence, we cannot exclude the possibility of a beta hydride elimination pathway via N-arm opening, although this route is calculated to have higher energy requirement than the stepwise outer-sphere pathway for the traditional Ru–PNNEt2 complex.9
Scheme 5. Importance of the Terminal N–H Moiety.

Energy values correspond to Gibbs free energies (kcal mol–1) with respect to the ethoxy complex + ethylamine at 298.15 K in a diethyl ether continuum. All reactant concentrations are 1 M, except for H2 which is at 1 atm. Mass balance is ensured throughout.
The hydride abstraction from the alkoxy and the hemiaminoxy complexes, leading to the formation of the aldehyde and amide, respectively, and the dihydride complex, is proposed to proceed via the formation of a high-energy intermediate where the hydrogen atom coordinates to the metal center (Scheme 5a, Int 1–2).9,18 This step is also associated with a high activation barrier, especially the amide formation step, which has been previously computed to be the highest energy-requiring step in the case of the PNNEt2 system.9 In the case of the PNNEt2 system, this hydride abstraction transition state can proceed via the involvement of the picolyl CH2 protons. Our computation suggests that in the case of the PNNH system, the terminal N–H moiety can allow the mechanism to proceed via a low-energy pathway. For example, the pathway involving the terminal N–H is 0.9 kcal/mol lower in energy for the aldehyde formation step than the PCH2 pathway. On the other hand, for the amide formation step, which is also a higher energy-demanding step compared to the aldehyde formation, the new pathway was found to be 8.5 kcal/mol lower in energy (Scheme 5a). This additional stabilization likely explains the high catalytic activities of the PNNH complexes at low reaction temperatures. Please note that although we compute here a stepwise outer-sphere reaction pathway, the possibility of a concerted outer-sphere reaction pathway involving simultaneous proton and hydride transfer from the substrate to the ligand and metal, respectively, similar to the mechanisms associated with many reactions with Noyori and Shvo’s catalysts, cannot be ruled out (Scheme S2).19 Nevertheless, the concerted mechanism can also be surmised to be more stabilized by the terminal N–H route as compared to the P–CH2 one.
Further support of the importance of the N–H proton and its acidity is obtained from comparing the catalysis rate of different Ru–PNNH complexes. As is seen in Table 1 and Scheme 5b, a significant increase in the catalytic activity was observed when the N substitution of the Ru–PNNH complex was changed from tert-butyl to the benzyl group. To understand whether this change in activity is likely due to steric or electronic factors, we synthesized the novel N-propyl derivative of Ru–PNNH complex 9 (synthesis methods and characterization data are in the Supporting Information). The catalytic activity of the n-Pr Ru–PNNH analogue was found to be in between those of the t-Bu and Bn analogues under similar reaction conditions (Scheme 5b). This suggests that the higher activity of the benzyl derivative likely results from the electronic properties of the benzyl group since n-Pr and the benzyl moiety display similar steric bulks around the N donor. In the case of the benzyl substitution, the terminal N–H is more acidic, likely providing the highest stabilization to the transition state of hydride elimination from alkoxy and hemiaminoxy complexes. In addition, based on the density functional theory calculations, an overall lower activation energy was computed for the amide formation in the PNNHBn system compared to the PNNHtBu system and the PNNEt2 system (Scheme 5b). Thus, the acidity of the N–H bond is an important factor influencing the low-temperature catalytic activity of these complexes. This also hints at a possible avenue toward developing more efficient catalysts, perhaps functioning under even ambient conditions.
We also synthesized several commercially available amide bond-containing pharmaceuticals via this method at low temperatures to demonstrate its utility (Scheme 6). Despite the activity of several complexes toward catalyzing the dehydrogenative formation of the amide bond, the synthesis of even simple pharmaceutical drugs via this atom-economical method has not been demonstrated until now, even at higher reaction temperatures.20 Moclobemide is a reversible inhibitor of monoamine oxidase A, approved as an antidepressant in United Kingdom and Canada.21 Using the dehydrogenative coupling method, moclobemide was conveniently synthesized from the corresponding alcohol and amine in 92% yield (isolated yield 85%) upon refluxing in MTBE with 1 as a catalyst.22 Notably, with H2 being the only byproduct of the reaction, the resulting moclobemide can be easily purified by recrystallization in the cold diethyl ether/pentane solvent mixture without the requirement of purification by column chromatography. Similarly, the antidyspeptic drug itopride (brand name: Ganaton)23 and the antiemetic trimethobenzamide were synthesized from the corresponding alcohol and amine with 92 and 94% yields, respectively, under similar reaction conditions, without the requirement of purification via column chromatography. In the case of the insect repellant diethyltoluamide, it was synthesized in 32% yield via this method at higher temperatures (reflux in 1,4-dioxane) and in the presence of 1 eq K3PO4 due to lower nucleophilicity of the secondary amine compared to the primary ones.6b Further investigation on the synthesis of more complex pharmaceuticals via this method through catalyst improvement and condition optimization is ongoing in our group.
Scheme 6. Synthesis of Various Pharmaceutical Drugs.

Conditions A: alcohol (0.5 mmol), amine (0.6 mmol), 2 (0.005 mmol), t-BuOK (0.01 mmol), MTBE (2 mL, bp 55.2 °C), reflux (bath temp. 70 °C), 60 h. Conditions B: alcohol (0.5 mmol), amine (5 mmol), 1 (0.005 mmol), t-BuOK (0.01 mmol), K3PO4 (0.5 mmol), dioxane (2 mL), reflux in a closed tube for 60 h (see the Supporting Information for details).
Yields in parentheses are isolated yields, and yields outside parentheses are 1H NMR yields.
Conclusions
In summary, we report the near-ambient-temperature dehydrogenative formation of the amide bond (reflux in Et2O) from alcohols and amines. The Ru–PNNH complexes, featuring a terminal N–H unit, showed remarkable catalytic activities at this low temperature. This high activity is presumably due to the new catalytic pathway involving the terminal N–H moiety, and the acidity of this terminal N–H heavily influenced the low-temperature catalytic rate. An unusual resting state of the complex during catalysis was observed, where the aldehyde binds between the N-side arm and Ru center of the complex via a reversible C–C bond formation. Using Ru–PNNH complex 2, we synthesized several amides under refluxing conditions in Et2O or MTBE. Furthermore, several amide bonds containing commercially available pharmaceuticals, including moclobemide, itopride, and trimethobenzamide, were prepared at low temperature via this atom-economical method. Our future efforts in this context will be aimed toward the development of even more efficient catalysts for this transformation based on the mechanistic insights gained during this study as well as toward the synthesis of more complex pharmaceuticals via this method.
Acknowledgments
Dedicated to Prof. Christian Bruneau for his outstanding contribution to catalysis. This research was supported by the European Research Council (ERC AdG 692775). D.M. holds the Israel Matz Professorial Chair of Organic Chemistry. S.K. acknowledges the Sustainability and Energy Research Initiative (SAERI) of the Weizmann Institute of Science for a research fellowship.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c00728.
Author Present Address
∥ Ningbo Institute of Materials Technology & Engineering, Chinese Academy of Sciences, Ningbo, 315201, P.R. China
Author Contributions
§ S.K. and Y.X. contributed equally.
The authors declare no competing financial interest.
Notes
Synthetic procedures, NMR spectra, and characterization data for all the new compounds are available within this article and its Supporting Information. The X-ray crystallographic coordinates for the structure reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number CCDC 2043153. The data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttp://www.ccdc.cam.ac.uk/data_request/cif. Any further relevant data are available from the authors upon reasonable request.
Supplementary Material
References
- a Cupido T.; Tulla-Puche J.; Spengler J.; Albericio F. The synthesis of naturally occurring peptides and their analogs. Curr. Opin. Drug Discovery Dev. 2007, 10, 768–783. [PubMed] [Google Scholar]; b Hu P.; Ben-David Y.; Milstein D. Rechargeable Hydrogen Storage System Based on the Dehydrogenative Coupling of Ethylenediamine with Ethanol. Angew. Chem., Int. Ed. 2016, 55, 1061–1064. 10.1002/anie.201505704. [DOI] [PubMed] [Google Scholar]; c Hu P.; Fogler E.; Diskin-Posner Y.; Iron M. A.; Milstein D. A novel liquid organic hydrogen carrier system based on catalytic peptide formation and hydrogenation. Nat. Commun. 2015, 6, 6859. 10.1038/ncomms7859. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Xie Y.; Hu P.; Ben-David Y.; Milstein D. A Reversible Liquid Organic Hydrogen Carrier System Based on Methanol-Ethylenediamine and Ethylene Urea. Angew. Chem., Int. Ed. 2019, 58, 5105–5109. 10.1002/anie.201901695. [DOI] [PubMed] [Google Scholar]
- a Carey J. S.; Laffan D.; Thomson C.; Williams M. T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 2006, 4, 2337–2347. 10.1039/b602413k. [DOI] [PubMed] [Google Scholar]; b Dunetz J. R.; Magano J.; Weisenburger G. A. Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals. Org. Process Res. Dev. 2016, 20, 140–177. 10.1021/op500305s. [DOI] [Google Scholar]
- a Han S.-Y.; Kim Y.-A. Recent development of peptide coupling reagents in organic synthesis. Tetrahedron 2004, 60, 2447–2467. 10.1016/j.tet.2004.01.020. [DOI] [Google Scholar]; b Valeur E.; Bradley M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 2009, 38, 606–631. 10.1039/b701677h. [DOI] [PubMed] [Google Scholar]; c de Figueiredo R. M.; Suppo J.-S.; Campagne J.-M. Nonclassical Routes for Amide Bond Formation. Chem. Rev. 2016, 116, 12029–12122. 10.1021/acs.chemrev.6b00237. [DOI] [PubMed] [Google Scholar]; d Montalbetti C. A. G. N.; Falque V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. 10.1016/j.tet.2005.08.031. [DOI] [Google Scholar]; e Sabatini M. T.; Boulton L. T.; Sneddon H. F.; Sheppard T. D. A green chemistry perspective on catalytic amide bond formation. Nat. Catal. 2019, 2, 10–17. 10.1038/s41929-018-0211-5. [DOI] [Google Scholar]
- a Constable D. J. C.; Dunn P. J.; Hayler J. D.; Humphrey G. R.; Leazer J. L.; Linderman R. J.; Lorenz K.; Manley J.; Pearlman B. A.; Wells A.; Zaks A.; Zhang T. Y. Key green chemistry research areas—a perspective from pharmaceutical manufacturers. Green Chem. 2007, 9, 411–420. 10.1039/b703488c. [DOI] [Google Scholar]; b Bandichhor R.; Bhattacharya A.; Diorazio L.; Dunn P.; Fraunhoffer K.; Gallou F.; Hayler J.; Hickey M.; Hinkley B.; Hughes D.; Humphreys L.; Kaptein B.; Mathew S.; Oh L.; Richardson P.; White T.; Wuyts S. Green Chemistry Articles of Interest to the Pharmaceutical Industry. Org. Process Res. Dev. 2013, 17, 1394–1405. 10.1021/op400263a. [DOI] [Google Scholar]
- Gunanathan C.; Ben-David Y.; Milstein D. Direct Synthesis of Amides from Alcohols and Amines with Liberation of H2. Science 2007, 317, 790. 10.1126/science.1145295. [DOI] [PubMed] [Google Scholar]
- a Lane E. M.; Uttley K. B.; Hazari N.; Bernskoetter W. Iron-Catalyzed Amide Formation from the Dehydrogenative Coupling of Alcohols and Secondary Amines. Organometallics 2017, 36, 2020–2025. 10.1021/acs.organomet.7b00258. [DOI] [Google Scholar]; b Srimani D.; Balaraman E.; Hu P.; Ben-David Y.; Milstein D. Formation of Tertiary Amides and Dihydrogen by Dehydrogenative Coupling of Primary Alcohols with Secondary Amines Catalyzed by Ruthenium Bipyridine-Based Pincer Complexes. Adv. Synth. Catal. 2013, 355, 2525–2530. 10.1002/adsc.201300620. [DOI] [Google Scholar]; c Gusev D. G. Rethinking the Dehydrogenative Amide Synthesis. ACS Catal. 2017, 7, 6656–6662. 10.1021/acscatal.7b02415. [DOI] [Google Scholar]; d Chen C.; Verpoort F.; Wu Q. Atom-economic dehydrogenative amide synthesis via ruthenium catalysis. RSC Adv. 2016, 6, 55599–55607. 10.1039/c6ra10643a. [DOI] [Google Scholar]; e Crochet P.; Cadierno V.. Ruthenium-Catalyzed Amide-Bond Formation. In Ruthenium in Catalysis; Dixneuf P. H., Bruneau C., Eds.; Springer International Publishing: Cham, 2014; pp 81–118. [Google Scholar]; f Schley N. D.; Dobereiner G. E.; Crabtree R. H. Oxidative Synthesis of Amides and Pyrroles via Dehydrogenative Alcohol Oxidation by Ruthenium Diphosphine Diamine Complexes. Organometallics 2011, 30, 4174–4179. 10.1021/om2004755. [DOI] [Google Scholar]; g Nordstrøm L. U.; Vogt H.; Madsen R. Amide Synthesis from Alcohols and Amines by the Extrusion of Dihydrogen. J. Am. Chem. Soc. 2008, 130, 17672–17673. 10.1021/ja808129p. [DOI] [PubMed] [Google Scholar]; h Zweifel T.; Naubron J.-V.; Grützmacher H. Catalyzed Dehydrogenative Coupling of Primary Alcohols with Water, Methanol, or Amines. Angew. Chem., Int. Ed. 2009, 48, 559–563. 10.1002/anie.200804757. [DOI] [PubMed] [Google Scholar]; i Ghosh S. C.; Muthaiah S.; Zhang Y.; Xu X.; Hong S. H. Direct Amide Synthesis from Alcohols and Amines by Phosphine-Free Ruthenium Catalyst Systems. Adv. Synth. Catal. 2009, 351, 2643–2649. 10.1002/adsc.200900482. [DOI] [Google Scholar]; j Muthaiah S.; Ghosh S. C.; Jee J.-E.; Chen C.; Zhang J.; Hong S. H. Direct Amide Synthesis from Either Alcohols or Aldehydes with Amines: Activity of Ru(II) Hydride and Ru(0) Complexes. J. Org. Chem. 2010, 75, 3002–3006. 10.1021/jo100254g. [DOI] [PubMed] [Google Scholar]; k Prades A.; Peris E.; Albrecht M. Oxidations and Oxidative Couplings Catalyzed by Triazolylidene Ruthenium Complexes. Organometallics 2011, 30, 1162–1167. 10.1021/om101145y. [DOI] [Google Scholar]; l Spasyuk D.; Vicent C.; Gusev D. G. Chemoselective Hydrogenation of Carbonyl Compounds and Acceptorless Dehydrogenative Coupling of Alcohols. J. Am. Chem. Soc. 2015, 137, 3743–3746. 10.1021/ja512389y. [DOI] [PubMed] [Google Scholar]; m Mondal A.; Subaramanian M.; Nandakumar A.; Balaraman E. Manganese-Catalyzed Direct Conversion of Ester to Amide with Liberation of H2. Org. Lett. 2018, 20, 3381–3384. 10.1021/acs.orglett.8b01305. [DOI] [PubMed] [Google Scholar]; n Kothandaraman J.; Kar S.; Sen R.; Goeppert A.; Olah G. A.; Prakash G. K. S. Efficient Reversible Hydrogen Carrier System Based on Amine Reforming of Methanol. J. Am. Chem. Soc. 2017, 139, 2549–2552. 10.1021/jacs.6b11637. [DOI] [PubMed] [Google Scholar]; o Fu Z.; Lee J.; Kang B.; Hong S. H. Dehydrogenative Amide Synthesis: Azide as a Nitrogen Source. Org. Lett. 2012, 14, 6028–6031. 10.1021/ol302915g. [DOI] [PubMed] [Google Scholar]; p Selvamurugan S.; Ramachandran R.; Prakash G.; Nirmala M.; Viswanathamurthi P.; Fujiwara S.; Endo A. Ruthenium(II) complexes encompassing 2-oxo-1,2-dihydroquinoline-3-carbaldehyde thiosemicarbazone hybrid ligand: A new versatile potential catalyst for dehydrogenative amide synthesis. Inorg. Chim. Acta 2017, 454, 46–53. 10.1016/j.ica.2015.12.024. [DOI] [Google Scholar]; q Li L.; Lei M.; Liu L.; Xie Y.; Schaefer H. F. Metal–Substrate Cooperation Mechanism for Dehydrogenative Amidation Catalyzed by a PNN-Ru Catalyst. Inorg. Chem. 2018, 57, 8778–8787. 10.1021/acs.inorgchem.8b00563. [DOI] [PubMed] [Google Scholar]; r Saha B.; Sengupta G.; Sarbajna A.; Dutta I.; Bera J. K. Amide synthesis from alcohols and amines catalyzed by a RuII–N-heterocyclic carbene (NHC)–carbonyl complex. J. Organomet. Chem. 2014, 771, 124–130. 10.1016/j.jorganchem.2013.12.051. [DOI] [Google Scholar]; s Cho D.; Ko K. C.; Lee J. Y. Catalytic Mechanism for the Ruthenium-Complex-Catalyzed Synthesis of Amides from Alcohols and Amines: A DFT Study. Organometallics 2013, 32, 4571–4576. 10.1021/om4005324. [DOI] [Google Scholar]; t Choi G.; Hong S. H. Selective N-Formylation and N-Methylation of Amines Using Methanol as a Sustainable C1 Source. ACS Sustainable Chem. Eng. 2019, 7, 716–723. 10.1021/acssuschemeng.8b04286. [DOI] [Google Scholar]
- Kumar A.; Espinosa-Jalapa N. A.; Leitus G.; Diskin-Posner Y.; Avram L.; Milstein D. Direct Synthesis of Amides by Dehydrogenative Coupling of Amines with either Alcohols or Esters: Manganese Pincer Complex as Catalyst. Angew. Chem., Int. Ed. 2017, 56, 14992–14996. 10.1002/anie.201709180. [DOI] [PubMed] [Google Scholar]
- a Shimizu K.-i.; Ohshima K.; Satsuma A. Direct Dehydrogenative Amide Synthesis from Alcohols and Amines Catalyzed by γ-Alumina Supported Silver Cluster. Chem.—Eur. J. 2009, 15, 9977–9980. 10.1002/chem.200901896. [DOI] [PubMed] [Google Scholar]; b Zhu J.; Zhang Y.; Shi F.; Deng Y. Dehydrogenative amide synthesis from alcohol and amine catalyzed by hydrotalcite-supported gold nanoparticles. Tetrahedron Lett. 2012, 53, 3178–3180. 10.1016/j.tetlet.2012.04.048. [DOI] [Google Scholar]; c Hakim Siddiki S. M. A.; Toyao T.; Shimizu K.-i. Acceptorless dehydrogenative coupling reactions with alcohols over heterogeneous catalysts. Green Chem. 2018, 20, 2933–2952. 10.1039/c8gc00451j. [DOI] [Google Scholar]
- Li H.; Wang X.; Huang F.; Lu G.; Jiang J.; Wang Z.-X. Computational Study on the Catalytic Role of Pincer Ruthenium(II)-PNN Complex in Directly Synthesizing Amide from Alcohol and Amine: The Origin of Selectivity of Amide over Ester and Imine. Organometallics 2011, 30, 5233–5247. 10.1021/om200620n. [DOI] [Google Scholar]
- Gunanathan C.; Milstein D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712. 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]
- a Fogler E.; Garg J. A.; Hu P.; Leitus G.; Shimon L. J. W.; Milstein D. System with Potential Dual Modes of Metal–Ligand Cooperation: Highly Catalytically Active Pyridine-Based PNNH–Ru Pincer Complexes. Chem.—Eur. J. 2014, 20, 15727–15731. 10.1002/chem.201405295. [DOI] [PubMed] [Google Scholar]; He T.; Buttner J. C.; Reynolds E. F.; Pham J.; Malek J. C.; Keith J. M.; Chianese A. R. Dehydroalkylative Activation of CNN- and PNN-Pincer Ruthenium Catalysts for Ester Hydrogenation. J. Am. Chem. Soc. 2019, 141, 17404–17413. 10.1021/jacs.9b09326. [DOI] [PubMed] [Google Scholar]; c Pham J.; Jarczyk C. E.; Reynolds E. F.; Kelly S. E.; Kim T.; He T.; Keith J. M.; Chianese A. R.. The key role of the latent N–H group in Milstein’s catalyst for ester hydrogenation. Chem. Sci. 2021, 10.1039/D1SC00703C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanaprakasam B.; Milstein D. Synthesis of Amides from Esters and Amines with Liberation of H2 under Neutral Conditions. J. Am. Chem. Soc. 2011, 133, 1682–1685. 10.1021/ja109944n. [DOI] [PubMed] [Google Scholar]
- Montag M.; Zhang J.; Milstein D. Aldehyde Binding through Reversible C–C Coupling with the Pincer Ligand upon Alcohol Dehydrogenation by a PNP–Ruthenium Catalyst. J. Am. Chem. Soc. 2012, 134, 10325–10328. 10.1021/ja303121v. [DOI] [PubMed] [Google Scholar]
- A minor isomer of complex 1d was also observed (9%) in the solution, with a chemical shift of 121 ppm in the 31P NMR spectrum and a hydride chemical shift of −15.1 ppm in the 1H NMR spectrum.
- Huff C. A.; Kampf J. W.; Sanford M. S. Reversible carbon–carbon bond formation between carbonyl compounds and a ruthenium pincer complex. Chem. Commun. 2013, 49, 7147–7149. 10.1039/c3cc43517b. [DOI] [PubMed] [Google Scholar]
- Similarly the benzaldehyde bound complex (1d′) also exchanges the bound benzaldehyde with free hexanal/p-Cl benzaldehyde when excess hexanal (10 equiv) or p-Cl benzaldehyde is added to a solution of 1d (Figures S24 and 25).
- Kar S.; Rauch M.; Kumar A.; Leitus G.; Ben-David Y.; Milstein D. Selective Room-Temperature Hydrogenation of Amides to Amines and Alcohols Catalyzed by a Ruthenium Pincer Complex and Mechanistic Insight. ACS Catal. 2020, 10, 5511–5515. 10.1021/acscatal.0c01406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alberico E.; Lennox A. J. J.; Vogt L. K.; Jiao H.; Baumann W.; Drexler H.-J.; Nielsen M.; Spannenberg A.; Checinski M. P.; Junge H.; Beller M. Unravelling the Mechanism of Basic Aqueous Methanol Dehydrogenation Catalyzed by Ru–PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 14890–14904. 10.1021/jacs.6b05692. [DOI] [PubMed] [Google Scholar]; b Zou Y. Q.; Wolff N.; Rauch M.; Feller M.; Zhou Q. Q.; Anaby A.; Diskin-Posner Y.; Shimon L. J. W.; Avram L.; Ben-David Y.; Milstein D. Homogeneous Reforming of Aqueous Ethylene Glycol to Glycolic Acid and Pure Hydrogen Catalyzed by Pincer-Ruthenium Complexes Capable of Metal–Ligand Cooperation. Chem.—Eur. J. 2021, 27, 4715–4722. 10.1002/chem.202005450. [DOI] [PubMed] [Google Scholar]; c Gusev D. G. Revised Mechanisms of the Catalytic Alcohol Dehydrogenation and Ester Reduction with the Milstein PNN Complex of Ruthenium. Organometallics 2020, 39, 258–270. 10.1021/acs.organomet.9b00542. [DOI] [Google Scholar]
- a Dub P. A.; Gordon J. C. The role of the metal-bound N–H functionality in Noyori-type molecular catalysts. Nat. Rev. Chem. 2018, 2, 396–408. 10.1038/s41570-018-0049-z. [DOI] [Google Scholar]; b Samec J. S. M.; Bäckvall J.-E.; Andersson P. G.; Brandt P. Mechanistic aspects of transition metal-catalyzed hydrogen transfer reactions. Chem. Soc. Rev. 2006, 35, 237–248. 10.1039/b515269k. [DOI] [PubMed] [Google Scholar]; c Johnson J. B.; Bäckvall J.-E. Mechanism of Ruthenium-Catalyzed Hydrogen Transfer Reactions. Concerted Transfer of OH and CH Hydrogens from an Alcohol to a (Cyclopentadienone)ruthenium Complex. J. Org. Chem. 2003, 68, 7681–7684. 10.1021/jo034634a. [DOI] [PubMed] [Google Scholar]; d Dub P. A.; Gordon J. C. The mechanism of enantioselective ketone reduction with Noyori and Noyori–Ikariya bifunctional catalysts. Dalton Trans. 2016, 45, 6756–6781. 10.1039/c6dt00476h. [DOI] [PubMed] [Google Scholar]; e Dub P. A.; Gordon J. C. Metal–Ligand Bifunctional Catalysis: The “Accepted” Mechanism, the Issue of Concertedness, and the Function of the Ligand in Catalytic Cycles Involving Hydrogen Atoms. ACS Catal. 2017, 7, 6635–6655. 10.1021/acscatal.7b01791. [DOI] [Google Scholar]
- For oxidative formation of amide-bond containing pharmaceuticals from corresponding alcohol and amine, see:Piszel P. E.; Vasilopoulos A.; Stahl S. S. Oxidative Amide Coupling from Functionally Diverse Alcohols and Amines Using Aerobic Copper/Nitroxyl Catalysis. Angew. Chem., Int. Ed. 2019, 58, 12211–12215. 10.1002/anie.201906130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moclobemide. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson J. K., Ed.; Elsevier: Oxford, 2016; pp 1076–1080. [Google Scholar]
- a For other alternative methods to synthesize moclobemide, see:Singha K.; Ghosh S. C.; Panda A. B. N-Doped Yellow TiO2 Hollow Sphere-Mediated Visible-Light-Driven Efficient Esterification of Alcohol and N-Hydroxyimides to Active Esters. Chem.—Asian J. 2019, 14, 3205–3212. 10.1002/asia.201900878. [DOI] [PubMed] [Google Scholar]; b Bantreil X.; Kanfar N.; Gehin N.; Golliard E.; Ohlmann P.; Martinez J.; Lamaty F. Iron-catalyzed benzamide formation. Application to the synthesis of moclobemide. Tetrahedron 2014, 70, 5093–5099. 10.1016/j.tet.2014.06.001. [DOI] [Google Scholar]
- https://www.drugbank.ca/drugs/DB04924 (accessed Feb 06, 2021).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.