

Abstract
Background
DNA polymerase epsilon (POLE) is encoded by the POLE gene, and POLE-driven tumors are characterized by high mutational rates. POLE-driven tumors are relatively common in endometrial and colorectal cancer, and their presence is increasingly recognized in ovarian cancer (OC) of endometrioid type. POLE-driven cases possess an abundance of TCT > TAT and TCG > TTG somatic mutations characterized by mutational signature 10 from the Catalog of Somatic Mutations in Cancer (COSMIC). By quantifying the contribution of COSMIC mutational signature 10 in RNA sequencing (RNA-seq) we set out to identify POLE-driven tumors in a set of unselected Mayo Clinic OC.
Methods
Mutational profiles were calculated using expressed single-nucleotide variants (eSNV) in the Mayo Clinic OC tumors (n = 195), The Cancer Genome Atlas (TCGA) OC tumors (n = 419), and the Genotype-Tissue Expression (GTEx) normal ovarian tissues (n = 84). Non-negative Matrix Factorization (NMF) of the mutational profiles inferred the contribution per sample of four distinct mutational signatures, one of which corresponds to COSMIC mutational signature 10.
Results
In the Mayo Clinic OC cohort we identified six tumors with a predicted contribution from COSMIC mutational signature 10 of over five mutations per megabase. These six cases harbored known POLE hotspot mutations (P286R, S297F, V411L, and A456P) and were of endometrioid histotype (P = 5e−04). These six tumors had an early onset (average age of patients at onset, 48.33 years) when compared to non-POLE endometrioid OC cohort (average age at onset, 60.13 years; P = .008). Samples from TCGA and GTEx had a low COSMIC signature 10 contribution (median 0.16 mutations per megabase; maximum 1.78 mutations per megabase) and carried no POLE hotspot mutations.
Conclusions
From the largest cohort of RNA-seq from endometrioid OC to date (n = 53), we identified six hypermutated samples likely driven by POLE (frequency, 11%). Our result suggests the clinical need to screen for POLE driver mutations in endometrioid OC, which can guide enrollment in immunotherapy clinical trials.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12920-021-01017-7.
Keywords: Hypermutation, Mutational signatures, Ovarian endometrioid cancer, POLE, RNA-seq
Background
DNA polymerase epsilon (POLE) is a key member of the DNA polymerase family and is involved in error correction during replication [1]. Somatic mutations in the exonuclease domain of POLE have been identified in 7–12% of uterine endometrial cancers, in 1–2% of colorectal cancers, and at low frequencies in stomach cancer, glioblastoma, breast cancer, and others [2–5]. Recurrent hotspot mutations include P286R, S297F, and V411L, which are characterized by ultramutation [5, 6]. Patients with POLE-driven endometrial cancer have a favorable prognosis, a higher neoantigen load, an increased number of tumor-infiltrating lymphocytes, and they may benefit from immunotherapy [6–8].
Somatic POLE mutations are uncommon in serous ovarian cancer (OC) [9, 10] but the presence of POLE mutations is increasingly recognized in ovarian endometrioid cancer (OEC) [10–14]. Patients with POLE-driven OEC have earlier disease onset, and an increased number of CD8 + intraepithelial tumor-infiltrating lymphocytes [11, 14]. The prevalence of POLE-driven tumors in different OEC studies ranges from 3 to 13% [10–12, 14–16], and is likely driven by sample size, cohort selection criteria, and type of mutation detection assay. Interestingly, in a Japanese cohort of concurrent ovarian and endometrial cancer, the frequency of POLE mutations was high (five of 8 cases; 62%) detected by Sanger sequencing [13]; however, the reported POLE mutations (Q292E, E396V, D287N, and N293D) do not correspond to known hotspot mutations. This result highlights the need to ascertain the effects of particular POLE mutations and whether they are accompanied by hypermutation.
POLE-driven tumors are associated with a distinct mutational signature found in whole-genome sequencing from tumor-normal pairs which is characterized by a high number of TCT > TAT and TCG > TTG mutations [4, 17]. Such distinct mutational profile is known as COSMIC mutational signature 10 [17]. Exome sequencing detected an abundance of COSMIC mutational signature 10 in OEC cases with known POLE hotspot mutations [16]. Previously, we developed a novel method for inferring and quantifying distinct mutational signatures using RNA sequencing (RNA-seq) data [18, 19]. We applied this method in tumor-only fresh-frozen RNA sequencing (RNA-seq) samples in endometrial cancer and colorectal cancer tumors from The Cancer Genome Atlas (TCGA) and identified POLE cases with high specificity and sensitivity [19].
Using previously published RNA-seq from a Mayo Clinic OC cohort (n = 195), which included the largest set of OEC transcriptomes to date (n = 53), we sought to leverage the mutational signatures approach to identify POLE-driven cases and to characterize their clinical characteristics [20, 21].
Methods
Mayo Clinic and public data selection and RNA-seq
Selected participants were patients who were at least 20 years old and were ascertained at Mayo Clinic from 1992 through 2009 within one year after receiving a pathologically confirmed diagnosis of primary invasive epithelial OC, fallopian tube cancer, or primary peritoneal cancer. (Table 1). Patients were treated using standard first line platinum-based chemotherapy. Tumors were snap frozen immediately after surgery and stored at − 80 °C. A gynecologic pathologist confirmed the clinical diagnoses and verified the tumor histology and grade and the presence of 70% tumor content before RNA extraction from fresh frozen tissue. As described previously [20, 21] transcriptomic sequencing was performed in four batches with TruSeq Library Preparation kits (Stranded Total RNA Library Preparation Kit or RNA Library Preparation Kit v2; Illumina, Inc) and sequenced on the Illumina HiSeq 2000 sequencer with 100–base pair paired-end reads. All patients gave informed consent; all protocols were approved by the Mayo Clinic Institutional Review Board.
Table 1.
Characteristics of the 195 patients in the Mayo Clinic ovarian cancer cohort
| Feature | Value |
|---|---|
| Age at diagnosis, mean (SD), y | 62 (12) |
| Histology, no. (%) | |
| Serous | 114 (58) |
| Endometrioid | 53 (27) |
| Clear cell | 14 (7) |
| Undifferentiated | 8 (4) |
| Mucinous | 3 (2) |
| Other | 3 (2) |
| Grade, no. (%) | |
| 1 | 20 (10) |
| 2 | 24 (12) |
| 3 | 146 (75) |
| Unknown | 5 (3) |
| Stage, no. (%) | |
| I | 45 (23) |
| II | 15 (8) |
| III | 100 (51) |
| IV | 33 (17) |
| Unknown | 2 (1) |
We also used OC RNA-seq from TCGA (n = 419) and from normal ovarian tissue from the Genotype-Tissue Expression (GTEx) project (Broad Institute) (n = 84) [9, 22].
Bioinformatics methods
RNA-seq of Mayo Clinic, TCGA, and GTEx data sequencing reads were processed through the Mayo Clinic MAP-RSeq v.2.1.5 computational workflow, and variants were calculated with RVboost 0.1 [18, 23]. We considered expressed single nucleotide variants from RVboost with a Q score greater than 10%, read depth greater than 10, a minor allelic frequency less than 2% in the 1000 Genomes Project [24], and not present in recurrent expressed single-nucleotide variants identified in RNA-seq from adjacent normal tissue. The RNA-seq capture region was defined as positions with 20 × coverage as calculated by the Genome Analysis Toolkit (GATK; Broad Institute). Samples with read depth over 20 × at less than five million positions were excluded from this analysis. Tumor mutation burden (TMB) was calculated as the number of considered expressed variants per capture region × 106.
We used the mutational signatures v2 from the Catalogue of Somatic Mutations in Cancer (COSMIC) [25] where the mutational profiles are represented as the proportion of each substitution type (C > A, C > G, C > T, T > A, T > C, and T > G) and its trinucleotide context (the nucleotide before and after each mutated base) [17]. For the detection of mutational signatures and their contribution to each sample, we used R version 3.4.2 (R Foundation) with the MutationalPatterns v1.4.3 package [26]. To measure the similarity between two mutational signatures we used the cosine similarity as implemented in the function cos_sim in the MutationalPatterns v1.4.3 package. The cosine similarity takes values between 0 and 1, with a value close to 1 if there is great similarity between signatures and close to 0 if the two signatures are dissimilar.
Statistical analysis
We used R version 3.4.2 with the tidyverse 1.2.1 package to perform statistical analyses and generate graphs. Whiskers in the boxplots correspond 75th (and 25th) Quantile ± 1.5 IQR (Interquantile range) and outliers correspond to values outside this range. Two-sided Mann–Whitney tests (Wilcoxon rank sum tests) were used for comparisons of contributions of POLE mutational signature across the Mayo OC cohort and TCGA/GTEx, and age across patients with and without POLE mutations. Histology findings from patients with and without POLE mutations were compared with the Fisher exact test.
Results
The Mayo Clinic OC cohort with existing RNA-seq consisted of 195 patients whose clinical characteristics are described in Table 1 [20, 21]. This cohort, contains an abundance of nonserous histologies (81 of 195) and includes 53 with OEC, which makes it the largest OEC RNAseq collection to date.
Mutational profiles from eSNV were calculated in the Mayo Clinic OC tumors (n = 195), TCGA OC tumors (n = 419), and the GTEx normal ovarian tissues (n = 84). Using Non-negative Matrix Factorization (NMF) [26] we were able to approximate each sample’s mutational profile as a combination of four distinct mutational signatures (Fig. 1a and Additional file 1: Fig. S1). Using the cosine similarity, we established a high degree of resemblance between the approximated mutational profiles from NMF and the original mutational profile (median cosine similarity, 0.92; interquartile range, 0.89–0.93) (Additional file 1: Fig. S2).
Fig. 1.
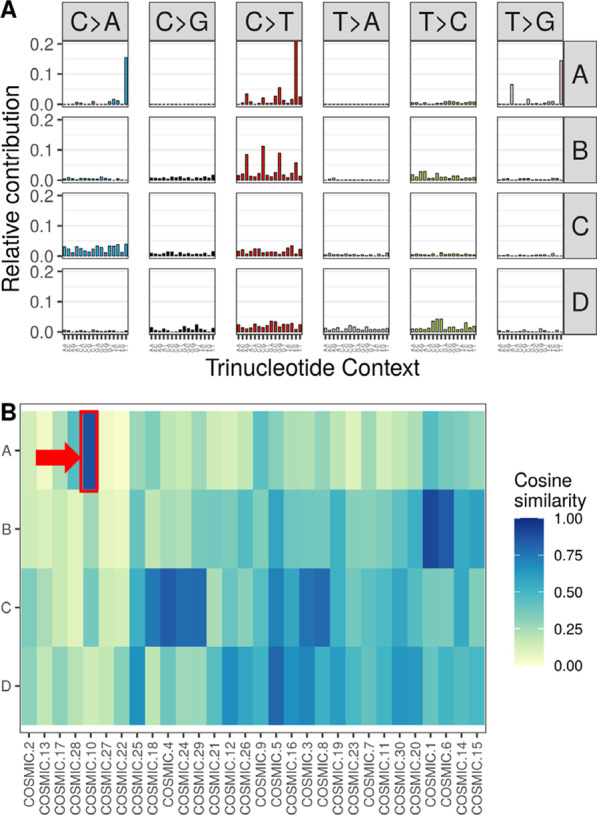
Mutational Signatures from RNA Sequencing of Tissues with and without Ovarian Cancer (OC). a The four mutational signatures according to trinucleotide context in the Mayo Clinic and TCGA OC tumors combined. b Heatmap of the cosine similarity of the four mutational signatures compared against the reference Catalogue of Somatic Mutations in Cancer (COSMIC) signatures. COSMIC signatures are grouped on the x-axis by using hierarchical clustering using the average method according to their cosine similarities. Mutational signature A has a high cosine similarity against COSMIC mutational signature 10 and it is denoted in red
Using the cosine similarity, the four mutational signatures were compared against the COSMIC mutational signatures catalog v2 (Fig. 1b) [17]. Signature B has a cosine similarity of 0.91 against COSMIC signature 1 which is associated with aging [27]. Signature C has cosine similarities of 0.85 and 0.77 against COSMIC signatures 4 and 3, which are associated with tobacco exposure and defective double strand repair respectively [17]. Signature D has a cosine similarity of 0.81 against COSMIC signature 5, a signature found in all cancer types [17].
Of note, signature A has a cosine similarity of 0.88 to COSMIC signature 10, which is associated with POLE defects [2]. Six samples in the Mayo Cohort have a similar mutational distribution characterized by an enrichment of the POLE contribution (Additional file 1: Fig. S1B and C).
The POLE signature contribution of such six samples (of 195) from our Mayo OC were deemed as outliers (> 5.74 Mut per megabase) (see Methods and Additional file 1: Fig. S3). Normal samples from GTEx and serous OC samples from TCGA had a median of 0.16 mutations per megabase; maximum 1.78 mutations per megabase of the POLE signature and can be used as negative POLE controls (Fig. 2a). The six outlier Mayo OC samples have a higher POLE signature contribution (median 46.34 mutations per megabase; P = 2.37E−05) when compared to the negative POLE controls (Fig. 2a). The individual mutational profiles for each of those six samples are shown in Fig. 2b.
Fig. 2.
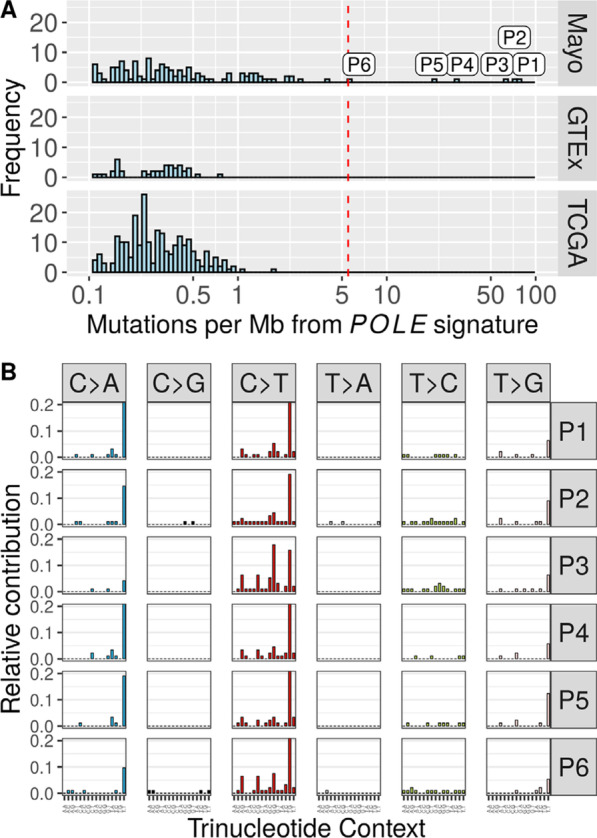
Identification of DNA Polymerase Epsilon (POLE)-Driven Ovarian Cancer Samples. a Distribution of the tumor mutation burden (TMB) attributed to the POLE signature across the cohorts from Mayo Clinic (Mayo), Genotype-Tissue Expression (GTEx), and The Cancer Genome Atlas (TCGA), which allows the identification of six samples (P1–P6) with high POLE contribution and more than five mutations per megabase. The red dashed line indicates a threshold used to separate between POLE positive and negative cases. b Mutational profiles of six POLE-driven Mayo Clinic OEC tumors according to the frequency of trinucleotide context for the six samples; their distinct C > A and C > T peaks are associated with the POLE signature. Red line denotes the threshold used for the outlier direction (5.7 Mut per Mb)
Within these six samples, we found expressed mutations corresponding to the POLE hotspot COSMIC mutations P286R, S297F, V411L, and A456P (Table 2 and on Additional file 1: Fig. S3) [28]. No other samples in the Mayo OC, the TCGA OC, or the normal ovarian GTEx cohort harbored expressed mutations in any of the POLE hotspots.
Table 2.
Clinical Characteristics of six POLE Samples from the Mayo Clinic Ovarian Cancer Cohort
| Sample | POLE mutation | POLE TMB | Age, years | Histology | Stage | Grade | Vital status |
|---|---|---|---|---|---|---|---|
| P1 | A456P | 75.2 | 46 | Endometrioid | III | 2 | Alive |
| P2 | S297F | 71.7 | 49 | Endometrioid | I | 3 | Alive |
| P3 | V411L | 64.1 | 49 | Endometrioid | III | 3 | Deceased |
| P4 | P286R | 28.6 | 46 | Endometrioid | I | 2 | Alive |
| P5 | P286R | 21.6 | 49 | Endometrioid | I | 1 | Alive |
| P6 | V411L | 5.8 | 51 | Endometrioid | I | 1 | Alive |
POLE DNA polymerase epsilon, TMB tumor mutation burden
All six samples were of endometrioid histotype (P = 5e−04) and constituted 11% of OEC cases. Average age at onset was earlier (48.33 years) than in the non-POLE OEC cohort (60.13 years; P = 0.008). Four of these six samples corresponded to stage 1, and two corresponded to stage 3 (Table 2).
Discussion
RNA-seq is routinely used for transcriptome quantification and fusion detection, as illustrated by our previous studies where we characterized gene expression and fusions across different OC histology types [20, 21]. This study constitutes a novel reuse of this OC RNA-seq cohort, along with multiple public RNA-seq datasets including TCGA and GTEx, to enable clinically significant discoveries of previously unidentified POLE altered cases. Furthermore, our bioinformatics approach can identify the distinct POLE mutational signatures in RNA-seq as well as confirm the expression of POLE hotspot mutations. This adds to the clinical utility of RNA-seq which can already detect fusions and calculate tumor mutational burden in a single clinical assay, as opposed to DNA-seq [29, 30].
POLE-driven tumors have a favorable prognosis, an increased number of tumor-infiltrating lymphocytes, and can benefit from immunotherapy. Despite these distinct characteristics of POLE-driven tumors, POLE mutation status is not routinely evaluated in a clinical setting for OEC. By using RNA-seq on the largest OEC cohort to our knowledge (n = 53), we found six of 53 OEC samples (11%) that were POLE-driven. Those six samples had a highly specific mutational profile corresponding to the well-characterized POLE COSMIC signature 10 profile, and POLE hotspot mutations (P286R, S297F, V411L, and A456P). Two of the 6 patients in the group of POLE cases had advanced-stage OEC with relatively high recurrence risks; therefore, OEC patients with POLE-driven tumors can be eligible for immunotherapy trials if their cancers progress or recur.
The tumor mutational burden (TMB) attributed to COSMIC signature 10 of our POLE cases spans from over five to more than 75 mutations per megabase. Patients with higher TMB attributed to COSMIC signature 10 have worse clinical characteristics and outcomes than among non-POLE OECs; the sample with the lowest TMB was from a patient with a low-grade, stage 1 cancer, and the three samples with the highest TMB correspond to higher-grade or higher stage cancers. However, the limited number of POLE cases in this study precluded any statistical analysis, and further independent studies with larger sample sizes are necessary to validate and confirm such a trend.
The smaller number of mutations in RNA-seq as compared to Whole Genomic Sequencing (WGS) reduces the statistical power to find known signatures present at lower levels and to distinguish the contribution of closely related and flat signatures. To illustrate this point, COSMIC signature 3 which is associated with defective homologous recombination and is prevalent in OC had only cosine similarity of 0.77 against signature C, while signature 4 which is associated with tobacco and not known to play a role in OC had a cosine similarity of 0.84 against signature C. Future studies to quantify the limit of detection of mutational signatures in RNA-seq are warranted.
Conclusions
Using RNA-seq mutational signatures from the largest OEC cohort to date (n = 53), we found that 6 hypermutated samples (11%) had evidence of POLE-driven tumors.
Supplementary Information
Additional file 1. Supplementary Figures.
Acknowledgements
We wish you to acknowledge English Language editing and review services at the department of Scientific Publications at Mayo Clinic, Rochester, MN. We thank the reviewers whose insightful comments improved and clarified the current manuscript.
Abbreviations
- COSMIC
Catalogue of Somatic Mutations in Cancer
- GTEx
Genotype-Tissue Expression
- MMR
mismatch repair
- MSK-IMPACT
Memorial Sloan Kettering integrated mutation profiling of actionable cancer targets
- OC
ovarian cancer
- OEC
ovarian endometrioid cancer
- POLE
DNA polymerase epsilon
- RNA-seq
RNA sequencing
- TCGA
The Cancer Genome Atlas
- TMB
tumor mutation burden
Authors' contributions
J.I.D. and C.W. performed and supervised bioinformatics analyses, participated in study design and interpretation of results, and manuscript drafting. P.C., V.S., and Z.C.F. participated in generation of results and performed bioinformatics analysis and manuscript drafting. E.L.G., S. J.W., and Y.H. participated in study design and manuscript drafting. All authors read and approved the final manuscript.
Funding
This study was supported by R21-CA222867, R01-CA122443, R01-CA248288 from the National Institutes of Health.
Availability of data and materials
The datasets and code generated during and/or analyzed during the current study are available in https://github.com/jdavilal/mutational_sig_oec.
Declarations
Ethics approval and consent to participate
All patients gave informed consent, and all protocols were approved by the Mayo Clinic Institutional Review Board. All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Consent for publication is not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jaime I. Davila, Email: davila3@stolaf.edu
Chen Wang, Email: wang.chen@mayo.edu.
References
- 1.Pursell ZF, Isoz I, Lundström E-B, Johansson E, Kunkel TA. Yeast DNA polymerase epsilon participates in leading-strand DNA replication. Science. 2007;317(5834):127–130. doi: 10.1126/science.1144067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shinbrot E, Henninger EE, Weinhold N, Covington KR, Göksenin AY, Schultz N, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. 2014;24(11):1740–1750. doi: 10.1101/gr.174789.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16(2):71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 6.Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, ter Haar N, et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst. 2015;107(1):402. doi: 10.1093/jnci/dju402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015;1(9):1319–1323. doi: 10.1001/jamaoncol.2015.2151. [DOI] [PubMed] [Google Scholar]
- 8.Mehnert JM, Panda A, Zhong H, Hirshfield K, Damare S, Lane K, et al. Immune activation and response to pembrolizumab in POLE-mutant endometrial cancer. J Clin Invest. 2016;126(6):2334–2340. doi: 10.1172/JCI84940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou Y, Liu FY, Liu H, Wang F, Li W, Huang MZ, et al. Frequent POLE1 p.S297F mutation in Chinese patients with ovarian endometrioid carcinoma. Mutat Res. 2014;761:49–52. doi: 10.1016/j.mrfmmm.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Hoang LN, McConechy MK, Köbel M, Anglesio M, Senz J, Maassen M, et al. Polymerase epsilon exonuclease domain mutations in ovarian endometrioid carcinoma. Int J Gynecol Cancer. 2015;25(7):1187–1193. doi: 10.1097/IGC.0000000000000492. [DOI] [PubMed] [Google Scholar]
- 12.Cybulska P, Paula ADC, Tseng J, Leitao MM, Jr, Bashashati A, Huntsman DG, et al. Molecular profiling and molecular classification of endometrioid ovarian carcinomas. Gynecol Oncol. 2019;154(3):516–523. doi: 10.1016/j.ygyno.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa M, Nakayama K, Nakamura K, Ono R, Yamashita H, Ishibashi T, et al. High frequency of POLE mutations in synchronous endometrial and ovarian carcinoma. Hum Pathol. 2019;85:92–100. doi: 10.1016/j.humpath.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Leskela S, Romero I, Rosa-Rosa JM, Caniego-Casas T, Cristobal E, Pérez-Mies B, et al. Molecular heterogeneity of endometrioid ovarian carcinoma: an analysis of 166 cases using the endometrial cancer subrogate molecular classification. Am J Surg Pathol. 2020;44(7):982–990. doi: 10.1097/PAS.0000000000001478. [DOI] [PubMed] [Google Scholar]
- 15.Parra-Herran C, Lerner-Ellis J, Xu B, Khalouei S, Bassiouny D, Cesari M, et al. Molecular-based classification algorithm for endometrial carcinoma categorizes ovarian endometrioid carcinoma into prognostically significant groups. Mod Pathol. 2017;30(12):1748–1759. doi: 10.1038/modpathol.2017.81. [DOI] [PubMed] [Google Scholar]
- 16.Teer JK, Yoder S, Gjyshi A, Nicosia SV, Zhang C, Monteiro ANA. Mutational heterogeneity in non-serous ovarian cancers. Sci Rep. 2017;7(1):9728. doi: 10.1038/s41598-017-10432-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Davila JI, Baheti S, Bhagwate AV, Wang X, Kocher JP, et al. RVboost: RNA-seq variants prioritization using a boosting method. Bioinformatics. 2014;30(23):3414–3416. doi: 10.1093/bioinformatics/btu577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jessen E, Liu Y, Davila J, Kocher J-P, Wang C. Determining mutational burden and signature using RNA-seq from tumor-only samples. BMC Med Genomics. 2021;14(1):65. doi: 10.1186/s12920-021-00898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Earp MA, Raghavan R, Li Q, Dai J, Winham SJ, Cunningham JM, et al. Characterization of fusion genes in common and rare epithelial ovarian cancer histologic subtypes. Oncotarget. 2017;8(29):46891–46899. doi: 10.18632/oncotarget.16781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fridley BL, Dai J, Raghavan R, Li Q, Winham SJ, Hou X, et al. Transcriptomic characterization of endometrioid, clear cell, and high-grade serous epithelial ovarian carcinoma. Cancer Epidemiol Biomark Prev. 2018;27(9):1101. doi: 10.1158/1055-9965.EPI-17-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lonsdale J, Thomas J, Salvatore M, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kalari KR, Nair AA, Bhavsar JD, O'Brien DR, Davila JI, Bockol MA, et al. MAP-RSeq: mayo analysis pipeline for RNA sequencing. BMC Bioinform. 2014;15:224. doi: 10.1186/1471-2105-15-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucl Acids Res. 2019;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blokzijl F, Janssen R, van Boxtel R, Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10(1):33. doi: 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47(12):1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucl Acids Res. 2018;47(D1):D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winters JL, Davila JI, McDonald AM, Nair AA, Fadra N, Wehrs RN, et al. Development and verification of an RNA sequencing (RNA-Seq) assay for the detection of gene fusions in tumors. J Mol Diagn. 2018;20(4):495–511. doi: 10.1016/j.jmoldx.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 30.DiGuardo MA, Davila JI, Jackson RA, Nair AA, Fadra N, Minn KT, et al. RNA-Seq reveals differences in expressed tumor mutation burden in colorectal and endometrial cancers with and without defective DNA-mismatch repair. J Mol Diagn. 2021;23(5):555–564. doi: 10.1016/j.jmoldx.2021.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplementary Figures.
Data Availability Statement
The datasets and code generated during and/or analyzed during the current study are available in https://github.com/jdavilal/mutational_sig_oec.