

Abstract
There is a clinical need for direct-acting antivirals targeting SARS-CoV-2, the coronavirus responsible for the COVID-19 pandemic, to complement current therapeutic strategies. The main protease (Mpro) is an attractive target for antiviral therapy. However, the vast majority of protease inhibitors described thus far are peptidomimetic and bind to the active-site cysteine via a covalent adduct, which is generally pharmacokinetically unfavorable. We have reported the optimization of an existing FDA-approved chemical scaffold, perampanel, to bind to and inhibit Mpro noncovalently with IC50s in the low-nanomolar range and EC50s in the low-micromolar range. Here, we present nine crystal structures of Mpro bound to a series of perampanel analogs, providing detailed structural insights into their mechanism of action and structure-activity relationship. These insights further reveal strategies for pursuing rational inhibitor design efforts in the context of considerable active-site flexibility and potential resistance mechanisms.
Keywords: SARS-CoV-2, 3CL main protease, 3CL Mpro, protease inhibitor, enzyme kinetics, X-ray crystallography, drug design
Graphical abstract
Deshmukh et al. investigate the structure-activity relationship of a novel pharmacophore targeting the SARS-CoV-2 main protease derived from the chemical scaffold of the FDA-approved drug perampanel. X-ray crystal structures of enzyme-inhibitor complexes and in vitro activity assays provide guidance for rational drug design.
Introduction
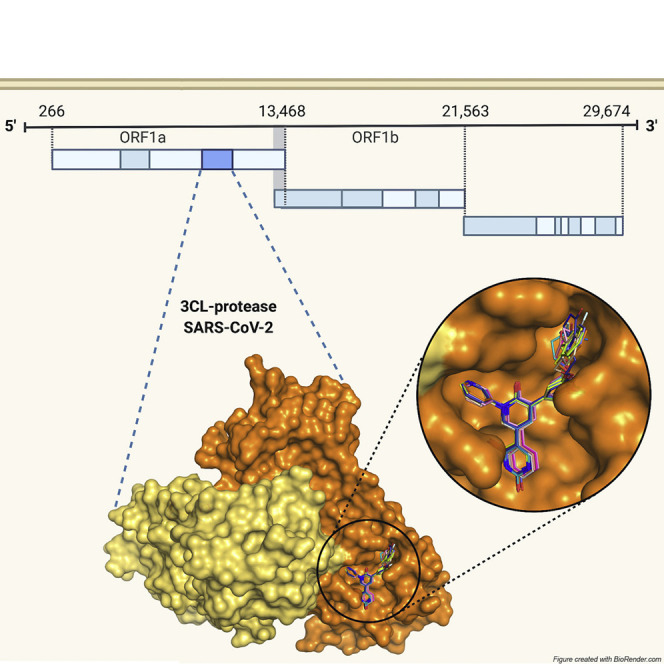
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19, is a positive-sense, single-stranded, enveloped RNA virus belonging to the betacoronavirus genus (Cui et al., 2019). The COVID-19 pandemic is responsible for 110.7 million cases and over 2.4 million deaths to date (World Health Organization, 2021). Alongside widespread global vaccination efforts, there is a need for improved direct-acting antivirals (DAAs) that decrease mortality and morbidity. Currently, the only antiviral with US Food and Drug Administration (FDA) Emergency Use Approval for treatment of COVID-19 is remdesivir, a repurposed RNA-dependent, RNA-polymerase inhibitor. While remdesivir has been shown to reduce recovery time in infected individuals, its impact on mortality when administered alone remains limited (Beigel et al., 2020). Thus, there is an urgent clinical need for the investigation and translation of antivirals.
The transcriptome of SARS-CoV-2 encodes three enzymes that have emerged as attractive targets for novel DAAs: the RNA-dependent RNA polymerase (RdRP or nsp12), the papain-like protease (PLpro or nsp3), and the 3C-like protease (3CLpro, Mpro, or nsp5) (Kim et al., 2020). Between the two proteases, Mpro is a preferred target for investigation of DAAs due to the putative promiscuity of substrate-mimetic inhibitors of PLpro, and the unique substrate specificity of Mpro and conservation of active-site residues (Ullrich and Nitsche, 2020). Mpro is autocatalytically cleaved and subsequently cleaves 11 sites along the overlapping viral polyproteins pp1a and p11ab, releasing nonstructural proteins (nsps) required for replication (Ullrich and Nitsche, 2020). Mpro is thus a key determinant of viral replication, and novel inhibitors have already demonstrated promising activity and safety in vitro (Mengist et al., 2020).
Despite recent advances in identifying novel Mpro inhibitors with attractive potency and safety in vitro, relatively few studies have reported pharmacokinetic properties (Mengist et al., 2020). Most inhibitors described thus far with promising properties in vitro bind covalently to the active-site cysteine (Cys145) via an electrophilic warhead, a feature generally associated with less favorable selectivity and pharmacokinetic properties compared with noncovalent inhibitors (Cannalire et al., 2020). While noncovalent, nonpeptidomimetic inhibitors are sought for their improved drug-likeness, the main challenge lies in optimizing active-site binding to achieve comparable activity. One strategy for designing novel, drug-like, noncovalent compounds is the optimization of low-affinity hits that are existing drugs with known pharmacokinetic properties. We have previously reported the optimization of the antiepileptic drug perampanel, initially identified in a virtual screen, from a weak inhibitor of Mpro (half-maximal inhibitory concentration [IC50] 100–250 μM), to several lead compounds with activities in the low-nanomolar range by means of an iterative approach complementing free-energy perturbation calculations and compound synthesis with structural characterization (Ghahremanpour et al., 2020; Zhang et al., 2021). Moreover, this lead optimization approach yielded compound 26, which showed promising antiviral activity (half-maximal effective concentration [EC50] 2.0 ± 0.7 μM) and cytotoxicity (half-maximal cytotoxic concentration [CC50] >100 μM) (Zhang et al., 2021). This effort provides the most extensive description to date of several noncovalent inhibitors of Mpro derived from an FDA-approved chemical scaffold with in vitro activities that improve upon activities of recently described covalent inhibitors (Dai et al., 2020; Zhang et al., 2020, 2021). Here, we present nine X-ray crystal structures of Mpro bound to perampanel analogs, providing insight into the structure-activity relationship for this pharmacophore and a framework for understanding how rational drug design efforts may be pursued in the context of conformational flexibility of key residues lining the active site of Mpro. In addition, this structural information offers guidance in the design of future analogs against potential drug-resistant variants.
Results
Structurally guided optimization of perampanel as an active-site inhibitor of Mpro
The high-resolution crystal structure of the free SARS-CoV-2 Mpro shows the overall structure to be strikingly similar to that of SARS-CoV-1 Mpro (Lee et al., 2020; Zhang et al., 2020). It is a dimer of protomers A and B that are related by crystallographic symmetry. Each protomer is composed of three domains: domains I and II, which are antiparallel β barrels that form the active site containing the Cys145-His41 catalytic dyad at their interface, and a helical domain III involved in dimerization (Anand et al., 2002; Dai et al., 2020; Zhang et al., 2020). The active site at the interface of domains I and II accommodates its peptide substrate in clefts S1′–S3′ and S1–S5, with cleavage occurring between P1 and P1′ in the substrate (corresponding to S1 and S1′ in the active site) (Cannalire et al., 2020) (Figure 1 ). The active site is malleable, with regions in the S2, S4, and S5 sites demonstrating considerable conformational flexibility upon binding diverse chemical groups (Kneller et al., 2020). The S1 site is formed by Phe140, Ser144, Asn142, His163, Glu166, and His172 and is further shaped by Ser1 of protomer B, which reaches into the active site of protomer A and interacts with Glu166 (Lee et al., 2020). The preference of S1 is for hydrophilic side-chain groups, with His163 poised to act as a hydrogen-bond donor (Cannalire et al., 2020; Kneller et al., 2020). S2 is a cleft formed by Met49 and the backbones of Asp187-Gln189 with a preference for mostly hydrophobic groups (Cannalire et al., 2020; Kneller et al., 2020). S3 and S4 extend toward the solvent and present residues important for substrate and ligand binding that can undergo significant conformational shifts upon interactions with ligands (Cannalire et al., 2020; Kneller et al., 2020). S4 is lined by the mobile, hydrophobic tip of the S4 β hairpin that is capped by a flexible S4–S5 loop of Arg188-Ala184 that can be displaced toward the solvent by bulky substituents (Kneller et al., 2020).
Figure 1.

Surface and cartoon representations of SARS-CoV-2 Mpro
Dimer (left) with selected residues of the active site shown as sticks (right). Domains of protomers A and B are colored in shades of blue and green, respectively. Ser1 of protomer B is shown as sticks in green (right). Met165, a key residue of the S4 β hairpin (residues 165–170), as well as Gln189 of the S5 loop (residues 189–194) are labeled. PDB: 7L14.
In an effort to design a chemical scaffold that inhibited Mpro noncovalently, we conducted a consensus virtual screen of 2,000 FDA-approved drugs from which the antiepileptic drug perampanel was identified as having a promising docked structure (Ghahremanpour et al., 2020). It possesses a cloverleaf motif projecting pyridinyl, phenyl, and cyanophenyl groups into S3/S2, S1, and S1′, respectively, and demonstrates modest in vitro inhibitory activity against Mpro (Ghahremanpour et al., 2020). A series of initial modifications aimed at optimizing the perampanel template for tighter binding resulted in a greater than 10-fold improvement in inhibitory activity in vitro to yield compound 2 (Figure 2 and Table 1 ) (Zhang et al., 2021). The rationale for these modifications is described in depth elsewhere (Zhang et al., 2021). In brief, the carbonyl of the pyridinone was moved from C2 to C6 to form a main-chain hydrogen bond with Glu166, the phenyl ring was replaced with a pyridinyl to form a hydrogen bond with Nε of His163, and the pyridinyl was replaced with a chlorophenyl to remove a noninteracting, solvent-exposed pyridinyl nitrogen and insert a chlorophenyl edge into S2 (Zhang et al., 2021). The cyanophenyl was retained for its attractive proximity to Cys145, but an alternative series replacing the cyanophenyl with a uracil was also pursued to maximize polar contacts in S1′ (Zhang et al., 2021). Here, we report the structural insights obtained from the crystal structures of the perampanel analogs bound to Mpro that further optimize interactions within the S1′, S2, and S3/S4 pockets of the enzyme.
Figure 2.

Structures of perampanel analogs
Perampanel and compounds 2 and 3 are shown in gray. Cyanophenyl derivatives of 2 building into S3–S4 are shown in lime. Uracil derivatives of 3 building into S3–S4 are shown in green. The benzyloxy uracil, 14, and its derivatives are shown in blue. The cyclopropyl uracil analog, 31, and piperazin-2-one uracil analog, 50, are shown in orange and red, respectively.
Table 1.
In vitro activities of perampanel analogs
| Compound | IC50 (μM) |
|---|---|
| Perampanel | 100–250 |
| 2 | 9.99 ± 2.50 |
| 3 | 6.38 ± 1.21 |
| 4 | 4.02 ± 1.36 |
| 5 | 0.14 ± 0.02 |
| 6 | 0.47 ± 0.02 |
| 11 | 0.120 ± 0.016 |
| 14 | 0.128 ± 0.015 |
| 15 | 0.110 ± 0.013 |
| 16 | 0.100 ± 0.035 |
| 19 | 0.037 ± 0.007 |
| 21 | 0.018 ± 0.002 |
| 23 | 0.020 ± 0.005 |
| 25 | 0.025 ± 0.003 |
| 26 | 0.170 ± 0.022 |
| 29 | 0.25–0.50 |
| 31 | 0.037 ± 0.004 |
| 50 | 0.25–0.50 |
Activities of perampanel and analogs 2–50 with previously reported IC50s of perampanel and compounds 2–26 (Zhang et al., 2021) with standard deviation calculated from triplicate measurements. While IC50 values could not be calculated for compounds 29 and 50 due to incomplete inhibition at maximum concentrations tested, in vitro activity data demonstrate that both compounds reduce Mpro activity to 50% between 0.25 μM and 0. 5 μM (Figure S2).
Optimizing binding in S1′, S1, and S2 facilitates noncovalent occlusion of the catalytic center of SARS-CoV-2 Mpro
In an attempt to optimize binding to the S1′ cleft of Mpro, an alternative series of inhibitors in which the cyanophenyl ring of perampanel was replaced with a uracil group was pursued (Figures 3A and 3B). Both the uracil oxygen of compound 11 and nitrile nitrogen of compound 5 are shown to hydrogen bond with the NH of Cys145, with distances of 3.14 Å and 3.55 Å for the nitrile and uracil, respectively (Figure 3B) (Zhang et al., 2021). The nitrile also engages in a 3.1-Å interaction with the NH of Gly143 but with a C≡N···H angle close to 90°, whereas the uracil is additionally stabilized by two main-chain hydrogen bonds with Thr26, which further straddles the oxyanion hole residues. The additional two interactions of the uracil reaching into S2′ may contribute to the increased inhibition observed for the uracil analog of compound 11 compared with 5 (Table 1). The positions of the uracil oxygen and nitrile nitrogen that hydrogen bond with Cys145 in the uracil and cyanophenyl analogs superpose to optimize hydrogen-bond geometry, resulting in a 0.8-Å shift in the uracil ring toward the oxyanion hole residues and minor displacements in the pyridinone, pyridine, and fluorophenyl moieties. The “oxyanion hole” formed by Cys145, Gly143, and Ser144 is occluded by both substituents bound to S1′.
Figure 3.

Crystal structures of complexes with perampanel analogs highlighting key active-site residues
(A) The crystal structure of 11 (teal) is shown in the Mpro active site (orange). Subsites are labeled in blue.
(B) The uracil and cyanophenyl moieties in S1′ are shown as sticks with hydrogen bonds to Cys145 and Thr26 shown as dashes.
(C) The hydrogen bond between His163 and the pyridine of 11 in S1 is shown as dashes.
(D) 23 (orange) and 21 (green) are shown as sticks with Arg188 and Met49.
All analogs described in this study retain the pyridine ring in the P1 position; alternative heteroaromatics were considered but none has emerged to yield greater binding (Figure 3C). The crystal structures of these analogs bound to Mpro uniformly show the pyridine binding to S1 of Mpro and forming a 2.9-Å hydrogen bond with His163 (Figure 3C). His163, conserved among CoV Mpro, hydrogen bonds with a glutamine in the native substrate and γ-lactam substituents in several reversible covalent inhibitors (Dai et al., 2020; Yang et al., 2003; Zhang et al., 2020). The placement of a pyridine in S1 has also been described in the crystal structure of Mpro bound to calpain inhibitor XII and the docked structure of an acrylamide-bearing inhibitor predicted by a generative model trained on a library of compounds screened against Mpro (Sacco et al., 2020; Santana et al., 2021). These interactions suggest the importance of His163 in determining substrate specificity in S1 and the utility in designing Mpro inhibitors that target this residue (Yang et al., 2003).
Together, the uracil/cyanophenyl and pyridine substituents in S1′ and S1, respectively, occlude the catalytic center of the active site by blocking substrate access to the oxyanion hole residues and His41. Further modifications of our inhibitor series aimed to extend substituents into the S2 and S3/S4 pockets to enhance binding affinity. In compounds 2–25 and 29–31, the chlorophenyl moiety is positioned with a chlorine atom pointing into S2 and a second position on the phenyl ring that points toward the S3 site. In the X-ray crystal structures of 9, 5, 11, 21, and 25, the chlorophenyl packs in S2 with the side chain of Met49 directed toward the chloro substituent. In 23, which harbors a fluoro group in the para position in addition to the chloro in the meta position, the ring is rotated relative to the central pyridinone ring to optimize close packing of the fluoro group against the backbone of Arg188, resulting in a minor displacement of the meta-Cl away from Met49 (Figure 3D). In the structure of 23, the flexible Met49 points away from the meta-Cl, diminishing the close packing observed in 21, the analog of 23 lacking the para-F substituent in S2. This loss may explain why there is little gain in activity from adding a fluoro group in the para position despite its improved packing in S2 and highlights the possibility of isolated conformational changes in the active site that can instruct rational design efforts (Kneller et al., 2020).
Structure-guided extension into the hydrophobic S3/S4 cavities drives inhibitory activity
A number of synthesized analogs aimed to optimize packing interactions with the hydrophobic residues lining the shallow, hydrophobic S4 cavity of Mpro through substitutions off of the chlorophenyl ring that point toward S3 and S4 sites (Figures 2 and 4A–4D). It has been noted that the S4 β hairpin can undergo conformational changes to accommodate a wide variety of ligands, underlying the need for an iterative approach guided by structural validation to exploit optimal packing in S4. Exploration of S4 began with extension of a propoxy group toward Met165 and Leu167 lining the S4 β hairpin. The structure of the cyanophenyl analog, 5, is described elsewhere (Zhang et al., 2021). The 1.78-Å crystal structure of the uracil analog, 11, superposes with 5 with minor displacements described above induced by a shift in the uracil toward the oxyanion hole residues in S1′ (Figures 4A and 5A ). In brief, the propoxy in 5 and 11 occupies a gauche conformation, packing its terminal methyl group into a cavity lined by the β-hairpin residues Met165 and Leu167 and S5 loop Gln192 (Zhang et al., 2021). The crystal structure for the methoxyethoxy analog, 6, shows the chain similarly adopting a gauche conformation with an oxygen placed between the hydrophobic side chains of Met165 and Leu167 (Figures 4B and 5C). The Leu167 side chain forces the terminal methyl group to point toward Gln192, introducing an unfavorable contact between the methoxyethoxy terminal methyl group and the Gln192 nitrogen and pushing Cβ of Gln192 away from the active site. The introduction of an unfavorable mismatch between hydrophobic and hydrophilic groups in this region explains the loss of activity of 6 relative to its propoxy analogs.
Figure 4.

Structures of perampanel analogs binding to the active site of Mpro
Inhibitors are shown as sticks and Mpro is shown in lilac. Compound numbers are shown in black in each panel (bottom left) and subsite labels are shown in blue. 11 (A) is shown in is shown in teal. 6 (B) is shown in purple with Gln192 of the complex with 5 (7L11) in cyan. 29 (C) is shown in magenta with the S5 loop of 11 shown in cyan. 25 (D) is shown in dark purple. 14 (E, 7L12) is shown in orange with Pro168 of the complex with 11 shown in cyan. 16 (F) is shown in yellow with Met165 and Leu167 of 14 (7L12) and 11 shown in magenta and cyan, respectively. 19 (G) is shown in dark purple with Met165 of 14 (7L12) shown in cyan. 21 (H, 7L13) is shown in green. 26 (I, 7L14) is shown in pink with the S5 loop of 19 shown in cyan. 50 (J) is shown in brown.
Figure 5.

Composite omit σA-weighted 2mFo-Fc electron density maps of the Mpro-inhibitor complexes
Maps for compounds (A) 11, (B) 23, (C) 6, (D) 29, (E) 25, (F) 15, (G) 16, (H) 19, and (I) 50 are shown. Protein residues are shown in green and inhibitors are shown in blue. Compound numbers are labeled in each panel. Inhibitor coordinates were excluded to calculate omit maps. Omit maps are contoured to 1.0σ.
The 1.79-Å crystal structure of 29 bound to Mpro shows the tert-butanol group of 29 to pack into the hydrophobic pocket formed by Met165 and Leu167 (Figures 4C and 5D). The tertiary hydroxyl group forms a 3.3-Å hydrogen bond with the backbone carbonyl of Thr190. The flexible S5 loop proximal to Thr190 slightly shifts away from the active site to accommodate a rotation of the Thr190 carbonyl to facilitate this interaction. To position the hydroxyl group for hydrogen bonding with Thr190, the proximity of a terminal methyl group induces an unfavorable contact with the Thr190 backbone, which may contribute to the greater than 2-fold decrease in activity of compound 29 compared with 11 (Table 1 and Figure S2). Structural insights from packing of 5, 11, 6, and 29 in S4 revealed a preference for a hydrophobic chain that positions a hydrophobic group between Met165 and Leu167.
The trifluoromethyl derivative of the propoxy analog 11, compound 25, shows improved inhibitory activity, potentially owing to increased packing interactions in S4 (Figures 4A and 4D; Table 1) The 1.95-Å resolution crystal structure of the complex with 25 shows the trifluoromethyl side chain adopting a gauche conformation, as in the complex with 11, but with the trifluoromethyl group directed toward the S5 loop (Figures 4D and 5E). The S5-directed conformation of the trifluoromethyl group positions fluorine atoms against Cγ of Gln192 and Gln189, with the highly mobile side chain of Gln189 bent toward the S5 loop forming a pocket. Moreover, 25 extends a fluorine atom toward Met165, which packs closely against the methionine terminal methyl group. These structures confirm that the active site of Mpro is significantly malleable upon binding of diverse ligands, and this structural information can further be exploited by building into flexible cavities, particularly S4, to fine-tune packing and maximize contacts to increase inhibitory activity (Kneller et al., 2020).
Negotiating active-site flexibility and ligand packing in S4 fine-tunes active-site interactions
Another series of inhibitors in our study aimed to retain packing in the Met165/Leu167 pocket while extending toward Pro168 through the introduction of aromatic ring substituents on the chlorophenyl ring (Figures 2 and 4E–4I). The crystal structure of the first of this series, the benzyloxy analog, 14, in complex with Mpro has been described previously (Zhang et al., 2021). In the crystal structure of the complex with 14, the benzyl group extends toward Pro168, and while it does not adopt a face-to-face orientation, it engages in a CH-π interaction with Hδ of Pro168 (Figure 4E). Furthermore, Pro168 undergoes a 1.4-Å displacement toward the benzyloxy group relative to its position in the structure in complex with 11 while the S5 loop is pushed away from the active site. Notably, the Cδ/γ Pro168 edge packs against the benzene ring in the structure of 14 in contrast to its pushed-out position in the crystal structure of Mpro in complex with the hepatitis C protease inhibitor boceprevir, in which it is displaced 3.2 Å relative to its position in the free enzyme by the bulky substituent in S5 (Kneller et al., 2020). Binding of the benzyloxy analog in the crystal structure also displaces a well-ordered water that forms a hydrogen bond with the backbone carbonyl of Glu166 that is present in the structures of 9, 5, 11, 29, 50, and 26. Despite the expansion toward the S4 hairpin, 14 shows little improvement in inhibitory activity compared with 11. The phenethyloxy analog, 15, similarly shows little improvement in activity from 14, despite having the potential to extend further toward the S4 hairpin. The 1.75-Å crystal structure of 15 shows the phenethyloxy group in a different conformation than that of the benzyloxy group of 14 (Figures 5F and S1). However, the compact conformation of 15 observed here may result from the need to avoid steric clashes between the chain of 15 and a crystallographic symmetry-related molecule of Mpro that packs close to the S3/S4 sites in the C2 crystal form (Figure S1).
The 1.96-Å crystal structure of the complex with 16, the ortho-methyl analog of 14, reveals the ortho-methyl group to insert into the Met165/Leu167 pocket in similar mode as 11 (Figures 4F and 5G). As observed in the complex structure of 11, the side chains of both Met165 and Leu167 rearrange to allow the terminal methyl of the chain of 16 to pack into this hydrophobic pocket. Despite the extension into this pocket, significant improvement in activity of 16 relative to 14 was not observed. 19, in contrast, which replaces the ortho-methyl group in 16 with a meta-fluoro group, shows a nearly 3-fold increase in activity (Table 1). The 2.44-Å resolution crystal structure of 19 bound to Mpro shows the fluoro group to pack between the S5 loop and Cγ of Pro168 (Figures 4G and 5H). Moreover, Met165, which can occupy multiple conformations both in free and liganded states of the enzyme, is pointed toward the ligand in 19, introducing packing that is not present in 16 due to the overlapping position of the ortho-methyl group, which pushes the terminal methyl of Met165 out of the active site to avoid steric clash. Compound 21, the mono-chloro analog of 16, shows an approximately 2-fold increase in activity compared with 19. The crystal structure of 21 bound to Mpro is described elsewhere (Zhang et al., 2021). 21 interacts with the S5 loop and Met165, which is pointed toward the active site as in the 19-bound structure but slightly away from the P4 substituent (Figure 4H). Additionally, the terminal methyl of Met165 packs against the ortho-chloro group.
It is interesting to compare the structures and activities of 19 and 21 with that of 31, which extends a cyclopropyl group into S4 and inhibits Mpro activity with an IC50 of 0.037 ± 0.004 μM, comparable with that of 19 (Zhang et al., 2021). The high-resolution structure of the less-active cyanophenyl analog of the cyclopropyl derivative, compound 26, is described elsewhere (Zhang et al., 2021). In brief, the cyclopropyl edge packs against Leu167, and while it does not engage in the additional close packing interaction with Met165 as do 19 and 21, it induces a 1.6- to 2.0-Å shift in the S5 loop toward the active site relative to the structures of 19 and 21 (Figure 4I).
Inhibitory activity is driven by hydrophobic packing in S4
The 2.19-Å resolution crystal structure of Mpro bound to 50 provides insights into the effects of inserting the more hydrophilic piperazin-2-one substituent, for which the electron density is clearly defined in the active site, into the S4 pocket (Figures 4J and 5I). Hydrophobic interactions with the S5 loop and Met165 are largely lost in the complex with 50. Instead, the piperazin-2-one ring is shown to make two polar contacts with less than optimal geometry with the backbone NH and carbonyl of Gln192 of 2.8 Å and 2.9 Å, respectively, and a hydrogen bond to a well-ordered water that bridges the piperazin-2-one and the Glu166 backbone carbonyl (Figure 4J). It is unsurprising that the flexible S4 accommodates this substituent owing to its observed induced flexibility (Kneller et al., 2020). It is notable, however, that while the S4 cavity is highly adaptable, optimized packing interactions in this pocket can significantly impact inhibitory activity. This is evident from the structure-activity relationship of 50, which inhibits Mpro activity with an IC50 of 0.25–0.50 μM in contrast to 19 and 21, which inhibit with IC50s in the low-nanomolar range, suggesting that hydrophobic packing in S4 drives formation of a stable complex and thus inhibitory activity (Table 1 and Figure S2).
Discussion
There remains a clinical need for DAAs against SARS-CoV-2 to combat mortality and morbidity associated with COVID-19. Therapeutic strategies currently target the surface-exposed spike protein and the RNA-dependent RNA polymerase, but no therapy targeting the main protease has received FDA approval for use against COVID-19 despite its attractiveness as a drug target and a tremendous repertoire of investigations into the design and repurposing of potential inhibitors. Two key obstacles in the search for clinically relevant Mpro inhibitors are the generally problematic pharmacokinetic properties of covalent and peptidomimetic protease inhibitors and the challenge of designing inhibitors with IC50s in the submicromolar range. Low-nanomolar IC50s have thus far been described for relatively few compounds including peptidomimetic aldehyde compounds 11a and 11b, GC-376, and boceprevir and telaprevir derivatives, which form covalent adducts with the active-site Cys145 (Dai et al., 2020; Ma et al., 2020; Qiao et al., 2021). One barrier to efficiently optimizing design of submicromolar inhibitors is likely the demonstrated conformational flexibility of active-site residues involved in substrate and ligand binding (Kneller et al., 2020). It has previously been suggested that repurposing protease inhibitors may be a viable strategy and that computationally guided design of active-site inhibitors may be challenging due to induced flexibility of the active site (Bzowka et al., 2020; Kneller et al., 2020). Our structural investigation into this pharmacophore elaborates on this previous work, demonstrating induced flexibility of the active site while offering an alternative framework for efficiently obtaining lead compounds guided by free-energy perturbation calculations (Zhang et al., 2021). The current work examining the Mpro-inhibitor complexes reveals the structural validation that compound binding capitalizes on optimal packing interactions with flexible residues (Zhang et al., 2021). We provide proof of concept for this framework in the context of active site flexibility of Mpro, describing optimized hits derived from the weak Mpro inhibitor, perampanel, with a roughly 5,000-fold increase in activity in kinetic assays and activity against viral replication in cell culture in the low-micromolar range (Zhang et al., 2021).
Owing to the cloverleaf motif preserved from perampanel, the perampanel analogs span the S1′ to S5 subsites of the active site and interact with key active-site residues, including Cys145, without forming a covalent adduct (Zhang et al., 2021). The “leaflets” of the analogs interact with Cys145 and Thr26 in S1′ and His163 in S1 while forming variable packing interactions along the S4 β hairpin and S5 loop, which are highly dependent on size and hydrophobicity of the chains extending into the S4 pocket. Optimal activity was achieved for hydrophobic groups that occupy S4 without inducing conformational changes in key residues including Met165, Pro168, and Gln192 that decrease opportunities for packing, requiring a detailed structural understanding of active-site residue movements upon binding of different ligands. Notably, we demonstrate that even compounds that extend chains with hydrophilic groups into S4 that present opportunities for hydrogen bonding, including 6, 29, and 50, form stable complexes amenable to structural determination but show in vitro activity in the high-nanomolar range. Moreover, minor changes to the packing of hydrophobic groups in S4, as discussed for six-membered ring series 14–21, can drive significant changes in side-chain interactions and, thus, activity. This demonstrates that while the active site is highly adaptable to binding a range of ligands, a simple repurposing approach is unlikely to yield inhibitors with low-nanomolar activities that will be relevant to clinical translation.
Another challenge associated with inhibition of viral targets is the accumulation of resistance mutations that could render therapies less effective. Analysis of mutational “coldspots” in SARS-CoV-2 Mpro reveal that the residues that constitute the catalytic dyad, Cys145 and His41, as well as Gln192 and Leu167, undergo mutations at lower rates or are less favorable to mutate relative to other active-site residues (Bzowka et al., 2020; Krishnamoorthy and Fakhro, 2021). While all analogs described in this study form a hydrogen bond with Cys145, the propoxy and trifluoromethyl analogs 5, 11, and 25 additionally engage in hydrophobic packing interactions with Gln192 and 5, 11, 16, and 26 form interactions with Leu167, indicating that several perampanel analogs form interactions that play key roles in driving activity with residues that undergo mutation at low rates. Furthermore, a study of the effects of resistance mutations in Mpro of a model coronavirus, the murine hepatitis coronavirus, reveal that while there is a relatively low threshold for accumulation of resistance mutations in Mpro, accumulation of resistance mutations is associated with a striking attenuation of viral replication (Deng et al., 2014).
There is a particular interest in the development of novel protease inhibitors that would be efficacious against strains that have developed or will develop resistance to different classes of protease inhibitors. Most Mpro inhibitors currently under investigation preclinically as well as the inhibitors currently in clinical trials target the active site, but interactions with distinct residues can be exploited by different classes of protease inhibitors to expand the repertoire of drugs that can be used in combination or in succession to proactively address resistance mechanisms. PF-00835231 is an active-site Mpro inhibitor currently in clinical trials that binds to Cys145 via a covalent warhead and is additionally stabilized by hydrogen bonds to His163, His164, and the Glu166 backbone, hydrophobic interactions in S2, and van der Waals interactions spanning the surface of a closed S3 (Hoffman et al., 2020). While some contacts are shared somewhat universally by Mpro inhibitors, such as those with Cys145 and His163, the hydrogen bonds with His164 and Glu166 as well as hydrophobic interactions of PF-00835231 reaching into S2 and electrostatic interactions on the surface of S3 represent important binding features that do not overlap with those of our perampanel-derived pharmacophore. Notably, this clinical candidate does not share any contacts in S4/S5 that drive inhibition of several of our lead compounds, suggesting a potential alternative route for Mpro inhibition in the context of PF-00835231-induced resistance. The recently announced oral protease inhibitor candidate, PF-07321332, similarly forms a covalent bond with Cys145 via a nitrile warhead and inserts a γ-lactam into S1, a trifluoroacetyl cap into S3/S4, and a hydrophobic substituent in S2 (Owen, 2021). It will be interesting to extensively compare the binding features of this inhibitor candidate with our perampanel-derived pharmacophore following the release of its crystal structure.
In summary, we present structural insights into the design and optimization of a perampanel-derived pharmacophore against Mpro that provides a framework for optimizing inhibitors in the context of a highly flexible active site by fine-tuning hydrophobic packing interactions in the S3–S5 subsites. In addition to pursuing lead compounds from this study as preclinical candidates, we present insights gained from the design and validation of this pharmacophore to guide future studies targeting the SARS-CoV-2 main protease and provide a structural framework for designing compounds in the context of potential resistance mutations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli | New England Biolabs, Inc. | Cat#C2530H |
| Chemicals, peptides, and recombinant proteins | ||
| 3-Bromo-5-chlorophenol | AA Blocks | Cat#AA0033JM |
| 4-Bromo-2-methylbutan-2-ol | Combi-Blocks | Cat#QH-9520 |
| 1,2-Dibromoethane | Sigma-Aldrich | Cat#240656 |
| Tert-butyl 2-oxopiperazine-1-carboxylate | Combi-Blocks | Cat#QB-3609 |
| Benzyl alcohol | ACROS | Cat#AC39688 |
| Sodium hydride | Sigma-Aldrich | Cat#452912 |
| 5-Bromo-2-fluoropyridine | Combi-Blocks | Cat#PY-7039 |
| (2,4-Dimethoxypyrimidin-5-yl)boronic acid | Combi-Blocks | Cat#BB-8331 |
| Bis(triphenylphosphine)palladium(II) chloride | Alfa Aesar | Cat#41245 |
| 3-Pyridylboronic acid | TCI | Cat#P1673 |
| N,N,N',N'-Tetramethylethylenediamine | AmericanBio | Cat#AB2020 |
| N-bromosuccinimide | Sigma-Aldrich | Cat#B81255-100G |
| Bis(pinacolato)diboron | Combi-Blocks | Cat#BB-2214 |
| Lithium chloride | Honeywell Fluka | Cat#73036 |
| p-Toluenesulfonic acid monohydrate | ACROS | Cat#139021000 |
| Recombinant SARS-CoV-2 Mpro | Zhang, et al., 2021 | N/A |
| Dabcyl-KTSAVLQ↓SGFRKM-E(Edans)-NH2 | GL Biochem | N/A |
| PEGRx HT Crystallization Screen | Hampton Research | Cat#HR2-086 |
| Deposited data | ||
| SARS-CoV-2 3CL Mpro-4 | Zhang, et al., 2021 | PDB ID: 7L10 |
| SARS-CoV-2 3CL Mpro-5 | Zhang, et al., 2021 | PDB ID: 7L11 |
| SARS-CoV-2 3CL Mpro-6 | This study | PDB ID: 7M8X |
| SARS-CoV-2 3CL Mpro-14 | Zhang et al., (2021) | PDB ID: 7L12 |
| SARS-CoV-2 3CL Mpro-11 | This study | PDB ID: 7M8M |
| SARS-CoV-2 3CL Mpro-29 | This study | PDB ID: 7M8Z |
| SARS-CoV-2 3CL Mpro-15 | This study | PDB ID: 7M8Y |
| SARS-CoV-2 3CL Mpro-16 | This study | PDB ID: 7M8N |
| SARS-CoV-2 3CL Mpro-19 | This study | PDB ID: 7M8O |
| SARS-CoV-2 3CL Mpro-21 | Zhang, et al., 2021 | PDB ID: 7L13 |
| SARS-CoV-2 3CL Mpro-50 | This study | PDB ID: 7M90 |
| SARS-CoV-2 3CL Mpro-26 | Zhang, et al., 2021 | PDB ID: 7L14 |
| SARS-CoV-2 3CL Mpro-25 | This study | PDB ID: 7M91 |
| SARS-CoV-2 3CL Mpro-23 | This study | PDB ID: 7M8P |
| Recombinant DNA | ||
| Plasmid pGEX-6p-1 containing SARS-CoV-2 Mpro | Zhang et al. (2020) | N/A |
| Software and algorithms | ||
| XDS | Kabsch (2010) | N/A |
| PHASER | Mccoy et al. (2007) | N/A |
| COOT | Emsley et al. (2010) | N/A |
| PHENIX | Adams et al. (2010) | N/A |
| Mnova NMR | Mestrelab | https://mestrelab.com/download/mno va/nmr |
| ChemDraw Professional 18.2 | PerkinElmer | https://www.perkinelmer.com/category/chemdraw |
| Other | ||
| Agilent DD2 400 MHz NMR spectrometer | Agilent Technologies | https://www.agilent.com/ |
| Agilent DD2 600 MHz NMR spectrometer | Agilent Technologies |
https://www.agilent.com/ |
| Agilent 1260 Infinity II HPLC | Agilent Technologies | https://www.agilent.com/en/products/l iquid-chromatography |
| Agilent prep-C18 scalar reversed-column (4.6 mm × 100 mm, 5 μm). | Agilent Technologies | https://www.agilent.com/en/product/s mall-molecule- columns/reversed-phase-hplc- columns |
| Advion Express mass spectrometer | Advion | https://www.advion.com/products/expression-cms/ |
Resource availability
Lead contact
Further information and requests for either resources or reagents should be directed to and will be fulfilled by the lead contact Karen Anderson (karen.anderson@yale.edu).
Materials availability
Requests for materials generated in this study should be directed to and will be fulfilled by the lead contact Karen Anderson (karen.anderson@yale.edu).
Data and code availability
All data generated in these studies are available upon request from lead contact Karen Anderson (karen.anderson@yale.edu). Structures from this study are deposited to the PDB with IDs 7M8X, 7M8M, 7M8Z, 7M8Y, 7M8N, 7M8O, 7M90, 7M91, and 7M8P and are publicly available (www.rcsb.org).
Experimental model and subject details
For expression of SARS-CoV-2 Mpro, E. coli bacterial strain BL21 (DE3) was used and grown at 37C in LB media. Additional details can be found in the Method Details.
Method details
Purification and crystallization of SARS-CoV-2 Mpro
Recombinant SARS-CoV-2 Mpro was expressed, purified and crystallized as previously described (Ghahremanpour et al., 2020; Zhang et al., 2020, 2021). A PGEX-6p-1 vector containing the gene for SARS-CoV-2 Mpro with a His6 tag and PreScission cleavage site was kindly provided by the Hilgenfeld lab (Zhang et al., 2020). This plasmid was transformed into BL21 (DE3) cells and a transformed clone was used to inoculate 50mL of LB media with ampicillin (100μg/mL) grown at 37C for 6 hours with shaking. The starter culture was used to inoculate 2L LB media with ampicillin (100μg/mL) which was grown at 37C until the OD600 reached 0.8. Isopropyl-D-thiogalactoside (IPTG, 0.5mM) was used to induce protein expression overnight at 16C with shaking. Cells were harvested at 10,000xg for 15 minutes at 4C and stored at -80C. Cells were resuspended in 30mL lysis buffer (Buffer A; 20mM Tris, 150mM NaCl, pH 7.8) and lysed with a cell disruptor. The lysate was centrifuged (20,000xg, 1h, 4C) and the supernatant was loaded onto a 5mL HisTrapFF column (GE Healthcare) and His-tagged protein was eluted on an Akta Pure (Cytiva) with elution buffer (Buffer B; 20mM Tris 150mM NaCl 500mM imidazole pH 7.8) in a linear gradient from 0% to 100% elution buffer over 100mL following washing with 30 column volumes of lysis buffer. The His6 tag was cleaved at 4C with overnight incubation with PreScission protease during dialysis into buffer C (20mM Tris, 150mM NaCl, 1mM DTT, pH 7.8). Dialyzed protein was loaded onto a 5mL GSTrap FF (GE Healthcare) and 5mL HisTrapFF connected in tandem and the flow-through containing cleaved protein was collected and exchanged into buffer D (20mM Tris, 1mM DTT, pH 8.0) using a 30kDa cutoff column (Amicon) at 3000xg at 4C. Mpro was further purified by loading onto a 5mL HiTrap Q FF column (GE Healthcare) and eluting along a linear gradient from 0% to 50% buffer E (20mM Tris, 1M NaCl, 1mM DTT, pH 8.0) over 100mL. Fractions containing Mpro were pooled, concentrated and exchanged into 20mM Tris, 150mM NaCl, 1mM EDTA, 1mM DTT, pH 7.8 and flash frozen. Samples were thawed, incubated with 2mM inhibitor at 37°C for 30 minutes and subsequently centrifuged at 10,000xg to remove precipitant. Crystallization screens were set up using commercially available screening kits with 20mg/mL Mpro (PEGRx2, Hampton Research). Crystallization was set up at a 1:1 protein:reservoir solution ratio in 2μL drops to equilibrate with 80μL reservoir solution at 18°C using sitting-drop vapor-diffusion. Crystals grew overnight in several conditions and were flash frozen in liquid nitrogen after brief exposure to a cryobuffer containing reservoir solution plus 15% glycerol. Co-crystals with compound 6 were grown in 0.1M sodium citrate tribasic dihydrate pH 5.0, 10% polyethylene glycol 6,000. Co-crystals with compound 11 were grown in 0.1M sodium malonate pH 8.0, 0.1M Tris pH 8.0, 30% w/v polyethylene glycol 1,000. Co-crystals with 29 were grown in 0.1M sodium citrate tribasic dihydrate pH 5.0, 10% w/v polyethylene glycol 6,000. Co-crystals with 15 grew in 0.1M imidazole pH 7.0, 20% v/v Jeffamine ED-2001 pH 7.0. Co-crystals with 16 were grown in 0.1M BIS-TRIS pH 6.5, 25% v/v polyethylene glycol 300. Co-crystals with 19 were grown in 0.1M MES monohydrate pH 6.0, 22% v/v polyethylene glycol 400. Co-crystals with 50 were grown in 0.1M BIS-TRIS pH 6.5, 20% w/v polyethylene glycol 1,500. Co-crystals with 25 were grown in 0.1M sodium citrate tribasic dihydrate pH 5.0, 10% polyethylene glycol 6,000. Co-crystals with 23 were grown in 0.1M succinic acid pH 7.0, 0.1M BICINE pH 8.5, 30% v/v polyethylene glycol monomethyl ether 550.
Enzyme kinetics of Mpro with inhibitors
Purified protein was diluted in reaction buffer (20mM Tris 100mM NaCl 1mM DTT pH 7.3) to a final concentration of 100nM in an opaque 96-well plate (Costar) and incubated with or without compound at varying concentrations for 15 minutes with shaking. The reaction was initiated with the addition of 50μM FRET substrate (Dabcyl-KTSAVLQ↓SGFRKM-E(Edans-NH2); GL Biochem) solubilized in reaction buffer. Cleavage of the substrate generates a product with a free Edans group for which fluorescence was monitored at an excitation wavelength of 360nm and emission wavelength of 460nm. Baseline subtraction was performed for every concentration and all experiments were performed in triplicate.
Structural determination of Mpro bound to inhibitors
X-ray data was collected at the National Synchrotron Light Source II (NSLS-II) and Advanced Photon Source (APS) on beamlines AMX and 24-ID-E/24-ID-C, respectively. Datasets were indexed using XDS (Kabsch,2010). PHASER was used for molecular replacement (Mccoy et al., 2007). COOT and Phenix Refine were used for model building and refinement, respectively (Adams et al., 2010; Emsley et al., 2010). Data processing statistics are in Table S1. All software was compiled by SBGrid (Morin et al., 2013). For molecular replacement, 6Y2G was used as the search model for structures of Mpro in complex with 11, 16, 19 and 23. 6Y2F was used as the search model for the structures with 6, 29, 50, and 25. 6Y2E was used as the search model for 15.
Synthesis of compounds 29 and 50
All reagents and solvents were obtained from commercial suppliers and used without further purification unless otherwise indicated. Column chromatography was carried out on silica gel (300 – 400 mesh). All reactions were monitored by thin-layer chromatography (TLC), and silica gel plates with fluorescence F-254 were used and visualized with UV light. All of the final compounds were purified to > 95% purity, as determined by high-performance liquid chromatography (HPLC). HPLC analysis was performed on an Agilent 1260 Infinity II HPLC system with the use of an Agilent prep-C18 scalar reversed-column (4.6 mm × 100 mm, 5 μm).
The binary solvent system was 0.1% formic acid in water (A) and acetonitrile (B), and eluted in a gradient manner from 5% to 100% (A/B) in 15 minutes. The absorbance was detected at 254 nm, and the flow rate was 1.5 mL/min. 1H NMR and 13C NMR spectra were recorded on an Agilent DD2 400 MHz or an Agilent DD2 600 MHz NMR spectrometer. Coupling constants (J) are expressed in hertz (Hz). Spin multiplicities are described as s (singlet), br. s (broad singlet), t (triplet), q (quartet), and m (multiplet). Chemical shifts (δ) are listed in parts per million (ppm) relative to internal standard tetramethylsilane (TMS) or solvent. Regular mass spectral (MS) data were acquired on an Advion Express mass spectrometer (Ithaca, NY, USA). High resolution MS data were acquired on a Shimadzu 9030 Quadrupole Time-of-Flight high-resolution mass spectrometer (Columbia, MD, USA).
Synthesis of 4-(3-Bromo-5-chlorophenoxy)-2-methylbutan-2-ol (S1a) (Figure 6). 3-Bromo-5-chlorophenol (0.517 g2.5 mmol, 1.0 eq), 4-bromo-2-methylbutan-2-ol (0.434 g, 2.6 mmol, 1.04 eq) and K2CO3 (0.691 g, 5 mmol, 2.0 eq) were suspended in DMF (30 mL) and stirred at 80°C for 1 h. Then the mixture was concentrated in vacuo and the residue was extracted with water (40 mL) and dichloromethane (2× 40 mL). The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography (gradient = 0–5% EtOAc/Hex) to yield the title compound S1a as a clear oil (0.63 g, yield 86%).1H NMR (400 MHz, CDCl3) δ 7.13 – 7.09 (m, 1H), 6.96 (s, 1H), 6.87 – 6.83 (m, 1H), 4.14 (t, J = 6.4 Hz, 2H), 1.98 (t, J = 6.4 Hz, 2H), 1.64 (s, 1H), 1.31 (s, 6H). MS m/z (ESI): 293.0 [M+H]+, 295.0 [M+H]+.
Figure 6.

Synthesis of 4-(3-bromo-5-chlorophenoxy)-2-methylbutan-2-ol (S1a)
Synthesis of 1-Bromo-3-(2-bromoethoxy)-5-chlorobenzene. 3-Bromo-5-chlorophenol (2.07 g, 10 mmol, 1.0 eq), 1,2-dibromoethane (3.76 g, 10 mmol, 2.0 eq) and K2CO3 (4.15 g, 30 mmol, 3.0 eq) were suspended in DMF (50 mL) and stirred at 80°C for 3 h. Then the mixture was concentrated in vacuo and the residue was extracted with water (40 mL) and dichloromethane (2× 40 mL). The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography (gradient = 100% petroleum ether) to yield the title compound as a clear oil (2.26 g, yield 36%).1H NMR (400 MHz, CDCl3) δ 7.18 – 7.09 (m, 1H), 6.97 (s, 1H), 6.90 – 6.81 (m, 1H), 4.25 (t, J = 6.1 Hz, 2H), 3.62 (t, J = 6.1 Hz, 2H).
Synthesis of Tert-butyl 4-(2-(3-bromo-5-chlorophenoxy)ethyl)-2-oxopiperazine-1-carboxylate (S1b) (Figure 7). 1- Bromo-3-(2-bromoethoxy)-5-chlorobenzene (0.86 g, 2.75 mmol, 1.1 eq), tert-butyl 2-oxopiperazine-1-carboxylate (0.5 g, 2.5 mmol, 1.0 eq) and K2CO3 (0.691 g, 5 mmol, 2.0 eq) were suspended in DMF (30 mL) and stirred at 80°C for 5 h. Then the mixture was concentrated in vacuo and the residue was extracted with water (40 mL) and dichloromethane (3× 40 mL).
Figure 7.

Synthesis of tert-butyl 4-(2-(3-bromo-5-chlorophenoxy)ethyl)-2-oxopiperazine-1-carboxylate (S1b)
The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography (gradient = 0–10% EtOAc/Hex) to yield the title compound as a white solid (0.46 g, yield 38.6%).1H NMR (400 MHz, CDCl3) δ 7.13 (t, J = 1.7 Hz, 1H), 6.96 (t, J = 1.9 Hz, 1H), 6.84 (t, J = 1.9 Hz, 1H), 4.07 (d, J = 4.9 Hz, 2H), 3.71 (t, J = 5.9 Hz, 2H), 3.39 (s, 2H), 2.86 (s, 4H), 1.53 (s, 9H). MS m/z (ESI): 433.1 [M+H]+, 435.1 [M+H]+.
2-(Benzyloxy)-5-bromopyridine (S2) (Figure 8). Benzyl alcohol (6.75 g, 62.48 mmol, 1.1 eq) was dissolved in anhydrous THF (120 mL) and cooled to 0°C, 60% sodium hydride (2.73 g, 68.2 mmol, 1.2 eq) was added and stirred for 30 min at 0°C. Then 5-bromo-2-fluoropyridine (10 g, 56.8 mmol, 1.0 eq) was added and the mixture was heated at 70°C overnight. The reaction was quenched by the dropwise addition of water at 0°C, then brine and more ethyl acetate were added for extraction. The organic layer was dried (Na2SO4) and the solvent was removed under reduced pressure, the residue was recrystallized in hexane to afford the title compound as a white solid (12 g, 80.1% yield). 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 2.3 Hz, 1H), 7.65 (dd, J = 8.8, 2.5 Hz, 1H), 7.44 (d, J = 7.2 Hz, 2H), 7.39 – 7.30 (m, 3H), 6.72 (d, J = 8.8 Hz, 1H), 5.34 (s, 2H). MS m/z (ESI): 264.0 [M+H]+, 266.0 [M+H]+.
Figure 8.

Synthesis of 29 and 50
5-(6-(Benzyloxy)pyridin-3-yl)-2,4-dimethoxypyrimidine (S3) (Figure 8). 2-(Benzyloxy)-5- bromopyridine (S2, 7 g, 26.5 mmol, 1.0 eq), (2,4-dimethoxypyrimidin-5-yl)boronic acid (6.34 g, 34.45 mmol, 1.3 eq), cesium carbonate (17.3 g, 53 mmol, 2.0 eq) and bis(triphenylphosphine)palladium(II) chloride (0.93 g, 1.3 mmol, 0.05 eq) were suspended in DMF (150 mL). The mixture underwent three cycles of vacuum/filling with N2, then stirred at 80°C for 5h. The mixture was concentrated in vacuo and the residue was resuspended in water (80 mL) and extracted with dichloromethane (2× 80 mL). The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography with an ethyl acetate / hexane gradient (0 –10%) to afford the title compound as a white solid (6.2 g, yield 72.1%). 1H NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 8.24 (s, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 7.7 Hz, 2H), 7.39 (t, J = 7.3 Hz, 2H), 7.34 (d, J = 6.6 Hz, 1H), 6.88 (d, J = 8.6 Hz, 1H), 5.42 (s, 2H), 4.04 (s, 3H), 4.03 (s, 3H). MS m/z (ESI): 324.2 [M+H]+.
5-(2,4-Dimethoxypyrimidin-5-yl)pyridin-2(1H)-one (S4) (Figure 8). 5-(6-(Benzyloxy)pyridin-3-yl)-2,4- dimethoxypyrimidine (S3, 6.2 g) and palladium on activated carbon (500 mg) were suspended in methanol (150 mL) and water (10 mL), the mixture underwent 3 cycles of vacuum/filling with H2 and stirred at 40°C for 4 h. After the reaction was complete, dichloromethane was added to dissolve the solid product, then the mixture was filtered, the filtrate was concentrated in vacuo to give the title compound as a gray solid (4.4 g, yield 99%). 1H NMR (400 MHz, CDCl3) δ 13.21 (br.s, 1H), 8.20 (s, 1H), 7.67 (dd, J = 9.4, 2.6 Hz, 1H), 7.63 (d, J = 2.2 Hz, 1H), 6.67 (d, J = 9.4 Hz, 1H), 4.04 (s, 3H), 4.03 (s, 3H). MS m/z (ESI): 234.1 [M+H]+. 5-(2,4-Dimethoxypyrimidin-5-yl)-2H-[1,3'-bipyridin]-2-one (S5). 5-(2,4-Dimethoxypyrimidin-5-yl)pyridin-2(1H)-one (S4, 4.4 g, 18.9 mmol, 1.0 eq), 3-pyridylboronic
acid (4.65 g, 37.8 mmol, 2.0 eq), cupric acetate (3.43 g, 18.9 mmol, 1.0 eq) and N,N,N',N'- Tetramethylethylenediamine (4.4 g, 37.8 mmol, 2.0 eq) were suspended in anhydrous DMF (120 mL), the mixture was bubbled with dry air and stirred at room temperature for 4 days. After the reaction completed, the mixture was concentrated in vacuo and the residue was diluted with ammonium water (5%, 40 mL) and dichloromethane (3× 40 mL) for extraction. The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography with a methanol/ dichloromethane gradient (0–5%) to yield the desired product as a white solid (4.3 g, 74% yield). 1H NMR (400 MHz, CDCl3) δ 8.70 (s, 2H), 8.23 (s, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.62 (dd, J = 9.6, 2.2 Hz, 1H), 7.54 (d, J = 2.5 Hz, 1H), 7.48 (dd, J = 8.1, 4.8Hz, 1H), 6.75 (d, J = 9.6 Hz, 1H), 4.05 (s, 3H), 4.03 (s, 3H). MS m/z (ESI): 311.2 [M+H]+. 3-Bromo-5-(2,4-dimethoxypyrimidin-5-yl)-2H-[1,3'-bipyridin]-2-one (S6). 5-(2,4-Dimethoxypyrimidin-5-yl)-2H-[1,3'-bipyridin]-2-one (S5, 4.3 g 13.9 mmol, 1.0 eq) was dissolved in anhydrous DMF (80 mL), the mixture underwent 3 cycles of vacuum/filling with N2, then N-bromosuccinimide (9.9 g, 55.6 mmol, 4 eq) was added and stirred at room temperature for 4 h. Then the reaction was quenched with aqueous sodium thiosulfate solution (1 M, 50 mL) at 0°C and stirred at this temperature for 2 h. The aqueous layer was extracted with dichloromethane (3× 60 mL) and then the combined organic layer was concentrated in vacuo at low temperature (20°C). The residue was further dried using a vacuum pump, then the crude product was purified using silica gel chromatography with a methanol/ dichloromethane gradient (0 - 5%) to afford an orange solid (3.7 g, 69% yield). 1H NMR (400 MHz, CDCl3) δ 8.72 (s, 2H), 8.22 (s, 1H), 8.04 (d, J = 2.3 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.53 (dd, J = 8.0, 3.7 Hz, 2H), 4.07 (s, 3H), 4.04 (s, 3H). MS m/z (ESI): 389.1 [M+H]+, 391.1 [M+H]+.
3-(3-Chloro-5-(3-hydroxy-3-methylbutoxy)phenyl)-5-(2,4-dimethoxypyrimidin-5-yl)-2H-[1,3'-bipyridin]-2-one (S7a) (Figure 8). 4-(3-Bromo-5-chlorophenoxy)-2-methylbutan-2-ol (S1a, 129.2mg, 0.44 mmol, 1.1 eq), KOAC (86.4 mg, 0.88 mmol, 2.0 eq), bis(pinacolato)diboron (114.3 mg, 0.45 mmol, 1.12 eq), and (PPh3)2PdCl2 (28 mg, 0.04 mmol, 0.1 eq) were suspended in anhydrous DMF (10 mL). The mixture underwent three cycles of vacuum/filling with N2, then stirred at 80°C for 1 h. After the reaction was complete, 1 mL of methanol was added to scavenge excess of pinacolborane, and the mixture was cooled to rt. Then 3-bromo-5-(2,4-dimethoxypyrimidin-5- yl)-2H-[1,3'-bipyridin]-2-one (S6, 155.7 mg, 0.4 mmol, 1.0 eq) and K2CO3(110.6 mg, 0.8 mmol, 2.0 eq) were added to the mixture, which was underwent three cycles of vacuum/filling with N2. The mixture was heated to 120°C for 1h. After the reaction was complete, the DMF was removed, water was added, and the solution was extracted with DCM (3× 40 mL). The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography with a dichloromethane / ethyl acetate / methanol (90% / 5% / 5%) gradient to afford the target compound as a white solid (119 mg, 57% yield). 1H NMR (400 MHz, CDCl3) δ 8.75 (s, 1H), 8.72 (d, J = 4.1 Hz, 1H), 8.28 (s, 1H), 8.00 – 7.93 (m, 1H), 7.76 (d, J = 2.5 Hz, 1H), 7.58 (d, J = 2.5 Hz, 1H), 7.53 (dd, J = 8.2, 5.1 Hz, 1H), 7.30 – 7.27 (m, 2H), 6.93 (t, J = 2.1 Hz, 1H), 4.20 (t, J = 6.3 Hz, 2H), 4.07 (s, 3H), 4.05 (s, 3H), 1.98 (t, J = 6.3 Hz, 3H), 1.30 (s, 6H). MS m/z (ESI): 523.2 [M+H]+.
Tert-butyl 4-(2-(3-chloro-5-(5-(2,4-dimethoxypyrimidin-5-yl)-2-oxo-2H-[1,3'-bipyridin]-3- yl) phenoxy)ethyl)-2-oxopiperazine-1-carboxylate (S7b) (Figure 8). Tert-butyl 4-(2-(3-bromo-5- chlorophenoxy) ethyl)-2-oxopiperazine-1-carboxylate (S1b, 190.8 mg, 0.44 mmol, 1.1 eq), KOAC (86.4 mg, 0.88 mmol, 2.0 eq), bis(pinacolato)diboron (114.3 mg, 0.45 mmol, 1.12 eq), and (PPh3)2PdCl2 (28 mg, 0.04 mmol, 0.1 eq) were suspended in anhydrous DMF (10 mL). The mixture underwent three cycles of vacuum/filling with N2, then stirred at 80°C for 1 h. After the reaction was complete, the mixture was cooled to rt. Then 3-bromo-5-(2,4-dimethoxypyrimidin- 5-yl)-2H-[1,3'-bipyridin]-2-one (S6, 155.7 mg, 0.4 mmol, 1.0 eq) and K2CO3 (110.6 mg, 0.8 mmol, 2.0 eq) were added to the mixture, which was underwent three cycles of vacuum/filling with N2. The mixture was heated to 120°C for 1h. After the reaction was complete, the DMF was removed, water was added, and the solution was extracted with DCM (3× 40 mL). The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography with a dichloromethane / ethyl acetate / methanol (90% / 5% / 5%) gradient to afford the target compound as a pale yellow solid (98 mg, 37% yield). 1H NMR (400 MHz, CDCl3) δ 8.74 (d, J = 2.2 Hz, 1H), 8.72 (d, J = 4.8 Hz, 1H), 8.27 (s, 1H), 7.92 (d, J = 8.9 Hz, 1H), 7.77 (d, J = 2.6 Hz, 1H), 7.57 (d, J = 2.5 Hz, 1H), 7.50 (dd, J = 8.1, 4.9 Hz, 1H), 7.29 (dd, J = 5.6, 1.8 Hz, 2H), 6.92 (t, J = 2.0 Hz, 1H), 4.18 – 4.12 (m, 2H), 4.06 (s, 3H), 4.04 (s, 3H), 3.73 (s, 2H), 3.43 (s, 2H), 2.89 (s, 4H), 1.53 (s, 9H). MS m/z (ESI): 563.2 [M-C5H9O2+H2]+.
5-(3-(3-Chloro-5-(3-hydroxy-3-methylbutoxy)phenyl)-2-oxo-2H-[1,3'-bipyridin]-5-yl) pyrimidine-2,4(1H,3H)-dione (29) (Figure 8). 3-(3-Chloro-5-(3-hydroxy-3-methylbutoxy)phenyl)-5-(2,4- dimethoxypyrimidin-5-yl)-2H-[1,3'-bipyridin]-2-one (S7a, 110 mg, 0.21 mmol, 1.0 eq) was dissolved in anhydrous DMF (10 mL), LiCl (89 mg, 2.1 mmol, 10 eq) and p-toluenesulfonic acid (361.6 mg, 2.1 mmol, 10 eq) were added and stirred at 80°C for 1 h. After the reaction was complete, the mixture was concentrated in vacuo, the residue was suspended with saturated aqueous NaHCO3 (15 mL), then the mixture was filtered, the solid was washed with NaHCO3, water and hexane, dried with a lyophilizer to yield the final compound as a pale yellow solid (92 mg, yield 88%). 1H NMR (600 MHz, DMSO-d 6) δ 11.19 (s, 2H), 8.72 (d, J = 1.9 Hz, 1H), 8.63 (d, J = 4.6 Hz, 1H), 8.04 (d, J = 2.1 Hz, 1H), 8.02 – 7.96 (m, 2H), 7.88 (s, 1H), 7.57 (dd, J = 8.0,4.9 Hz, 1H), 7.37 (s, 1H), 7.27 (s, 1H), 6.97 (s, 1H), 4.36 (s, 1H), 4.09 (t, J = 7.1 Hz, 2H), 1.81(t, J = 7.1 Hz, 2H), 1.13 (s, 6H). 13C NMR (151 MHz, DMSO-d 6) δ 163.79, 159.66, 159.62,152.04, 149.58, 148.07, 141.09, 140.14, 139.54, 138.12, 136.54, 135.28, 133.65, 128.18, 124.31,120.90, 114.38, 113.88, 112.97, 107.37, 68.43, 65.58, 42.42, 30.16. HRMS m/z (ESI) calcd for C25H24ClN4O5 [M+H]+ 495.1430 found: 495.1432.
5-(3-(3-Chloro-5-(2-(3-oxopiperazin-1-yl)ethoxy)phenyl)-2-oxo-2H-[1,3'-bipyridin]-5-yl) pyrimidine-2,4(1H,3H)-dione (50) (Figure 8). Tert-butyl 4-(2-(3-chloro-5-(5-(2,4-dimethoxypyrimidin-5- yl)-2-oxo-2H-[1,3'-bipyridin]-3-yl) phenoxy)ethyl)-2-oxopiperazine-1-carboxylate (S7b, 90 mg, 0.136 mmol, 1.0 eq) was dissolved in anhydrous DMF (5 mL), LiCl (60 mg, 1.4 mmol, 10 eq) and p-toluenesulfonic acid (241 mg, 1.4 mmol, 10 eq) were added and stirred at 80°C for 1 h. After the reaction was complete, the mixture was concentrated in vacuo, then the residue was redissolved in DCM (5 mL) and trifluoroacetic acid (2 mL) and stirred at room temperature for 1h. The mixture was concentrated in vacuo, the residue was suspended with saturated aqueous NaHCO3 (15 mL), then the mixture was extracted with DCM (3× 30 mL), The combined organic layer was concentrated in vacuo and the crude product was purified using silica gel chromatography with a methanol / dichloromethane (0-5%) gradient to afford the target compound as a pale yellow solid (40 mg, 55% yield). 1H NMR (600 MHz, DMSO-d 6) δ 11.30 (s, 2H), 8.72 (s, 1H), 8.64 (d, J = 4.3 Hz, 1H), 8.04 (s, 1H), 8.00 (d, J = 7.7 Hz, 2H), 7.88 (s, 1H), 7.70 (s, 1H), 7.57 (dd, J = 7.9, 4.8 Hz, 1H), 7.40 (s, 1H), 7.29 (s, 1H), 7.01 (s, 1H), 4.13 (t, J = 5.4 Hz, 2H), 3.12 (s, 2H), 3.01 (s, 2H), 2.75 (t, J = 5.3 Hz, 2H), 2.64 (t, J = 5.3 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.08, 163.67, 159.68, 159.38, 151.34, 149.61, 148.06, 140.21, 139.73, 139.47, 138.07, 136.85, 135.29, 133.66, 128.11, 124.32, 121.12, 114.43, 113.98, 112.60, 107.86, 66.38, 57.42, 55.80, 49.53, 40.78. HRMS m/z (ESI) calcd for C26H24ClN6O5 [M+H]+ 535.1491 found: 535.1493.
Quantification and statistical analysis
Kinetic analysis was performed in GraphPad Prism 8. Raw data was plotted and initial rate (V0) was calculated by the slope of the linear part of the kinetic curve using linear regression analysis in Prism. Vi was determined as the initial velocity of the reaction of Mpro and substrate alone (without inhibitor). Vi/V0 was plotted on the Y axis and log(inhibitor concentration) was plotted on the X axis. IC50 curves were generated for each replicate using nonlinear regression (log(agonist) vs. response) in Prism and triplicate IC50 values were averaged and standard deviation values were calculated. IC50 values and standard deviation values are found in Table 1.
Acknowledgments
The SARS-CoV-2 Mpro plasmid was provided by the Hilgenfeld lab (Zhang et al., 2020). This work was supported by the US National Institutes of Health (NIH) (GM32136, AI087925, R35 GM132092, T32GM136651) and by a CoReCT Pilot Grant from the Yale University School of Medicine. E.A.S. acknowledges support for the NSF Graduate Research Fellowship Program. This research used resources AMX of the National Synchrotron Light Source II, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract no. DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the NIH, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010). This work is also based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the NIGMS from the NIH (P30 GM124165). The Eiger 16M detector on the 24-ID-E beamline is funded by an NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a US DOE Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357.
Author contributions
M.G.D. and J.A.I. carried out expression and purification, X-ray crystallography, enzyme kinetics, and data analysis. C.-H.Z. and E.A.S. carried out synthesis and analysis of compounds. Writing was done by M.G.D. J.A.I., R.A.R., S.J.M., E.A.S., C.-H.Z., W.L.J., and K.S.A. provided guidance and revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.str.2021.06.002.
Supplemental information
References
- Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha- helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J.H., Tomashek K.M., Dodd L.E., Mehta A.K., Zingman B.S., Kalil A.C., Hohmann E., Chu H.Y., Luetkemeyer A., Kline S., et al. Remdesivir for the treatment of Covid-19—final report. N. Engl. J. Med. 2020;383:1813–1826. doi: 10.1056/nejmoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzowka M., Mitusinska K., Raczynska A., Samol A., Tuszynski J.A., Gora A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020;21:3099. doi: 10.3390/ijms21093099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannalire R., Cerchia C., Beccari A.R., Di Leva F.S., Summa V. Targeting SARS-CoV-2 proteases and polymerase for COVID-19 treatment: state of the art and future opportunities. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.0c01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., Shi Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019;17:181–192. doi: 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., stjohn S.E., Osswald H.L., O'Brien A., Banach B.S., Sleeman K., Ghosh A.K., Mesecar A.D., Baker S.C. Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. J. Virol. 2014;88:11886–11898. doi: 10.1128/JVI.01528-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghahremanpour M.M., Tirado-Rives J., Deshmukh M., Ippolito J.A., Zhang C.H., Cabeza de Vaca I., Liosi M.E., Anderson K.S., Jorgensen W.L. Identification of 14 known drugs as inhibitors of the main protease of SARS-CoV-2. ACS Med. Chem. Lett. 2020;11:2526–2533. doi: 10.1021/acsmedchemlett.0c00521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., et al. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The architecture of SARS-CoV-2 transcriptome. Cell. 2020;181:914–921 e910. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D.W., Galanie S., Phillips G., O'Neill H.M., Coates L., Kovalevsky A. Malleability of the SARS-CoV-2 3CL M(pro) active-site cavity facilitates binding of clinical antivirals. Structure. 2020;28:1313–1320 e1313. doi: 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy N., Fakhro K. Identification of mutation resistance coldspots for targeting the SARS-CoV2 main protease. IUBMB Life. 2021;73:670–675. doi: 10.1002/iub.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Worrall L.J., Vuckovic M., Rosell F.I., Gentile F., Ton A.T., Caveney N.A., Ban F., Cherkasov A., Paetzel M., Strynadka N.C.J. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020;11:5877. doi: 10.1038/s41467-020-19662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengist H.M., Mekonnen D., Mohammed A., Shi R., Jin T. Potency, safety, and pharmacokinetic profiles of potential inhibitors targeting SARS-CoV-2 main protease. Front. Pharmacol. 2020;11:630500. doi: 10.3389/fphar.2020.630500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin A., Eisenbraun B., Key J., Sanschagrin P.C., Timony M.A., Ottaviano M., Sliz P. Collaboration gets the most out of software. eLife. 2013;2:e01456. doi: 10.7554/elife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. American Chemical Society; 2021. Oral Inhibitors of the SARS-CoV-2 Main Protease for the Treatment of COVID-19. [Google Scholar]
- Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B., Shen C., et al. SARS-CoV-2 M(pro) inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco M.D., Ma C., Lagarias P., Gao A., Townsend J.A., Meng X., Dube P., Zhang X., Hu Y., Kitamura N., et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against M(pro) and cathepsin L. Sci. Adv. 2020;6:eabe0751. doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana M.V.S., Silva F.P., Jr. De novo design and bioactivity prediction of SARS- CoV-2 main protease inhibitors using recurrent neural network-based transfer learning. BMC Chem. 2021;15:8. doi: 10.1186/s13065-021-00737-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020;30:127377. doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2021 World Health Organization (2021). Coronavirus Disease (COVID-19) Situation Report, https://www.who.int/publications/m/item/weekly-epidemiological-update---23-february-2021.
- Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U S A. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.-H., Stone E.A., Deshmukh M., Ippolito J.A., Ghahremanpour M.M., Tirado-Rives J., Spasov K.A., Zhang S., Takeo Y., Kudalkar S.N., et al. Potent noncovalent inhibitors of the main protease of SARS-CoV-2 from molecular sculpting of the drug perampanel guided by free energy perturbation calculations. ACS Cent. Sci. 2021;7:467–475. doi: 10.1021/acscentsci.1c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoC-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated in these studies are available upon request from lead contact Karen Anderson (karen.anderson@yale.edu). Structures from this study are deposited to the PDB with IDs 7M8X, 7M8M, 7M8Z, 7M8Y, 7M8N, 7M8O, 7M90, 7M91, and 7M8P and are publicly available (www.rcsb.org).