

ABSTRACT
Antimicrobial resistance (AMR) remains one of the most challenging phenomena of modern medicine. Machine learning (ML) is a subfield of artificial intelligence that focuses on the development of algorithms that learn how to accurately predict outcome variables using large sets of predictor variables that are typically not hand selected and are minimally curated. Models are parameterized using a training data set and then applied to a test data set on which predictive performance is evaluated. The application of ML algorithms to the problem of AMR has garnered increasing interest in the past 5 years due to the exponential growth of experimental and clinical data, heavy investment in computational capacity, improvements in algorithm performance, and increasing urgency for innovative approaches to reducing the burden of disease. Here, we review the current state of research at the intersection of ML and AMR with an emphasis on three domains of work. The first is the prediction of AMR using genomic data. The second is the use of ML to gain insight into the cellular functions disrupted by antibiotics, which forms the basis for understanding mechanisms of action and developing novel anti-infectives. The third focuses on the application of ML for antimicrobial stewardship using data extracted from the electronic health record. Although the use of ML for understanding, diagnosing, treating, and preventing AMR is still in its infancy, the continued growth of data and interest ensures it will become an important tool for future translational research programs.
KEYWORDS: antibiotic resistance, antimicrobial stewardship, drug discovery, machine learning, mechanisms of action, whole-genome sequencing
INTRODUCTION
Use of broad-spectrum antibiotics are a routine part of medical care for a significant proportion of hospitalized patients globally. Unsurprisingly, this practice has led to the emergence and global spread of multidrug-resistant pathogens. According to the Centers for Disease Control and Prevention (CDC), antimicrobial resistance (AMR) was responsible for the deaths of 35,900 people in the United States in 2018 (1), with projected increases as the population ages. The death toll worldwide is estimated to be a staggering 700,000 per year (2). The arrival of a new generation of antibiotics has provided hope to patients and providers, but bacterial evolution demands continuous innovation in the realm of drug development to ensure that gains are preserved. Furthermore, the cost of new treatments and the lack of access in resource-limited settings points to an urgent need for generalizable approaches to prevent AMR.
Drug resistance arises from the multiscale interaction between evolutionary forces, microbiology, the built environment, and human behavior, making it one of the great challenges of the 21st century. Machine learning (ML) is a subfield of artificial intelligence that has emerged as one potential avenue by which to address this complex phenomenon. ML is primarily concerned with the development of algorithms that are able to build a predictive model using a training data set, with little to no human input (3). Algorithms are broadly categorized into supervised ML, where algorithms train on data that contain the outcome of interest; unsupervised ML, where algorithms learn structures within the data set de novo; and reinforcement learning, where algorithms learn to optimize for a desired outcome as new data are continually incorporated. Relative to traditional statistical modeling, ML algorithms are typically less concerned with understanding data-generating processes and more focused on performing prediction on unseen data, referred to as a test data set (4). However, in recent years there has also been considerable progress on developing ML algorithms for causal inference with observational data (5, 6). Figure 1A provides an overview of the data processing steps involved in ML analyses, and Fig. 1B illustrates the three main categories of models.
FIG 1.

General overview of machine learning analyses. (A) ML analyses are capable of integrating a wide variety of data types. These include raw images or instrument traces, pathogen genomic sequences, data obtained from wet lab experiments, and information contained in the electronic health record. The latter encompasses clinical pathology results, free text notes, and structured data such as demographics, comorbidities, procedures, allergies, medication exposures, and hospital encounters. These inputs must be carefully cleaned and the relevant features extracted or engineered. Multiple validation checks are often necessary to ensure the data remain accurate after preprocessing. Next, the data are split into a training set used to define the model parameters and a remaining portion is held out for testing model performance. (B) There are three broad categories of ML analyses. The first two are supervised and unsupervised learning. In supervised learning, training data contains labels denoting the outcome of interest (i.e., antibiotic resistance phenotypes). The model trains on these data and then predicts the predefined outcomes of interest on test data. Unsupervised models are trained on data that does not contain labels for the outcome of interest. The model therefore searches on its own for relationships between variables and then predicts these relationships on unlabeled test data. A typical use for unsupervised learning involves clustering high dimensional data and outlier detection. The final category of ML analyses are reinforcement learning models. These models comprise an “agent” which interacts with its environment over time. The state of the environment is provided to the agent, and the agent then chooses an action, e.g., an antibiotic treatment choice, from a set of available options. It then assesses the impact of that action on the environment through a reward function. The purpose of the reinforcement learning agent is to learn a set of actions for different states (i.e., a “policy”) that maximizes the cumulative reward.
Interest in applying ML to health care has intensified over the past decade, reflecting the exponential increase in biological and medical data availability, massive improvements in computational power, and critical breakthroughs in algorithm development. Applications to the practice of infectious diseases have encompassed the diagnosis of infection (7, 8), the early identification of sepsis (9–11), and targeting preexposure prophylaxis for people living with human immunodeficiency virus (12, 13). In this review, we provide an overview of the current state of research into the applications of ML for understanding, treating, and preventing AMR. We structure this review into three sections: the prediction of resistance phenotypes from pathogen genomic data; the understanding of antibiotic mechanisms of action and its corollary, novel drug development; and lastly, antimicrobial stewardship using ML-driven clinical decision support. While the field is still young, we propose that ML will come to play an important supporting role in the ongoing effort to reduce the burden of AMR worldwide.
ML FOR PREDICTING AMR FROM PATHOGEN GENOMIC SEQUENCES
The increasing prevalence of AMR necessitates routine antimicrobial susceptibility testing (AST) to ensure adequate treatment. Phenotypic testing is the gold standard for AST, but the process of bacterial isolation and subsequent culture in the presence of antibiotics typically takes 2 days for nonfastidious bacterial pathogens and up to several weeks for slow-growing organisms like Mycobacterium tuberculosis. It is not routinely performed for some pathogens such as Neisseria gonorrhoeae, despite well-documented increases in resistance. An alternative method that is gaining traction due to decreasing sequencing costs and improvements in analytic tools uses the microbial genotype, rather than the phenotype, to determine AMR (14, 15). Genotypic methods promise to not only be faster than phenotypic methods by bypassing laboratory culture but may also provide insight into the mechanisms driving AMR, enable early detection of transmission events, and provide important ancillary information such as the bacterial strain and virulence factors (16).
Genotypic AMR prediction methods require an organism to be sequenced and for that sequence to be translated into a prediction using either a rule-based or an ML approach, as recently reviewed (17). Rule-based approaches use prior knowledge to interpret the presence or absence of known AMR genes (e.g., mecA and vanA) or resistance-causing mutations (e.g., gyrA S83L conferring fluoroquinolone resistance in Escherichia coli), resulting in excellent performance in well-studied organisms with a small number of highly penetrant resistance loci such as Staphylococcus aureus and M. tuberculosis (18, 19). However, since rule-based approaches require a comprehensive knowledge of organism-specific resistance mechanisms and manual curation, they are difficult to scale to encompass the breadth of organisms routinely encountered in clinical practice and tend to have difficulty predicting complex AMR mechanisms, such as polymyxin resistance in Klebsiella pneumoniae (20).
ML models have increasingly been shown to provide accurate predictions of AST when trained on sufficiently large training data sets despite the models not having prior knowledge of resistance mechanisms. The foundational training data are high dimensional, given an average genome size of 4 Mb, and yet are relatively few in number due to the limited size of public databases with linked AST phenotypes and the expense of obtaining new sequences. Thus, the number of input features can vastly outnumber the training examples, leading to the risk of overfitting (21). Here, we describe recent advances that have begun to overcome these challenges, specifically focusing on (i) the training data requirements, (ii) the features that ML models have been trained to classify, and (iii) the ML models that have been explored.
Importance of training data.
Genotypic ML models typically require large databases of high-quality sequencing data from diverse isolates that are labeled with phenotypic AST data (Fig. 2A). The sequencing data input for genotypic ML models is most commonly shotgun DNA sequences from isolates, but shotgun metagenomic DNA sequences (22) or sequenced amplicons can also be used. Alternatively, rather than basing predictions solely on genomic DNA sequences, one can use antibiotic treatment-induced transcriptional responses, which have the advantage of incorporating both phenotypic and genotypic data for the prediction of AMR (23, 24).
FIG 2.
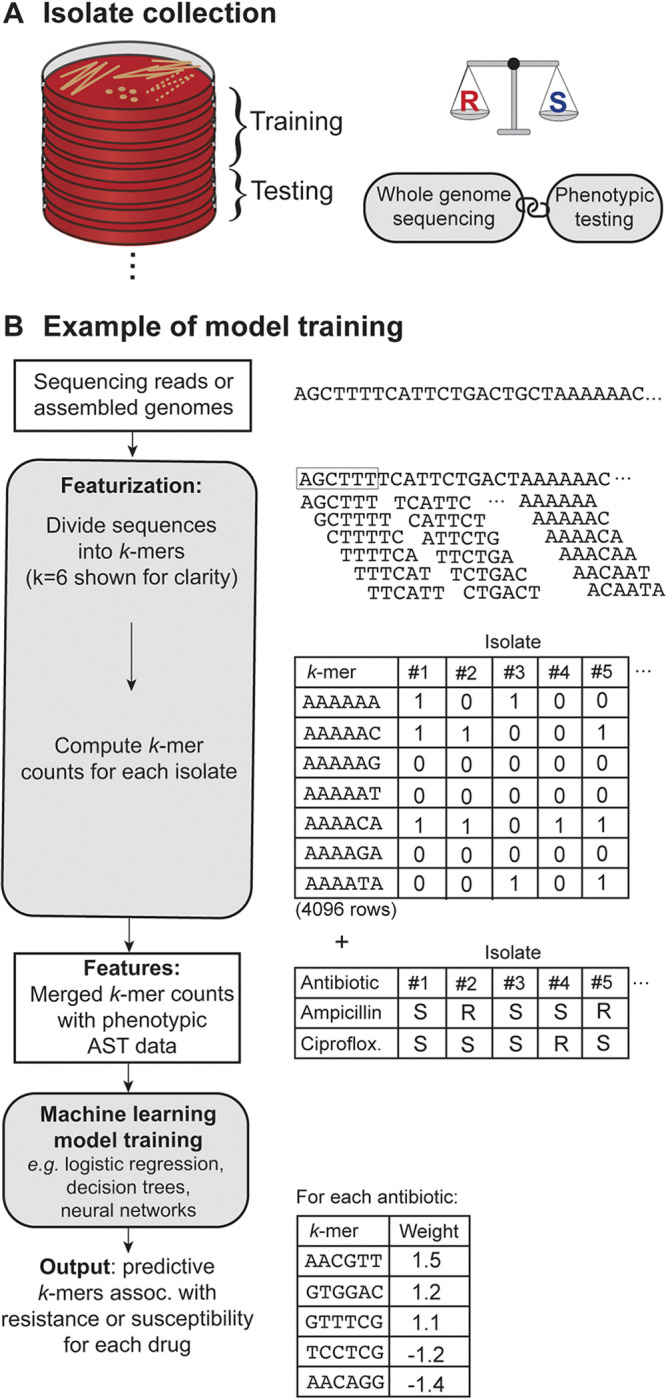
Schematic overview of the process of training a machine learning (ML) model to predict antimicrobial susceptibility testing (AST) results from whole-genome sequences. (A) ML models rely on data sets containing tens to thousands of isolates with paired whole-genome sequences and phenotypic AST results. Data sets are divided into training and test sets, where the training set is used to fit the parameters of the model and the test set evaluates the model accuracy. An optimal data set contains a balance of resistant and susceptible examples for each organism-drug combination. (B) The data inputs are usually quality-controlled sequencing reads or assembled genomes, which are transformed into overlapping subsequences of length k, referred to as a k-mer. A typical length for a k-mer feature is 13 to 31 nucleotides, but a length of 6 nucleotides is shown here for clarity. There are 4k k-mer possibilities (e.g., 4,096 when k = 6), and the counts of each k-mer present in a given sequence are tallied for each isolate in the data set. The selected features are then merged with the phenotypic AST data, and this matrix is used as the input for a supervised machine learning model. The model analyzes the matrix to find the features, e.g., k-mers, that best predict resistance or susceptibility to a given antibiotic.
Phenotypic AST data are used as the ML model’s “ground truth” and ensuring accuracy is critical to ensure model validity. However, clinical microbiologists are well aware of the variability inherent in AST results even when using standardized operating protocols. Breakpoints vary among guidelines and can change over time, an example of “data set shift” (25). Certain drugs may be tested by some labs but not others depending on local prescribing practices, leading to missing data. MICs can vary by one to two 2-fold dilutions across laboratories, contributing noise to the phenotypic output upon which the model is trained (26, 27). Subsequently, ML models tend to have higher accuracy for species-drug combinations with clear-cut bimodal MIC distributions, where the wild-type and resistant populations are clearly distinguished and the impact of phenotypic measurement variability is the least. Conversely, species-drug combinations with significant overlap between the wild-type and resistant distributions can be particularly difficult to classify, and isolates with MICs near the breakpoint or in the “intermediate” category are sometimes excluded from predictions (26, 28, 29). Hicks et al. tested a variety of ML models to predict gonococcal resistance to ciprofloxacin (well-separated bimodal distribution) and azithromycin (significant overlap in wild-type and resistant distributions). The ciprofloxacin model accuracy was >95% regardless of the clinical breakpoint or ML model used. Conversely, the azithromycin model accuracy ranged from 77 to 88% when trained on all isolates. When they removed strains with azithromycin MICs within two doubling dilutions of the nonsusceptible breakpoint, the model accuracy improved to 88 to >95%, demonstrating a contribution of the MIC distribution to model performance. More commonly, intermediate isolates are grouped with resistant isolates rather than eliminating them all together. Despite these challenges, accurate MIC predictions have been demonstrated for numerous antibiotics with N. gonorrhoeae, Salmonella, and K. pneumoniae (30–32), and accurate binary predictions have been demonstrated more widely (17).
Given the cost and difficulty of obtaining data, how many training examples are needed? There is no universal rule. Models for exceedingly simple mechanisms such as methicillin resistance in S. aureus require fewer than 100 examples to achieve >99% accuracy (33). Species such as Pseudomonas aeruginosa with high genomic variability and numerous antibiotic resistance mechanisms may require thousands of examples to capture the long tail of resistance determinants and nonlinear genetic interactions (34). In a nontyphoidal Salmonella data set, increasing the training set size from 500 to 4,500 genomes improved the average model accuracy from 90 to 95% (30). In addition to collecting a sufficient number of training examples, it is also important to acquire a balance of susceptible and nonsusceptible examples that ideally cover a broad range of MICs. Compared to models trained on balanced data sets, models trained on skewed data sets comprised mostly of susceptible examples tend to have lower sensitivity and higher specificity, and vice versa (26), with an overrepresentation of resistant examples. If generalizability is desired, isolate diversity is also needed to capture resistance mechanisms that vary in prevalence geographically and to avoid training on phylogenetic confounders (26, 30), although strategies to control for population structure have been recently demonstrated (35). The CRyPTIC Consortium collected over 10,000 M. tuberculosis isolates globally to include all major M. tuberculosis lineages and obtain sufficient examples of rare ethambutol or pyrazinamide-resistant isolates (19, 26).
General strategies for prediction.
Most ML models train on continuous or binary vectors of fixed width. Transforming complex input data into this form is known as “feature extraction.” A common method for deriving features from genomic shotgun sequencing data is to divide the sequencing reads or contigs into subsequences of length k, also called k-mers (Fig. 2B) (33, 36–40). The k-mers within a given sample can then be counted and transformed into a vector by marking the presence/absence or frequency of each k-mer, of which there are 4k possibilities (though only a fraction of these, approximately five million, are present in a given training set). A typical k-mer length is from 13 to 31 nucleotides, where longer k-mers are potentially more specific but prone to sequencing error and require more training data to sufficiently sample the larger feature space (33, 37, 38, 40, 41). Others have extracted features by mapping reads to antibiotic resistance genes, functional orthologs, or even pangenomes, which can capture variations in novel genes and regulatory sequences, and deriving features based on the presence or absence of genes, single nucleotide variants, or indels (42–46). An orthogonal approach extracted features from protein sequences based on the proteins’ function, primary structure, secondary structure, and physicochemical properties (47).
After featurization, the prediction of AMR becomes a routine supervised learning problem. Many off-the-shelf ML models have been applied, including logistic regression, random forests, decision trees (e.g., XGBoost and AdaBoost), and neural networks. For this particular application, the choice of ML model tends to have less of an effect on model accuracy than the characteristics of the underlying training data (26). Drouin et al. took a unique approach by focusing on model interpretability, which is the ability for a user to understand how a model arrived at its prediction (38). They applied two algorithms—classification and regression trees, to learn decision trees, and set covering machines, to learn logical operators—to determine a simple set of rules that reveal resistance mechanisms and predict phenotypes with fairly high accuracy. More typical ML models can also provide interpretability, e.g., by mapping predictive k-mers back to their genomic locus and quantifying the locus’ importance in the overall prediction (20). Generally, ML models that are interpretable rather than “black-box” are likely to gain greater acceptance in clinical practice and have the added benefits of generating testable hypotheses for novel mechanisms and identifying spurious associations (48).
Comparing model accuracy for a given organism across publications is challenging due to differing validation data sets, evaluation metrics (e.g., area under the curve, F1 score, or accuracy), and drug selection. Head-to-head comparisons of ML algorithms on well-curated test sets would provide valuable insight into optimal feature and model selection. For example, Mason et al. recently compared three non-ML WGS-based phenotypic prediction methods on a test set of 1,379 S. aureus isolates and found 98.3% concordance between genotypic predictions and laboratory phenotypes, with 99.6% concordance among the three genotypic methods (49).
Expanding the breadth and depth of genotypic AMR prediction.
Significant advances have been made in the field of genotypic AMR prediction using ML, but there are important limitations related to comprehensiveness. Studies to date have primarily focused on predicting the most commonly used antibiotics in a few of the most commonly encountered bacterial pathogens. The use of ML-based prediction models in routine clinical practice will require expanding training data sets to include the non-aureus staphylococci, streptococci other than S. pneumoniae, and Enterobacterales other than E. coli and K. pneumoniae. More focused investigations will also be required for organisms such as Pseudomonas aeruginosa that have phenotypic plasticity and complex AMR patterns that can confound genotypic predictions (29, 37). Greater effort should also be placed on sequencing and predicting unusual but highly clinically significant organisms such as vancomycin-nonsusceptible S. aureus. It is also a challenge to obtain sufficient data to predict resistance to new antibiotics, such as ceftazidime-avibactam, ceftolozane-tazobactam, cefiderocol, and eravacycline, due to a lack of tested isolates and the paucity of resistant strains. Initiatives such as the CDC and the U.S. Food and Drug Administration (FDA) Antibiotic Resistance Isolate Bank and the FDA-ARGOS database that collect, validate, and sequence organisms with important resistance patterns will be tremendously valuable to fill the current gap.
ML FOR UNDERSTANDING MECHANISMS OF AMR AND ANTIMICROBIAL DISCOVERY
While the clinical need for effective antimicrobials is growing, the pipeline for discovering new and effective antimicrobials is shrinking (50). Traditional compound screening methods used in the pharmaceutical industry commonly fail to arrive at chemically distinct compound classes, driving innovation for the discovery of new indications for existing antimicrobials and for novel antimicrobial classes. ML is particularly well suited to support both of these goals. In this section, we review the growing use of ML approaches for uncovering antimicrobial mechanisms of action and for the discovery of novel antimicrobial agents.
Understanding antimicrobial mechanisms of action.
Antimicrobial agents are generally understood to function either by inhibiting the activity of essential microbial gene products or by destroying the mechanical integrity of essential microbial components such as membranes or DNA. However, antimicrobial efficacy is highly context dependent, and several aspects of microbial physiology can enhance or inhibit antimicrobial treatment outcomes (51) indicating that drug-target interactions alone are insufficient for explaining why antimicrobials succeed or fail in the clinic. ML-based approaches are now beginning to meet this knowledge gap, enabling the rapid identification of physiological processes impacted by antimicrobial agents with unknown function, pathway mechanisms for antibiotic-induced cell death, and resistance mechanisms from sequenced genomes.
Experimental platforms are increasing in throughput and quantitative sophistication, permitting deep characterization of the cellular phenotypes induced by antimicrobial agents. Applications of supervised ML to data from platform technologies such as high-throughput fluorescence microscopy (52) and mass spectrometry-based metabolomics (53) are now enabling the inference of antimicrobial mechanisms of action based on morphology or biochemical fingerprint. These techniques work by first characterizing the cellular responses to well-characterized antimicrobials and then applying ML algorithms to these measurements to train an antimicrobial classifier. Such approaches can be effective if a poorly characterized antimicrobial shares a target with well-studied antimicrobials, but these techniques are limited by the diversity of the training data.
Modern ML algorithms are now capable of assembling highly predictive models when given sufficiently large training data sets; however, the associations between features identified by ML algorithms rarely translate to specific biomolecular entities which can be experimentally perturbed. The result is a so-called “black-box” that precludes biological understanding. Recent efforts have focused on developing interpretable “white-box” ML methods that can provide direct insight into biological mechanisms. However, this domain of ML remains in a nascent stage due to limitations in how training data are often presented to ML algorithms. One of the earliest demonstrations of interpretable ML sought to identify metabolic pathways contributing to antibiotic-induced lethality (54). Instead of training a predictive model directly on the metabolites used as biochemical perturbations in a bactericidal antibiotic response screen, Yang et al. used a literature-curated network model of bacterial metabolism to first transform screening perturbations into metabolic network states corresponding to the screening conditions and then applied these metabolic states as training data alongside the antibiotic response measurements. The resulting ML model directly identified purine biosynthesis as a novel metabolic pathway involved in the lethal processes of bactericidal antibiotics, that is activated by antibiotic-induced nucleotide pool disruptions (55) and that drives lethal stress-induced metabolic dysfunction (56). These results were validated genetically and biochemically, demonstrating how interpretable ML may enable the discovery of causal biological mechanisms.
Interpretable ML approaches are now also beginning to shed light on novel biological mechanisms directly from the whole-genome sequences of clinical isolates. As described above, conventional ML approaches are sufficient for predicting antibiotic susceptibility phenotypes from genotypic data. However, biological understanding for how these phenotypes emerge requires additional information on how gene products have evolved to function together in microbial physiology. Although AMR is commonly understood to be explained only by mechanisms that either reduce the effective intracellular concentration of an antimicrobial agent or inhibit the interaction between an antimicrobial and its target, the diversity in antimicrobial treatment failure phenotypes suggest that some AMR mechanisms may be found in aspects of microbial physiology beyond the drug-target interaction (57). Metabolic network models are now being applied to powerfully address this knowledge gap. Kavvas et al. used a genome-scale metabolic model of M. tuberculosis to transform whole-genome sequences from drug-resistant clinical strains into interpretable ML training data and identified novel resistance mechanisms that are specific to different antituberculosis antibiotics (58). To achieve this, these authors first developed strain-specific metabolic models of drug-susceptible and drug-resistant clinical isolates, applying SNPs in metabolic genes as modeling constraints, and then developed a ML classifier based on the resulting model simulations to identify metabolic reactions that would be predictive for AMR against each antituberculosis antibiotic. These analyses identified both direct targets of these antibiotics and several metabolic enzymes as drivers of AMR, demonstrating the power of interpretable ML to uncover novel AMR mechanisms.
Interpretable ML approaches have the potential to transform our understanding of how AMR arises, and unlike traditional genome-wide association studies, prior knowledge about genes harboring mutations is directly incorporated into the overall ML framework, providing a direct link between model results and pathway mechanisms. Elucidating the underlying biological mechanisms responsible for AMR is an important step toward the development of novel therapeutic regimens that may block the formation of AMR (59).
Discovering novel antimicrobial agents.
ML is poised to fundamentally transform pharmaceutical drug discovery and development by providing tools that are accelerating target identification, lead discovery, preclinical development and clinical development (60). In the case of antimicrobial discovery, recent studies demonstrate how ML can be applied to learn small molecule structural features from screens which include existing antimicrobials to design novel antimicrobials. Innovation in this area has focused on both the development of novel screening strategies that leverage genetically engineered microbial strains to improve screening sensitivity (61) and the application of novel ML algorithms that utilize new chemical structure representations to improve algorithmic learning of chemical properties (62). Johnson et al. developed a screening strategy for discovering biochemical inhibitors of essential genes in M. tuberculosis by first generating a genetic library consisting of hypomorph knockdowns for these essential genes and then screening 50,000 chemical compounds against these hypomorphs (61). Using known antituberculosis antibiotics as a reference ground-truth, these authors applied supervised ML classification analyses to identify novel classes of chemical inhibitors of both existing drug targets (such as DNA gyrase, mycolic acid synthesis, and folate metabolism) and new drug targets (such as the EfpA efflux pump), which they validated in wild-type cells.
Stokes et al. performed a biochemical screen of ∼2,300 chemically diverse compounds for antimicrobial activity against E. coli and used the resulting data to train a deep learning ML model to predict antimicrobial activity from chemical structures alone (62). These authors applied the resulting ML model to the Drug Repurposing Hub and discovered SU3327 (a c-Jun N-terminal kinase inhibitor), which they validated as a potent inhibitor of ESKAPE pathogens and multidrug-resistant pathogens. Applying this ML model to the >107 million molecules in the ZINC15 database, the authors discovered eight putative antimicrobial compounds that are structurally distinct from known antimicrobials, demonstrating how ML can drive lead discovery. Lead compounds emerging from both these studies powerfully illustrate how ML approaches can fuel drug discovery and development.
ML approaches have also been applied to aid in the design (63) and optimization (64) of antimicrobial peptides (AMPs), with demonstrated efficacy against drug-resistant pathogens. AMPs are natural substrates for ML algorithms since they can be fully represented by their peptide sequences. Moreover, AMPs are typically short (<30 amino acids), making them tractable for oligopeptide synthesis and enabling more comprehensive screens of the chemical structure landscape than would be possible for small molecules. Wu et al. designed DP7, a novel 12-amino-acid AMP with activity against S. aureus, by training an ML model with multiple 12-amino-acid AMPs to estimate the contribution of amino acids at each position to overall antimicrobial activity (63). These authors then synthesized this AMP and demonstrated that this had in vivo efficacy against both drug-susceptible and drug-resistant S. aureus (65). Similarly, Porto et al. applied an ML genetic algorithm to peptides derived from the guava plant to design optimized plant-templated AMPs with antimicrobial activity (64). These analyses led to the discovery of the novel AMP Guavanin 2, which the authors demonstrated had in vivo efficacy against diverse pathogens. As ML approaches improve in their ability to aid peptide and protein engineering (66), AMPs are likely to become important tools for combating AMR.
Prospects for overcoming antimicrobial resistance.
Collectively, ML approaches are revealing important insights into the biological mechanisms underlying antimicrobial treatment failure and providing novel candidates for accelerating antimicrobial discovery. Although ML algorithms are becoming increasingly democratized for basic and translational research, continued innovation at the interface between ML and AMR requires the availability of large, high-quality data sets. Continued progress hinges upon advances in screening and sequencing methods that capture the enormous diversity of clinical pathogens, sequenced strains, and antimicrobial compounds. As microbial knowledge bases become increasingly complete, new ML formalisms will also be required to efficiently and effectively improve the interpretability of the predictive models generated by ML.
ML FOR ANTIMICROBIAL STEWARDSHIP
Antimicrobial stewardship programs (ASPs) seek to align antibiotic use with appropriate indications and to assist in the selection of treatment regimens that are of the narrowest spectrum and of the shortest duration without compromising efficacy (67). This is a critically important endeavor as the prescription of unnecessary or inappropriately broad-spectrum antibiotics by health care providers is a major driver of AMR. ASPs are very effective at the local scale but most require significant investment in human resources for manual data curation and provider feedback, which limit the ability to scale up. Clinical decision support systems (CDSSs) for antibiotic stewardship using probabilistic models exist (68–71) but have not yet been widely adopted since most are highly specialized to the health systems in which they were trained.
The electronic health record (EHR) is a rich source of information for clinicians, as well as for ML models that seek to augment clinician decisions. The application of ML-based CDSSs for antimicrobial stewardship offers a pathway for scaling up ASP interventions while maintaining highly personalized recommendations. However, very few studies have used this approach to date. In this section, we review the challenges associated with the use of EHR data, integrating early examples of ML-based tools aiming to support stewardship efforts and provide guidance around best practices for future work.
Challenges in the analysis of EHR data.
The EHR contains many important risk factors for AMR, including prior infections and resistance profiles, recent exposures to hospitals and antibiotics, laboratory values, imaging and pathological reports, and prior responses to treatment. Data come in both structured and unstructured (free-text) formats. Newer ML approaches are bypassing human-entered data altogether by analyzing primary images and waveforms. A unique aspect of clinical microbiologic data is the existence of susceptibility phenotypes not only for the antibiotic chosen by the clinician but also for other potential treatment candidates. Susceptibility profiles allow an ML algorithm to evaluate the impact of an array of potential treatment decisions in parallel, a situation referred to as the counterfactual, and is critical for developing policies that are optimal for a chosen task while accounting for off-target effects (72).
The use of EHR data, however, presents many challenges. First, it is highly prone to containing erroneous or indecipherable data due to entry error. Second, the complexity of AMR requires pulling data across multiple databases, significantly increasing the skill and time necessary to validate data cleaning processes (73). Third, EHR data are subject to nonrandom missing observations, confounding by indication and selection bias (74). Fourth, susceptibility data are subject to changes in clinical breakpoints, as well as shifts in the underlying distribution over time, potentially reducing the accuracy of models trained on historical data (75). Fifth, representations of data are often specific to the institution from whence they arose, a potential barrier to generalizability. It is necessary to pay careful attention to each of these pitfalls to ensure an ML CDSS produces accurate recommendations specific to the local environment.
Measurements for clinical evaluation.
Much of the focus of ML applications in health care has been on performing prediction for various clinical tasks. Popular metrics for model performance include sensitivity, specificity and the Area Under the Receiver Operating Curve (AUROC) characteristic. In the realm of AMR, Ghosh et al. describe the use of an ensemble of ML models to predict susceptibility phenotypes across common pathogen-antibiotic combinations in septic intensive care unit (ICU) patients (76). Similarly, Feretzakis et al. used an ensemble method to predict AMR in a single center study of ICU patients in Greece (77), and Goodman et al. reported on an ML-derived decision tree for identification of patients with a bloodstream infection due to an extended-spectrum beta-lactamase-producing organism (78). Each study reported AUROCs, but no evaluations were made to compare the performance of the model to that of clinicians using unseen test data.
To understand the clinical impact of a CDSS, it is necessary to translate probabilities into decisions using cutoffs (79) and then evaluate against the decisions made by providers either retrospectively using a test data set (preferably from a different health care system) or prospectively in a trial format. In the case of antibiotic stewardship, a CDSS must choose thresholds that balance the tension between reducing the spectrum of therapy and preserving clinical efficacy. Thresholds that intentionally bias decisions away from broad-spectrum therapy may lead to unacceptably high rates of inappropriate therapy, defined as the choice of an agent that is shown to be resistant in vitro. Conversely, thresholds that bias decisions to ensure clinical success could lead to high levels of broad-spectrum therapy. Sensitivity analyses over a range of clinically relevant thresholds may be necessary to better evaluate tradeoffs. Kanjilal et al. provide an example of this approach in their evaluation of an empirical treatment decision algorithm for patients with uncomplicated urinary tract infection (UTI) (80). A wide range of probability cutoffs were evaluated, with the final selection based on their ability to maximize reductions in the use of second-line therapies while accepting rates of inappropriate therapy that were no greater than that of clinicians. Using this approach, they were able to build an ML policy optimization approach that reduced fluoroquinolone usage by >67% relative to the retrospective decisions of clinicians, while also reducing inappropriate therapy by 18%. In practice, it may be beneficial to allow end users to adjust thresholds to fit their personal tolerance for risk since this is known to vary considerably between providers, provided it is within the bounds of practice guidelines. Subsequent work using the same cohort utilized a direct learning approach rather than prediction of resistance to individual antibiotics and was able to achieve similar outcomes relative to the initial study but allowed for a deeper understanding of factors influencing antibiotic treatment decisions (72).
Considerations for deployment.
Deploying a ML CDSS at the point-of-care entails integration into clinical workflows and development of a front-end interface that provides interpretable recommendations with measures of uncertainty. CDSSs should appear to end users at the appropriate moment in the treatment decision process and provide rapid recommendations that are specific to the patient. It is important for any ML-based model to provide the logic behind a recommendation to ensure it can be adequately assessed by the treating clinician. Furthermore, sustainability will require that ML models to retrain on a periodic basis using new data that has accrued in databases to ensure recommendations reflect the local patient population.
There are several examples of CDSSs for antibiotic stewardship that have successfully deployed at the point of care, though none utilize ML. The TREAT CDSS uses a set of hand-picked features to predict the probability of bacterial infection and antibiotic resistance and then provides recommendations on which treatments to choose using a weighting algorithm. TREAT was evaluated using a cluster randomized control trial and found to significantly reduce inappropriate antibiotic therapy by 9% (69). The ongoing INSPIRE-ASP UTI (70) and PNA (71) trials have deployed similar probabilistic prediction models using a small set of hand-picked variables to provide stewardship around empirical treatment decisions for UTI and lower respiratory tract infections. The INSPIRE trials are being conducted as a cluster randomized trial across 59 hospitals in the United States, with the primary completion date anticipated in 2022.
Existing CDSSs are trained on EHR data that are specific to a hospital or health care system and have limited generalizability to new environments where data collection systems and representations may differ greatly. However, over the past decade considerable effort has gone into building universal medical ontologies and backend software infrastructure that allows for interoperability of EHR data (81). The goal of such efforts is to create a marketplace for decision support applications similar to that of an “application store” for smartphones, where developers can rely on consistent input data that is agnostic to EHR vendors (82). The maturation of such a marketplace would be a major development for ML CDSSs designed for antimicrobial stewardship.
Designing for equity.
The majority of ML-CDSS applications for clinical infectious diseases focus on ICU patients in high-income countries, reflecting the relative abundance of data in this population (83). However, the vast majority of antibiotic prescribing occurs in outpatient settings, where data sets are relatively sparse. The mismatch between data and clinical need is even more striking for minority populations and in resource-limited settings in sub-Saharan Africa and Asia (84), where disparities in outcomes for infections and the burden of AMR are the highest.
The inequality in the amount of data available in disadvantaged populations reflects fundamental inequalities in access to health care, but there are important steps that researchers can take to help ensure the benefits of ML are more equitably distributed. For instance, studies seeking to target vulnerable populations can build their data collection systems to gather relevant features for the population of interest in a manner that integrates into larger training databases. This facilitates the generalization of models developed in higher income or in socioeconomically advantaged communities to more diverse populations. Researchers can also design models to be computationally efficient and rely on as few features as possible (77), reducing the barriers to deployment in settings where the IT infrastructure may be less developed.
Three studies describe the application of ML to antibiotic stewardship in outpatient and resource-limited settings. Yelin et al. developed an ML CDSS to predict resistance in outpatients presenting with UTI using insurance claims, susceptibility, and a small set of clinical metadata. Using logistic regression and gradient boosted decision trees, they were able to retrospectively achieve a 30% reduction in the selection of inappropriate antibiotic therapy (85). The previously mentioned study by Kanjilal et al. similarly focused on outpatients with uncomplicated UTI (80). A study by Oonsivilai et al. compared eight different ML models for predicting resistance in Cambodian children with bacteremia and showed AUROCs ranging from 0.7 to 0.8 (86). The paucity of studies in this area highlight the urgent need for researchers to invest more effort in designing ML applications for underserved populations.
Elements for success.
Building an effective and sustainable ML-derived decision support tool for antimicrobial stewardship requires close collaboration between the analytic team, clinicians, IT engineers, behavioral scientists, and experts in implementation science. The analytic team is responsible for developing highly accurate and computationally efficient models that can use data inputs “as is” and provide outputs that can easily interface with EHR systems independent of vendor. Provider insight is fundamental to identifying data features that are clinically relevant to AMR, for evaluating inconsistencies in the data, interpreting unexpected results, selecting models that are acceptable to end users and designing randomized trials to evaluate utility. IT engineers are necessary for building and maintaining the infrastructure necessary for deployment at the point of care. Behavioral scientists can provide an understanding of the drivers for current prescription practice, identify strategies for behavior modification using CDSSs and perform field testing. Implementation science will be critical to carry research findings into clinical practice. Finally, designing models and CDSSs with an explicit intent to ensure equity is critical for success.
CONCLUSIONS
A new model for translational research is emerging, which utilizes advanced ML algorithms to improve AMR diagnostics, provide deeper insight into biologic mechanisms of resistance, expand pathways for antibiotic development, and develop personalized clinical decision support for rational treatment. These activities hold promise for overcoming the challenges posed by AMR. The factors limiting growth in the field include the relative paucity of high-quality training data sets and the small number of groups working at the intersection of ML and AMR. However, there are no signs that the exponential growth in data generation, computational capacity, and algorithm development is abating. Similarly, interest from governmental institutions and public-private partnerships in understanding, treating, and preventing AMR has never been higher. The confluence of these two trends ensures that the future of the field is bright. Importantly, a significant and sustained reduction in the burden of disease will require a strong emphasis on building interdisciplinary research teams and an intentional focus on equity.
Contributor Information
Sanjat Kanjilal, Email: skanjilal@bwh.harvard.edu.
Alexander J. McAdam, Boston Children’s Hospital
REFERENCES
- 1.Centers for Disease Control and Prevention. 2019. Antibiotic resistance threats in the United States, 2019. Centers for Disease Control and Prevention, Atlanta, GA. [Google Scholar]
- 2.O’Neill J. 2016. Tackling drug-resistant infections globally: final report and recommendations. Analysis and Policy Observatory, Hawthorn, Australia. https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf. [Google Scholar]
- 3.Burkov A. 2019. The hundred-page machine learning book. Andriy Burkov. [Google Scholar]
- 4.Bzdok D, Altman N, Krzywinski M. 2018. Statistics versus machine learning. Nat Methods 15:233–234. 10.1038/nmeth.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuler MS, Rose S. 2017. Targeted maximum likelihood estimation for causal inference in observational studies. Am J Epidemiol 185:65–73. 10.1093/aje/kww165. [DOI] [PubMed] [Google Scholar]
- 6.Díaz I. 2019. Machine learning in the estimation of causal effects: targeted minimum loss-based estimation and double/debiased machine learning. Biostatistics 21:353–358. 10.1093/biostatistics/kxz042. [DOI] [PubMed] [Google Scholar]
- 7.Naydenova E, Tsanas A, Howie S, Casals-Pascual C, Vos MD. 2016. The power of data mining in diagnosis of childhood pneumonia. J R Soc Interface 13:20160266. 10.1098/rsif.2016.0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiens J, Campbell WN, Franklin ES, Guttag JV, Horvitz E. 2014. Learning data-driven patient risk stratification models for Clostridium difficile. Open Forum Infect Dis 1:ofu045. 10.1093/ofid/ofu045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komorowski M, Celi LA, Badawi O, Gordon AC, Faisal AA. 2018. The artificial intelligence clinician learns optimal treatment strategies for sepsis in intensive care. Nat Med 24:1716–1720. 10.1038/s41591-018-0213-5. [DOI] [PubMed] [Google Scholar]
- 10.Nemati S, Holder A, Razmi F, Stanley MD, Clifford GD, Buchman TG. 2018. An interpretable machine learning model for accurate prediction of sepsis in the ICU. Crit Care Med 46:547–553. 10.1097/CCM.0000000000002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsoukalas A, Albertson T, Tagkopoulos I. 2015. From data to optimal decision making: a data-driven, probabilistic machine learning approach to decision support for patients with sepsis. JMIR Med Inform 3:e11. 10.2196/medinform.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krakower DS, Gruber S, Hsu K, Menchaca JT, Maro JC, Kruskal BA, Wilson IB, Mayer KH, Klompas M. 2019. Development and validation of an automated HIV prediction algorithm to identify candidates for pre-exposure prophylaxis: a modeling study. Lancet HIV 6:e696–e704. 10.1016/S2352-3018(19)30139-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcus JL, Hurley LB, Krakower DS, Alexeeff S, Silverberg MJ, Volk JE. 2019. Use of electronic health record data and machine learning to identify candidates for HIV pre-exposure prophylaxis: a modeling study. Lancet HIV 6:e688–e695. 10.1016/S2352-3018(19)30137-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schürch AC, Schaik W. 2017. Challenges and opportunities for whole-genome sequencing-based surveillance of antibiotic resistance. Ann N Y Acad Sci 1388:108–120. 10.1111/nyas.13310. [DOI] [PubMed] [Google Scholar]
- 15.Cohen KA, Manson AL, Desjardins CA, Abeel T, Earl AM. 2019. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: progress, promise, and challenges. Genome Med 11:45. 10.1186/s13073-019-0660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Didelot X, Bowden R, Wilson DJ, Peto TEA, Crook DW. 2012. Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13:601–612. 10.1038/nrg3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su M, Satola SW, Read TD. 2018. Genome-based prediction of bacterial antibiotic resistance. J Clin Microbiol 57:e01405-18. 10.1128/JCM.01405-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon NC, Price JR, Cole K, Everitt R, Morgan M, Finney J, Kearns AM, Pichon B, Young B, Wilson DJ, Llewelyn MJ, Paul J, Peto TE, Crook DW, Walker AS, Golubchik T. 2014. Prediction of Staphylococcus aureus antimicrobial resistance by whole-genome sequencing. J Clin Microbiol 52:1182–1191. 10.1128/JCM.03117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.CRyPTIC Consortium and the 100,000 Genomes Project. 2018. Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencing. New Engl J Med 379:1403–1415. 10.1056/NEJMoa1800474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macesic N, Walk OIBD, Pe’er I, Tatonetti NP, Peleg AY, Uhlemann AC. 2020. Predicting phenotypic polymyxin resistance in Klebsiella pneumoniae through machine learning analysis of genomic data. mSystems 5:e00656-19. 10.1128/mSystems.00656-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romero A, Carrier PL, Erraqabi A, Sylvain T, Auvolat A, Dejoie E, Legault M-A, Dubé M-P, Hussin JG, Bengio Y. 2016. Diet networks: thin parameters for fat genomics. arXiv 1611.09340.
- 22.Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, Zhang L. 2018. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6:23. 10.1186/s40168-018-0401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharyya RP, Bandyopadhyay N, Ma P, Son SS, Liu J, He LL, Wu L, Khafizov R, Boykin R, Cerqueira GC, Pironti A, Rudy RF, Patel MM, Yang R, Skerry J, Nazarian E, Musser KA, Taylor J, Pierce VM, Earl AM, Cosimi LA, Shoresh N, Beechem J, Livny J, Hung DT. 2019. Simultaneous detection of genotype and phenotype enables rapid and accurate antibiotic susceptibility determination. Nat Med 25:1858–1864. 10.1038/s41591-019-0650-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki S, Horinouchi T, Furusawa C. 2014. Prediction of antibiotic resistance by gene expression profiles. Nat Commun 5:5792. 10.1038/ncomms6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelly CJ, Karthikesalingam A, Suleyman M, Corrado G, King D. 2019. Key challenges for delivering clinical impact with artificial intelligence. BMC Med 17:195. 10.1186/s12916-019-1426-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hicks AL, Wheeler N, Sanchez-Buso L, Rakeman JL, Harris SR, Grad YH. 2019. Evaluation of parameters affecting performance and reliability of machine learning-based antibiotic susceptibility testing from whole-genome sequencing data. PLoS Comput Biol 15:e1007349. 10.1371/journal.pcbi.1007349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mouton JW, Meletiadis J, Voss A, Turnidge J. 2018. Variation of MIC measurements: the contribution of strain and laboratory variability to measurement precision. J Antimicrob Chemother 73:2374–2379. 10.1093/jac/dky232. [DOI] [PubMed] [Google Scholar]
- 28.Davies TJ, Stoesser N, Sheppard AE, Abuoun M, Fowler P, Swann J, Quan TP, Griffiths D, Vaughan A, Morgan M, Phan HTT, Jeffery KJ, Andersson M, Ellington MJ, Ekelund O, Woodford N, Mathers AJ, Bonomo RA, Crook DW, Peto TEA, Anjum MF, Walker AS. 2020. Reconciling the potentially irreconcilable? Genotypic and phenotypic amoxicillin-clavulanate resistance in Escherichia coli. Antimicrob Agents Chemother 64:e02026-19. 10.1128/AAC.02026-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khaledi A, Weimann A, Schniederjans M, Asgari E, Kuo T, Oliver A, Cabot G, Kola A, Gastmeier P, Hogardt M, Jonas D, Mofrad MR, Bremges A, McHardy AC, Häussler S. 2020. Predicting antimicrobial resistance in Pseudomonas aeruginosa with machine learning-enabled molecular diagnostics. EMBO Mol Med 12:e10264. 10.15252/emmm.201910264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen M, Long SW, McDermott PF, Olsen RJ, Olson R, Stevens RL, Tyson GH, Zhao S, Davis JJ. 2018. Using machine learning to predict antimicrobial minimum inhibitory concentrations and associated genomic features for nontyphoidal Salmonella. J Clin Microbiol 57:e01260-18. 10.1128/JCM.01260-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eyre DW, Silva DD, Cole K, Peters J, Cole MJ, Grad YH, Demczuk W, Martin I, Mulvey MR, Crook DW, Walker AS, Peto TEA, Paul J. 2017. WGS to predict antibiotic MICs for Neisseria gonorrhoeae. J Antimicrob Chemother 72:1937–1947. 10.1093/jac/dkx067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen M, Brettin T, Long SW, Musser JM, Olsen RJ, Olson R, Shukla M, Stevens RL, Xia F, Yoo H, Davis JJ. 2018. Developing an in silico minimum inhibitory concentration panel test for Klebsiella pneumoniae. Sci Rep 8:421. 10.1038/s41598-017-18972-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis JJ, Boisvert S, Brettin T, Kenyon RW, Mao C, Olson R, Overbeek R, Santerre J, Shukla M, Wattam AR, Will R, Xia F, Stevens R. 2016. Antimicrobial resistance prediction in PATRIC and RAST. Sci Rep 6:27930. 10.1038/srep27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freschi L, Jeukens J, Kukavica-Ibrulj I, Boyle B, Dupont M-J, Laroche J, Larose S, Maaroufi H, Fothergill JL, Moore M, Winsor GL, Aaron SD, Barbeau J, Bell SC, Burns JL, Camara M, Cantin A, Charette SJ, Dewar K, Déziel É, Grimwood K, Hancock REW, Harrison JJ, Heeb S, Jelsbak L, et al. 2015. Clinical utilization of genomics data produced by the international Pseudomonas aeruginosa consortium. Front Microbiol 6:1036–1038. 10.3389/fmicb.2015.01036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Earle SG, Wu CH, Charlesworth J, Stoesser N, Gordon NC, Walker TM, Spencer CCA, Iqbal Z, Clifton DA, Hopkins KL, Woodford N, Smith EG, Ismail N, Llewelyn MJ, Peto TE, Crook DW, McVean G, Walker AS, Wilson DJ. 2016. Identifying lineage effects when controlling for population structure improves power in bacterial association studies. Nat Microbiol 1:16041. 10.1038/nmicrobiol.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bradley P, Gordon NC, Walker TM, Dunn L, Heys S, Huang B, Earle S, Pankhurst LJ, Anson L, Cesare M.d, Piazza P, Votintseva AA, Golubchik T, Wilson DJ, Wyllie DH, Diel R, Niemann S, Feuerriegel S, Kohl TA, Ismail N, Omar SV, Smith EG, Buck D, McVean G, Walker AS, Peto TEA, Crook DW, Iqbal Z. 2015. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun 6:10063. 10.1038/ncomms10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferreira I, Beisken S, Lueftinger L, Weinmaier T, Klein M, Bacher J, Patel R, Haeseler A.v, Posch AE. 2020. Species identification and antibiotic resistance prediction by analysis of whole-genome sequence data by use of ARESdb: an analysis of isolates from the Unyvero Lower Respiratory Tract Infection Trial. J Clin Microbiol 58:e00273-20. 10.1128/JCM.00273-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drouin A, Letarte G, Raymond F, Marchand M, Corbeil J, Laviolette F. 2019. Interpretable genotype-to-phenotype classifiers with performance guarantees. Sci Rep 9:4071. 10.1038/s41598-019-40561-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lees JA, Vehkala M, Välimäki N, Harris SR, Chewapreecha C, Croucher NJ, Marttinen P, Davies MR, Steer AC, Tong SYC, Honkela A, Parkhill J, Bentley SD, Corander J. 2016. Sequence element enrichment analysis to determine the genetic basis of bacterial phenotypes. Nat Commun 7:12797. 10.1038/ncomms12797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aun E, Brauer A, Kisand V, Tenson T, Remm M. 2018. A k-mer-based method for the identification of phenotype-associated genomic biomarkers and predicting phenotypes of sequenced bacteria. PLoS Comput Biol 14:e1006434. 10.1371/journal.pcbi.1006434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camp P-JV, Haslam DB, Porollo A. 2020. Prediction of antimicrobial resistance in Gram-negative bacteria from whole-genome sequencing data. Front Microbiol 11:1013. 10.3389/fmicb.2020.01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moradigaravand D, Palm M, Farewell A, Mustonen V, Warringer J, Parts L. 2018. Prediction of antibiotic resistance in Escherichia coli from large-scale pan-genome data. PLoS Comput Biol 14:e1006258. 10.1371/journal.pcbi.1006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Her HL, Wu YW. 2018. A pan-genome-based machine learning approach for predicting antimicrobial resistance activities of the Escherichia coli strains. Bioinformatics 34:i89–i95. 10.1093/bioinformatics/bty276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Greenberg DE, Pifer R, Jiang S, Xiao G, Shelburne SA, Koh A, Xie Y, Zhan X. 2020. VAMPr: VAriant Mapping and Prediction of antibiotic resistance via explainable features and machine learning. PLoS Comput Biol 16:e1007511. 10.1371/journal.pcbi.1007511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lees JA, Mai TT, Galardini M, Wheeler NE, Corander J. 2019. Improved inference and prediction of bacterial genotype-phenotype associations using pangenome-spanning regressions. Biorxiv:852426. [DOI] [PMC free article] [PubMed]
- 47.Chowdhury AS, Khaledian E, Broschat SL. 2019. Capreomycin resistance prediction in two species of Mycobacterium using a stacked ensemble method. J Appl Microbiol 127:1656–1664. 10.1111/jam.14413. [DOI] [PubMed] [Google Scholar]
- 48.FDA. 2020. Artificial intelligence (AI) and machine learning (ML) in medical devices. U.S. Food and Drug Administration, Washington, DC. [Google Scholar]
- 49.Mason A, Foster D, Bradley P, Golubchik T, Doumith M, Gordon NC, Pichon B, Iqbal Z, Staves P, Crook D, Walker AS, Kearns A, Peto T. 2018. Accuracy of different bioinformatics methods in detecting antibiotic resistance and virulence factors from Staphylococcus aureus whole-genome sequences. J Clin Microbiol 56:e01815-17. 10.1128/JCM.01815-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown ED, Wright GD. 2016. Antibacterial drug discovery in the resistance era. Nature 529:336–343. 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 51.Yang JH, Bening SC, Collins JJ. 2017. Antibiotic efficacy: context matters. Curr Opin Microbiol 39:73–80. 10.1016/j.mib.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith TC, Pullen KM, Olson MC, McNellis ME, Richardson I, Hu S, Larkins-Ford J, Wang X, Freundlich JS, Ando DM, Aldridge BB. 2020. Morphological profiling of tubercle bacilli identifies drug pathways of action. Proc Natl Acad Sci U S A 117:18744–18753. 10.1073/pnas.2002738117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zampieri M, Szappanos B, Buchieri MV, Trauner A, Piazza I, Picotti P, Gagneux S, Borrell S, Gicquel B, Lelievre J, Papp B, Sauer U. 2018. High-throughput metabolomic analysis predicts mode of action of uncharacterized antimicrobial compounds. Sci Transl Med 10:eaal3973. 10.1126/scitranslmed.aal3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang JH, Wright SN, Hamblin M, McCloskey D, Alcantar MA, Schrübbers L, Lopatkin AJ, Satish S, Nili A, Palsson BO, Walker GC, Collins JJ. 2019. A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell 177:1649–1661. 10.1016/j.cell.2019.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belenky P, Ye JD, Porter CBM, Cohen NR, Lobritz MA, Ferrante T, Jain S, Korry BJ, Schwarz EG, Walker GC, Collins JJ. 2015. Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep 13:968–980. 10.1016/j.celrep.2015.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schrader SM, Vaubourgeix J, Nathan C. 2020. Biology of antimicrobial resistance and approaches to combat it. Sci Transl Med 12:eaaz6992. 10.1126/scitranslmed.aaz6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kavvas ES, Yang L, Monk JM, Heckmann D, Palsson BO. 2020. A biochemically interpretable machine learning classifier for microbial GWAS. Nat Commun 11:2580. 10.1038/s41467-020-16310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Domenech A, Brochado AR, Sender V, Hentrich K, Henriques-Normark B, Typas A, Veening JW. 2020. Proton motive force disruptors block bacterial competence and horizontal gene transfer. Cell Host Microbe 27:544–555. 10.1016/j.chom.2020.02.002. [DOI] [PubMed] [Google Scholar]
- 60.Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, Li B, Madabhushi A, Shah P, Spitzer M, Zhao S. 2019. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 18:463–477. 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johnson EO, LaVerriere E, Office E, Stanley M, Meyer E, Kawate T, Gomez JE, Audette RE, Bandyopadhyay N, Betancourt N, Delano K, Silva ID, Davis J, Gallo C, Gardner M, Golas AJ, Guinn KM, Kennedy S, Korn R, McConnell JA, Moss CE, Murphy KC, Nietupski RM, Papavinasasundaram KG, Pinkham JT, Pino PA, Proulx MK, Ruecker N, Song N, Thompson M, Trujillo C, Wakabayashi S, Wallach JB, Watson C, Ioerger TR, Lander ES, Hubbard BK, Serrano-Wu MH, Ehrt S, Fitzgerald M, Rubin EJ, Sassetti CM, Schnappinger D, Hung DT. 2019. Large-scale chemical-genetics yields new M. tuberculosis inhibitor classes. Nature 571:72–78. 10.1038/s41586-019-1315-z. [DOI] [PubMed] [Google Scholar]
- 62.Stokes JM, Yang K, Swanson K, Jin W, Cubillos-Ruiz A, Donghia NM, MacNair CR, French S, Carfrae LA, Bloom-Ackerman Z, Tran VM, Chiappino-Pepe A, Badran AH, Andrews IW, Chory EJ, Church GM, Brown ED, Jaakkola TS, Barzilay R, Collins JJ. 2020. A deep learning approach to antibiotic discovery. Cell 180:688–702. 10.1016/j.cell.2020.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu X, Wang Z, Li X, Fan Y, He G, Wan Y, Yu C, Tang J, Li M, Zhang X, Zhang H, Xiang R, Pan Y, Liu Y, Lu L, Yang L. 2014. In vitro and in vivo activities of antimicrobial peptides developed using an amino acid-based activity prediction method. Antimicrob Agents Chemother 58:5342–5349. 10.1128/AAC.02823-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Porto WF, Irazazabal L, Alves ESF, Ribeiro SM, Matos CO, Pires ÁS, Fensterseifer ICM, Miranda VJ, Haney EF, Humblot V, Torres MDT, Hancock REW, Liao LM, Ladram A, Lu TK, Fuente-Nunez C.dl, Franco OL. 2018. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat Commun 9:1490. 10.1038/s41467-018-03746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang R, Wang Z, Tian Y, Yin Q, Cheng X, Lian M, Zhou B, Zhang X, Yang L. 2019. Efficacy of antimicrobial peptide DP7, designed by machine-learning method, against methicillin-resistant Staphylococcus aureus. Front Microbiol 10:1175. 10.3389/fmicb.2019.01175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang KK, Wu Z, Arnold FH. 2019. Machine-learning-guided directed evolution for protein engineering. Nat Methods 16:687–694. 10.1038/s41592-019-0496-6. [DOI] [PubMed] [Google Scholar]
- 67.Dyar OJ, Huttner B, Schouten J, Pulcini C, ESGAP (ESCMID Study Group for Antimicrobial stewardshiP) . 2017. What is antimicrobial stewardship? Clinical. Clin Microbiol Infect 23:793–798. 10.1016/j.cmi.2017.08.026. [DOI] [PubMed] [Google Scholar]
- 68.Leibovici L, Kariv G, Paul M. 2013. Long-term survival in patients included in a randomized controlled trial of TREAT, a decision support system for antibiotic treatment. J Antimicrob Chemother 68:2664–2666. 10.1093/jac/dkt222. [DOI] [PubMed] [Google Scholar]
- 69.Paul M, Andreassen S, Tacconelli E, Nielsen AD, Almanasreh N, Frank U, Cauda R, Leibovici L. 2006. Improving empirical antibiotic treatment using TREAT, a computerized decision support system: cluster randomized trial. J Antimicrob Chemother 58:1238–1245. 10.1093/jac/dkl372. [DOI] [PubMed] [Google Scholar]
- 70.Clinicaltrials.gov. 2018. The INSPIRE-ASP UTI Trial. U.S. National Library of Medicine, Washington, DC. https://clinicaltrials.gov/ct2/show/NCT03697096. [Google Scholar]
- 71.Clinicaltrials.gov. 2018. The INSPIRE-ASP PNA Trial. U.S. National Library of Medicine, Washington, DC. https://clinicaltrials.gov/ct2/show/NCT03697070. [Google Scholar]
- 72.Boominathan S, Oberst M, Zhou H, Kanjilal S, Sontag D. 2020. Treatment policy learning in multiobjective settings with fully observed outcomes, p 1937–1947. In Proceedings of the 26th ACM SIGKDD Conference on Knowledge Discovery and Data Mining (KDD) (to appear), 2020. 10.1145/3394486.3403245. [DOI] [Google Scholar]
- 73.Sculley D, Holt G, Golovin D, Davydov E, Phillips T, Ebner D, Chaudhary V, Young M. 2014. Machine learning: the high interest credit card of technical debt. https://static.googleusercontent.com/media/research.google.com/en//pubs/archive/43146.pdf.
- 74.Paxton C, Niculescu-Mizil A, Saria S. 2013. Developing predictive models using electronic medical records: challenges and pitfalls. AMIA Annu Symp Proc 2013:1109–1115. [PMC free article] [PubMed] [Google Scholar]
- 75.Subbaswamy A, Saria S. 2019. From development to deployment: dataset shift, causality, and shift-stable models in health AI. Biostatistics 21:345–352. [DOI] [PubMed] [Google Scholar]
- 76.Ghosh D, Sharma S, Hasan E, Ashraf S, Singh V, Tewari D, Singh S, Kapoor M, Sengupta D. 2019. Machine learning based prediction of antibiotic sensitivity in patients with critical illness. medRxiv 19007153. [Google Scholar]
- 77.Feretzakis G, Loupelis E, Sakagianni A, Kalles D, Martsoukou M, Lada M, Skarmoutsou N, Christopoulos C, Valakis K, Velentza A, Petropoulou S, Michelidou S, Alexiou K. 2020. Using machine learning techniques to aid empirical antibiotic therapy decisions in the intensive care unit of a general hospital in Greece. Antibiotics 9:50. 10.3390/antibiotics9020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Goodman KE, Lessler J, Harris AD, Milstone AM, Tamma PD. 2019. A methodological comparison of risk scores versus decision trees for predicting drug-resistant infections: a case study using extended-spectrum beta-lactamase (ESBL) bacteremia. Infect Control Hosp Epidemiol 40:400–408. 10.1017/ice.2019.17. [DOI] [PubMed] [Google Scholar]
- 79.MacFadden DR, Daneman N, Coburn B. 2018. Optimizing empiric antibiotic selection in sepsis: turning probabilities into practice. Clin Infect Dis 66:479–479. 10.1093/cid/cix775. [DOI] [PubMed] [Google Scholar]
- 80.Kanjilal S, Oberst M, Boominathan S, Zhou H, Hooper DC, Sontag D. 2020. A decision algorithm to promote outpatient antimicrobial stewardship for uncomplicated urinary tract infection. Sci Transl Med 12:eaay5067. 10.1126/scitranslmed.aay5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mandel JC, Kreda DA, Mandl KD, Kohane IS, Ramoni RB. 2016. SMART on FHIR: a standards-based, interoperable apps platform for electronic health records. J Am Med Inform Assoc 23:899–908. 10.1093/jamia/ocv189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mandl KD, Kohane IS. 2017. A 21st-century health IT system: creating a real-world information economy. N Engl J Med 376:1905–1907. 10.1056/NEJMp1700235. [DOI] [PubMed] [Google Scholar]
- 83.Peiffer-Smadja N, Rawson TM, Ahmad R, Buchard A, Georgiou P, Lescure F-X, Birgand G, Holmes AH. 2020. Machine learning for clinical decision support in infectious diseases: a narrative review of current applications. Clin Microbiol Infect 26:584–595. 10.1016/j.cmi.2019.09.009. [DOI] [PubMed] [Google Scholar]
- 84.Bebell LM, Muiru AN. 2014. Antibiotic use and emerging resistance how can resource-limited countries turn the tide? Glob Heart 9:347–358. 10.1016/j.gheart.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yelin I, Snitser O, Novich G, Katz R, Tal O, Parizade M, Chodick G, Koren G, Shalev V, Kishony R. 2019. Personal clinical history predicts antibiotic resistance of urinary tract infections. Nat Med 25:1143–1123. 10.1038/s41591-019-0503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Oonsivilai M, Mo Y, Luangasanatip N, Lubell Y, Miliya T, Tan P, Loeuk L, Turner P, Cooper BS. 2018. Using machine learning to guide targeted and locally-tailored empiric antibiotic prescribing in a children’s hospital in Cambodia. Wellcome Open Res 3:131. 10.12688/wellcomeopenres.14847.1. [DOI] [PMC free article] [PubMed] [Google Scholar]