

Abstract
Myocardial infarction (MI) and osteoporotic fracture are leading causes of morbidity and mortality, and epidemiological evidence linking their incidence suggests possible crosstalk. MI can exacerbate atherosclerosis through the sympathetic nervous system (SNS) activation and β3 adrenoreceptor-mediated release of hematopoietic stem cells, leading to monocytosis. We hypothesized that this same pathway initiates systemic bone loss following MI, since osteoclasts differentiate from monocytes. In this study, MI was created with left anterior descending artery ligation in 12-week-old male mice (n = 24) randomized to β3-adrenergic receptor (AR) antagonist (SR 59230A) treatment or no treatment for 10 days postoperatively. Additional mice (n = 21, treated and untreated) served as unoperated controls. Bone mineral density (BMD), bone mineral content (BMC), and body composition were quantified at baseline and 10 days post-MI using dual-energy x-ray absorptiometry; circulating monocyte levels were quantified and the L5 vertebral body and femur were analyzed with microcomputed tomography 10 days post-MI. We found that MI led to circulating monocyte levels increases, BMD and BMC decreases at the femur and lumbar spine in MI mice (−6.9% femur BMD, −3.5% lumbar BMD), and trabecular bone volume decreases in MI mice compared with control mice. β3-AR antagonist treatment appeared to diminish the bone loss response (−5.3% femur BMD, −1.2% lumbar BMD), though these results were somewhat inconsistent. Clinical significance: These results suggest that MI leads to systemic bone loss, but that the SNS may not be a primary modulator of this response; bone loss and increased fracture risk may be important clinical comorbidities following MI or other ischemic injuries.
Keywords: β3-adrenergic receptor, μCT, bone loss, DXA, myocardial infarction
1 |. INTRODUCTION
Osteoporotic fractures and myocardial infarction (MI) are two of the leading cause of morbidity and mortality worldwide.1 Over 20% of patients aged 60 and over will die within 1 year following a hip fracture,2,3 and over 30% will die within 5 years.4,5 Similarly, within 5 years of the first MI, 36% of men and 47% of women will die due to MI-related complications.6 Interestingly, there is strong epidemiological evidence showing that MI is associated with an increased risk of subsequent fracture. For example, Gerber et al.7 found that fracture incidence rates increased markedly over time (hazard ratio [HR] = 1.32) among those with previous MI compared with control patients. A possible interpretation of these findings is that the incidence of fracture and MI is reflective of advanced stages of underlying chronic diseases such as osteoporosis and atherosclerosis, which are etiologically linked.8 However, another contributing factor may be that MI initiates an adaptive healing response that actively initiates bone loss systemically, thus increasing the subsequent risk of fracture.
Our previous study described a positive feedback loop between fracture and systemic bone loss in mice, which could lead to increased risk of subsequent fractures.9 In that study, fracture led to significant decreases in whole-body bone mineral density (BMD) in both young (3 months old) and middle-aged (12 months old) mice within 2 weeks postinjury. Fracture also resulted in ~11%–18% losses of trabecular bone volume in the L5 vertebral body at the same time point. These changes in BMD and bone microstructure were associated with decreased voluntary activity, increased systemic inflammation, and increased osteoclast number and activity at 3 days postinjury. These data demonstrate that acute skeletal injury (femur fracture) initiates a systemic response leading to loss of bone at distant skeletal sites. However, it is not known whether non-musculoskeletal injuries, such as MI, could also lead to systemic bone loss.
A similar positive feedback loop has been described for the cardiovascular system, wherein the systemic inflammatory response following MI in mice exacerbates underlying atherosclerosis.10 This study demonstrated that the activation of the sympathetic nervous system (SNS) after MI initiated the release of hematopoietic progenitor cells from the bone marrow, ultimately leading to monocytosis, accumulation of monocytes within atherosclerotic lesions, and exacerbated lesion formation. Treatment of mice with a β3-adrenergic receptor (AR) antagonist after MI lowered protease activity and myeloid cell content, ultimately decreasing the severity of monocytosis. It is possible that a similar SNS-mediated pathway may also be a key mediator of systemic bone loss following a fracture or other acute injuries, since osteoclasts have a hematopoietic lineage and differentiate from monocytes. However, the role of the SNS in systemic bone loss following a fracture or other injuries has not been described.
It is well-established that increased monocyte concentrations can lead to increased osteoclast activity and subsequent bone loss.11 However, the SNS can also directly affect bone cells, which could confound the effects of monocytosis on the bone. The SNS acts through signaling three types of β-ARs (β1, β2, and β3). Osteoclasts and osteoblasts primarily express β1 and β2 receptors,12–20 both which have a direct effect on bone cell activity.12 In contrast, little is known about the role of β3 in the skeletal system. Activation of β3 has been shown to increase osteoclastogenesis and subsequent bone resorption in vitro, but there is no consensus of its effect in vivo.13,21
In the current study, we sought to determine if acute ischemic injury (MI) leads to a loss of bone systemically and whether the SNS-induced monocytosis is a key regulator of this response. We used the same β3-AR antagonist (SR 59230A) that was shown to lower protease activity and inhibit monocytosis in post-MI mice,10 thus allowing us to determine if this same underlying mechanism contributed to bone loss following MI, while minimizing the direct effects of SNS inhibition on bone cells. We hypothesized that systemic bone loss would occur after MI, and that blockade of β3-AR would diminish or prevent this bone loss, implicating the SNS as a mediator of systemic bone adaptation following acute injury. These findings would describe a novel and potentially critical comorbidity associated with MI and other ischemic injuries and could inform future treatments that aim to preserve skeletal health in these patients.
2 |. METHODS
2.1 |. Animals
Forty-four male C57BL/6 mice were obtained from the Jackson Laboratory at 10 weeks of age and were acclimated to the housing vivarium for 2 weeks before the start of experiments. At 12 weeks of age, mice were randomized to MI surgery (n = 24) or anesthetized, unoperated controls (n = 21). Twenty-one mice (11 MI, 10 control) were administered a selective β3-AR antagonist (SR 59230A, Sigma-Aldrich; twice daily IP injection, 5 mg/kg body weight) for 10 days postoperatively starting immediately postsurgery. All mice were euthanized 10 days post-MI; this time point was selected based on our previous studies of bone loss following anterior cruciate ligament (ACL) rupture and femur fracture in mice, in which we observed peak trabecular bone loss approximately 1–2 weeks after injury.9,22 All animals were maintained and used in accordance with the National Institutes of Health guidelines on the care and use of laboratory animals, and all procedures were approved by the UC Davis Institutional Animal Care and Use Committee.
2.2 |. Myocardial infarction surgery
The left anterior descending (LAD) coronary artery was permanently ligated as previously described.23 Briefly, mice were anesthetized with isoflurane, intubated, and continuously monitored with a 3-lead electrocardiogram (ECG). A small incision was made, oblique muscles were bluntly separated to expose the ribs, and a small opening was created in the muscle of the fourth intercostal space. The ribs were then separated, and the pericardium was opened. The LAD was identified and permanently ligated using an 8–0 Prolene suture. LAD ligation was confirmed by ST segment elevation on the ECG. The ribs and oblique muscles were closed using a 6–0 Ethilon suture and the skin was closed using wound clips. Approximately 150 μl of sterile saline and 0.1 mg/kg buprenorphine were injected subcutaneously before allowing the mouse to recover in its cage on a 35°C warmer for ~1 h. Standard postoperative procedures were followed for 7 days, including analgesia (0.1 mg/kg buprenorphine) twice per day for 48 h. Wound clips were removed after 7 days. Unoperated control animals were subjected to anesthesia for 30 min and followed the same analgesia schedule.
2.3 |. Measurement of infarct size
After euthanasia, hearts were removed and placed immediately into cardioplegic solution (composition in mmol/L: NaCl 110, CaCl2 1.2, KCl 16, MgCl2 16, and NaHCO3 10) to prevent continued electrical activity and subsequent ischemic injury to myocytes. Hearts were frozen for 15 min, then sliced into 1 mm thick sections (Mouse Heart Slicer Matrix with 1.0 mm coronal section, Zivic Instruments). Heart slices were stained with 1% 2,3,5-triphenyltetrazolium in phosphate-buffered saline (PBS) for 15 min at 35°C after which the slices were stored in PBS for 24 h. Heart sections were gently blotted with a Kimwipe, then imaged with an office scanner (EPSON Perfection 4990 Photo). Individual color images were taken at 1200 dpi resolution for each heart section, and images were analyzed using ImageJ.24,25 To determine the area of ischemic tissue, a color filter was placed on the image to exclude all colors except for white. The filter was then manually adjusted until only the unstained ischemic tissue was highlighted. The total size of ischemic injury was quantified as the total area of ischemic (unstained) tissue in all transverse slices for each heart normalized by the total area of all slices (6–8 sections for each heart).
2.4 |. White blood cell analysis
Whole blood was collected from the peritoneal cavity at the time of euthanasia for differential white blood cell count to determine the percentage of monocytes and neutrophils in the blood. Approximately 0.5–0.75 ml of blood was slowly collected through the inferior vena cava using a 30-gauge needle and a 1 ml syringe. The needle tip was removed before the blood was placed directly from the syringe into the collection tube. All blood samples were placed in tubes coated with K2EDTA(BD Microtainer®) and were gently inverted 10 times before storing at 4°C. Blood samples were transferred to the UC Davis Veterinary Clinical Labs (UC Davis, Veterinary Medical Teaching Hospital) for differential white blood cell count within 24 h of collection.
2.5 |. Dual-energy x-ray absorptiometry analysis
Whole-body dual-energy x-ray absorptiometry (DXA) imaging was performed at baseline (1 day before surgery) and 9 days postsurgery (1 day before euthanasia) to determine body composition, BMD, and bone mineral content (BMC) of the whole body, lumbar spine, femoral diaphysis, and whole femur. Mice were anesthetized with isoflurane and placed in a cabinet x-ray system (Mozart®, Kubtec Medical Imaging). The whole-body analysis automatically excluded the head and wound clips, and BMD, BMC, bone area, lean mass area, and adipose tissue area were calculated using the manufacturer′s software. For the lumbar spine region of interest, the L4 through L6 vertebrae were manually selected; the whole femur and femoral diaphysis were analyzed using the same method. The femoral diaphysis was determined as the middle third of the femur. The imaging system was calibrated before each use to ensure consistent data.
2.6 |. Micro-computed tomography analysis
L5 vertebrae and both legs were collected following euthanasia and fixed in 4% paraformaldehyde for 3–4 days before preservation in 70% ethanol. L5 vertebrae and right femora were imaged with microcomputed tomography (SCANCO μCT 35) to determine the trabecular bone microstructure of the L5 vertebral body and distal femoral metaphysis and cortical bone microstructure of the femoral mid-diaphysis. All bones were imaged according to the guidelines for μCT of rodent bone (energy = 55 kVP, intensity = 114 mA, nominal voxel size = 6 μm, and integration time = 900 ms).26 Analysis of trabecular bone in the L5 vertebral body was performed by manually contouring 2D transverse slices in the region between the cranial and caudal growth plates and excluded the vertebral processes. Analysis of the femoral metaphysis was similarly performed with manual contouring beginning at the convergence of the distal femoral growth plate and extending 1500 μm (250 slices) proximal. Trabecular bone volume fraction (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and other microstructural parameters were determined using the manufacturer’s analysis software. Analysis of cortical bone in the femoral diaphysis was performed by contouring transverse slices centered on the midpoint of the femur including a total of 600 μm (100 slices). Bone area (B.Ar), cortical thickness (Ct.Th), bone tissue mineral density, and other microstructural parameters were determined using the manufacturer’s analysis software.
2.7 |. Three-point bending mechanical testing of femora
Mechanical testing was performed on femurs using three-point bending to determine bone structural and material properties using a materials testing system (ELF 3200, TA Instruments). Following μCT imaging, femurs were rehydrated for 10–15 min in PBS solution before mechanical testing. The span length of the lower supports was 8 mm, and the femur was positioned so that the posterior aspect of each bone was downward (loaded in tension). The bone was preloaded to 1–2 N to ensure contact with the upper platen. Loading was applied at a displacement rate of 0.01 mm/s until fracture, and displacement and resultant force were recorded at 50 Hz. Whole-bone structural properties were determined from force–displacement curves using standard methods.27 Stiffness was calculated as the slope of the linear preyield region. Postyield displacement was determined as the displacement difference between the yield and fracture displacements. Material properties were calculated using previously established beam theory equations.27 Elastic modulus, yield stress, and ultimate stress were determined using the bending moment of inertia (I) and bone radius (c) determined from μCT analysis of the femoral mid-diaphysis.
2.8 |. Statistical analysis
All results are expressed as mean ± standard deviation. Cross-sectional data were analyzed by two-way analysis of variance (ANOVA) stratified by operation (MI or control) and treatment (β3-AR antagonist or untreated) to determine the main effects and interactions. DXA data were longitudinal and were analyzed using repeated measures ANOVA to determine differences in the time course of outcomes. Post hoc analyses were performed using Tukey’s Honest Significant Difference test. Two-way ANOVA was performed using JMP Pro 14.2.0 (SAS Institute Inc.). Repeated measures ANOVA values were through jamovi (version 0.9). Statistically significant differences were identified at p ≤ .05; trends were noted at p ≤ .10.
3 |. RESULTS
3.1 |. Measurement of infarct size
The presence of ischemic (unstained) tissue was consistently observed in the left ventricle and areas inferior to the ligation site, confirming successful MI (Figure 1B). When infarct areas were normalized by total heart area (IA/TA), there was no significant difference between the infarct sizes of the β3-AR antagonist treated mice and that of untreated mice (Figure 1A).
FIGURE 1.
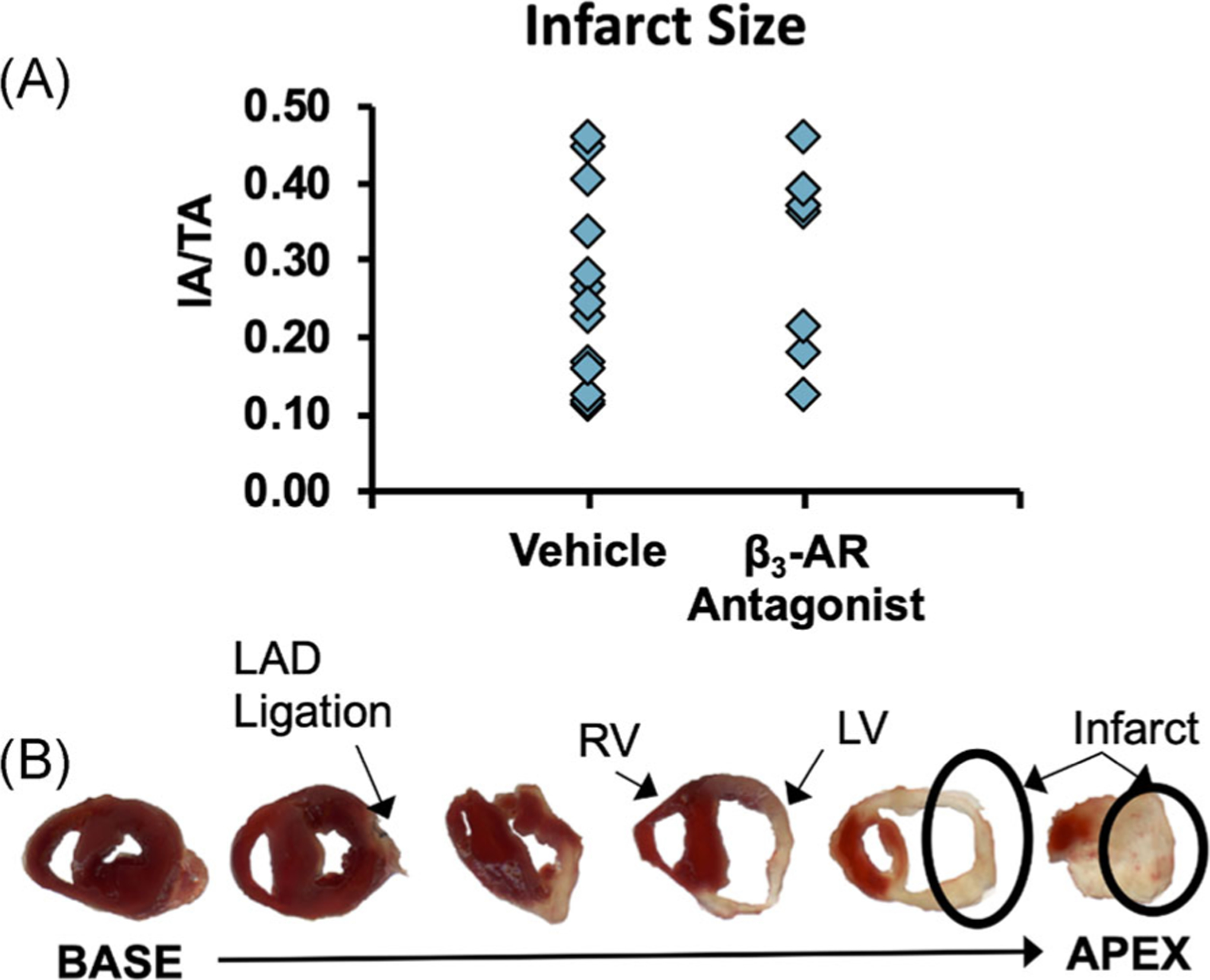
(A) Infarct sizes normalized by total heart area for mice following the surgical creation of MI. No differences were observed between untreated mice and mice treated with a β3-adrenergic receptor antagonist (IA/TA = infarct area/total area). (B) Representative 1 mm thick cross-sections of a mouse heart 10 days after MI surgery. β3-AR, beta 3-adrenergic receptor; MI, myocardial infarction
3.2 |. White blood cell analysis
At 10 days post-MI, circulating monocyte levels were significantly greater in MI mice than in control mice (Figure 2A; main effect of MI: p = .007), and mice treated with the β3-AR antagonist had a higher percentage of monocytes in the serum than untreated mice (main effect of treatment: p = .033). Similarly, neutrophil levels in MI mice were higher than in control mice (Figure 2B; main effect of MI: p = .0003), though there was no significant effect of treatment on neutrophil levels. No significant interaction was observed between MI and β3-AR antagonist treatment for either monocyte or neutrophil levels.
FIGURE 2.
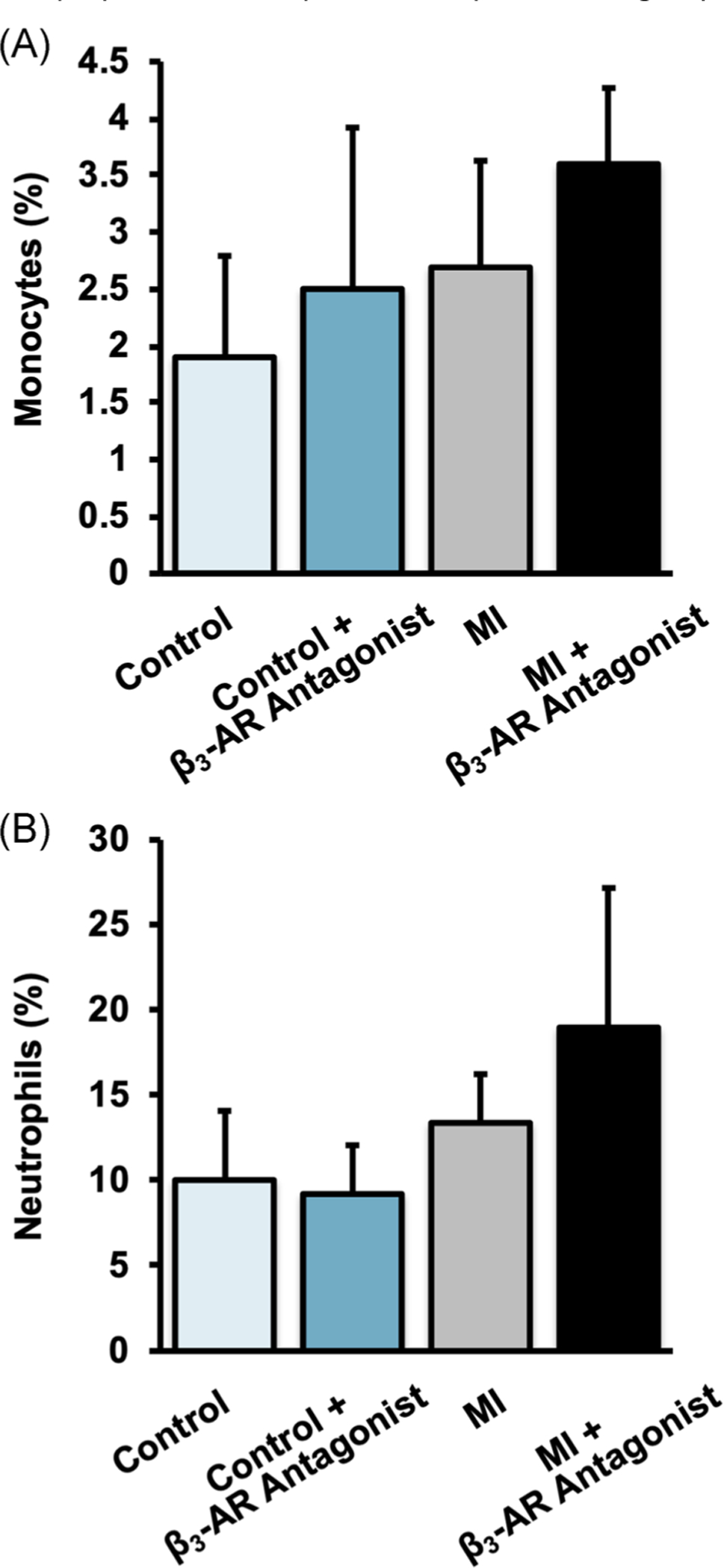
Percent of (A) monocytes and (B) neutrophils in blood 10 days post-MI. Monocyte levels were significantly increased by MI (main effect of MI: p = .007), and by treatment with a β3-AR antagonist (main effect of treatment: p = .033). Similarly, neutrophil levels were significantly increased by MI (main effect of MI: p = .0003). β3-AR, beta 3-adrenergic receptor; MI, myocardial infarction
3.3 |. Dual-energy x-ray absorptiometry analysis
Whole-body DXA of mice revealed few significant changes from baseline to 9 days post-MI in any of the experimental groups and no statistically significant differences based on MI or β3-AR antagonist treatment (Figure 3). Generally, whole-body BMD decreased in MI mice from baseline to 9 days post-MI (though not statistically significant), while BMD of control mice increased significantly during this time period. Whole-body BMC followed similar trends, with MI mice exhibiting less of an increase from baseline than control mice on average.
FIGURE 3.
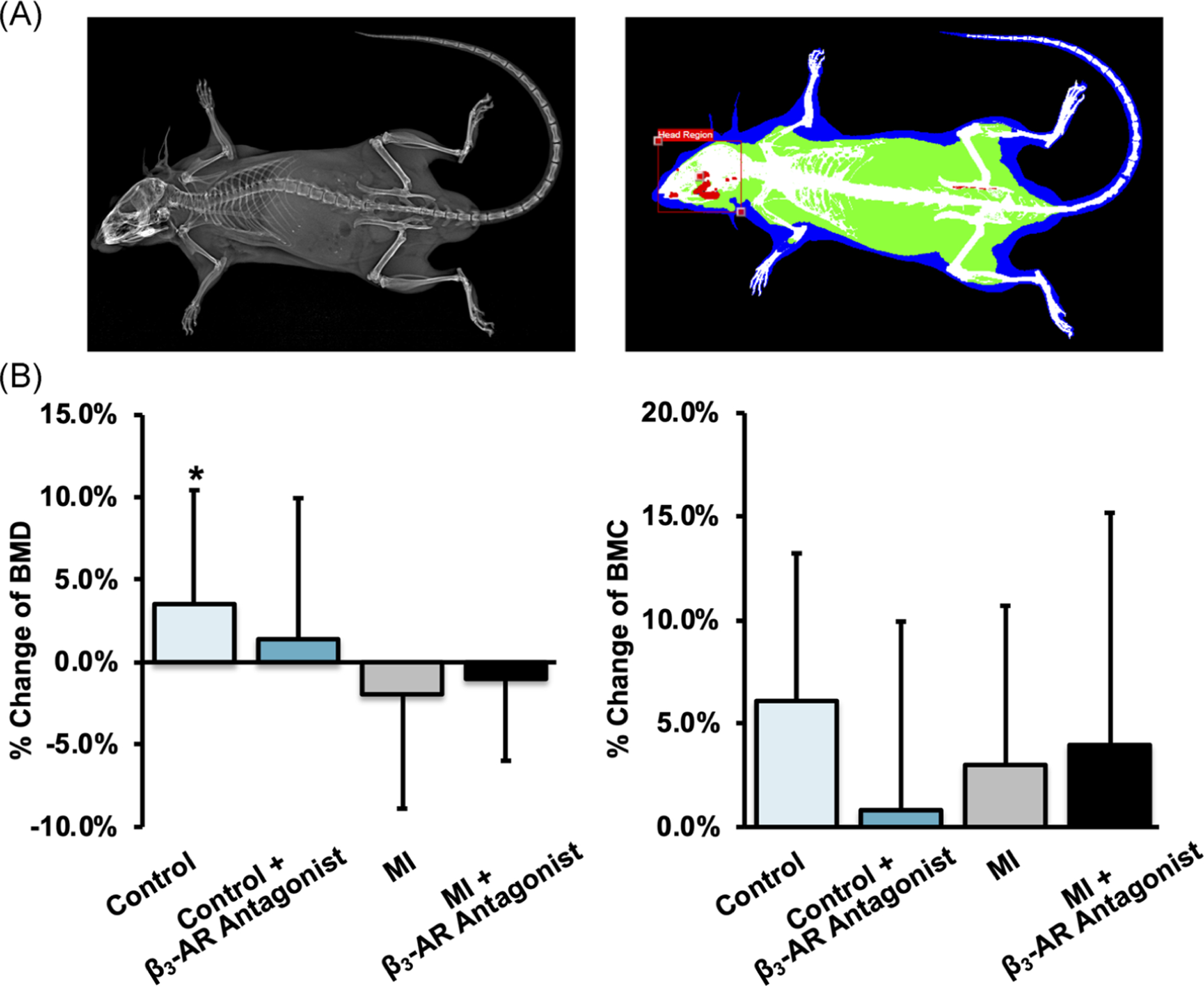
(A) X-ray (left) and DEXA (right) images of the whole mouse. (B) The average change of whole-body BMD (left) and BMC (right) from baseline to 9 days post-MI. * denotes significant change (p ≤ .05) from baseline to 9 days post-MI. β3-AR, beta 3-adrenergic receptor; BMC, bone mineral content; BMD, bone mineral density; DEXA, dual-energy x-ray absorptiometry; MI, myocardial infarction
Results from the analysis of BMD and BMC in the lumbar spine were more definitive (Figure 4). At this skeletal site, control mice generally showed an increase in BMD and BMC, while MI mice showed decreases in BMD and BMC from baseline to 9 days post-MI. Additionally, untreated mice exhibited greater changes from baseline (+2.5% BMD and +6.0% BMC for control mice, −3.5% BMD and −4.2% BMC for MI mice) compared with β3-AR antagonist treated mice (+0.4% BMD and +3.7% BMC for control mice, −1.2% BMD and −2.5% BMC for MI mice).
FIGURE 4.
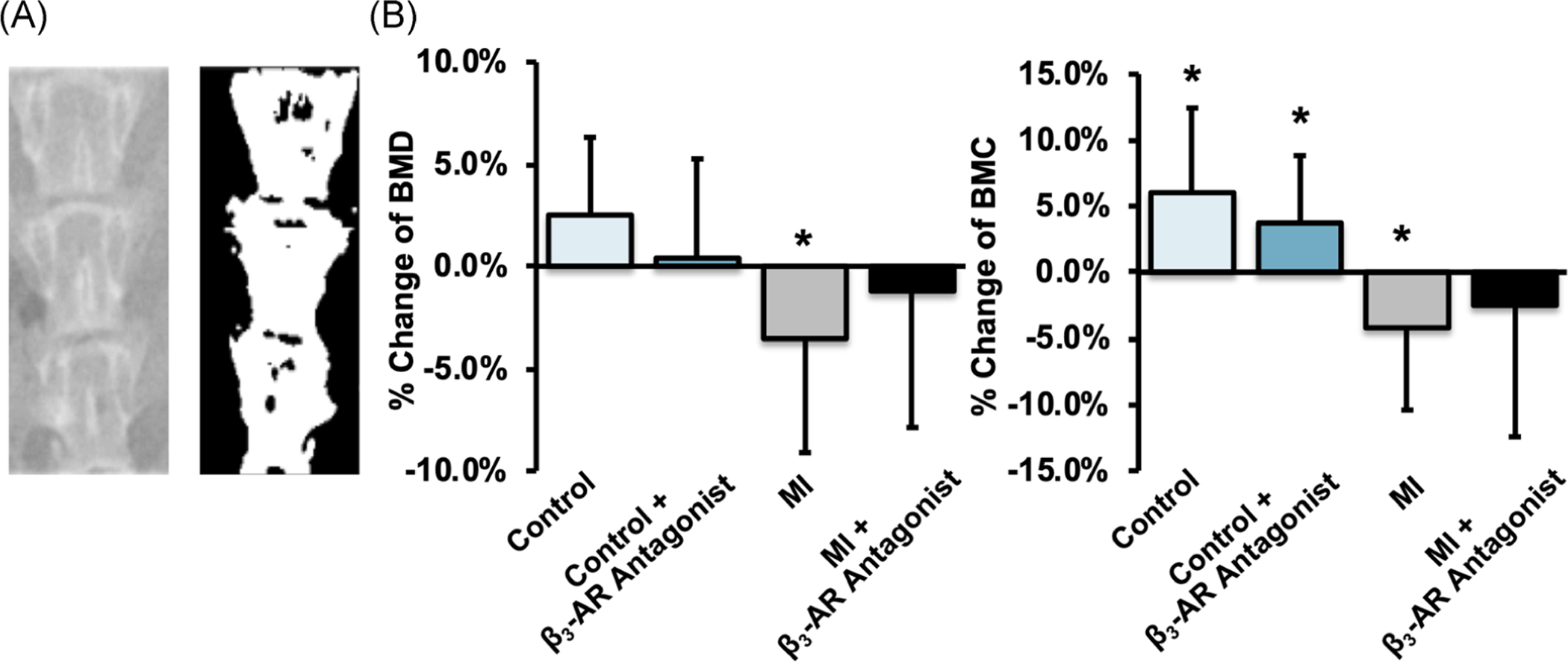
(A) X-ray (left) and thresholded (right) images of the lumbar spine (L4–L6). (B) The average change of lumbar BMD (left) and BMC (right) from baseline to 9 days post-MI. Overall, average lumbar BMD and BMC of MI mice decreased from baseline to 9 days post-MI, and untreated mice exhibited greater changes than β3-AR antagonist treated mice. * denotes significant change (p ≤ .05) from baseline to 9 days post-MI. β3-AR, beta 3-adrenergic receptor; BMC, bone mineral content; BMD, bone mineral density; MI, myocardial infarction
Results from the analysis of BMD and BMC of the femur followed similar trends to those from the lumbar spine. For both the whole femur and femoral diaphysis (Figure 5A and C), BMD in the MI groups decreased significantly and this decrease was greater for untreated mice (−6.9% whole femur, p = .017; −5.6% diaphysis, p = .040) than for β3-AR antagonist treated mice (−5.3% whole femur, p = .034; −4.5% diaphysis, p = .081). No significant differences were observed between β3-AR antagonist treated control mice and untreated control mice. BMC of the whole femur showed similar results, with BMC of untreated MI mice decreasing from baseline to 9 days post-MI (−5.2% whole, p = .018; −6.5% diaphysis, p = .012); this change in BMC was less in β3-AR antagonist treated MI mice.
FIGURE 5.
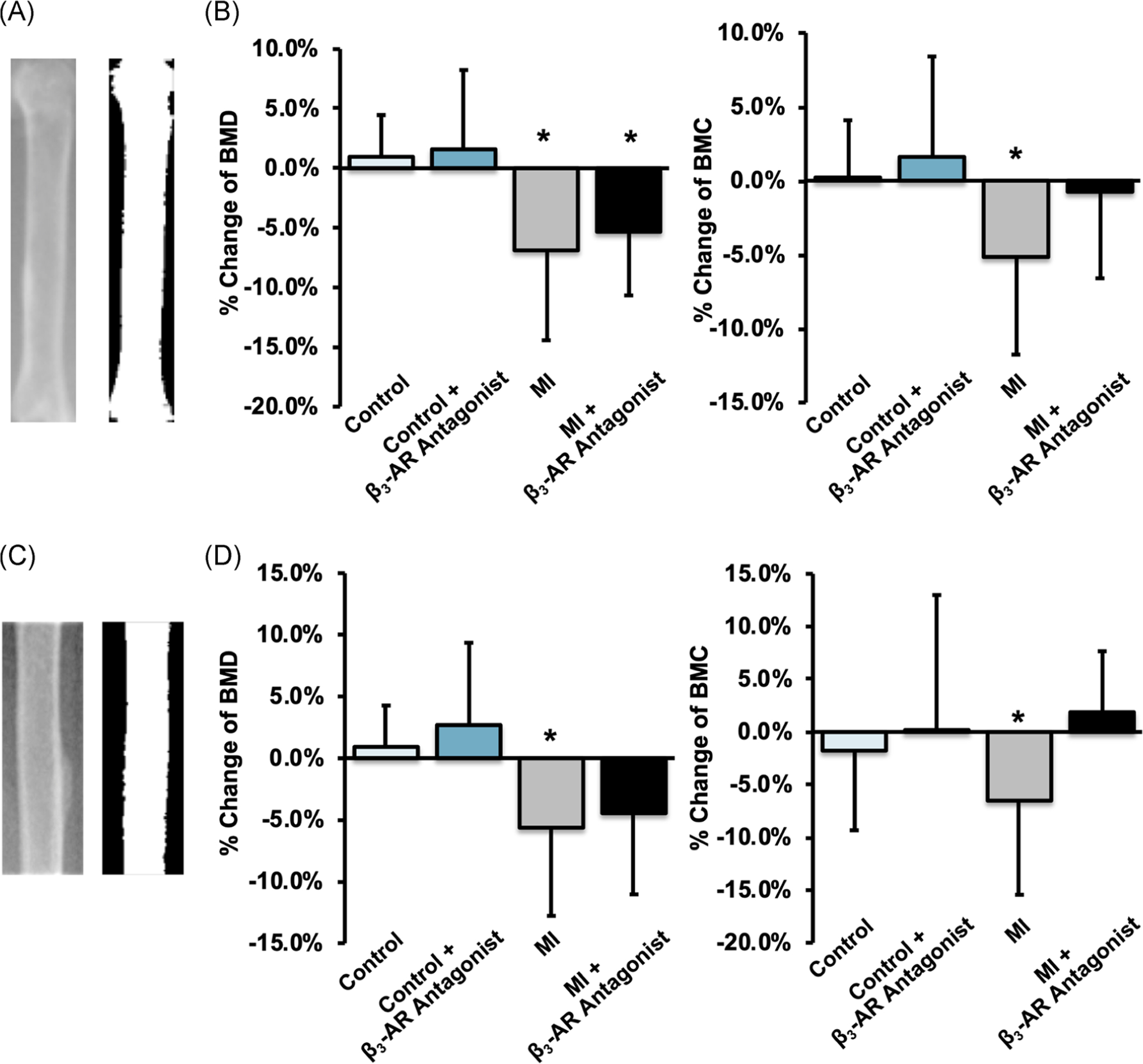
(A,C) X-ray and thresholded images of the whole femur (A) and the femoral diaphysis (C). (B,D) The average change of femoral BMD (left) and BMC (right) from baseline to 9 days post-MI for the whole femur and femoral diaphysis. Both BMD and BMC decreased significantly in MI mice, and this decrease was greater for untreated mice than for β3-AR antagonist treated mice. * denotes p ≤ .05 between baseline and 9 days post-MI. β3-AR, beta 3-adrenergic receptor; BMC, bone mineral content; BMD, bone mineral density; MI, myocardial infarction
Whole-body adipose tissue area decreased from baseline to 9 days for all groups (Figure 6A), though this change was not statistically significant for untreated control mice. Whole-body lean tissue area, however, decreased only in MI mice (main effect of MI: p < .001), and this change was greatest for β3-AR antagonist treated MI mice (−18.3%, p ≤ .001).
FIGURE 6.
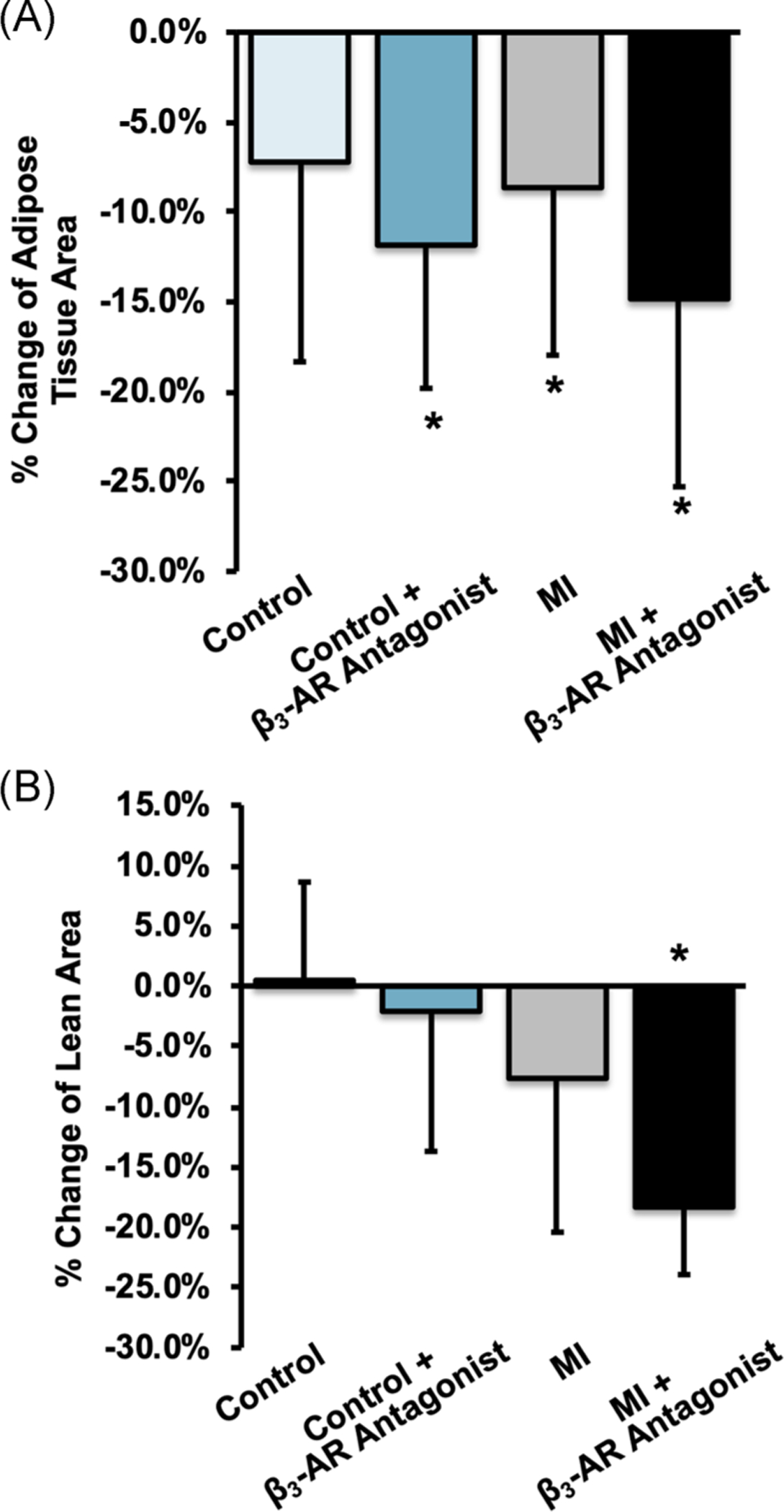
Average change in (A) whole-body fat area and (B) whole-body lean tissue area between baseline and 9 days post-MI. Fat area decreased all groups, but lean tissue area decreased only in MI mice (main effect of MI: p < .001), and this change was greatest for β3-AR antagonist treated MI mice (−18.3%, p ≤ .001). * denotes p ≤ .05 between baseline and 9 days post-MI. β3-AR, beta 3-adrenergic receptor; MI, myocardial infarction
3.4 |. Micro-computed tomography analysis
Trabecular bone analysis of the axial (L5 vertebral body) and appendicular (distal femoral metaphysis) skeleton yielded results that were somewhat consistent with those from DXA analysis (Figure 7). Untreated MI mice exhibited an 8.2% lower BV/TV in the L5 vertebral body than untreated control mice (p = .103); untreated MI mice also exhibited an 11.4% lower BV/TV (p = .051) and a 6.6% lower Tb.Th (p = .094) in the distal femoral metaphysis than untreated control mice. These MI-associated differences were smaller in β3-AR antagonist treated mice, though this may be in part due to β3-AR antagonist treated control mice having lower BV/TV and Tb.Th than untreated control mice. No significant differences were observed between any experimental groups for cortical bone microstructural outcomes at the femoral diaphysis.
FIGURE 7.
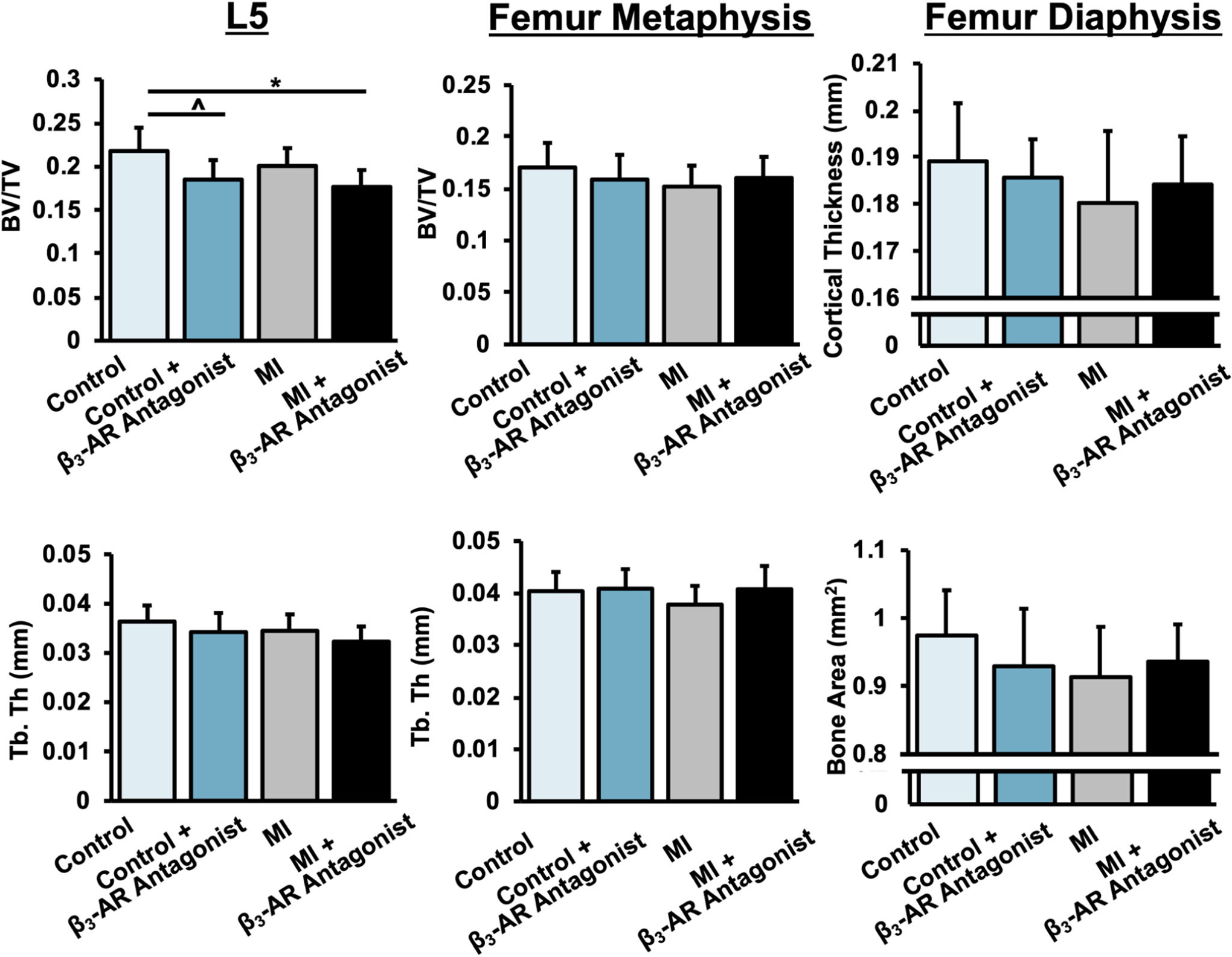
Trabecular bone volume fraction (BV/TV) and trabecular thickness (Tb.Th) of trabecular bone in the L5 vertebral body (left) and distal femoral metaphysis (center); cortical thickness and cortical bone area of the femoral mind-diaphysis (right) 10 days post-MI. BV/TV and Tb.Th were lower in MI mice, and these MI-associated differences were largely mitigated in β3-AR antagonist treated mice. p < .05 are marked by lines between groups. β3-AR, beta 3-adrenergic receptor; MI, myocardial infarction
3.5 |. Three-point bending mechanical testing of femora
Mechanical testing of femurs in three-point bending revealed no significant differences between groups in tissue material properties such as modulus of elasticity and ultimate stress (Table S1). Similarly, we observed no significant differences in structural properties such as stiffness, ultimate force, and postyield displacement. However, we observed significant interactions between MI and β3-AR antagonist treatment for yield force (p = .004) and yield stress (p = .046).
3.6 |. Correlations of bone outcomes with infarct size
No significant correlations were observed between infarct size and monocyte or neutrophil levels, or between infarct size and DXA data for whole-body or regional measurements. However, we observed significant negative correlations between infarct size and trabecular microstructure in the L5 vertebral body and femoral metaphysis (Figure S1). In the L5 region, untreated MI mice had significant correlations for BV/TV (R2 = .414, p = .018) and apparent BMD (R2 = .453, p = .018) and nearly significant correlation for Tb.Th (R2 = .300, p = .053). In the femoral metaphysis, β3-AR antagonist treated MI mice showed similar significance with BV/TV (R2 = .561, p = .020), apparent BMD (R2 = .582, p = .017), and Tb.Th (R2 = .474, p = .040).
3.7 |. Correlations of areal BMD (DXA) with volumetric μCT outcomes
In both the L5 vertebrae and the femur, correlations between areal BMD measured by DXA and μCT outcomes were positive and statistically significant (Figure S2). Lumbar spine BMD from DXA positively correlated with BV/TV (R2 = .414, p < .001) and Tb.Th (R2 = .385, p < .001) in the L5 vertebral body. Similarly, there was a positive correlation between whole femur BMD from DXA and BV/TV (R2 = .380, p < .001) and Tb.Th (R2 = .149, p = .011) in the trabecular bone in the femoral metaphysis. Femoral diaphysis BMD from DXA was significantly correlated with Ct.Th (R2 = .281, p < .001) and cortical bone area (R2 = .455, p < .001) in the femoral diaphysis from μCT (Figure S2).
4 |. DISCUSSION
In this study, we investigated bone loss in the whole-body and at axial and appendicular skeletal sites following MI in mice. Consistent with our hypothesis, MI mice had a lower BMD and BMC in the axial and appendicular skeletal sites and lower BV/TV and Tb.Th at the same skeletal sites compared with control mice. BMD and BMC change in β3-AR antagonist treated mice showed a relatively muted effect compared with untreated mice. This study is one of the first to show a causative effect of MI leading to systemic bone loss, and these data suggest that the SNS may play a role in this response.
The magnitude of systemic bone loss we observed in this study is consistent with our previous study of bone loss following femoral fracture in mice.9 In both cases, whole-body BMD and BMC decreased in injured mice compared with age-matched control mice within 2 weeks postinjury. The trabecular bone microstructure was also diminished in the L5 vertebral body and distal femur in both injured groups. While μCT results were not statistically significant in the current study, a power analysis showed that increasing our group sizes from n = 11–13 to n = 23–33 would provide sufficient power to show statistically significant differences. The lumbar vertebrae and the femur were chosen as representative skeletal sites for the axial and appendicular skeleton, respectively, where we are able to evaluate trabecular bone. These skeletal sites have different mechanical loading and bone microarchitecture; therefore, it would be reasonable to expect differing responses between axial and appendicular sites, though we observed relatively consistent responses in the current study. Our previous study also included quantification of other variables such as bone formation rate, osteoclast number and activity, voluntary activity, and levels of interleukin 6 (IL-6) in serum. These parameters were not measured in the current study, but we anticipate that we would observe similar trends in these outcomes following MI in mice. Overall, these results suggest that systemic bone loss following MI is comparable to that of femoral fracture. These results are further confirmed by a recent study on apolipoprotein E-deficient (ApoE−/−) mice by Kanazawa et al.28 In this study, ApoE−/− mice underwent MI and were placed into sedentary or exercised groups. Sedentary mice had decreased BMD in the femur, and this effect was mitigated in the exercise group. While it is difficult to determine the role of exercise or SNS-initiated monocytosis in these skeletal changes, it is clearly established that MI can initiate bone loss.
Our findings using a β3-AR antagonist to mitigate bone loss in mice following MI yielded inconsistent results. In the study by Dutta et al.,10 injections with the same β3-AR antagonist decreased the inflammatory response through the increased withdrawal of stem cell retention factors by β3 expressing cells. Four days post-MI, blood HSPC’s levels and inflammatory markers were lower in treated groups.10 In contrast, we observed greater monocyte and neutrophil levels intreated mice, although this could be largely due to the time point when blood was collected (day 4 in Dutta et al.10 vs. day 10 in the current study). Additionally, our DXA results suggest that the β3-AR antagonist led to small decreases in bone volume and mildly diminished systemic bone loss after MI. However, the treatment effect noted by DXA does not fully agree with data from μCT and three-point bending, thus it is difficult to make a definitive conclusion about the effect of the β3-AR antagonist on bone loss. We also observed the effects of β3 inhibition on changes in lean tissue area and adipose tissue area. While it was expected that MI may decrease lean tissue and adipose tissue mass due to postoperation recovery, the addition of the β3-AR antagonist exacerbated the loss of both lean and adipose tissue. These findings are contrary to previous studies that showed that β3 agonists promote weight loss, especially in obese mice and rats.29–32 Further investigation using more direct methodologies would be needed to investigate this discrepancy. Altogether, these inconsistent results of the β3-AR antagonist suggest that SNS-initiated monocytosis does not play a key role in bone loss following MI. Our study did not produce convincing data regarding β3-AR inhibition in preventing bone loss, suggesting that other mechanisms are primarily responsible for MI-induced bone loss.
This study established, for the first time, a novel mechanistic relationship between acute cardiac injury and systemic skeletal remodeling. Despite some epidemiological evidence supporting a link between fracture and MI, this is one of the first studies to show a causal relationship between these two seemingly unrelated events. Interestingly, the epidemiological evidence linking the incidence of fracture and MI suggests this link is bidirectional; therefore, fracture may also exacerbate atherosclerosis, leading to increased risk of subsequent MI. This is supported by a study by Chiang et al.,33 which reported a significantly higher risk of subsequent MI following a hip fracture (HR = 1.29), and a study by Weber et al.,34 which reported structural and functional changes in the heart following long bone fracture associated with complement activation, the release of extracellular histones, and systemic tumor necrosis factor elevation. Further studies are required to further establish this relationship mechanistically.
The current study has some limitations that must be acknowledged. First, quantification of monocytosis was performed by drawing blood through a needle, which allows for the possibility of cell lysing and subsequent inaccuracy in results. Furthermore, our method of measuring monocytosis was a more general assessment of blood cell composition than in Dutta’s study, which looked at many markers of inflammation in more specific areas such as the bone marrow and the spleen, two important areas for monocyte proliferation. Second, the β-blocker we used was specific to β3 receptors, where its interaction with the skeletal system is not as well known as the other types of β-ARs. As a result, it is possible the effects we see with the β3-AR antagonist are partially due to direct interaction with β3 receptors in bone cells, rather than SNS-initiated monocytosis. Third, many of our variables were measured at only one time point (10 days post-MI). We chose this time point because we were initially concerned with the survival of the MI-operated mice, and we have previously shown that peak trabecular bone loss after ACL rupture or femur fracture in mice occurs between 7 and 14 days after injury.9,22 However, it is possible that 10 days is not the time point when peak bone loss occurs following MI. Further studies incorporating multiple time points are needed to fully establish the time course of bone loss and recovery post-MI. Fourth, animals used for this study were young, male mice. Although MI occurs more commonly in the older population and there is evidence that bone remodeling after fracture differs between young and middle-aged mice,9 we chose to study young mice as it allows for minimal confounding factors from existing comorbidities and for their higher survival rate following MI. There are also potential sex-based differences to consider both for MI and bone response. There is evidence that female patients have a less severe apoptotic response to MI, resulting in less ischemic damage.35 This protective advantage may also present in bone remodeling, as estrogen has been known to protect against bone loss.36 Finally, this study did not include direct measures of bone formation, osteoclast differentiation, bone resorption, or other biological outcomes. These analyses will be integral in future studies investigating mechanisms underlying the post-MI changes in bone mass and microstructure. Despite these limitations, this study was able to establish a causal link between MI and bone loss, present a thorough analysis of bone density and microstructural changes initiated by MI at multiple skeletal sites, and correlate these findings to infarct size and other outcomes.
5 |. CONCLUSION
This study is the first to establish a causal relationship between MI and bone loss at multiple skeletal sites and suggests that the SNS may have a limited role in this adaptation. This injury-induced response may also be operative in human subjects after MI, and maybe potentially catastrophic comorbidity in post-MI patients. Further delineating the relationship and mechanisms governing this crosstalk could inform future treatments aimed at preventing injuries and preserving skeletal health following ischemic injuries.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Heike Wulff for her valuable input on this project. The research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under Award Number AR071459. The content is solely the responsibility of the authors. The funding bodies were not involved with design, collection, analysis, or interpretation of data; or in the writing of the manuscript.
Funding information
National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant/Award Number: AR071459
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- 1.Murray CJ, Richards MA, Newton JN, et al. UK health performance: findings of the Global Burden of Disease Study 2010. Lancet. 2013; 381(9871):997–1020. [DOI] [PubMed] [Google Scholar]
- 2.Jiang HX, Majumdar SR, Dick DA, et al. Development and initial validation of a risk score for predicting in-hospital and 1-year mortality in patients with hip fractures. J Bone Miner Res. 2005;20(3):494–500. [DOI] [PubMed] [Google Scholar]
- 3.Schnell S, Friedman SM, Mendelson DA, Bingham KW, Kates SL. The 1-year mortality of patients treated in a hip fracture program for elders. Geriatr Orthop Surg Rehabil. 2010;1(1):6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee YK, Lee YJ, Ha YC, Koo KH. Five-year relative survival of patients with osteoporotic hip fracture. J Clin Endocrinol Metab. 2014; 99(1):97–100. [DOI] [PubMed] [Google Scholar]
- 5.Peterson MG, Cornell CN, Paget SA, Allegrante JP. Five-year survival in a cohort of hip fracture patients: the predictive role of pre-fracture health status. HSS J. 2008;4(1):43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart disease and stroke statistics—2017 update: a report from the American Heart Associate. Circulation. 2017;135(10):e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerber Y, Melton LJ 3rd, Weston SA, Roger VL. Association between myocardial infarction and fractures: an emerging phenomenon. Circulation. 2011;124(3):297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farhat GN, Cauley JA. The link between osteoporosis and cardiovascular disease. Clin Cases Miner Bone Metab. 2008;5(1):19–34. [PMC free article] [PubMed] [Google Scholar]
- 9.Emami AJ, Toupadakis CA, Telek SM, Fyhrie DP, Yellowley CE, Christiansen BA. Age dependence of systemic bone loss and recovery following femur fracture in mice. J Bone Miner Res. 2019; 34(1):157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dutta P, Courties G, Wei Y, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohtsuji M, Lin Q, Okazaki H, et al. Anti-CD11b antibody treatment suppresses the osteoclast generation, inflammatory cell infiltration, and autoantibody production in arthritis-prone FcgammaRIIB-deficient mice. Arthritis Res Ther. 2018;20(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeda S, Elefteriou F, Levasseur R, et al. Leptin regulates bone formation via the sympathetic nervous system. Cell. 2002;111(3):305–317. [DOI] [PubMed] [Google Scholar]
- 13.Moore RE, Smith CK II, Bailey CS, Voelkel EF, Tashjian AH Jr. Characterization of beta-adrenergic receptors on rat and human osteoblast-like cells and demonstration that beta-receptor agonists can stimulate bone resorption in organ culture. Bone Miner. 1993; 23(3):301–315. [DOI] [PubMed] [Google Scholar]
- 14.Togari A, Arai M, Mizutani S, Mizutani S, Koshihara Y, Nagatsu T. Expression of mRNAs for neuropeptide receptors and β-adrenergic receptors in human osteoblasts and human osteogenic sarcoma cells. Neurosci Lett. 1997;233(2–3):125–128. [DOI] [PubMed] [Google Scholar]
- 15.Kellenberger S, Muller K, Richener H, Bilbe G. Formoterol and isoproterenol induce c-fos gene expression in osteoblast-like cells by activating β2-adrenergic receptors. Bone. 1998;22(5):471–478. [DOI] [PubMed] [Google Scholar]
- 16.Majeska R, Minkowitz B, Bastian W, Einhorn T. Effects of β-adrenergic blockade in an osteoblast-like cell line. J Orthop Res. 1992;10(3):379–384. [DOI] [PubMed] [Google Scholar]
- 17.Elefteriou F, Ahn JD, Takeda S, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434(7032):514–520. [DOI] [PubMed] [Google Scholar]
- 18.Kondo H, Takeuchi S, Togari A. β-Adrenergic signaling stimulates osteoclastogenesis via reactive oxygen species. Am J Physiol Endocrinol Metab. 2012;304(5):E507–E515. [DOI] [PubMed] [Google Scholar]
- 19.Whitsett JA, Burdsall J, Workman L, Hollinger B, Neely J. β-adrenergic receptors in pediatric tumors: uncoupled β1-adrenergic receptor in Ewing’s Sarcoma23. J Natl Cancer Inst. 1983;71(4):779–786. [PubMed] [Google Scholar]
- 20.Nuntapornsak A, Wongdee K, Thongbunchoo J, Krishnamra N, Charoenphandhu N. Changes in the mRNA expression of osteoblast-related genes in response to β3-adrenergic agonist in UMR106 cells. Cell Biochem Funct. 2010;28(1):45–51. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi T, Tsuboi T, Arai M, Togari A. Adrenergic stimulation of osteoclastogenesis mediated by expression of osteoclast differentiation factor in MC3T3-E1 osteoblast-like cells. Biochem Pharmacol. 2000;61(5):579–586. [DOI] [PubMed] [Google Scholar]
- 22.Christiansen BA, Anderson MJ, Lee CA, Williams JC, Yik JH, Haudenschild DR. Musculoskeletal changes following non-invasive knee injury using a novel mouse model of post-traumatic osteoarthritis. Osteoarthr Cartil. 2012;20(7):773–782. [DOI] [PubMed] [Google Scholar]
- 23.De Jesus NM, Wang L, Herren AW, et al. Atherosclerosis exacerbates arrhythmia following myocardial infarction: role of myocardial inflammation. Heart Rhythm. 2015;12(1):169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rueden CT, Schindelin J, Hiner MC, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017;18(1):529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7): 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using microcomputed tomography. J Bone Miner Res. 2010; 25(7):1468–1486. [DOI] [PubMed] [Google Scholar]
- 27.Jepsen KJ, Silva MJ, Vashishth D, Guo XE, van der Meulen MCH. Establishing biomechanical mechanisms in mouse models: practical guidelines for systematically evaluating phenotypic changes in the diaphyses of long bones. J Bone Miner Res. 2015;30(6):951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanazawa M, Matsumoto Y, Takahashi K, et al. Treadmill exercise prevents reduction of bone mineral density after myocardial infarction in apolipoprotein E-deficient mice. Eur J Prev Cardiol. 2020;27(1):28–35. [DOI] [PubMed] [Google Scholar]
- 29.Collins S, Daniel KW, Petro AE, Surwit RS. Strain-specific response to beta 3-adrenergic receptor agonist treatment of diet-induced obesity in mice. Endocrinology. 1997;138(1):405–413. [DOI] [PubMed] [Google Scholar]
- 30.de Souza CJ, Burkey BF. Beta 3-adrenoceptor agonists as anti-diabetic and anti-obesity drugs in humans. Curr Pharm Des. 2001; 7(14):1433–1449. [DOI] [PubMed] [Google Scholar]
- 31.White CL, Ishihara Y, Dotson TL, Hughes DA, Bray GA, York DA. Effect of a beta-3 agonist on food intake in two strains of rats that differ in susceptibility to obesity. Physiol Behav. 2004;82(2–3): 489–496. [DOI] [PubMed] [Google Scholar]
- 32.Xiao C, Goldgof M, Gavrilova O, Reitman ML. Anti-obesity and metabolic efficacy of the β3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22°C. Obesity. 2015;23(7):1450–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiang CH, Liu CJ, Chen PJ, et al. Hip fracture and risk of acute myocardial infarction: a nationwide study. J Bone Miner Res. 2013; 28(2):404–411. [DOI] [PubMed] [Google Scholar]
- 34.Weber B, Lackner I, Knecht D, et al. Systemic and cardiac alterations after long bone fracture [published online ahead of print March 19, 2020]. Shock. 10.1097/SHK.0000000000001536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunlay SM, Roger VL. Gender differences in the pathophysiology, clinical presentation, and outcomes of ischemic heart failure. Curr Heart Fail Rep. 2012;9(4):267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen JR, Haley RL, Hidestrand M, et al. Estradiol protects against ethanol-induced bone loss by inhibiting up-regulation of receptor activator of nuclear factor-kappaB ligand in osteoblasts. J Pharmacol Exp Ther. 2006;319(3):1182–1190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.