

Abstract
Alterations in muscle mitochondrial substrate preference have been postulated to play a major role in the pathogenesis of muscle insulin resistance. In order to examine this hypothesis, we assessed the ratio of mitochondrial pyruvate oxidation (VPDH) to rates of mitochondrial citrate synthase flux (VCS) in muscle. Contrary to this hypothesis we found that high fat diet (HFD)-fed insulin resistant rats did not manifest altered muscle substrate preference (VPDH/VCS) in soleus or quadriceps muscles in the fasting state. Furthermore, hyperinsulinemic-euglycemic (HE) clamps increased VPDH/VCS in both muscles in normal and insulin resistant rats. We then examined muscle VPDH/VCS flux in insulin sensitive and insulin resistant humans and found similar relative rates of VPDH/VCS following an overnight fast (~20%) and similar increases in VPDH/VCS fluxes during a HE clamp. Taken together, these findings demonstrate that alterations in mitochondrial substrate preference are not an essential step in the pathogenesis of muscle insulin resistance.
eTOC
Lipid-induced hepatic insulin resistance is mediated by plasma membrane bound sn-1,2-diacylglycerols, which promote PKCε translocation to the plasma membrane. PKCε is both necessary and sufficient for mediating lipid-induced hepatic insulin resistance through phosphorylation of insulin receptor Thr1160.
Graphical Abstract
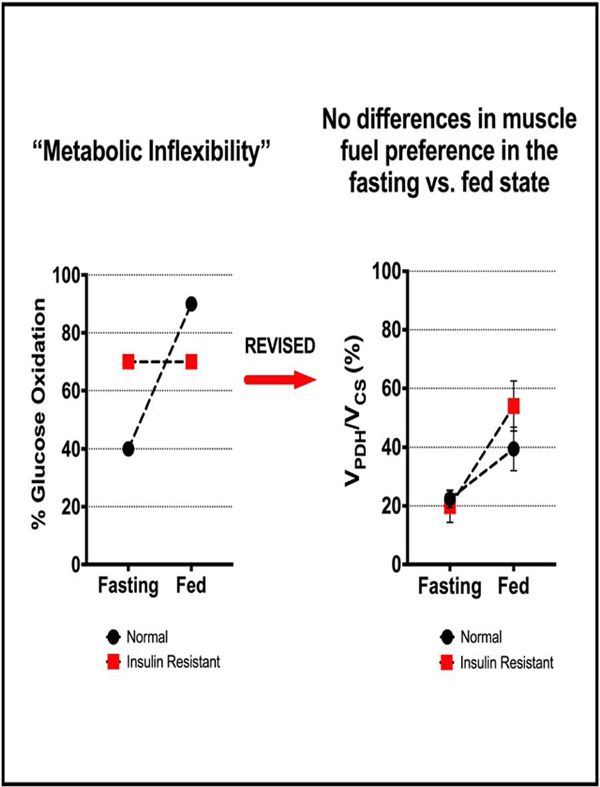
Introduction
Type 2 diabetes (T2D) is characterized by defects in insulin’s ability to suppress hepatic glucose production and stimulate glucose uptake by peripheral tissues (Shulman 2000). Skeletal muscle, which accounts for the majority of insulin-stimulated peripheral glucose uptake, plays a major role in the development of whole body insulin resistance and progression to T2D, and intramyocellular ectopic lipid accumulation and associated impairments in insulin-stimulated muscle glucose uptake have been observed to precede the onset of T2D (Jacob et al., 1999; Krssak et al., 1999; Perseghin et al., 1999). In addition, exercise has been shown to bypass impairments in insulin-mediated GLUT4 translocation, improving peripheral glucose disposal (Kennedy et al., 1999; Perseghin et al., 1996; Ren et al., 1994). Thus, there is considerable interest in determining the precise mechanism by which skeletal muscle insulin resistance develops.
In this setting, alterations in metabolic substrate preference and oxidation have been hypothesized as essential derangements in skeletal muscle insulin resistance. In a seminal study, Randle et al. proposed that increased mitochondrial fat oxidation could directly inhibit glycolysis, leading to a build-up of glycolytic intermediates, which in turn would lead to decreased cellular glucose uptake (Randle et al., 1963).
More recently, the concept of ‘metabolic inflexibility’ has been proposed to cause muscle insulin resistance. The metabolic inflexibility hypothesis, coined by Kelley et al., was based on in vivo experiments in humans, which found through leg-specific indirect calorimetry techniques that the limb of normal individuals relied predominantly on fat oxidation in the fasting state and on glucose oxidation in the insulin-stimulated state. In contrast insulin resistant individuals: 1) had paradoxically increased preference for muscle glucose oxidation in the fasting state and 2) could not modulate their muscle substrate preference in response to insulin (Kelley et al., 1992, 1999). These investigators went on to conclude that insulin resistance was a manifestation of underlying cellular ‘metabolic inflexibility,’ or the inability to alter muscle substrate oxidation in varying physiologic states (Kelley, 2005). Kelley and others have further hypothesized that defects in the mitochondrial enzymatic machinery that regulates both basal substrate metabolism and transitions between glucose and fat oxidation lead to “metabolic gridlock” and were mechanistically responsible for the onset of muscle insulin resistance and cardiometabolic disease (Muoio, 2014; Muoio et al., 2012). The concept of metabolic inflexibility has gained considerable attention in the field of metabolism, and recent studies have proposed aberrations in pyruvate dehydrogenase (PDH) (Zhang et al., 2014), pyruvate dehydrogenase kinase (PDK) isoforms (Holness et al., 2000), carnitine-palmitoyl transferase-1 (CPT1) (Koves et al., 2008), and carnitine acetyltransferase (CrAT) (Muoio, 2014; Muoio et al., 2012) as possible mediators of altered substrate handling in insulin resistant muscle and, therefore, as potential therapeutic targets in cardiometabolic disease and T2D.
Despite the concept’s widespread acceptance, whether alterations in muscle mitochondrial substrate oxidation explain muscle insulin resistance in vivo remains to be confirmed. In fact, the indirect calorimetric technique employed in Kelley’s original studies has significant methodological shortcomings that preclude making definitive conclusions about tissue-specific substrate oxidation and changes therein. For one, using arterio-venous (A-V) to measure respiratory quotient (RQ) across the leg are not truly muscle-specific but also reflect glucose and fat oxidation in additional tissues that share the same venous drainage in the leg such as fat, skin and bone. Further, indirect calorimetry captures substrate disappearance rather than oxidation and does not account for interconversions between metabolic substrates that occur in vivo and thus, is an imperfect measurement of true substrate oxidation in the cell (Simonson and DeFronzo, 1990). To date, no conclusive in vivo evidence exists that insulin resistant muscles have altered substrate preference in the fasting state and that the blunting of insulin’s ability to increase muscle glucose oxidation is a result of differences in muscle mitochondrial substrate oxidation rather than a primary defect in insulin-mediated glucose transport.
To directly assess whether alterations in muscle mitochondrial substrate preferences are responsible for muscle insulin resistance and avoid the limitations of the A-V indirect calorimetry methodology we applied a combined stable isotope tracer/liquid chromatography tandem mass spectrometry (LC-MS/MS) approach to directly quantify muscle-specific mitochondrial substrate preference and examine whether ‘metabolic inflexibility’, as described by Kelley et al., is operating in an in vivo rat model of insulin resistance as well as in insulin resistant humans. By infusing [13C6]glucose and analyzing 13C enrichments of alanine and glutamate isotopologues in muscles, we measured mitochondrial pyruvate dehydrogenase flux (VPDH) as a proportion of mitochondrial citrate synthase flux (VCS), a tissue-specific index for the relative contribution of glucose oxidation to total mitochondrial oxidation (VPDH/VCS). Using this method, we studied how induction of muscle insulin resistance in rats by high fat diet (HFD) feeding affects muscle mitochondrial substrate preference in the fasted state as well as during a hyperinsulinemic-euglycemic (HE)-clamp. In addition, we tested whether acute infusion of lipid during a HE clamp, thus putting glucose and fatty acids in direct metabolic competition, in normal rats could modulate muscle mitochondrial substrate oxidation in vivo and whether these modulations in muscle oxidative substrate preference would result in changes in whole body insulin sensitivity. Finally, in order to determine whether these results would translate to humans we applied a similar 13C stable isotope/LC-MS/MS methodology in muscle biopsy samples to assess basal and insulin-stimulated rates of VPDH/VCS along with in vivo 13C MRS measurements performed at 7-Tesla of insulin-stimulated mitochondrial citrate synthase flux (VCS) and rates of glycogen synthesis (VGLY) in skeletal muscle of insulin sensitive and insulin resistant humans.
Results
In comparison to normal chow fed rats, rats fed 4 weeks of a HFD exhibited increased body weight (Fig. S1A) and became insulin resistant, as manifested by higher plasma insulin concentrations required to maintain the same plasma glucose concentrations in the fasting state (Fig. 1A-B) as well as by reduced glucose infusion rate required to maintain euglycemia during a HE clamp (Fig. S1E). Furthermore, insulin-stimulated rates of muscle glucose uptake and muscle glucose transport, as measured by [14C]2-deoxyglucose uptake, were significantly impaired during the HE clamp (Fig. 1F-G), confirming the presence of muscle insulin resistance in HFD fed-rats.
Figure 1. HFD feeding induces muscle insulin resistance and defects in insulin signaling but these changes are not associated with alterations in muscle mitochondrial substrate preference.

A-B. HFD-fed rats have normal fasting blood glucose but higher plasma insulin levels. C. Muscle of insulin-resistant rats exhibit impaired IRK tyrosine1162 phosphorylation. D. Muscle of insulin-resistant rats exhibit impaired IRS-1-associated PI3K activity. E. Muscle of insulin-resistant rats has impaired insulin signaling as measured by AKT serine473 phosphorylation. F. HFD-fed rats have impaired muscle glucose uptake as measured by peripheral glucose disposal (Rd). G. HFD-fed rats have impaired muscle glucose transport as measured by 2-[14C]deoxyglucose (2-DG) uptake. H-I. HFD-fed insulin resistant rats do not exhibit altered VPDH/VCS fluxes in soleus and quad muscles in the fasting state. Insulin stimulation during a clamp increases VPDH/VCS in muscle in both groups, but is blunted in HFD-fed rats. 2-DG uptake measured in soleus, other muscle measures in quadriceps muscle due to limitation in the amount of tissue. Data are represented as mean ± SEM.
To test whether the metabolic inflexibility hypothesis could explain the development of muscle insulin resistance in these HFD-fed rats, we examined whether the HFD-fed rats had altered oxidative substrate preference in skeletal muscle in the basal state. We used [13C6] glucose as a tracer during the clamp and used tissue-specific 13C enrichments of alanine and glutamate to measure in vivo VPDH/VCS in soleus and quadriceps muscles (Alves et al., 2011; Ferris et al., 2017; Perry et al., 2018).
Using this approach, we found that, contrary to what Kelley et al. had observed in humans using A-V indirect calorimetry, skeletal muscle mitochondria of overnight fasted regular chow fed rats relied predominantly (>90%) on substrates other than pyruvate for oxidation, which was assumed to be largely fatty acids and to a lesser extent ketogenic amino acids (Fig. 1H-I) (Kelley et al., 1992, 1999). Furthermore, in contrast to what the metabolic inflexibility hypothesis would predict, we found that this strong preference for fat oxidation in both soleus and quad muscle during the fasting state did not change in insulin resistant HFD fed-rats (Fig. 1H-I). In response to insulin during a HE clamp, regular chow fed rats increased VPDH/VCS in soleus muscle to ~50% and in quad muscles, which contain a lower proportion of slow twitch (oxidative) to fast twitch (glycolytic) fibers, to ~25% (Fig. 1H-I). In HFD-fed rats, however, this increase in VPDH/VCS in response to insulin was blunted in both skeletal muscles (Fig. 1H-I).
This impairment in the ability of HFD-fed rats to increase VPDH/VCS in response to hyperinsulinemia could have resulted either from a defect in the mitochondrial oxidative capacity or from limited substrate availability secondary to a defect in insulin-mediated glucose transport. To differentiate between these possibilities we first measured phosphorylation of PDH (s232, s293, s300) in both fasting and clamped states between the two groups, and observed no differences in PDH phosphorylation between groups suggesting that HFD-fed rats did not have major impairments in PDH activity and capacity to oxidize glucose (Fig. 2A). Next we assessed muscle concentrations of tricarboxylic acid cycle metabolites (Fig. 2B) and acylcarnitines (Supplemental Table S1) and found that the intramyocellular concentrations of these metabolites were not altered with HFD feeding, making it unlikely that the mitochondria of insulin resistant rats had ‘metabolic overload’ that might impede smooth transitions between fat and glucose oxidation (Muoio, 2014; Muoio et al., 2012). Finally we assessed basal expression levels of key oxidative enzymes and found that there were no differences in the protein expression of PDH, CPT1b, and CrAT, a regulator of the mitochondrial acetyl-CoA pool that was suggested as a candidate for altered substrate handling in the metabolic inflexibility hypothesis of insulin resistance (Fig. 2C). Taken together, we did not find any evidence that the mitochondria of HFD-fed rats acquired defects in mitochondrial oxidative capacity that could explain the impairments in insulin-stimulated glucose oxidation we observed in skeletal muscle. To test whether impairments in glucose transport could provide an alternative explanation, we measured previously reported mediators of ectopic lipid-induced inhibition of insulin signaling and found that HFD-fed rats displayed increased baseline translocation of protein kinase Cθ (PKCθ) and PKCε, (Fig. 2D). Furthermore, consistent with activation of PKCθ and PKCε leading to proximal defects in insulin signaling, HFD-fed rats manifested significantly reduced insulin-induced insulin receptor kinase phosphorylation (Fig. 1C), reduced insulin receptor substrate-1 (IRS-1)-associated phosphoinositide 3-kinase (PI3-K) activity (Fig. 1D) and impaired insulin-stimulated phosphorylation of AKT (Fig. 1 D). Taken together, these results are consistent with previously reported observations that the major defect in the early stages of the pathogenesis of muscle insulin resistance is DAG-nPKC inhibition of insulin signaling (Szendroedi et al., 2014; Yu et al., 2002), rather than alterations in mitochondrial substrate preference.
Figure 2. HFD feeding induces translocation of PKCθ and PKCε but is unassociated with alterations in pPDH/PDH, mitochondrial metabolites or expression of key oxidative enzymes.

A. Phosphorylation of muscle PDH Serine293, Serine232, and Serine300 is unchanged in HFD-fed insulin resistant rats compared to regular chow fed rats in both basal and clamped states. B. Muscle concentrations of mitochondrial metabolites are unchanged in muscles of HFD-fed insulin resistant rats. C. Expression of key oxidative enzymes PDH, CPT1b, and CrAT are unchanged in muscles of insulin-resistant rats. D. Insulin-resistant rats have increased PKCθ and PKCε translocation (n=5 for each group). All data measured in quadriceps muscle. Data are represented as mean ± SEM.
Given that insulin resistant HFD-fed rats failed to display alterations in basal substrate use, we assessed whether acute modulation of substrate delivery could influence muscle VPDH/VCS in response to hyperinsulinemia in RC fed rats and if so whether this alteration in VPDH/VCS would result in impairment in insulin-stimulated peripheral glucose metabolism. HE clamps suppressed fasting white adipocyte tissue (WAT) lipolysis as exhibited by reductions in non-esterified fatty acid (NEFA) from ~1.0 mM to ~0.2 mM, but concomitant infusion of intralipid during a clamp raised NEFA back to approximately fasting plasma concentrations (~1.2 mM) (Fig. S2F). Acutely increasing plasma NEFA concentrations under HE conditions led to ~50% reductions in VPDH/VCS in both soleus and quadriceps muscle (Fig. 3C-D), which occurred independently of changes in phosphorylation of PDH (Fig. S2H). Interestingly, this acute infusion of intralipid tended to promote an increase in intramyocellular acetyl-CoA but did not cause an increase in intramuscular glucose-6-phosphate or citrate concentrations (Fig. 3F). This constellation of changes in intramyocellular metabolites with increased fat oxidation is consistent with acetyl-CoA-mediated allosteric inhibition of PDH activity, but not with Randle’s proposed mechanism of fat oxidation-induced inhibition of key glycolytic steps and glucose uptake, which predicts increases in glucose-6-phosphate and citrate concentrations with increased fat oxidation (Randle et al., 1963, 1970). Furthermore, we found that the significant reduction in muscle glucose oxidation seen in the intralipid group was not accompanied by any changes in measures of in vivo muscle insulin response such as whole-body glucose uptake, muscle glucose transport, and muscle AKT phosphorylation at the end of the 2 hr clamp (Fig. 3A-B, E), dissociating alterations in substrate oxidation from skeletal muscle insulin sensitivity.
Figure 3. Short term (90-min) lipid infusion in normal chow fed rats during a HE clamp alters muscle substrate preference (VPDH/VCS) but does not affect whole body or muscle-specific insulin sensitivity.

A. Lipid infusion during a HE clamp in normal chow fed rats does not alter insulin-stimulated muscle glucose uptake. B. Lipid infusion does not alter insulin-stimulated muscle glucose transport (2-DG uptake), measured in soleus. C-D. Lipid infusion lowers insulin-stimulated VPDH/VCS by 50% in soleus and quad muscles. E. Lipid infusion does not change muscle AKT phosphorylation, measured in quad. Muscle acetyl-CoA content trends to an increase with lipid infusion but other metabolites are unchanged. Data are represented as mean ± SEM.
In order to determine whether these results would translate to humans we performed two sets of studies in young, lean, sedentary insulin resistant (IR) individuals and age-weight-activity matched insulin sensitive (IS). In the first set of studies, we applied a similar 13C stable isotope/LC-MS/MS methodology as we utilized in the rodent studies to assess basal and insulin-stimulated relative rates of VPDH/VCS in skeletal muscle biopsies obtained from IR subjects and IS subjects. In the second set of studies, we assessed insulin-stimulated rates of mitochondrial VCS, VPDH/VCS flux, and glycogen synthesis (VGLY) in muscle by in vivo 13C MRS performed at 7 Tesla. Using these complementary approaches, we found that IR subjects had a marked reduction in insulin-stimulated peripheral glucose metabolism that could mostly be attributed to reductions in insulin-stimulated muscle glycogen synthesis assessed by in vivo MRS, consistent with prior studies (Shulman et al. 1990, Perseghin et al. 1996) (Fig. 4A-B). In addition, there was a marked reduction in insulin-stimulated VCS, which could mostly be attributed to reductions in the utilization of non-glucose substrates (e.g. NEFAs) (Fig. 4C). In contrast, rates of insulin-stimulated muscle glucose oxidation were similar between the two groups. Similar to what we observed in rat skeletal muscle mitochondrial VPDH/VCS was 0.22±0.03 in the insulin sensitive individuals demonstrating that non-glucose substrates accounted for ~80% of the substrate for muscle mitochondrial oxidation. Furthermore, in contrast to the metabolic inflexibility hypothesis proposed by Kelley et al., we observed no difference in muscle mitochondrial VPDH/VCS flux in the IR subjects fast compared to the IS individuals following an overnight, and insulin stimulation during the hyperinsulinemic clamp study promoted similar increases in muscle mitochondrial VPDH/VCS in both groups (Fig. 4D). Furthermore when corrected for number of carbons (triose versus hexose) the flux from plasma glucose into muscle glycogen synthesis was approximately 5-fold higher than into the TCA cycle via PDH in the IS group and 3-fold higher than into the TCA cycle in the IR group, consistent with muscle glycogen synthesis being the dominant pathway for insulin-stimulated muscle glucose metabolism in IS individuals and the major factor responsible for whole body insulin resistance in IR prediabetic, obese and T2D patients under these conditions.
Figure 4. Insulin resistant humans have impaired insulin-stimulated glucose disposal and muscle glycogen synthesis but do not manifest alterations in basal or insulin-stimulated muscle mitochondrial substrate preference (VPDH/VCS).

A. Insulin resistant subjects have impaired insulin-stimulated peripheral glucose uptake (Rd) compared to insulin sensitive subjects (IS: n=6, IR: n=8). B. Insulin resistant subjects have impaired insulin-stimulated muscle glycogen synthesis (IS: n=6, IR: n=8). C. Measurement of citrate synthase flux by in vivo MRS shows impaired muscle mitochondrial oxidation (VCS) in insulin resistant subjects; estimation of absolute glucose and fat oxidation rates using VPDH/VCS as a multiplier for VCS shows a decrease in both (IS: n=6, IR, n=8). Data are represented as mean ± SEM. D. Skeletal muscle VPDH/VCS in the fasting state is not different between insulin sensitive and insulin resistant subjects. Insulin-stimulation during a hyperinsulinemic-euglycemic clamp significantly increases VPDH/VCS to a similar degree in both groups (IS: n=9, IR: n=9).
Discussion
The precise mechanism leading to the development of muscle insulin resistance associated with obesity has not yet been defined, but alterations in mitochondrial substrate preference have emerged as an attractive possibility. Based on in vitro studies Randle was the first to hypothesize a path by which altered muscle substrate utilization, namely, increased muscle mitochondrial fatty acid oxidation, could inhibit not only glucose oxidation but glucose uptake in response to insulin (Randle et al., 1963, 1970). Building on this hypothesis Kelley and others have suggested that muscle insulin resistance is a result of ‘metabolic inflexibility’ that results in impairments in the muscle to make smooth transitions between utilizing glucose or fatty acids as substrates depending on varying physiological needs, which would in turn lead to “metabolic gridlock” (Kelley et al., 1992, 1999;Muoio, 2014;Muoio et al., 2012). These hypotheses have located mitochondrial fuel preference and switching as the nexus of muscle insulin resistance, in contrast to competing hypotheses that center on defects in insulin signaling leading to reductions in insulin-mediated glucose transport. The Kelley hypothesis of ‘metabolic inflexibility’ necessarily entails two predictions about insulin resistant muscle: first, that mitochondrial substrate preference is altered in insulin resistant muscle in the fasting state, where glucose oxidation is increased relative to fat oxidation (i.e. VPDH/VCS is increased in insulin resistant muscle compared to insulin sensitive muscle under fasting conditions); and second, that insulin resistant muscle cannot switch fuels between fasting and fed states (i.e. VPDH/VCS in insulin resistant muscle does not increase during the transition from fasted to fed state) (Kelley et al., 1992, 1999; Muoio, 2014). In accordance with the metabolic inflexibility hypothesis, the accumulation of intramyocellular lipid which is invariably observed in insulin resistant human muscle can therefore be attributed to increased glucose oxidation and decreased fat oxidation in the basal state.
While the ‘metabolic inflexibility’ hypothesis has become a widespread concept, the original A-V studies performed by Kelley and coworkers have significant methodological shortcomings that leave the precise relationship between muscle mitochondrial substrate oxidation and muscle insulin response hitherto unresolved (Kelley et al., 1992, 1999).
In order to circumvent these limitations, we applied a stable-isotope LC-MS/MS method to directly assess relative rates of muscle mitochondrial VPDH/VCS fluxes in a well-established HFD-fed rat model of lipid-induced muscle insulin resistance. We applied this method in both the soleus, a slow twitch muscle consisting of mostly of type 1 (slow twitch-oxidative) fiber types, as well as the quadriceps muscle, consisting of a mixture of type 1 (slow twitch-oxidative) and type II (fast twitch-glycolytic) fiber types (Ariano et al., 1973). Using these methods, we sought to adjudicate between two competing hypotheses for the pathogenesis of obesity-induced muscle insulin resistance: the first in which mitochondrial derangements lead to altered substrate preference and prevent insulin-mediated muscle glucose uptake by a backlog mechanism; and the second in which excess intramyocellular lipid accumulation interferes with insulin signaling and inhibits insulin-mediated glucose transport and indirectly modulates substrate utilization.
We found that our results contradict the predictions of the ‘metabolic inflexibility’ hypothesis in three major ways. First, while Kelley et al. observed that relative glucose oxidation in insulin sensitive fasting muscle contributed to 40% of total mitochondrial oxidation (Kelley et al., 1999), we found relative rates of VPDH/VCS in the overnight fasted state to be less than 10% in both rat soleus and quadriceps muscles. Second, while the ‘metabolic inflexibility’ hypothesis predicts that fasting substrate use in insulin resistant muscle would be significantly altered in favor of increased glucose oxidation, we found that the muscles of overnight fasted HFD fed-insulin resistant rats did not differ from muscles of overnight fasted normal chow fed rats in their mitochondrial substrate preference (VPDH/VCS) and also relied overwhelmingly on non-glucose (presumably predominantly fat) oxidation (Fig. 1H-I). This lack of altered fasting mitochondrial substrate preference was corroborated by the absence of significant changes in the expression of key mitochondrial oxidative enzymes such as PDH, CPT1b, and CrAT (Fig. 2C). Furthermore, in contrast to predictions of some proponents of metabolic flexibility, we found no evidence for mitochondrial metabolic congestion in insulin resistant muscle, as shown by unchanged concentrations of mitochondrial TCA cycle metabolites (Fig. 2B) or acylcarnitines (Supplementary Table #2) in HFD-fed rats. Finally, whereas Kelley et al. had observed that insulin resistant muscle could not upregulate relative rates of glucose oxidation to fat oxidation in response to insulin (i.e. were metabolically inflexible), we found that muscles of insulin resistant rats were able to increase VPDH/VCS in response to hyperinsulinemia (i.e. were metabolically flexible), though the increase in VPDH/VCS was blunted in the insulin resistant muscle compared to normal muscle (Fig. 1H-I). This blunting of increased VPDH/VCS in the insulin resistant rats was likely secondary to defects in insulin signaling (Fig. 1C-E) leading to reduced insulin-mediated glucose transport (Fig. 1F-G). Taken together these data demonstrate that the muscles of IR rats were in fact metabolically flexible, as reflected by increased insulin-stimulated VPDH/VCS, but that this response was constrained by defects in insulin-mediated glucose transport activity.
If muscle insulin resistance is not characterized by alterations in muscle substrate use, we asked conversely whether modulations in muscle substrate preference in normal rats could affect whole body and muscle insulin sensitivity. We found that acute changes in muscle VPDH/VCS were dissociated from muscle insulin sensitivity. Infusion of intralipid for 90 minutes in normal rats during a HE clamp raised plasma NEFA concentrations and significantly reduced insulin-stimulated VPDH/VCS in skeletal muscle, but did not significantly alter insulin-stimulated muscle glucose uptake, muscle glucose transport, or muscle insulin signaling (Fig. 3A-E). It is important to note that this result is very different from what occurs during a more prolonged (>180 min) lipid infusion, which results in severe insulin-stimulated muscle insulin resistance due to lipid-induced defects in insulin-stimulated glucose transport activity in both rodents and humans (Dresner et al., 1999; Roden et al., 1996; Yu et al., 2002). This delay in lipid-induced muscle insulin resistance can be attributed to the lag time (~180 min) in the accumulation of intramuscular DAGs, derived from the exogenous NEFAs, to reach a critical concentration to trigger the translocation of PKCθ and PKCε to the plasma membrane and inhibit insulin signaling (Szendroedi et al., 2014; Yu et al., 2002). Furthermore, we did not find evidence for Randle’s proposed glucose-fatty acid cycle in which increased fatty acid oxidation in the myocyte could inhibit glucose oxidation and glucose uptake through inhibition of key regulated steps in glycolysis. While Randle had observed that increased fat oxidation in muscle led to increased levels of acetyl CoA, citrate, and glucose-6-phosphate in vitro, we found that only acetyl CoA concentrations showed a tendency to be increased in vivo (Fig. 3F), suggesting that fatty acid oxidation could inhibit glucose oxidation through allosteric inhibition of PDH (and not through inhibition of key glycolytic steps) but that this reduction in PDH flux was ultimately inconsequential in the larger framework of muscle insulin response in vivo. These results are consistent with a previous study demonstrating that acutely increasing PDH flux with dichloroacetate did not affect muscle insulin sensitivity in vivo (Small et al., 2018). Taken together these results dissociate changes in muscle mitochondrial substrate preference from muscle insulin sensitivity and suggest that attempts to develop therapeutics to treat insulin resistance and T2D by targeting mitochondrial substrate preference would likely prove ineffective.
In order to determine whether these findings would translate to humans we applied a similar 13C stable isotope/LC-MS/MS approach to assess basal and insulin-stimulated rates of mitochondrial VPDH/VCS in skeletal muscle of insulin sensitive and insulin resistant humans. In addition, we also assessed insulin-stimulated rates of muscle VGLY and VCS by in vivo MRS in a parallel study. Using these combined methodological approaches, we found that IR subjects had a marked reduction in insulin-stimulated peripheral glucose metabolism, which could mostly be attributed to reductions in insulin-stimulated muscle VGLY (Fig. 4A-B). Similar to our observations in rodent skeletal muscle, non-pyruvate oxidation accounted for ~80% of total muscle mitochondrial oxidation (Fig. 4D), following an overnight fast, indicating that fatty acids were the preferred substrate in this state. In contrast to the metabolic inflexibility hypothesis there was no difference in mitochondrial VPDH/VCS in the basal fasting state in the IR subjects compared to the IS individuals (Fig. 4D). Furthermore, hyperinsulinemic clamps significantly increased VPDH/VCS in both groups (Fig. 4D) by approximately the same factor, suggesting that IR individuals remain capable of responding to substrate availability and insulin stimulation by switching substrate utilization to glucose (i.e. they are metabolically flexible). Interestingly, total insulin-stimulated VCS flux in skeletal muscle, assessed by in vivo 13C MRS, was decreased in IR subjects indicating that absolute rates of insulin-stimulated glucose and fat oxidation were also blunted in these individuals (Fig. 4C). Impaired insulin-stimulated VCS flux without corresponding alterations in VPDH/VCS suggests the presence of reduced mitochondrial function in insulin resistant human subjects is dissociated from any alterations in mitochondrial substrate preference.
Based on these data, we propose a clarification of the definition of “metabolic inflexibility” which has thus far been used imprecisely to describe both tissue specific and whole body alterations in the respiratory quotient in response to insulin and other physiologic perturbations. To avoid these ambiguities we have utilized the term “muscle mitochondrial substrate preference”. Our data demonstrate that in a HFD fed, insulin resistant rat model as well as in insulin resistant humans, muscle mitochondrial substrate preference is not impaired and that muscle VPDH/VCS can be manipulated without significantly altering whole body insulin-stimulated glucose disposal. These findings question the importance of alterations of mitochondrial substrate preference (i.e. metabolic inflexibility) as an explanation for the development of muscle insulin resistance, and suggest that ex vivo or in vitro studies of muscle mitochondrial substrate oxidation may demonstrate phenomena that are physiologically or mechanistically inconsequential.
Although we dispute a central role of alterations in mitochondrial substrate preference (metabolic inflexibility) as a major factor in the pathogenesis of muscle insulin resistance, robust evidence exists that reductions in mitochondrial function may be factor predisposing individuals towards intramyocellular lipid accumulation and muscle insulin resistance in the lean IR offspring of parents with T2D (Befroy et al., 2007; Morino et al., 2005; Petersen et al., 2004) and in the elderly (Petersen et al., 2003). These effects, however, could be attributed to reductions in rates of total mitochondrial oxidation (VCS), which would predispose these individuals to increased ectopic lipid accumulation rather than specific alterations in VPDH/VCS. Consistent with overall reductions in skeletal muscle mitochondrial function previously documented by both in vivo 31P MRS studies (Petersen et al., 2004) and 13C MRS studies (Befroy et al., 2007b), we also observed a reduction in insulin-stimulated muscle mitochondrial VCS flux in these studies (Fig. 4C). Thus, it is important to distinguish alterations in mitochondrial substrate preference (VPDH/VCS) per se, from reductions in overall rates of mitochondrial oxidation (VCS). These data demonstrate that VPDH/VCS has little impact on the development of muscle insulin resistance whereas modulation of overall mitochondrial oxidation rates (VCS) may lead to an imbalance between energy delivery to the myocyte vs. energy expenditure resulting in DAGs-PKCθ/PKCε-mediated inhibition of insulin signaling, and muscle insulin resistance. In support of this hypothesis Knowles et al. have recently identified a gene variant in N-acetyl transferase-2 (Nat2) in humans that is associated with insulin resistance and T2D (Knowles et al., 2015). Furthermore, recent studies have demonstrated that Nat1 (mouse Nat2 homologue) knockout mice display reduced whole-body energy metabolism (Camporez et al., 2017; Knowles et al., 2015), reduced mitochondrial function, increased liver and muscle TAG/DAG content and liver and skeletal muscle insulin resistance (Camporez et al., 2017).
In summary, these studies demonstrate that in contrast to the tenets of the metabolic inflexibility hypothesis of Kelley et al. there are no alterations in the basal rates of mitochondrial VPDH/VCS flux or impairments in the response of VPDH/VCS to insulin stimulation in lean, IR human or rodent skeletal muscle and that muscle mitochondrial pyruvate oxidation accounts for only ~20% of muscle energy metabolism following an overnight fast in both IR and IS humans. Furthermore, in humans the dominant pathway of insulin-stimulated muscle glucose uptake is glycogen synthesis and VPDH flux represents a relatively minor fraction of total muscle glucose metabolism. Taken together these results provide strong evidence against alterations in muscle mitochondrial substrate preference in having a major role in the pathogenesis of muscle insulin resistance in both humans and rodent models of lipid-induced muscle insulin resistance, contrary to the “metabolic inflexibility” hypothesis.
Limitations of Study
Limitations of our study include the relatively small number of IR and IS volunteers included in each study. In addition, due to limitations in the amount of muscle tissue that we were able to obtain from the human biopsy studies we only had enough tissue to assess 13C metabolite enrichments for the calculation of VPDH/VCS flux.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Gerald I. Shulman (gerald.shulman@yale.edu).
Materials Availability
Reagents generated in this study will be made available on request, but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
The study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Studies
All animal studies were approved by the Yale University Institutional Animal Care and Use Committee and were performed in accordance with all regulatory standards. 250 g male Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA) and were group housed (3 per cage) while they were fed either a high fat diet (Dyets #112245, Bethlehem, PA; 59% calories from fat, 26% from carbohydrate, 15% from protein) or a chow diet (Harlan Teklad #2018, Madison, WI; 18% calories from fat, 58% from carbohydrate, 24% from protein), ad lib for 3 weeks. All rats then underwent surgery under general isoflurane anesthesia for placement of polyethylene catheters in the common carotid artery (PE50 tubing, Instech Solomon, Plymouth Meeting, PA) and the jugular vein (PE90 tubing, Instech), after which they were singly housed. The rats were fed their respective diets ad lib for 1 more week, after which they were studied and sacrificed. All in vivo studies were performed following a 16 h overnight fast. At the conclusion of each study, rats were euthanized by IV pentobarbital.
Human Studies
Healthy, normal weight young individuals (n=18, Age: 25±1 years) were recruited by local advertising and prescreened to confirm that they were in excellent health, lean, and non-smoking. All subjects underwent a complete medical history, a physical examination with blood tests to verify normal blood and platelet counts; concentrations of aspartate aminotransferase, alanine aminotransferase, blood uric acid, cholesterol, and triglycerides, prothrombin time and partial-thromboplastin time. All had a normal birth weight (above 2.3 Kg) and a sedentary lifestyle. Habitual physical activity was measured over three consecutive days by step counting using a pedometer (Sportline, Inc.). Only subjects who were not participating in regular physical activity and had an average daily physical activity of less than walking 4.8 km (~10,000 steps) were eligible. The subjects were pre-screened with a three-hour oral glucose-tolerance test to assure normal glucose tolerance and 1H MRS studies to measure hepatic triglyceride, EMCL and IMCL content (Petersen et al., 2003) The initial insulin sensitivity was calculated from the OGTT plasma glucose and insulin levels and an insulin sensitivity index (ISI) as previously described (Matsuda and DeFronzo, 1999). Subject characteristics for both the muscle biopsy study and the in vivo 7T MRS study are shown in Supplemental Table 2. The protocol was approved by the Yale Human Investigation Committee and written consent was obtained from each subject after the purpose, nature, and potential complications of the studies had been explained.
METHOD DETAILS
Animal Studies
In all studies, tracers were infused through a catheter placed ~1 week prior in the carotid artery, and blood was obtained from a catheter in the jugular vein. Unless otherwise specified, all blood was drawn from the jugular vein. All studies began 1 hr after catheters were connected, reducing any impact of stress from handling on the physiology assessed.
Flux Analysis
To measure VPDH/VCS flux in the basal fasted state, rats were infused with [13C6]glucose (prime 3 mg/[Kg-min] for 5 min, continuous infusion rate 1 mg/[Kg-min]) for a total of 120 min, after which blood (600 μl whole blood) was obtained from the venous catheter and immediately centrifuged and the plasma obtained, and rats were sacrificed and their soleus and quadriceps muscles snap-frozen in situ using metal tongs pre-chilled in liquid N2. The VPDH/VCS flux was measured as the ratio of [4,5-13C2]glutamate / [13C3]alanine in soleus and quadriceps after a 2 hr infusion of [13C6]glucose (16.7 μmol/[Kg-min] prime for 5 min, 5.6 μmol/[Kg-min] continuous infusion) based on the assumptions we have described previously (Alves et al., 2011). Although calculation of true VPDH/VCS requires the comparison of intracellular pyruvate and acetyl CoA enriched by the [13C6]glucose tracer, these metabolites undergo rapid ex vivo degradation and are difficult to measure. Under conditions of steady state, intracellular pyruvate [m+3] was assumed to equilibrate with alanine [m+3], and acetyl CoA [m+2] was assumed to equilibrate with C4C5 glutamate [m+2]. Both alanine and glutamate are more stable ex vivo relative to their respective counterparts and were more amenable to reliable and consistent measurement of their intracellular enrichments. A rigorous comparison study of the 13C enrichments of these metabolites (performed in rat tissues freeze-clamped in situ to minimize ischemic degradation) showed highly significant correlations between alanine (m+3) and pyruvate (m+3) enrichments (R2=0.95, P<0.001) as well as between C4C5 glutamate (m+2) and acetyl CoA (m+2) enrichments (R2=0.89, P<0.001) under a variety of experimental conditions (Fig. S3A-B), validating our equilibration assumptions and indicating that alanine and C4C5 glutamate enrichments could be reliably used to approximate in vivo VPDH/VCS. Alanine enrichment was measured by GC/MS: samples were deproteinized with 5 volumes of methanol, dried under vacuum, derivatized with 1.) 3 volumes of n-butanol 4N HCl (65°C for 60 min, evaporated under N2 gas), and 2.) 100 μL of trifluoroacetic acid:methylene chloride (1:7). GC-MS was then used to determine the m+4 alanine enrichment (retention time ~4.1 min, m/z 242 [m0], 243 [m+1], 244 [m+2], 245 [m+3], 246 [m+4]). Glutamate enrichment was measured by LC-MS/MS: the samples were homogenized in 500 μL ice-cold methanol using a TissueLyser and filtered through a Nanosep filter. LC-MS/MS (AbSCIEX 6500 QTRAP with a Shimadzu ultrafast liquid chromatography system, negative ion mode) was used to monitor the relevant ion pairs: Total 13C-enrichment in glutamate was determined from the change from baseline distribution of mass isotopologues of the parent ion (m/z 146 to 151) and daughter ion (m/z 128 to 133: parent – H2O) pairs for m0 (146/128) to m+5 (151/133). The 13C enrichment (m+1 and m+2) of glutamate C4-C5 was determined from the change from baseline distribution of mass isotopologues corresponding to all permutations of the parent ions of 146->151 (m0 to m+5) and daughter ions 41->43 (m0 to m+2).
To measure muscle glucose uptake, a bolus of [14C] 2-deoxyglucose was administered in all rats. The harvested soleus and quadriceps muscles were processed to determine glucose uptake in both tissues by comparing the plasma [14C] specific activity decay curve to tissue [14C] specific activity, both measured using a scintillation counter (Jurczak et al., 2012).
Hyperinsulinemic Glucose Clamps
Rats were given a 40 mU/Kg bolus of Regular insulin followed by a 2 hr insulin infusion at a rate of 4.0 mU/(kg-min), while euglycemia was maintained with variable infusion of [13C6] glucose. Blood samples were drawn from the venous catheter at 0, 15, 30, 45, 60, 70, 80, 90, 100, 120 min of the clamp, with the samples from the 120 min time point used to measure clamp glucose turnover. Plasma insulin was measured by radioimmunoassay by the Yale Diabetes Research Center at the 0 and 120 min time points of the clamp. Glucose turnover in the clamp was calculated as above , and the rate of whole-body glucose disposal was calculated as the sum of the glucose infusion rate plus the glucose turnover rate measured in the clamp.
For combined lipid infusion and hyperinsulinemic-euglycemic clamps, rats were infused with Intralipid 20% emulsion (Baxter Inc.) at the rate 40 μL/[Kg-min] in addition to variable infusions of [13C6] glucose tracer for 2 hrs. Plasma samples from the 0 and 120 min time points for the 2 hrs lipid infusions were used to measure plasma non-esterified fatty acid levels with the method described below.
Biochemical Analysis
Plasma glucose concentrations were measured enzymatically using the YSI Glucose Analyzer (Yellow Springs, OH). Plasma NEFA concentrations were measured spectrophotometrically using a Wako reagent (Wako Diagnostics, Mountain View, CA).
Tissue Analyses
Acetyl-, malonyl-CoA and acyl-carnitines were measured as previously described (Perry et al., 2015; Perry et al., 2017). Muscle PKCθ and PKCε membrane/cytosol ratio, insulin receptor phosphorylation relative to total insulin receptor, and Akt phosphorylation relative to total Akt were measured by western blot as previously described (Samuel et al., 2004) using antibodies from Cell Signaling. PDH phosphorylation relative to total PDH was measured by western blot using antibodies from Calbiochem (pPDH s300, s232, s293) and Abcam (PDH). Glucose-6-phosphate concentrations were measured using an enzymatic assay from Sigma-Aldrich. Pyruvate concentrations were measured using GC/MS after derivatization with o-phenylene-diamine (Sigma-Aldrich) dissolved in 4M HCl, extraction with ethyl acetate, and further derivatization of the dried organic layer with a 1:1 BSTFA (Sigma-Aldrich) and pyridine (Sigma-Aldrich) mixture. Concentrations of TCA cycle intermediates (citrate, malate, succinate) were measured as reported previously (Perry et al., 2018). IRS-1-associated PI3-kinase activity was measured as reported previously (Yu et al., 2002). CPT1b was measured using antibodies from Santa Cruz. CrAT was measured using antibodies from ProteinTech. PDH, CPT1b, and CrAT levels were normalized to GAPDH levels, which was measured with antibodies from Cell Signaling. Muscle concentrations of G-6-P, pyruvate, acetyl-CoA, malonyl-CoA, acyl-carnitines and TCA cycle intermediates were measured in tissues freeze-clamped in situ to minimize changes due to post-mortem ischemia.
Human Studies
Diet and Study Preparation
For three days before the studies the subjects were instructed to eat a regular, weight-maintenance diet containing at least 150 g of carbohydrate and not to perform any exercise other than normal walking for the three days before the study (Petersen et al., 2003). Subjects were admitted to the Yale–New Haven Hospital Research Unit (HRU) the evening before the clamp study, and remained fasting from 10 PM until the completion of the study the following day the subjects continued to fast while having free access to regular drinking water until the end of the studies. Subject characteristics for the muscle biopsy studies for assessment of VPDH/VCS and the 7T in vivo MRS studies are shown in Supplemental Table 2.
1. Measurement of basal and insulin-stimulated VPDH/VCS flux by muscle biopsy
Hyperinsulinemic–Euglycemic Clamp Studies with Muscle Biopsies to Assess Basal and Insulin-Stimulated VPDH/VCS flux
After an overnight fast a two-hour infusion of [1-13C]glucose (388 mmol/L, 99% APE), Cambridge Isotopes, Andover, MA) was initiated as a primed/continuous infusion at a low-dose rate of 0.3 mg/(Kg-min). At the end of this two-hour period basal muscle biopsies were collected for measurements of basal 13C enrichments of [4-13C]glutamate and [3-13C]alanine and a three-hour euglycemic-hyperinsulinemic clamp was started with a primed-continuous insulin infusion at a rate of 40 mU/(m2-min) (U-100 regular insulin, Novo Nordisk, New Jersey) while keeping plasma glucose concentrations constant at euglycemia with a variable infusion of [1-13C] glucose (1.11 M, 25% APE, Cambridge Isotopes, Andover, MA). During the final 30 minutes of this hyperinsulinemic-euglycemic clamp muscle repeat biopsies were obtained for determination of 13C enrichments in [4-13C]glutamate and [3-13C]alanine following insulin-stimulation.
Muscle Biopsies
Muscle biopsies were collected after the skin over the vastus lateralis muscle was sterilely prepared with betadine, and 1% lidocaine was injected subcutaneously. A 2-cm incision was made using a scalpel, and a punch muscle biopsy was extracted using suction and a 5-mm Bergstrom biopsy needle (Warsaw, Indiana, USA). A piece of muscle tissue was dissected with a scalpel and immediately fixed in glutaraldehyde buffer for electron microscopy studies as described below. The remainder of the muscle tissue was blotted, snap frozen and stored in liquid nitrogen until assay (Morino et al., 2005).
2. In vivo 13C MRS measurement of insulin-stimulated rates of muscle metabolism
Participants [n=6 IS (3m/3f) and n=8 IR (4m/4f)] commenced fasting at 10 PM the evening before the study and remained fasting, with free access to drinking water, until the completion of the study the following day. Subjects were admitted to the Yale Magnetic Resonance Research Center in the morning prior to the study, and each individual then underwent an in vivo 13C-MRS assessment of insulin-stimulated muscle substrate utilization.
In vivo 13C MRS
In vivo 13C MRS experiments were performed at 7Tesla using an Agilent DirectDrive spectrometer (Agilent Technologies Inc., Santa Clara, CA) and a custom-built 13C / 1H probe, consisting of a 5 cm diameter 13C surface coil, with a pair of elliptical 9.5 × 7.5 cm 1H surface coils arrayed in quadrature (Befroy et al., 2010). After insertion of two antecubital IV lines for infusions and blood collections the subject was positioned supine with the right leg in a holder and the medial gastrocnemius muscle located directly over the 13C coil. Scout multi-slice, gradient-echo images were obtained to ensure correct positioning and to define regions of interest (ROI) for shimming and 13C MR spectroscopy. B0 field homogeneity was determined using an adiabatic FASTMAP sequence (Shen et al., 1997) and optimized using 1st and 2nd order shims. Typical 1H2O linewidths from a (20×20×20) mm voxel located within the medial gastrocnemius were 19Hz. Muscle metabolite content and 13C-enrichment were assessed using custom-written localized 13C spectroscopy sequences (Befroy et al., 2010). Baseline muscle glycogen content was determined from the [1-13C]glycogen peak (at 100.5ppm) acquired using an adiabatic pulse-acquire sequence, with WALTZ16 decoupling (Shaka et al., 1983) and 3-dimensional outer volume suppression (OVS) to select a ~75ml volume within the medial gastrocnemius. Natural abundance signals of muscle [5-13C]glutamate and [1-13C]glutamate (at 182.1ppm and 175.6ppm, respectively) were determined using the localized, adiabatic pulse-acquire sequence, as above, with nuclear Overhauser enhancement (nOe) to increase sensitivity. Muscle glutamate concentration was assessed using an adiabatic [1H]-13C polarization transfer (INEPT) sequence (Burum and Ernst, 1980) with WALTZ16 decoupling and 1D-ISIS (image-selected-in-vivo-spectroscopy) localization to select a 3cm thick slice through the medial gastrocnemius muscle. Transmitter offsets and INEPT echo-times were optimized to detect [2-13C]-glutamate (at 55.5ppm).
Measurement of insulin-stimulated rates of muscle glycogen-synthesis and citrate synthase flux
After a short 10 min break a 120–150 min hyperinsulinemic (40 mU m2-min) / euglycemic clamp was started using a variable infusion of 99% enriched [1-13C]glucose to maintain plasma glucose concentrations at ~5mM. Muscle 13C1-glycogen content was determined at ~60 minute intervals throughout the clamp to assess the incorporation of [1-13C]glucose into [1-13C]glycogen.
Sixty minutes into the clamp an infusion of [1-13C]acetate (99% APE, 350 mM) was commenced at a rate of 3 mg/Kg-min. 13C spectra were acquired at regular intervals to monitor the incorporation of 13C-label into [5-13C]glutamate and [1-13C]glutamate as acetate was oxidized via the TCA cycle (Fig. S4A). Citrate-synthase flux (VCS) was estimated using a modification of our previous approach using [1-13C]acetate in liver (Befroy et al., 2014), and described briefly below.
Data Analysis
All spectra were analyzed using software written in-house for Matlab (Mathworks Inc., Natick, MA) by Dr. Robin de Graaf (Yale University) and Dr. Befroy (this software is available on request from Dr.s de Graaf and Befroy via https://medicine.yale.edu/mrrc/). Concentrations of muscle glycogen and glutamate were determined from the baseline, natural abundance [1-13C]glycogen and [2- 13C]glutamate spectra, relative to spectra from phantoms of these metabolites with appropriate corrections for coil-loading and voxel position:
Mmuscle = the muscle 13C1-Glycogen or 13C2-Glutamate signal; Mphantom = the corresponding signal from a 150 mmolL−1 glycogen or 100 mmolL−1 glutamate phantom, respectively; Mformate(muscle) and Mformate(phantom) are the 13C signals of a formate point reference phantom (to establish coil-loading / sensitivity), acquired during the muscle or phantom study; and #scans indicates the number of transients acquired for the corresponding scan.
Rates of insulin-stimulated muscle glycogen synthesis (VGLY-syn) were estimated from the increment in the 13C1 signal over the final 60–90 min of the clamp as a function of the plasma [1-13 C] glucose enrichment:
where 13C1-Glycogen(t) = Muscle 13C-glycogen content at time, t; 13C1-Glucose = plasma glucose enrichment; Glucose = plasma glucose concentration; Δt = interval between successive plasma glucose measurements.
Subjects were classified as muscle insulin-sensitive (n=6, 4m/2f) or muscle insulin-resistant (n=8, 4m/4f) based on their rate of insulin-stimulated VGLY-syn; individuals with VGLY-syn < 0.2 mmol/L/min were considered to have muscular insulin-resistance.
Insulin-stimulated muscle citrate synthase flux (VCS) was determined by metabolic modelling analysis of the kinetics of [5-13C]- (and [1-13C]) glutamate turnover using plasma [1-13C]-acetate as the input function (Befroy et al., 2014). Time courses of muscle C5- and C1-glutamate enrichment for each subject were fitted to a metabolic model of the TCA cycle (Fig. S4B) using CWave software (developed by Dr. Mason). This model consists of a series of isotopic and mass balance differential equations (Fig. S4C) that describe the metabolism of acetate and was developed from previous schemes used to examine metabolism of [2-13C]-acetate in muscle (Befroy et al., 2007, Befroy et al., 2008) and [1-13C]-acetate in liver (Befroy et al., 2014). VCS was estimated by numerical simulation of the differential equations that contribute to the metabolic model using a non-linear least-squares (Levenberg-Marquardt) algorithm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons were performed using the 2-tailed Student’s t-test, unpaired unless otherwise specified in the figure legends, with significance defined as a p-value <0.05. GraphPad Prism 7.0 (San Diego, CA) was used for all statistical analysis. In most cases, n=8–10 rats per group, unless otherwise indicated in the figure legends. Data are presented as the mean±S.E.M.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PDH-E1α | Abcam | CAT#AB110330 |
| Phospho-PDH (s293) | Cell Signaling | CAT#31866S |
| Phospho-PDH (s232) | Millipore | CAT#AP1063 |
| Phospho-PDH (s300) | Sigma Aldrich | CAT#ABS2082 |
| CPT1b | Abcam | CAT#AB134988 |
| CrAT | ProteinTech | CAT#15170–1-AP |
| PKCθ | Cell Signaling | CAT#2059 |
| IRS-1 | Cell Signaling | CAT#2390 |
| AKT | Cell Signaling | CAT#2920 |
| Phospho-AKT (s473) | Cell Signaling | CAT#4060 |
| Insulin Receptor β | Cell Signaling | CAT#3025 |
| Phospho-IRK (Y1162) | Cell Signaling | CAT#3918 |
| GAPDH | Cell Signaling | CAT#5174S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Regular insulin | Sigma Aldrich | CAT#NDC 0002–8215-01 |
| 20% glucose | Hospira | CAT #NDC 0409–7935-19 |
| [1,2,3,4,5,6-13C6] glucose | Cambridge Isotopes | CAT#CLM-1396 |
| 2-[1-14C]-Deoxy-D-glucose | Perkin Elmer | CAT#NEC495A001MC |
| [1-13C] D-glucose | Cambridge Isotopes | CAT#CLM-420-PK |
| [1-13C] sodium acetate | Cambridge Isotopes | CAT#CLM-156-PK |
| Intralipid 20% emulsion | Baxter Inc. | CAT#2B6061 |
| Critical Commercial Assays | ||
| HR Series NEFA-HR(2) | Wako | CAT#999–34691 |
| HR Series NEFA-HR(2) | Wako | CAT#995–34791 |
| HR Series NEFA-HR(2) | Wako | CAT#991–34891 |
| HR Series NEFA-HR(2) | Wako | CAT#993–35191 |
| Glucose-6-phosphate Assay Kit | Sigma Aldrich | CAT#MAK014–1KT |
| Experimental Models: Organisms/Strains | ||
| Sprague-Dawley rats | Charles River | Strain code: 400 |
| Other | ||
| Regular chow diet | Harlan Teklad | CAT#2018 |
| High-fat diet | Dyets | CAT#112245 |
Highlights
Plasma membrane bound sn-1,2-diacylglycerols cause hepatic insulin resistance
PKCε is necessary and sufficient for mediating lipid-induced hepatic insulin resistance
PKCε promotes hepatic insulin resistance via phosphorylating insulin receptor Thr1160
Ceramides do not consistently track with hepatic insulin resistance
Context and Significance
Nonalcoholic fatty liver disease (NAFLD) affects 1 in 3 Americans and is strongly associated with hepatic insulin resistance and type 2 diabetes. However, the specific lipid molecules, their intracellular location, and the cellular mechanisms for lipid-induced insulin resistance are widely debated. Here Lyu et al. show that sn-1,2-diacylglycerols, bound to the plasma membrane, are the key lipid species responsible for activation of PKCε and hepatic insulin resistance. Furthermore, PKCε activation is the critical molecular link between hepatic lipid accumulation and hepatic insulin resistance: PKCε is both necessary and sufficient for the development of lipid-induced hepatic insulin resistance. These findings provide important new insights into the pathogenesis of hepatic insulin resistance associated with NAFLD, NASH and type 2 diabetes.
Acknowledgments
The authors thank Jianying Dong, Mario Kahn, Gina Butrico, and Irina Smolgovsky for their technical contributions. This study was funded by grants from the NIH (R01 DK113984, R01 DK49230, P30 DK059635, T32 DK101019, R00 CA215315, R01 NS087568, UL1TR000142) and the Novo-Nordisk Foundation CBMR.
Footnotes
Declaration of Interests
The authors have no relevant competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alves TC, Befroy DE, Kibbey RG, Kahn M, Codella R, Carvalho RA, Falk Petersen K, and Shulman GI (2011). Regulation of hepatic fat and glucose oxidation in rats with lipid-induced hepatic insulin resistance. Hepatology 53, 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariano MA, Edgerton VR, and Armstrong RB (1973). Hindlimb Muscle Fiber Populations of Five Mammals. J. Histochem. Cytochem. 21, 51–55. [DOI] [PubMed] [Google Scholar]
- Befroy DE, Petersen KF, Dufour S, Mason GF, Graaf R.A. de, Rothman DL, and Shulman GI (2007). Impaired Mitochondrial Substrate Oxidation in Muscle of Insulin-Resistant Offspring of Type 2 Diabetic Patients. Diabetes 56, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Befroy DE, Brown PB, Petersen KF, Shulman GI, and Rothman DL (2010). 1H decoupled 13C MRS in human muscle at 7T. In Proc Intl Soc Magn Reson Med (Stockholm, Sweden), p. #3295. [Google Scholar]
- Befroy DE, Perry RJ, Jain N, Dufour S, Cline GW, Trimmer JK, Brosnan J, Rothman DL, Petersen KF, and Shulman GI (2014). Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13C magnetic resonance spectroscopy. Nat Med 20, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camporez JP, Wang Y, Faarkrog K, Chukijrungroat N, Petersen KF, and Shulman GI (2017). Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl. Acad. Sci. 114, E11285–E11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline GW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, et al. (1999). Effects of free fatty acids on glucose transport and IRS-1–associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 103, 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris HA, Perry RJ, Moreira GV, Shulman GI, Horton JD, and Kahn CR (2017). Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. 114, 1189–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holness MJ, Kraus A, Harris RA, and Sugden MC (2000). Targeted upregulation of pyruvate dehydrogenase kinase (PDK)-4 in slow-twitch skeletal muscle underlies the stable modification of the regulatory characteristics of PDK induced by high-fat feeding. Diabetes 49, 775–781. [DOI] [PubMed] [Google Scholar]
- Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen CD, et al. (1999). Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 48, 1113–1119. [DOI] [PubMed] [Google Scholar]
- Jurczak MJ, Lee A-H, Jornayvaz FR, Lee H-Y, Birkenfeld AL, Guigni BA, Kahn M, Samuel VT, Glimcher LH, and Shulman GI (2012). Dissociation of Inositol-requiring Enzyme (IRE1α)-mediated c-Jun N-terminal Kinase Activation from Hepatic Insulin Resistance in Conditional X-box-binding Protein-1 (XBP1) Knock-out Mice. J. Biol. Chem. 287, 2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE (2005). Skeletal muscle fat oxidation: timing and flexibility are everything. J. Clin. Invest. 115, 1699–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, and Mandarino LJ (2000). Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49, 677–683. [DOI] [PubMed] [Google Scholar]
- Kelley DE, Mokan M, and Mandarino LJ (1992). Intracellular defects in glucose metabolism in obese patients with NIDDM. Diabetes 41, 698–706. [DOI] [PubMed] [Google Scholar]
- Kelley DE, Goodpaster B, Wing RR, and Simoneau J-A (1999). Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol.-Endocrinol. Metab. 277, E1130–E1141. [DOI] [PubMed] [Google Scholar]
- Kennedy JW, Hirshman MF, Gervino EV, Ocel JV, Forse RA, Hoenig SJ, Aronson D, Goodyear LJ, and Horton ES (1999). Acute exercise induces GLUT4 translocation in skeletal muscle of normal human subjects and subjects with type 2 diabetes. Diabetes 48, 1192–1197. [DOI] [PubMed] [Google Scholar]
- Knowles JW, Xie W, Zhang Z, Chennemsetty I, Assimes TL, Paananen J, Hansson O, Pankow J, Goodarzi MO, Carcamo-Orive I, et al. (2015). Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J. Clin. Invest. 125, 1739–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JRB, Newgard CB, et al. (2008). Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 7, 45–56. [DOI] [PubMed] [Google Scholar]
- Krssak M, Petersen KF, Dresner A, DiPietro L, Vogel SM, Rothman DL, Shulman GI, and Roden #1. (1999). Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42, 113–116. [DOI] [PubMed] [Google Scholar]
- Li S, An L, Yu S, Ferraris Araneta M, Johnson CS, Wang S, and Shen J (2016). (13)C MRS of human brain at 7 Tesla using [2-(13)C]glucose infusion and low power broadband stochastic proton decoupling. Magn Reson Med 75, 954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason GF, Falk Petersen K, de Graaf RA, Kanamatsu T, Otsuki T, and Rothman DL (2003). A comparison of 13C NMR measurements of the rates of glutamine synthesis and the tricarboxylic acid cycle during oral and intravenous administration of [1–13C]glucose. Brain Res. Protoc. 10, 181–190. [DOI] [PubMed] [Google Scholar]
- Matsuda M, and DeFronzo RA (1999). Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22, 1462–1470. [DOI] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, et al. (2005). Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Invest. 115, 3587–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM (2014). Metabolic Inflexibility: When Mitochondrial Indecision Leads to Metabolic Gridlock. Cell 159, 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Noland RC, Kovalik J-P, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, et al. (2012). Muscle-Specific Deletion of Carnitine Acetyltransferase Compromises Glucose Tolerance and Metabolic Flexibility. Cell Metab. 15, 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Camporez J-PG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang X-M, et al. (2015). Hepatic Acetyl CoA Links Adipose Tissue Inflammation to Hepatic Insulin Resistance and Type 2 Diabetes. Cell 160, 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Cline GW, Butrico GM, Wang Y, Zhang X-M, Rothman DL, Petersen KF, and Shulman GI (2017). Non-invasive assessment of hepatic mitochondrial metabolism by positional isotopomer NMR tracer analysis (PINTA). Nat. Commun. 8, 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Abulizi A, Kennedy L, Cline GW, and Shulman GI (2017). Mechanism for leptin’s acute insulin-independent effect to reverse diabetic ketoacidosis. The Journal of clinical investigation 127, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Wang Y, Cline GW, Rabin-Court A, Song JD, Dufour S, Zhang XM, Petersen KF, and Shulman GI (2018). Leptin Mediates a Glucose-Fatty Acid Cycle to Maintain Glucose Homeostasis in Starvation. Cell 172, 234–248.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perseghin G, Price TB, Petersen KF, Roden M, Cline GW, Gerow K, Rothman DL, and Shulman GI (1996). Increased Glucose Transport–Phosphorylation and Muscle Glycogen Synthesis after Exercise Training in Insulin-Resistant Subjects. N. Engl. J. Med. 335, 1357–1362. [DOI] [PubMed] [Google Scholar]
- Perseghin G, Scifo P, Cobelli FD, Pagliato E, Battezzati A, Arcelloni C, Vanzulli A, Testolin G, Pozza G, Maschio AD, et al. (1999). Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 48, 1600–1606. [DOI] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, and Shulman GI (2003). Mitochondrial Dysfunction in the Elderly: Possible Role in Insulin Resistance. Science 300, 1140–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Dufour S, Befroy D, Garcia R, and Shulman GI (2004). Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 350, 664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randle PJ, Garland PB, Hales CN, and Newsholme EA (1963). THE GLUCOSE FATTY-ACID CYCLE ITS ROLE IN INSULIN SENSITIVITY AND THE METABOLIC DISTURBANCES OF DIABETES MELLITUS. The Lancet 281, 785–789. [DOI] [PubMed] [Google Scholar]
- Randle PJ, England PJ, and Denton RM (1970). Control of the tricarboxylate cycle and its interactions with glycolysis during acetate utilization in rat heart. Biochem. J. 117, 677–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren JM, Semenkovich CF, Gulve EA, Gao J, and Holloszy JO (1994). Exercise induces rapid increases in GLUT4 expression, glucose transport capacity, and insulin-stimulated glycogen storage in muscle. J. Biol. Chem. 269, 14396–14401. [PubMed] [Google Scholar]
- Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, and Shulman GI (1996). Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 97, 2859–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Liu Z-X, Qu X, Elder BD, Bilz S, Befroy D, Romanelli AJ, and Shulman GI (2004). Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 279, 32345–32353. [DOI] [PubMed] [Google Scholar]
- Shaka A, Keeler J, Frenkiel T, and Freeman R (1983). An Improved Sequence for Broadband Decoupling: WALTZ-16. J Magn Reson 52, 335–338. [Google Scholar]
- Shen J, Rycyna RE, and Rothman DL (1997). Improvements on an in vivo automatic shimming method [FASTERMAP]. Magn Reson Med 38, 834–839. [DOI] [PubMed] [Google Scholar]
- Shulman GI (2000) Cellular Mechanisms of Insulin Resistance J. Clin. Invest. 106, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG (1990) Quantitation of Muscle Glycogen Synthesis in Normal Subjects and Subjects with Non-Insulin-Dependent Diabetes by 13C Nuclear Magnetic Resonance Spectroscopy 322:223–228. [DOI] [PubMed] [Google Scholar]
- Simonson DC, and DeFronzo RA (1990). Indirect calorimetry: methodological and interpretative problems. Am. J. Physiol.-Endocrinol. Metab. 258, E399–E412. [DOI] [PubMed] [Google Scholar]
- Small L, Brandon AE, Quek L-E, Krycer JR, James DE, Turner N, and Cooney GJ (2018). Acute activation of pyruvate dehydrogenase increases glucose oxidation in muscle without changing glucose uptake. Am. J. Physiol. Endocrinol. Metab. [DOI] [PubMed] [Google Scholar]
- Szendroedi J, Yoshimura T, Phielix E, Koliaki C, Marcucci M, Zhang D, Jelenik T, Müller J, Herder C, Nowotny P, et al. (2014). Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. 111, 9597–9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, et al. (2002). Mechanism by Which Fatty Acids Inhibit Insulin Activation of Insulin Receptor Substrate-1 (IRS-1)-associated Phosphatidylinositol 3-Kinase Activity in Muscle. J. Biol. Chem. 277, 50230–50236. [DOI] [PubMed] [Google Scholar]
- Zhang S, Hulver MW, McMillan RP, Cline MA, and Gilbert ER (2014). The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The study did not generate any unique datasets or code.