

Abstract
From an efficiency standpoint, microdroplet reactors enable significant improvements in the preparation of radiopharmaceuticals due to the vastly reduced reaction volume. To demonstrate these advantages, we adapt the conventional (macroscale) synthesis of the clinically-important positron-emission tomography tracer [18F]FDOPA, following the nucleophilic diaryliodonium salt approach, to a newly-developed ultra-compact microdroplet reaction platform. In this first microfluidic implementation of [18F]FDOPA synthesis, optimized via a high-throughput multi-reaction platform, the radiochemical yield (non-decay-corrected) was found to be comparable to macroscale reports, but the synthesis consumed significantly less precursor and organic solvents, and the synthesis process was much faster. In this initial report, we demonstrate the production of [18F]FDOPA in 15 MBq [400 μCi] amounts, sufficient for imaging of multiple mice, at high molar activity.
Keywords: Microdroplet reactor, Microfluidics, Green chemistry, Radiochemistry, Positron emission tomography
1. Introduction
Radiopharmaceuticals, including the imaging tracers used in positron-emission tomography (PET), are generally produced in automated radiosynthesizers to protect personnel from radiation exposure during the preparation of these compounds. Due to the short half-life of many useful radionuclides (e.g. fluorine-18, 110 min), radiopharmaceuticals are prepared in relatively small batches and preparation is performed geographically close to the imaging site and just prior to use. Automated synthesizers facilitate this repetitive production at many sites around the world1–3.
Typically, each synthesis carried out in such systems requires 10s of mg of the precursor material and/or other reagents, which results in high cost, and the need for complicated purification methods such as semi-preparative HPLC. Furthermore, relatively large amounts of solvents are needed, especially in the purification step, which typically uses hundreds of mL of solvent (mobile phase), including the time for column equilibration and cleaning. If one considers the repetitive need for radiopharmaceutical production each day, which is carried out at multiple locations throughout the world, the combined consumption of reagents and solvents becomes quite substantial.
The development of microfluidic-based reactors for radiochemistry4 could significantly reduce the consumption of reagents and solvent in the production of radiopharmaceuticals. Batch mode microscale syntheses can be performed with 2 to 3 orders of magnitude less reagents, which can significantly reduce the production cost and increase the molar activity (by reduction of [19F]fluoride contamination)5. High molar activity can improve signal to noise ratio and minimize avoid pharmacologic affect by minimizing occupancy when imaging targets with low tissue density6. Moreover, rapid purification is possible via analytical-scale HPLC instead of semi-preparative HPLC due to the reduced quantity of unreacted reagents and side products that need to be removed. Based on typically-used column sizes (i.e. 4.6 × 250 mm versus 10 × 250 mm), the consumption of organic solvents is reduced ~5-fold during column equilibration and cleaning. With shorter product retention times and flow rates of ~1 mL/min instead of ~5 mL/min, the consumption of solvents during the separation process can be reduced by an order of magnitude or more as well.
Recently, our group developed a high-throughput reactor7 and an automated microdroplet reactor8, both of which are based on the Teflon-coated silicon chip patterned with hydrophilic traps serving as reaction sites. The inexpensive silicon chips provide much higher thermal conductivity compared to the glass vial used in macroscale syntheses. Combined with the small reagent and reaction volumes (1–10 μL droplet versus 1 mL conventional reaction vessel), heating, cooling, and evaporation processes are greatly accelerated. In the previous work, we have successfully optimized the 1-step, 1-pot synthesis of the radiopharmaceutical [18F]fallypride, which is used for imaging of dopamine D2/D3 neuroreceptors with PET9. The droplet synthesis exhibited higher decay-corrected radiochemical yield (RCY; 78% versus 68 ± 1.6% (n=42)) and much shorter synthesis time (30 min versus 51 min), compared to the conventional macroscale method, giving a (uncorrected) activity yield of 65% versus 49%. Despite the small reaction volumes, a radionuclide concentrator10 can be used to perform the synthesis at clinically-relevant scales such as the production of multiple patient doses in a single batch11. In addition, these microdroplet reactors and their inherent advantages can readily be extended to parallel reaction systems for applications like synthesis optimization7.
In this work, using the amino acid PET tracer 3,4-dihydroxy-6-[18F]fluoro-L-phenylalanine ([18F]FDOPA) as an example, we show that the droplet radiochemistry platform can also be used to implement more complicated (multi-step, multi-pot) and lengthy synthesis pathways, and highlight the efficiency improvements afforded by microscale radiochemical reactors. [18F]FDOPA is mainly applied to the clinical imaging of Parkinson’s disease12, brain tumors13,14 and diseases related to the dopaminergic system. Recently, there has also been increasing interest in the use of [18F]FDOPA to image various neuroendocrine tumors (NETs)15, in which the uptake of [18F]FDOPA through the transmembrane amino acid transporter systems is upregulated; for example, [18F]FDOPA has exhibited high sensitivity and precision for imaging of carcinoid tumors, medullary thyroid cancer, etc.16–18.
Despite the utility of [18F]FDOPA, its usage has been limited due to its complicated synthesis. The earliest methods relied on a straightforward synthesis pathway involving electrophilic radiofluorination and deprotection19, but the low availability of [18F]F2 and other issues have led radiochemists to explore alternative production pathways based on nucleophilic radiofluorination using [18F]fluoride20. High radiochemical yield (RCY ~40%) has been reported using two-step copper-mediated approaches21, but these methods require a high amount (~40 mg) of the expensive precursor, which can complicate purification; the toxicity of copper also complicates purification and testing for clinical use. Libert et al. described a five-step radiosynthesis employing a chiral phase-transfer catalyst and successfully produced more than 45 GBq [1.2 Ci] [18F]FDOPA in a 65 min synthesis with 36 ± 3% (n=8) (decay corrected) RCY22. However, the preparation of the catalyst is difficult and expensive, and the synthesis can be difficult to implement on some synthesizers because two reaction steps are performed on solid supports. Kuik et al. reported a simple 2-step diaryliodonium salt synthesis method to produce [18F]FDOPA with RCY of 14 ± 4%23. The overall synthesis time was still relatively long (~117 min reported by Kuik et al., ~71 min reported by Collins et al.24) due to the need for an intermediate purification step.
To overcome the limitations of current synthesis methods for [18F]FDOPA, we explored the feasibility of synthesizing [18F]FDOPA in the microscale. Here, we adapt the diaryliodonium salt based synthesis of [18F]FDOPA23 to microdroplet format. Though other synthesis routes are available20, we focused on this particular method due to the commercial availability of the precursor. We optimized the synthesis protocol by testing various parameters, including concentrations of base and precursor, and reaction temperature. In addition, we investigated the use of the radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) to increase yield through prevention of precursor decomposition during the reaction25. Furthermore, we automated the synthesis on a newly developed ultra-compact microdroplet reactor8 (similar in size to a small (355 mL) coffee cup).
2. Materials and Methods
2.1. Materials
Anhydrous acetonitrile (MeCN, 99.8%), methanol (MeOH, 99.9%), ethanol (EtOH, 99.5%), diethylene glycol dimethyl ether (diglyme, 99.8%), TEMPO (98%), potassium carbonate (K2CO3, 99%), 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8]hexacosane (K222, 98%), hydrochloric acid (HCl, 37%), sulfuric acid (H2SO4, 99.99%), ethylenediaminetetraacetic acid (EDTA, 99%), acetic acid (99%), L-ascorbic acid and perchloric acid (HClO4) were purchased from Sigma-Aldrich. Both 6-Fluoro-L-DOPA hydrochloride (reference standard for L type [18F]FDOPA) and 6-Fluoro-D,L-DOPA hydrochloride (reference standard for mixture of D and L type [18F]FDOPA) were purchased from ABX Advanced Biochemical Compounds (Radeberg, Germany). ALPDOPA precursor was obtained from Ground Fluor Pharmaceuticals (Lincoln, NB, USA). DI water was obtained from a Milli-Q water purification system (EMD Millipore Corporation, Berlin, Germany). No-carrier-added [18F]fluoride in [18O]H2O was obtained from the UCLA Ahmanson Biomedical Cyclotron Facility.
Prior to synthesis of [18F]FDOPA, several stock solutions were prepared. Base stock solution was prepared by dissolving K222 (22.8 mg) and K2CO3 (4.08 mg) in a 9:1 (v/v) mixture of DI water and MeCN (600 μL). [18F]fluoride stock solution (containing 8.4mM K222 and 4.1mM K2CO3) was prepared by mixing [18F]fluoride/[18O]H2O (10 μL, ~220 MBq [~6.0 mCi]), base solution (10 μL) and DI water (100 μL). Precursor stock solution (containing 9mM ALDOPA) was prepared by dissolving ALDOPA (0.96 mg) in diglyme (120 μL, 75 mol% TEMPO). Finally, a collection solution to dilute the crude product prior to collection from the chip was prepared from a 4:1 (v/v) mixture of MeOH and DI water (500 μL).
2.2. Apparatus
Experiments were first performed on multi-reaction microfluidic chips to optimize the protocol in a more high-throughput fashion, and then the synthesis with optimal conditions was automated. Optimization experiments were performed on microfluidic chips comprising arrays of circular hydrophilic reaction sites patterned in a hydrophobic substrate (25 mm × 27.5 mm) (Fig. 1A). The patterned chips of low cost were prepared as described previously26 by coating silicon wafers with Teflon AF® (Chemours, Wilmington, DE, USA), and then etching away the coating to leave exposed silicon regions. The microfluidic chip was affixed atop of a heater platform to control temperature, and reagent addition and crude product collection were performed with a micro-pipette. Each chip was used once and then discarded.
Figure 1:
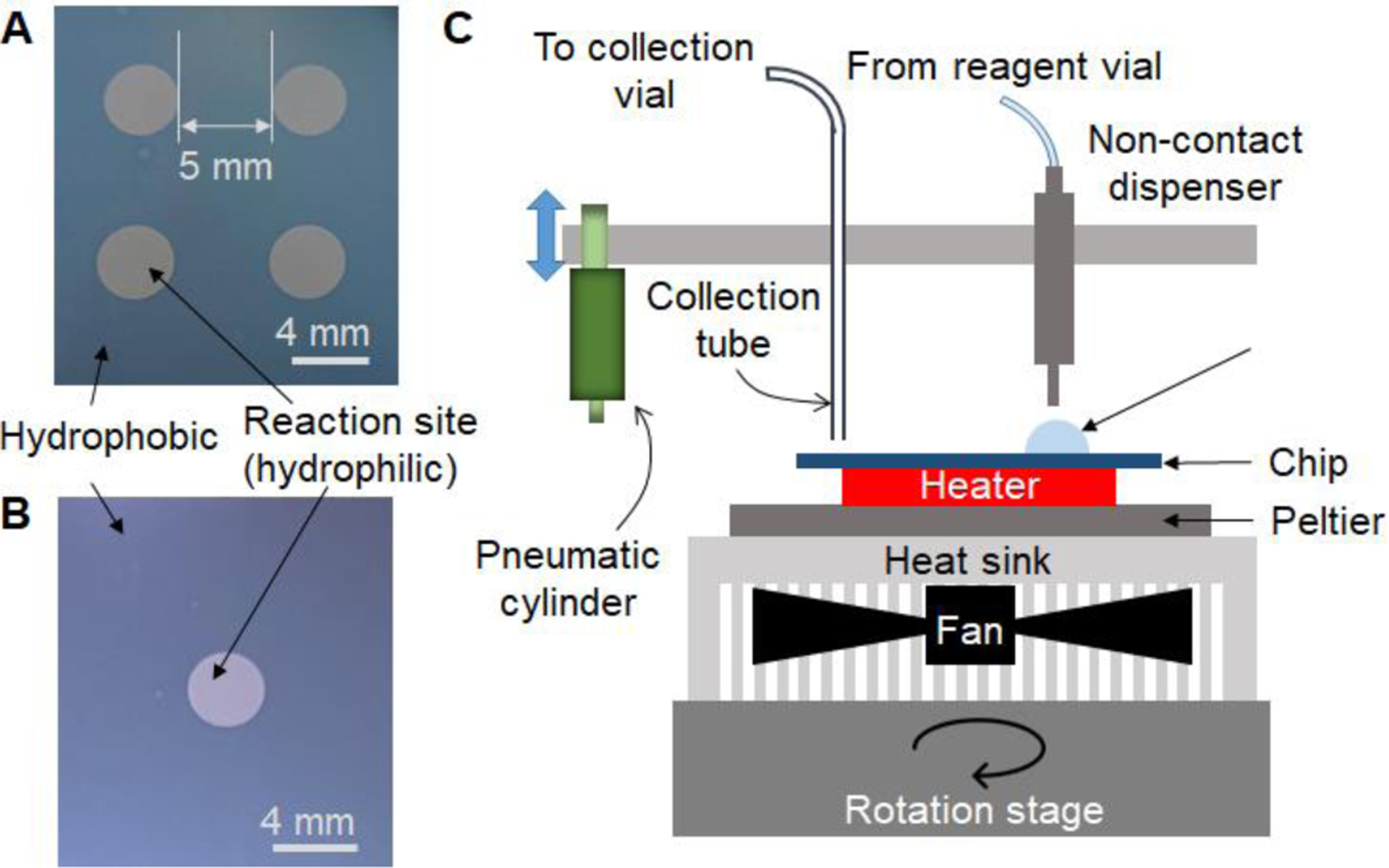
(A) Photograph of the microfluidic chip with four hydrophilic liquid traps serving as the reaction sites. Diameter of the site is 4 mm and pitch is 9 mm. (B) Photograph of the microfluidic chip comprising a Teflon-coated silicon wafer with one hydrophilic liquid trap serving as the reaction site. Diameter of the site is 4 mm. (C) Schematic of the side view of the experimental setup for the microdroplet reactor.
To increase safety and to facilitate routine production, we next automated the synthesis. Automated syntheses were conducted on chips with a single reaction site (Fig. 1B) operated using a custom-built compact framework (Fig. 1C), consisting of a rotating, temperature-controlled platform, a set of reagent dispensers, and a collection system to remove the reaction droplet from the chip at the end of the synthesis8. The rotation moves the reaction site beneath the desired reagent dispenser or product collection tube. Prior to synthesis, reagent vials connected to the reagent dispensers were loaded with the [18F]fluoride/base solution, precursor solution, replenishing solution (diglyme), deprotection solution (6M H2SO4) and collection solution, at the positions indicated in Fig. 3A. The LabView program was used to activate rotations, reagent dispensers, and vacuum for product collection at the appropriate times.
Figure 3.
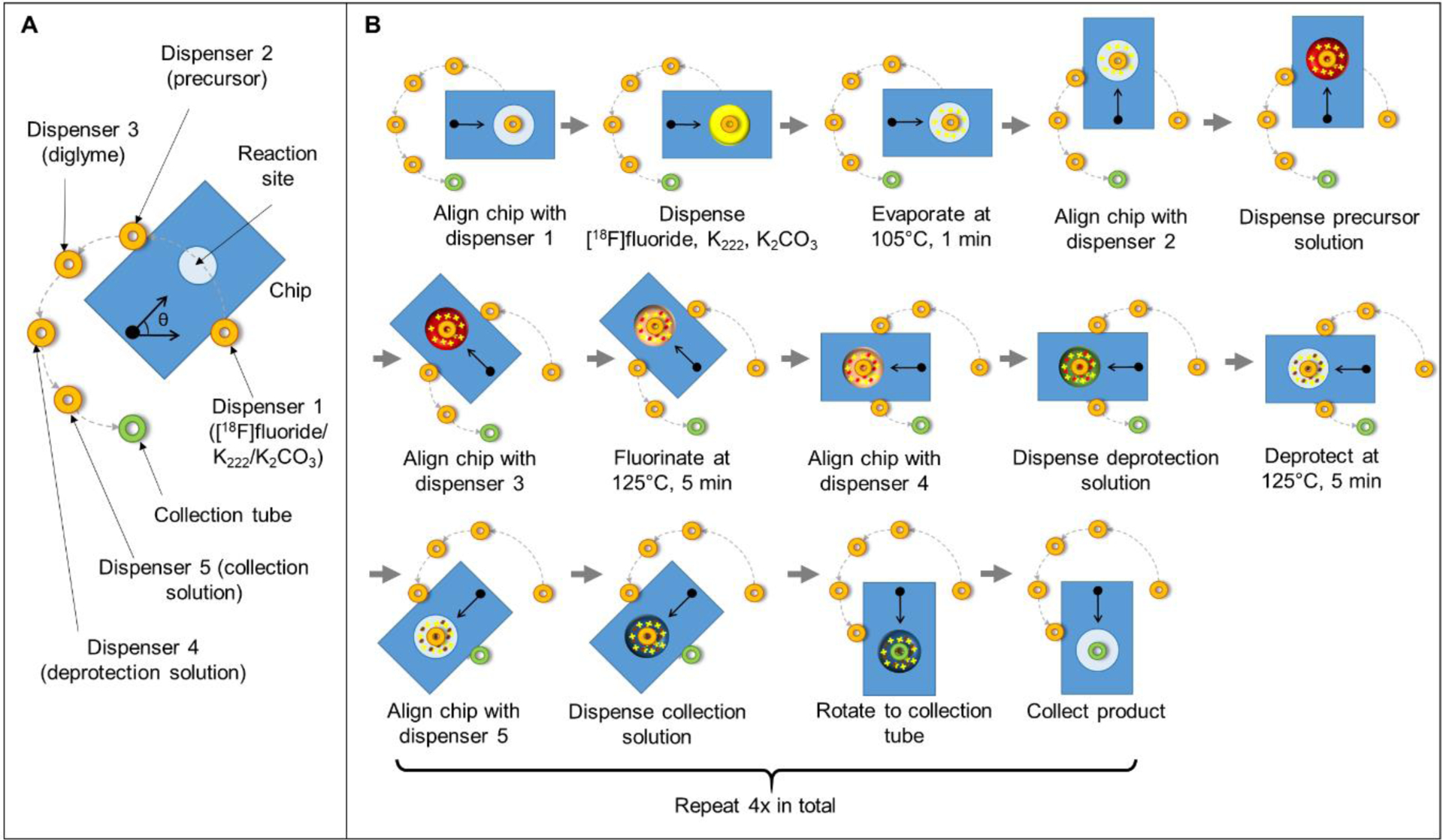
(A) Top view schematic of the microfluidic chip mounted on the rotating heating platform and the fixed locations of reagent dispensers and the collection tube above the chip. (B) Schematic of automated [18F]FDOPA synthesis process with the microdroplet reactor setup.
2.3. [18F]FDOPA synthesis
2.3.1. Optimization of [18F]FDOPA synthesis on the high-throughput chip
The synthesis scheme is shown in Fig. 2A and details of the microscale synthesis process on the high-throughput chip are elaborated in Fig. 2B. Briefly, a 10 μL droplet of [18F]fluoride stock solution (~11MBq, 84 nmol K222 / 41 nmol K2CO3) was first loaded on each reaction site, and the chip was heated to 105°C for 1 min to form the dried [18F]KF/K222 complex at each site. Then, a 10 μL droplet of precursor solution was added to reach reaction site and the chip was heated to 100°C to perform the fluorination step (Note that we prefer to radiofluorination as fluorination through the paper). During the 5 min reaction, the solvent was replenished at all sites by adding droplets (~7 μL) of diglyme every 30 s. Next, a 10 μL droplet of H2SO4 (6M) was added to each reaction site and the mixtures were heated to 125°C for 5 min to perform the deprotection step. Finally, for each individual reaction site, a 20 μL droplet of collection solution (4:1 v/v MeOH:H2O) was loaded to dilute the crude product, which was then recovered via pipette. The dilution and collection process was repeated 4x in total (for each site) to maximize the radioactivity recovery.
Figure 2.
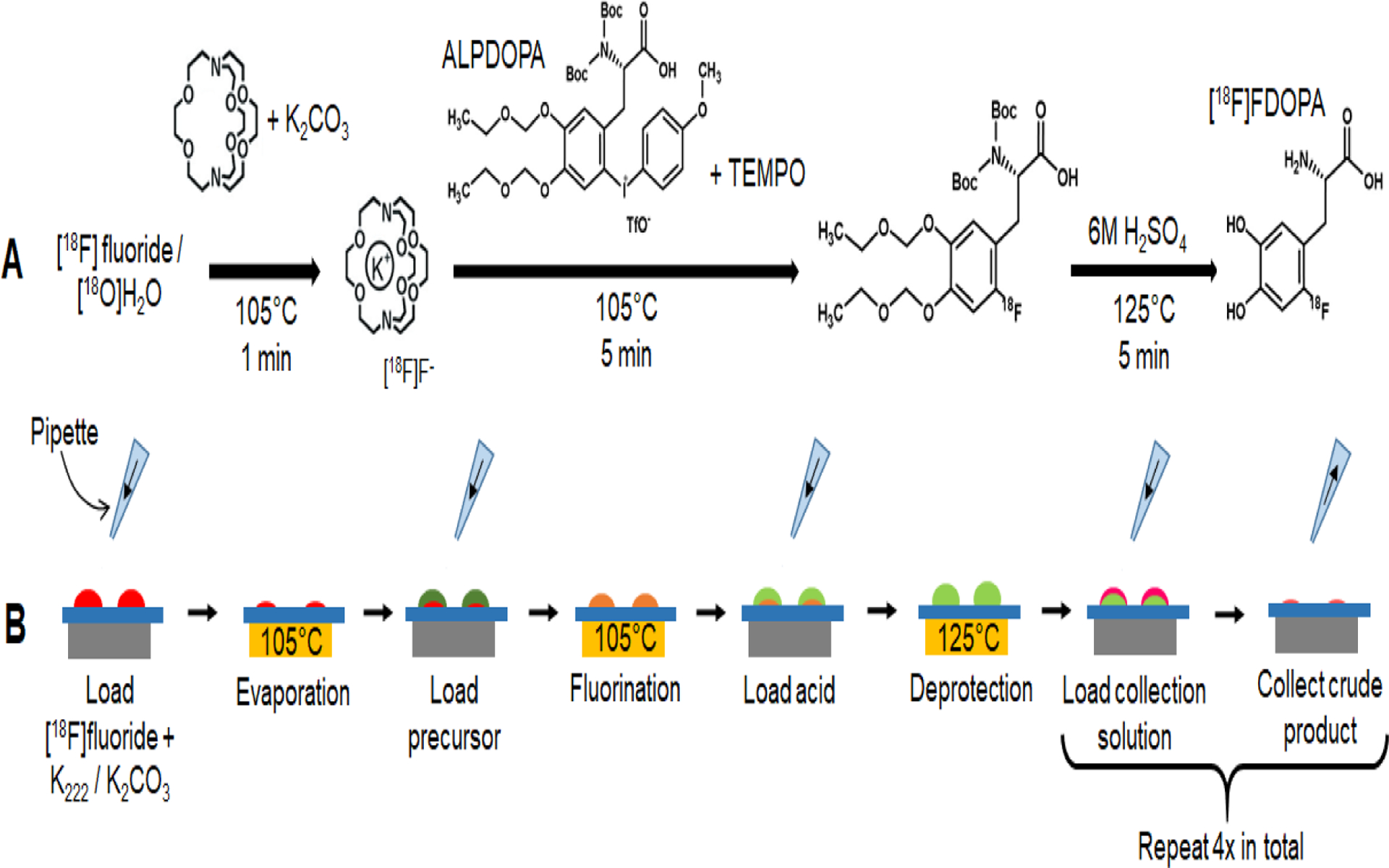
Multi-step radiochemical synthesis of [18F]FDOPA. (A) Synthesis scheme. (B) Schematic of manual [18F]FDOPA synthesis process using the multi-reaction chip.
2.3.2. Automated synthesis of [18F]FDOPA using the microdroplet reactor
Dispensers were filled with reagents as described in Fig. 3A and pressurized to 8 psi, and details of the microscale synthesis process using the microdroplet reactor is illustrated in Fig. 3B. To perform the synthesis of [18F]FDOPA, the chip was first rotated to position the reaction site below the dispenser 1 for [18F]fluoride stock solution and ten 1 μL droplets of [18F]fluoride stock solution (totaling ~18.5 MBq; ~0.5 mCi) were sequentially loaded onto the chip (total time < 10s). The chip was rotated 45° counterclockwise (CCW) and heated to 105°C for 1 min to evaporate the solvent and leave a dried residue of the [18F]KF/K222 complex at the reaction site. Then, the chip was rotated 45° CCW to position the reaction site under the precursor dispenser and ten 1 μL droplets of precursor solution were loaded to dissolve the dried residue. Next, the chip was rotated 45° CCW to position the reaction site under the replenishing dispenser (diglyme) and heated to 100°C for 5 min to perform the fluorination reaction. Solvent was replenished by adding a 1 μL droplet of diglyme every 10 s. Afterwards, the chip was rotated 45° CCW to position the reaction site under the deprotection solution dispenser, twenty 0.5 μL droplets of deprotection solution were loaded on the reaction site and the chip was heated to 125°C for 5 min to perform deprotection step. Finally, the chip was rotated 45° CCW to position the reaction site under the collection solution dispenser, and twenty 1 μL droplets of collection solution were deposited to dilute the crude product. After rotating the chip 45° CCW to position the reaction site under the collection tube, the diluted solution was transferred into the collection vial by applying vacuum. The collection process was repeated a total of four times to minimize the residue on the chip (i.e. by rotating the chip 45° CW back to the collection solution dispenser, loading more collection solution, etc.).
To perform the synthesis of [18F]FDOPA at higher activity levels, the same procedure was used, except that 20 droplets of more concentrated [18F]fluoride stock solution were sequentially loaded in the first step (totaling ~370 MBq [~10 mCi]), and solvent evaporation was performed for 2 min instead of 1 min. In addition, 0.25 M ascorbic acid was added into the deprotection solution to mitigate potential radiolysis.
2.4. Analytical methods
Performance of the fluorination step was assessed through measurements of radioactivity using a calibrated dose calibrator (CRC-25R, Capintec, Florham Park, NJ, USA) at various stages of the synthesis process, and measurements of fluorination efficiency using radio thin-layer chromatography (radio-TLC). All radioactivity measurements were corrected for decay. Radioactivity recovery was calculated as the ratio of radioactivity of the collected crude product to the starting radioactivity loaded onto the chip. Residual activity on the chip was the ratio of radioactivity on the chip after collection to the starting radioactivity on the chip. Fluorination yield (decay-corrected) was defined as the radioactivity recovery times the fluorination efficiency.
To accelerate the analysis, radio-TLC was performed using recently-developed parallel analysis methods. Groups of 4 samples were spotted via pipette (1 μL each, 1 mm pitch) onto each TLC plate (silica gel 60 F254 TLC plate, aluminum backing(Merck KGaA, Darmstadt, Germany)). TLC plates were dried in air and developed in the mobile phase (95:5 v/v MeCN : DI water). After separation, the multi-sample TLC plate was read out by imaging (5 min exposure) with a custom-made Cerenkov luminescence imaging (CLI) system27. To determine the fluorination efficiency, regions of interest (ROIs) were drawn on the final image (after image corrections and background subtraction) to enclose the radioactive regions/spots. Each ROI was integrated, and then the fraction of the integrated signal in that ROI (divided by the sum of integrated signal in all ROIs) was computed. Two radioactive species were separated in the samples: [18F]fluoride (Rf = 0.0) and the fluorinated intermediate (Rf = 1.0).
Analysis of samples (crude reaction mixture or purified product) was performed on a Smartline HPLC system (Knauer, Berlin, Germany) equipped with a degasser (Model 5050), pump (Model 1000), a UV detector (Eckert & Ziegler, Berlin, Germany) and a gamma-radiation detector and counter (B-FC- 4100 and BFC-1000; Bioscan, Inc., Poway, CA, USA). Injected samples were separated with a C18 column (Luna, 5 μm pore size, 250 × 4.6 mm, Phenomenex, Torrance, CA, USA). The mobile phase consisted of 1 mM EDTA, 50 mM acetic acid, 0.57 mM L-ascorbic acid and 1% v/v EtOH in DI water. The flow rate was 1.5 mL/min and UV absorbance detection was performed at 280 nm. The retention times of [18F]fluoride, [18F]FDOPA and the fluorinated intermediate were 2.4, 6.2, and 25.8 min, respectively. [18F]FDOPA conversion was determined via dividing the area under the [18F]FDOPA peak by the sum of areas under all three peaks. Crude radiochemical yield (crude RCY) was defined as the radioactivity recovery multiplied by the [18F]FDOPA conversion.
For purification, the collected crude product (~ 80 μL) was first diluted with 80 μL of the mobile phase, and then separated under the same conditions as above. To formulate the isolated product fraction (~2 mL, pH = 2), NaOH (1M) was added to result in a final product solution with pH ~6 (as recommended by Kuik et al.23). Isolated RCY was defined as the ratio of radioactivity of the formulated [18F]FDOPA to the starting radioactivity loaded on the chip.
For some experiments, The enantiomeric purity was verified by coinjecting the purified product and mixture of D and L type reference standard and separated using a chiral column (Crownpack CR(+), 5 μm, 150 × 4 mm, Chiral Technologies, West Chester, PA, USA) using a mobile phase of HClO4 solution (pH = 2) at a flow rate of 0.8 mL/min. Retention times of L-DOPA and D-DOPA were 9.5 and 12.1 min, respectively.
3. Results and discussion
3.1. Optimization of [18F]FDOPA synthesis on the high-throughput chip
Before developing our multi-reaction microfluidic chips, we performed some initial studies of the fluorination step with varied reaction conditions to establish a baseline upon which further fine-grained optimizations could be made. The initial studies examined reaction temperature (85 – 125 °C), reaction time (5 – 15 min), reaction solvent (DMF, MeCN, DMSO, diglyme), precursor concentration (9 – 71 mM), base amount (21 – 168 nmol of K222 and 10 – 82 nmol of K2CO3). The highest fluorination yield was observed using 84 nmol K222 / 41 nmol K2CO3, 9 mM precursor (in digylme), 5 min reaction at 105°C, but the yield (~7%) exhibited poor day to day consistency.
Based on the report of Carroll et al. that the yield and reproducibility of fluorination of diaryliodonium salts could be improved by adding TEMPO (to improve the stability of the diaryliodonium salt precursor)25, we investigated whether this approach could be used to improve the yield and consistency of [18F]FDOPA synthesis.
Initially we added 20 mol% TEMPO into the precursor solution, and performed a detailed study of the effect of precursor concentration on the fluorination yield (Fig. 4A) with 5 min reaction time at 105°C. The highest yields were obtained with moderate precursor concentrations: at 9 and 18 mM, the fluorination yields were 12.0 ± 1.7 % (n=3) and 11.6 ± 0.3 % (n=3), respectively. We chose 12 mM for subsequent experiments to study of the effect of TEMPO concentration (Fig. 4B). The fluorination yield was only 6.5 ± 0.1 % (n=2) without TEMPO but nearly tripled (18.8 ± 0.2 % (n=2)) when 80 mol% TEMPO was added. The improvement was mainly due to an increase in fluorination efficiency from 23 ± 1 % (n=2) to 53 ± 2 % (n=2) though slight improvement in radioactivity recovery was also observed. Next, we studied the effect of the amount of base, keeping the ratio of K222 to K2CO3 fixed at 2.05. (Fig. 4C). As the amount of base was increased, starting from 21 nmol K222 / 10 nmol K2CO3, the fluorination yield rose sharply and reached the maximum, 21.89 ± 0.02 % (n=2) at 84 nmol K222 / 41 nmol K2CO3). The fluorination yield remained relatively constant up to ~250 nmol K222 / 120 nmol K2CO3, and then began to drop significantly with higher base amount. The effect of those reaction variables on the fluorination efficiency and radioactivity recovery were summarized in Fig. S1 (ESI). Thus, for the later deprotection study, fluorination conditions were 9 mM precursor, 75 mol% TEMPO, and 84 nmol K222 / 41 nmol K2CO3.
Figure 4.
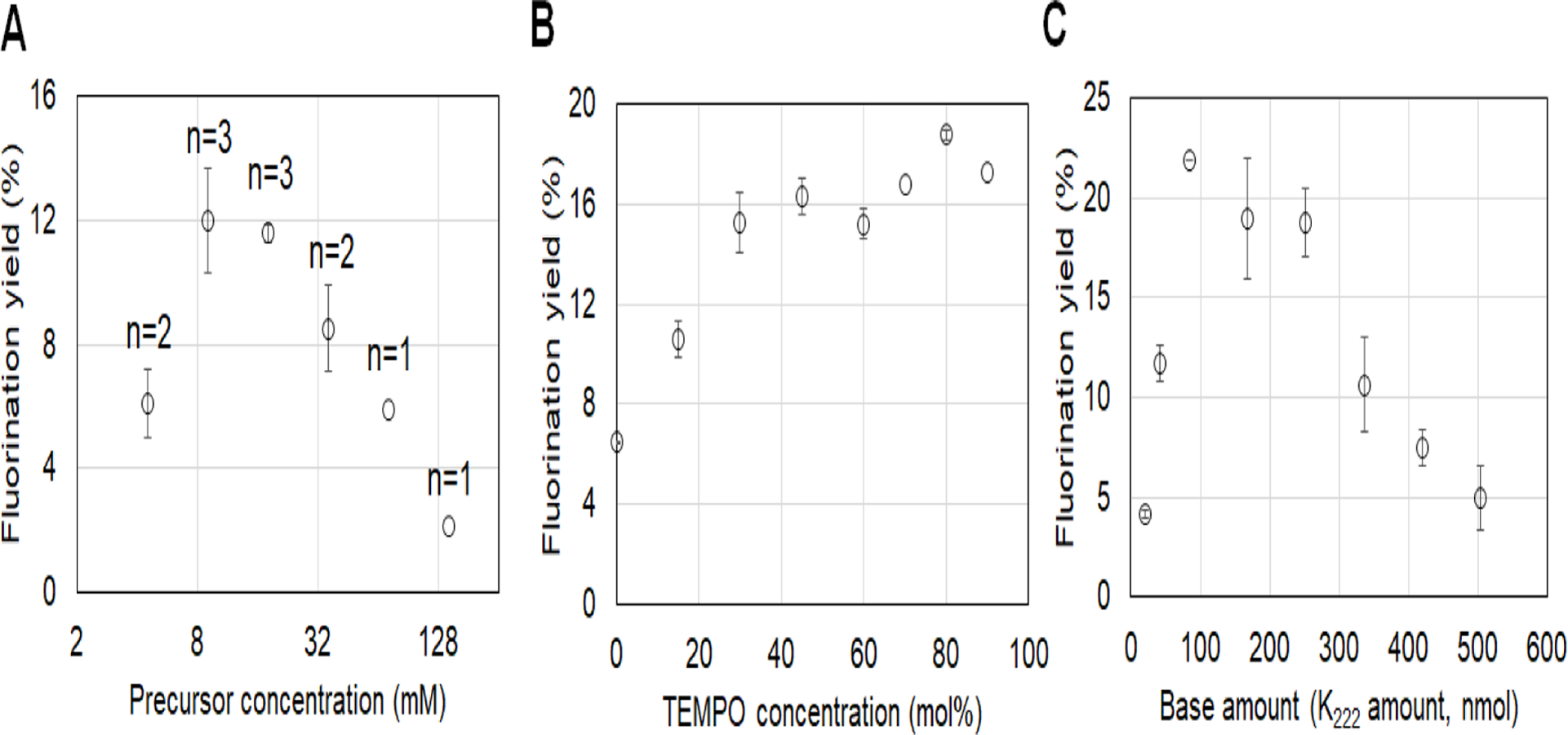
Optimization of microdroplet synthesis of [18F]FDOPA using the manual setup. (A) Effect of precursor concentration. (B) Effect of TEMPO concentration. n=2 for all points except 70 and 90% mol% (where n=1). (C) Effect of base amount, represented by K222 amount (K2CO3 amount is 2.05x lower). n=2. In all plots, data points represent averages and error bars represent standard deviations.
Deprotection was performed immediately after fluorination, with no intermediate purification step. Preliminary optimization (of type and concentration of deprotection reagent, and reaction temperature and time) are summarized in Table S1 (ESI). Even though the overall crude RCY was <10% due to performing these experiments starting with non-optimal fluorination conditions, comparative conclusions could still be drawn. Combining the optimal deprotection conditions (6 M H2SO4 at 120°C for 5 min) with the optimal fluorination conditions, [18F]FDOPA could be produced on the chip with crude RCY of 11% (n=1). By adding a cover plate over the droplet during deprotection (Fig. S2, ESI), the crude RCY could be further increased to 14.3 ± 0.5 % (n=2) (Table S2, ESI). Noting that the [18F]FDOPA conversion was only 84±5%(n=2) at 120°C, indicating the deprotection reaction was not complete, we increased the deprotection temperature to 130°C and the conversion improved to 95% (n=1).
Finally, we performed full (manual) syntheses including analytical-scale HPLC purification and formulation. The fluorination conditions were 75 mol% TEMPO, 9 mM precursor solution, 84 nmol K222/41 nmol K2CO3 at 105°C for 5 min, and the deprotection conditions were 6M H2SO4 at 130°C for 5 min (with cover plate). In these experiments, the resulting crude RCY and isolated RCY were 20.5 ± 3.5 % (n=3) and 15.1 ± 1.6 % (n=3), respectively (Table 1). An example of a radio-HPLC chromatogram of the crude product is shown in Fig. 5A, and a co-injection with L-DOPA and D-DOPA reference standards to determine enantiomeric purity (98.0 ± 0.2 (n=3)) is shown in Fig. 5B. The retention time of [18F]FDOPA was ~6 min, and the chromatogram was relatively clean with no nearby side-product peaks, despite omission of the intermediate cartridge purification between fluorination and deprotection steps23.
Table 1.
Comparison of microscale [18F]FDOPA synthesis performance using manual and automated approaches. All values are averages ± standard deviations for the indicated number of replicates. All percentages are decay-corrected unless otherwise indicated. N.M. indicates the quantity was not measured.
| Manual synthesis (n=3) | Automated synthesis-low starting activity (n=4) | Automated synthesis-higher starting activity (n=3) | |
|---|---|---|---|
| Starting activity (MBq) | 4.4 ~ 12 | 13 ~ 23 | 350 ~ 380 |
| Synthesis time including purification (min) | ~33 | ~30 | ~31 |
| [18F]FDOPA conversion (%) | 95.6 ± 0.4 | 78 ± 4 | 54.9 ± 1.9 |
| Crude RCY (%) | 20.5 ± 3.5 | 15.2 ± 2.1 | 5.2 ± 0.7 |
| Isolated RCY (%) | 15.1 ± 1.6 | 10.3 ± 1.4 | 4.7 ± 1.0 |
| Isolated RCY (%, non-corrected) | 12.3 ± 1.3 | 8.5 ± 1.2 | 3.9 ± 1.8 |
| Enantiomeric purity (%) | 98.0 ± 0.2 | N.M. | N.M. |
| Total activity loss during synthesis (%) | 50 ± 5 | 78 ± 2 | 85 ± 2 |
| Unrecoverable activity on cover chip (%) | 24.7 ± 0.3 | cover not used | cover not used |
| Unrecoverable activity on bottom chip (%) | 2.1 ± 0.4 | 2.9 ± 0.2 | 5.9 ± 2.9 |
| Radioactivity recovery (%) | 21 ± 4 | 20 ± 2 | 10 ± 2 |
Figure 5.
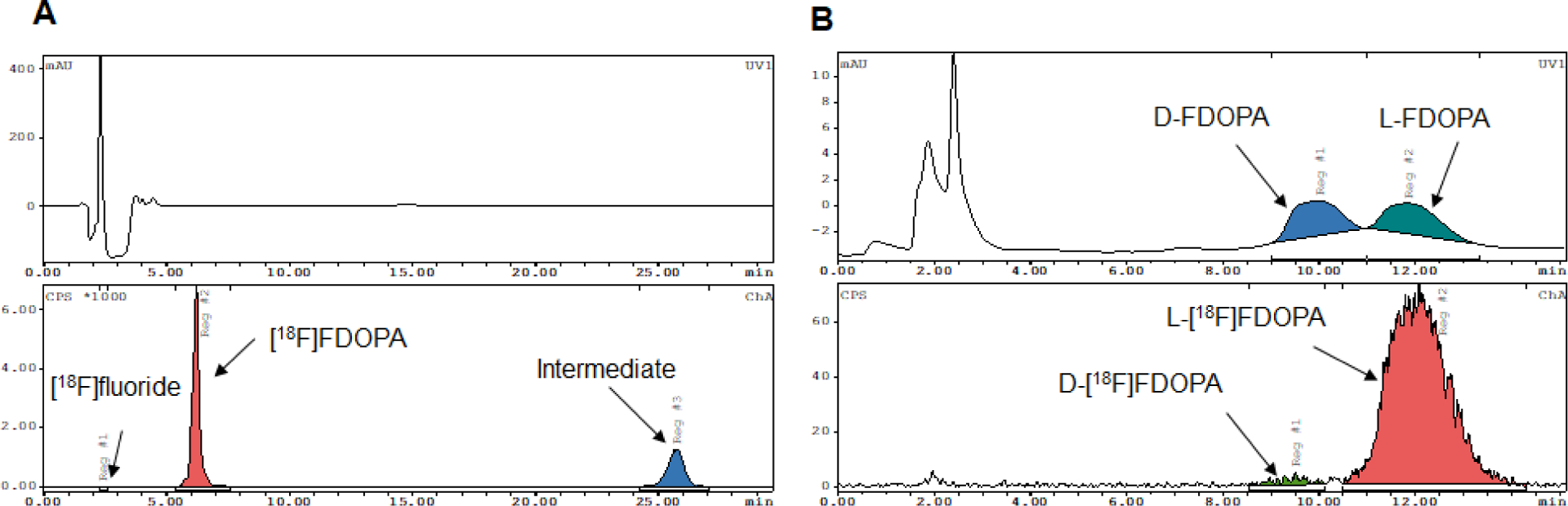
Examples of analytical radio-HPLC chromatograms of (A) crude [18F]FDOPA product, (B) purified [18F]FDOPA product coinjected with a mixture of reference standards of both D-FDOPA and L-FDOPA.
3.2. Automated synthesis of [18F]FDOPA using the microdroplet reactor
Considering the accuracy of droplet volume dispensed by the dispensers (~10%) studied previously, we adjusted some concentrations so the overall synthesis would be tolerant of volume errors and thus more robust and repeatable. The optimal conditions were selected where the slopes of the optimization curves (in Fig. 4) were close to zero. Automated syntheses were performed with 80 mol% TEMPO, 12 mM precursor solution and 101 nmol K222 / 49 nmol K2CO3.
Benefiting from the automated dispensing system, the frequency of replenishing solvent during heated reactions could be increased (up to several droplets per second, compared to one droplet per ~7s via manual dispensing), and we therefore briefly explored higher fluorination temperatures. Despite an increase in fluorination efficiency with the increase of reaction temperature from 100°C to 140°C (Fig. 6), there is reduction in radioactivity recovery across this temperature range, resulting in relatively small variation in overall fluorination yield. The maximum value (26.9 ± 1.3%, n=2) was achieved at a fluorination temperature of 120°C (with diglyme replenished every 4s). For the full automated synthesis, including the deprotection step, the crude RCY and isolated RCY were 15.2 ± 2.1% (n=3) and 10.3 ± 1.4% (n=3), respectively (Table 1). Both are slightly lower than the manual synthesis. Much of the reduction can be explained by lower observed [18F]FDOPA conversion for the automated synthesis (i.e., 78 ± 4% (n=3) compared to 95.6 ± 0.4% (n=3) for the manual synthesis), perhaps due to the absence of the cover plate. (The radioactivity recovery of both methods were comparable, i.e. 20 ± 2% (n=3) and 21 ± 4% (n=3), for the automated and manual procedures, respectively.) While in principle placement and removal of the cover plate could be automated, it would add considerable complexity to the setup and we considered the reduced yield a reasonable tradeoff. To further increase the [18F]FDOPA conversion, we attempted performing the deprotection step at even higher temperature (140°C), but significant side products appeared. The automated synthesis took ~22 min, which was slightly faster than the manual synthesis (~25 min). The on-chip method was considerably faster than the macroscale syntheses, and, in fact, the isolated non-decay-corrected yield of the microscale method (8.5 ± 1.2% (n=3)) was higher than both macroscale approaches, i.e. 2.9 ± 0.8 % (n=3)24 and 6.7 ± 1.9 % (n =?)23.
Figure 6.
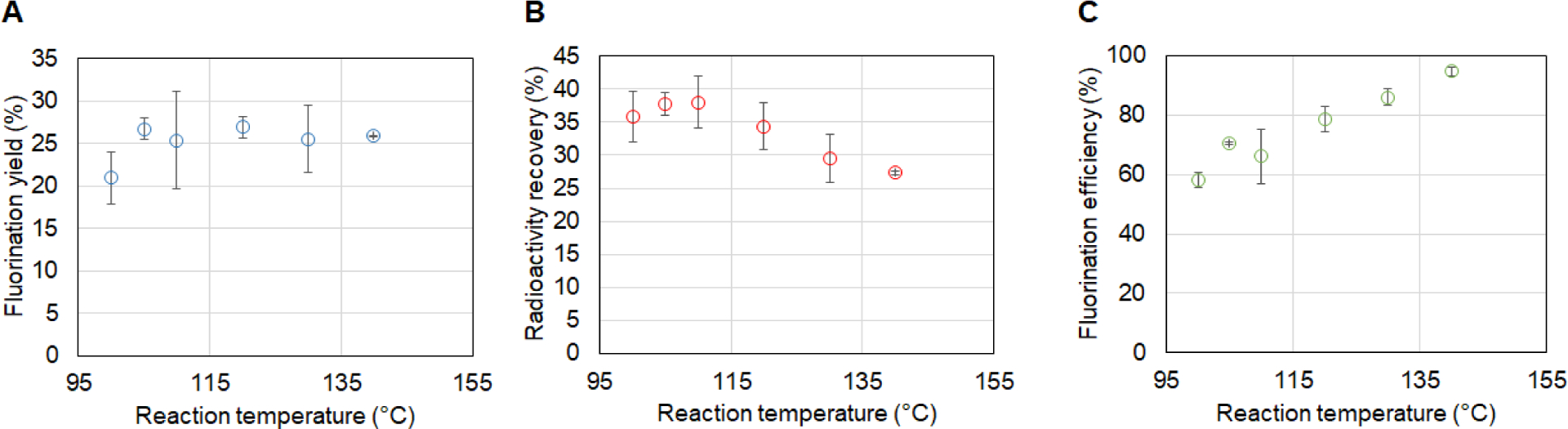
Optimization of reaction temperature. (A) Effect on the fluorination yield. (B) Effect on the radioactivity recovery. (C) Effect on the fluorination efficiency. Datapoints represent average values and error bars represent standard deviations. For 100, 105, 110, 120, 130, and 140°C datapoints, the number of replicates is n = 3, 2, 3, 3, 2, 2, respectively.
When we increased the starting activity to ~370 MBq, we observed a somewhat lower isolated radiochemical yield (non- corrected) of 3.9 ± 1.8% (n=3), which is still higher than one of the reported macroscale methods24, but lower than the other23. The reduction in crude RCY arises predominantely from a reduction in radioactivity recovery from 20 ± 2 % (n=4) to 10 ± 2 % (n=3) as the yield increases, a phenomenon we are continuing to investigate. The activity yield in these cases was ~15 MBq [400 μCi], which is sufficient for imaging of multiple (e.g., 4–8) mice, depending on the sensitivity of the preclinical PET scanner.
Molar activity of a radiopharmaceutical can generally be estimated by dividing the radioactivity of the compound injected into HPLC by the total molar amount (radioactive and non-radioactive forms), determined using the area under the UV absorbance peak in conjunction with an appropriate calibration curve (Fig. S3, ESI). In the case of the samples here, the UV peak was not detectable, but we can conservatively estimate an upper bound on the molar amount of FDOPA based on the lowest calibration point (0.2 nmol), and then compute a lower bound on the molar activity of >75 GBq/μmol [>2.0 Ci/μmol] (end of synthesis). Notably, this is significantly higher than the macroscale reports from Collins et al. (30–33 GBq/μmol [0.8–0.9 Ci/μmol], n=3)24 and Kuik et al. (35 ± 4 GBq/μmol [0.9 ± 0.1 Ci/μmol], n=?)23, despite starting with lower activity, which, in macroscale systems, typically results in lower molar activity5. We should note that due to scheduling constraints in our radiochemistry facility, our higher activity experiments were started ~3 h after the end of bombardment (EOB); had we performed these experiments immediately at EOB to avoid radioactive decay, even higher molar activities (i.e., >230 GBq/μmol) would likely have been observed.
3.3. Improvements in efficiency
Compared to macroscale methods for producing batches of [18F]FDOPA using the same precursor and synthesis route, the microscale method, with only 10 μL reaction volume, used more than two orders of magnitude less precursor, i.e. 0.12 μmol, compared to 16.8 μmol24 or 13.4 μmol23. The amounts of other reagents (i.e. K2CO3, K222 and H2SO4) were also reduced accordingly. However, we also produced less activity (i.e. ~370 MBq [10 mCi]) than the macroscale synthesis reported by Collins et al. (i.e., 15 GBq [400 mCi])24. (The synthesis scale was not reported in Kuik et al.23, making a comparison impossible.) Factoring in this difference in activity level produced, the microscale method is still more precursor efficient: the microscale method consumed 8.0 nmol/MBq of product, while the macroscale synthesis of Collins et al. consumed 39 nmol/MBq of product24. If focusing on the production of small batches of [18F]FDOPA (e.g. to support preclinical imaging studies, or to perform reaction optimization), the microscale synthesis would have an even greater advantage of precursor efficiency over macroscale methods; this is because in the macroscale, the full amount of precursor is still needed, even when the starting activity is drastically reduced.
Perhaps as a result of using small reagent masses, we also found it possible to eliminate the intermediate purification step used in macroscale methods between the fluorination and hydrolysis reactions. This step, consisting of solid phase extraction (trapping and eluting using a C18 cartridge), followed by a solvent evaporation step in the second reaction vessel before proceeding with hydrolysis, is generally utilized to remove the unreacted [18F]fluoride, salts, and side products. Omission of this step in the macroscale results in poor performance of hydrolysis and a difficult purification step. However, despite omitting this purification step in the microscale synthesis, the deprotection reaction proceeded efficiently, and there were very few peaks in the crude product and final product purification was straightforward.
Due to the smaller volumes used during [18F]fluoride drying, fluorination and deprotection reactions, the time needed for heating, cooling, and evaporations could be greatly reduced. The time needed for the microdroplet synthesis (up to the purification step) was only 23 min compared to the ~58 min24 or ~101 min23 reported for the macroscale synthesis. Furthermore, the small mass of reagents and small volume collected from the chip (~80 μL) facilitated the use of analytical-scale HPLC to perform purification. This enabled rapid purification (~7 min) and needed only a short time for formulation (~1 min). Overall the synthesis time with the microdroplet reactor was ~31 min, compared to ~71 min24, or ~117 min23 in conventional radiosynthesizers (Table 2).
Table 2.
Comparison of automated [18F]FDOPA synthesis performance in microscale (at the higher activity level tested) and macroscale. All values are averages ± standard deviations for the indicated number of replicates. Abbreviations: EOS = at end of synthesis
| This work (microdroplet reactor) (n=3) | Kuik et al. (conventional synthesizer) (n=?) | Collins et al. (conventional synthesizer) (n=3) | |
|---|---|---|---|
| Overall synthesis time (min) | ~ 31 | ~ 117 | ~ 71 |
| Precursor amount (μmol [mg]) | 0.12 [0.11] | 13.4 [12] | 16.8 [15] |
| Reaction volume (μL) | 10 | 750 | 1000 |
| Starting activity (GBq) | ~0.37 | Not reported | ~15 |
| Isolated RCY (%, decay corrected) | 4.7 ± 1.0 | 14 ± 4 | 4.5 ± 1.3 |
| Isolated RCY (%, non-decay corrected) | 3.9 ± 1.8 | 6.7 ± 1.9 | 2.9 ± 0.8 |
| Activity yield, EOS (GBq) | ~0.015 | Not reported | ~0.43 |
| Molar activity, EOS (GBq/μmol) | >75 | 30–33 | 35 ± 4 |
| Solvent volume for synthesis (mL) | 0.17 | 6.35 | 13.2 |
| Mobile phase volume for HPLC purification (mL) | 9.3 | 32 | 65 |
| Estimated mobile phase volume for HPLC column equilibration and cleaning (mL) | 48 | 240 | 240 |
In addition, volume reduction of the microscale syntheses contributes to a greener process (Table 2). The volume of solvents needed for all microscale synthesis steps (up to the purification step) was only 0.17 mL, compared to the solvent volumes needed for the macroscale syntheses (~6.4 mL23, or ~13 mL24). Also, with the use of analytical-scale HPLC for purification, the product was isolated with the consumption of 9.3 mL of mobile phase, while 32 mL23 or 65 mL24 of mobile phase were needed for separating products via semi-preparative HPLC in the macroscale syntheses. Furthermore, the amounts of solvents consumed while performing HPLC column equilibration and column cleaning are reduced about 5-fold when using an analytical column (4.6 mm × 250 mm) compared to a semi-preparative column (10 mm × 250 mm).
4. Conclusion
A straightforward synthesis for [18F]FDOPA using the diaryliodonium salt-based route was implemented in microscale (manually and automatically) for the first time. Optimization of various parameters (base amount, precursor concentration, reaction temperature, etc.…) and adding a radical scavenger (i.e. TEMPO), was performed using a high-throughput platform we recently developed based on multi-reaction droplet chips7, and the optimized synthesis was automated using a novel ultra-compact synthesizer based on a droplet microreactor8. The radiochemical yield (uncorrected) was comparable to macroscale reports and we demonstrated, via modest scaleup (20x) the production of ~15 MBq [400 μCi] which is sufficient for imaging in multiple mice. It is likely that higher scale synthesis can be achieved by integration with a radionuclide concentrator10 and by studying the factors that contributed to the slight reduction in yield when the activity was scaled up. As has been observed previously for microscale syntheses5, molar activity was high (>75 GBq /μmol [>2 Ci/μmol]). Though the molar activity has been shown in some cases not to impact the in vivo imaging of [18F]FDOPA23, many other tracers do benefit from production at high molar activity, especially those targeting low tissue density receptors5.
Notably, the tedious and time-consuming solid-phase extraction and evaporative solvent exchange (for intermediate purification between fluorination and deprotection) could be eliminated, and the crude product could be easily purified with analytical-scale HPLC. Due to these changes and the rapid heating (5 – 10 s), cooling (1.0 – 1.5 min), and evaporation of small volumes (10 μL is dried completely in < 1 min), the overall synthesis time was shortened significantly (~31 min vs ~71 min24 or ~117 min23).
Another prominent benefit brought by this microscale synthesis is its contribution to green chemistry by substantially reducing the amount of reagents (precursor), which could also significantly lower the cost per batch of [18F]FDOPA (currently hundreds of $USD per batch). Compared to the macroscale synthesis, precursor consumption was reduced by ~5x when normalized by the quantity of [18F]FDOPA produced; however, if one considers the situation of preparing only small batches of [18F]FDOPA (e.g., for preclinical imaging or for reaction optimization), the microscale method reduces precursor consumption by more than two orders of magnitude. The microscale synthesis also reduced by 5-fold the consumption of organic solvents. Furthermore, the synthesis apparatus is very simple and is likely to be less expensive than current macroscale radiosynthesizers, and the compact size potentially can enable self-shielding and avoid the need for hot cells and other infrastructure typical of a radiochemistry laboratory.
Supplementary Material
6. Acknowledgement
We thank Prof. Stephen DiMagno for providing precursor for these studies, Dr. Roger Slavik and the staff of the UCLA Biomedical Cyclotron facility for generously providing [18F]fluoride ion, and Gaoyuan Ma for helpful discussions. This work was funded in part by the National Institute on Aging (R21 AG049918), the National Cancer Institute (R21 CA212718), and the National Institute of Mental Health (R44 MH097271).
Footnotes
Conflict of interest disclosure
The Regents of the University of California have licensed technology to Sofie, Inc. that was invented by Dr. van Dam, and have taken equity in Sofie, Inc. as part of the licensing transaction. Dr. van Dam is a founder and consultant of Sofie, Inc. The remaining authors declare no conflicts of interest.
8 References
- 1.Sachinidis JI, Poniger S and Tochon-Danguy HJ, Curr. Radiopharm, 2010, 3, 248–253. [Google Scholar]
- 2.Keng PY, Esterby M and van Dam RM, in Positron Emission Tomography - Current Clinical and Research Aspects, ed. Hsieh C-H, InTech, Rijeka, Croatia, 2012, pp. 153–182. [Google Scholar]
- 3.Boschi S, Lodi F, Malizia C, Cicoria G and Marengo M, Appl. Radiat. Isot, 2013, 76, 38–45. [DOI] [PubMed] [Google Scholar]
- 4.Pascali G, Watts P and Salvadori P, Nucl. Med. Biol, 2013, 40, 776–787. [DOI] [PubMed] [Google Scholar]
- 5.Sergeev M, Lazari M, Morgia F, Collins J, Javed MR, Sergeeva O, Jones J, Phelps ME, Lee JT, Keng PY and van Dam RM, Commun. Chem, 2018, 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madsen K, Marner L, Haahr M, Gillings N and Knudsen GM, Nucl. Med. Biol, 2011, 38, 1085–1091. [DOI] [PubMed] [Google Scholar]
- 7.Rios A, Wang J, Chao PH and van Dam RM, RSC Adv, 2019, 9, 20370–20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Chao PH and van Dam RM, Lab. Chip, 2019, 2415–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchsbaum MS, Christian BT, Lehrer DS, Narayanan TK, Shi B, Mantil J, Kemether E, Oakes TR and Mukherjee J, Schizophr. Res, 2006, 85, 232–244. [DOI] [PubMed] [Google Scholar]
- 10.Chao PH, Lazari M, Hanet S, Narayanam MK, Murphy JM and van Dam RM, Appl. Radiat. Isot, 2018, 141, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chao PH, Wang J and Van Dam RM, in Proceedings of the 22nd International Conference on Miniaturized Systems for Chemistry and Life Sciences, Royal Society of Chemistry, Kaohsiung, Taiwan, 2018, pp. 1155–1158. [Google Scholar]
- 12.Fischman AJ, Radiol. Clin. North Am, 2005, 43, 93–106. [DOI] [PubMed] [Google Scholar]
- 13.Schiepers C, Chen W, Cloughesy T, Dahlbom M and Huang S-C, J. Nucl. Med, 2007, 48, 1651–1661. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Silverman DHS, Delaloye S, Czernin J, Kamdar N, Pope W, Satyamurthy N, Schiepers C and Cloughesy T, J. Nucl. Med, 2006, 47, 904–911. [PubMed] [Google Scholar]
- 15.Becherer A, Szabó M, Karanikas G, Wunderbaldinger P, Angelberger P, Raderer M, Kurtaran A, Dudczak R and Kletter K, J. Nucl. Med, 2004, 45, 1161–1167. [PubMed] [Google Scholar]
- 16.Jager PL, Chirakal R, Marriott CJ, Brouwers AH, Koopmans KP and Gulenchyn KY, J. Nucl. Med, 2008, 49, 573–586. [DOI] [PubMed] [Google Scholar]
- 17.Minn H, Kauhanen S, Seppänen M and Nuutila P, J. Nucl. Med, 2009, 50, 1915–1918. [DOI] [PubMed] [Google Scholar]
- 18.Koopmans KP, de Vries EG, Kema IP, Elsinga PH, Neels OC, Sluiter WJ, van der Horst-Schrivers AN and Jager PL, Lancet Oncol, 2006, 7, 728–734. [DOI] [PubMed] [Google Scholar]
- 19.de Vries EFJ, Luurtsema G, Brüssermann M, Elsinga PH and Vaalburg W, Appl. Radiat. Isot, 1999, 51, 389–394. [Google Scholar]
- 20.Pretze M, Wängler C and Wängler B, BioMed Res. Int, 2014, 2014, e674063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zischler J, Kolks N, Modemann D, Neumaier B and Zlatopolskiy BD, Chem. – Eur. J, 2017, 23, 3251–3256. [DOI] [PubMed] [Google Scholar]
- 22.Libert LC, Franci X, Plenevaux AR, Ooi T, Maruoka K, Luxen AJ and Lemaire CF, J. Nucl. Med, 2013, 54, 1154–1161. [DOI] [PubMed] [Google Scholar]
- 23.Kuik W-J, Kema IP, Brouwers AH, Zijlma R, Neumann KD, Dierckx RAJO, DiMagno SG and Elsinga PH, J. Nucl. Med, 2015, 56, 106–112. [DOI] [PubMed] [Google Scholar]
- 24.Collins J, Waldmann CM, Drake C, Slavik R, Ha NS, Sergeev M, Lazari M, Shen B, Chin FT, Moore M, Sadeghi S, Phelps ME, Murphy JM and van Dam RM, Proc. Natl. Acad. Sci, 2017, 114, 11309–11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll MA, Nairne J, Smith G and Widdowson DA, J. Fluor. Chem, 2007, 128, 127–132. [Google Scholar]
- 26.Wang J, Chao PH, Hanet S and van Dam RM, Lab. Chip, 2017, 17, 4342–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dooraghi AA, Keng PY, Chen S, Javed MR, “CJ” Kim C-J, Chatziioannou AF and van Dam RM, Analyst, 2013, 138, 5654–5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.