

Abstract
Nucleic acid delivery provides effective options to control intracellular gene expression and protein production. Efficient delivery of nucleic acid typically requires delivery vehicles to facilitate the entry of nucleic acid into cells. Among non-viral delivery vehicles, cationic materials are favored because of their high loading capacity of nucleic acids and prominent cellular uptake efficiency through electrostatic interaction. However, cationic moieties at high dosage tend to induce severe cytotoxicity due to the interference on cell membrane integrity. In contrast, non-cationic materials present alternative delivery approaches with less safety concerns than cationic materials. In this Progress Report, principles of non-cationic material design for nucleic acid delivery are discussed. Examples of such non-cationic platforms are highlighted, including complexation or conjugation with nucleic acids and self-assembled nucleic acid structures.
Keywords: Nucleic acid delivery, non-viral, non-cationic, material design
Graphical Abstract
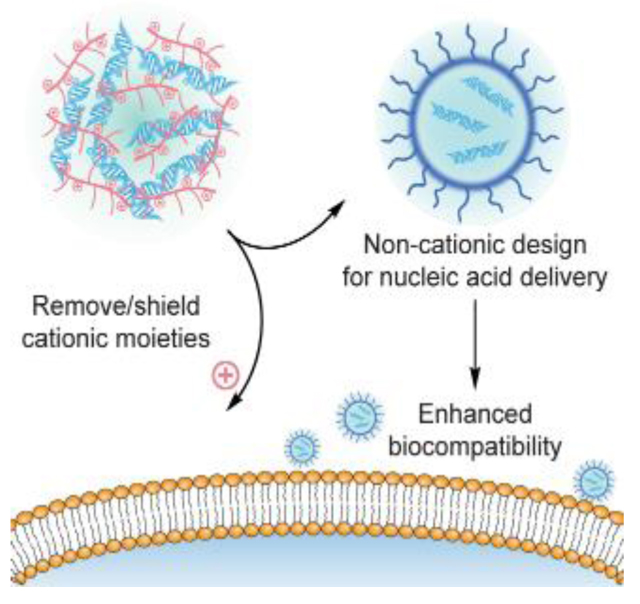
Non-cationic design provides a class of materials for nucleic acid delivery with reduced toxicity profiles. These materials are designed via either neutralizing the cationic moieties within the materials, or directly formulating with non-cationic materials. The high biocompatibility of non-cationic materials allows more flexibility on material dosage, broadening their applicability to resolve challenges in therapeutic delivery.
1. Introduction
Exogenous nucleic acids provide versatile options for the development of therapeutics. Efficient intracellular delivery of nucleic acids initiates specific biological process based on the type of nucleic acids.[1] Common choices of nucleic acid-based therapeutics include plasmid DNA, messenger RNA (mRNA), and small interfering RNA (siRNA). Briefly, successful delivery of either plasmid DNA into the nucleus[2] or mRNA into the cytosol[3] results in the production of the protein of interest. Delivery of siRNA into the cytosol introduces the process of RNA interference (RNAi), achieving gene specific knockdown.[4] Overall, nucleic acid delivery offers a robust toolkit for gene regulation, as well as contributing to the progress of research areas including small-molecule[5] and protein-based therapeutics.[6]
Nucleic acids generally require carriers to facilitate their delivery into cells. Because of the hydrophilic nature and large molecular weight, nucleic acid itself can hardly pass across the cell membrane.[7] Moreover, viral vectors[8] and synthetic materials[9] as carriers protect nucleic acids against nuclease degradation in the extracellular environment. Among these delivery carriers, viral vectors such as lentivirus[10] and adeno-associated virus[11] have been engineered to efficiently pack and deliver nucleic acids into cells. However, due to the possible immunogenic issues arising from the viral capsid proteins,[12] synthetic materials are introduced as alternative approaches for nucleic acid delivery. Since nucleic acids have phosphate-containing anionic backbones, the most straightforward material design for delivery carriers is to complex nucleic acids with cationic materials through electrostatic interaction. Cationic materials based on liposomes,[13] polymers,[14] and inorganic nanoparticles[15] have been extensively explored for nucleic acid delivery.
Cationic materials also facilitate nucleic acid delivery by interacting with cell membrane via electrostatic interaction. Since a majority of cell types shows net negative surface charge due to the patches on their glycocalyx,[16] positively charged materials are thus favored for therapeutic delivery applications. However, such electrostatic interaction is a “double-edged sword”. The dynamically assembled cell membrane can be disrupted when the dosage of cationic materials increases, directly causing significant cytotoxicity.[17] Apart from the damage on cell membrane, cationic materials have also been shown to affect the integrity of mitochondrial membrane,[18] as well as inducing genotoxicity and pulmonary inflammation.[19] Thus, developing delivery platforms with minimal safety concern is beneficial for the development of nucleic acid delivery.
Non-cationic materials are promising candidates with reduced toxicity profiles. These materials are designed via either neutralizing the cationic moieties within the materials, or directly formulating with non-cationic materials. With minimized net positive charge, non-cationic materials possess high biocompatibility comparing to cationic materials, allowing more flexibility on the material dosage and broadened applicability. While enhancing their biocompatible range, the removal or shielding of cationic moieties in non-cationic materials may result in decreased interaction with cell membrane, potentially reducing the efficiency of the delivery system. The design of non-cationic materials requires structural optimizations to take the above pros and cons into account. In this Progress Report, we will focus on the non-viral material design for nucleic acid delivery, specifically in non-cationic platforms.
2. Complexation with synthetic materials
The interaction between nucleic acids and synthetic materials is an essential aspect for the material design to formulate non-cationic delivery systems. Electrostatic interaction is mainly utilized to complex nucleic acids with synthetic materials, with additional steps to ensure the overall non-cationic feature of the resultant complexes. The general design principles of available techniques for such complexation will be discussed in this section.
2.1. Shielding cationic moieties
Limiting the exposure of cationic moieties will reduce the possibility of cell membrane damage that caused by positively charged materials. When cationic materials are involved in the formulation, the simplest approach to achieve the non-cationic feature is to shield these cationic moieties by anionic or charge-neutral molecules, including poly(ethylene glycol) (PEG) derivatives, anionic polysaccharides, and nucleic acids themselves.
PEG has been widely integrated into material design to increase their hydrophilicity, as well as reduce their non-specific interactions with surrounding biological components.[20] Such a stealth effect attributed to PEGylation can be utilized to develop carriers for nucleic acid delivery. In an early example from Kataoka and coworkers, they designed PEG-poly(L-lysine) block copolymers for plasmid DNA (pDNA) delivery (Figure 1a).[21] After forming the polyelectrolytes between the lysine residues on polymers and phosphate groups on pDNA, the PEG block was exposed on the surface to build up the shell of the complex, shielding the positive charges inside. Note that cationic moieties were not completely removed from these complexes. The PEG shell improved the stability of the complex, meanwhile reducing the polymer cytotoxicity comparing to poly(L-lysine) (PLL) homopolymers without the PEG block. Similarly, the PEG shell has also been incorporated for a non-cationic formulation through a sequential addition step. Wang and coworkers complexed siRNA with protonated doxorubicin through electrostatic interaction, followed by a coating step with PEG-polylactide (PLA) block copolymers (Figure 1b).[22] The PEG shell was attached on the siRNA-containing complex presumably through the hydrophobic interaction between PLA and doxorubicin. Such design resulted in a non-cationic platform for the co-delivery of siRNA and hydrophobic drugs. Since PEG is charge neutral and hydrophilic, non-covalently supplementing PEG as the shell material of delivery vehicles usually requires the driving force that provided by an additional material segment, including cationic or hydrophobic moieties.
Figure 1.
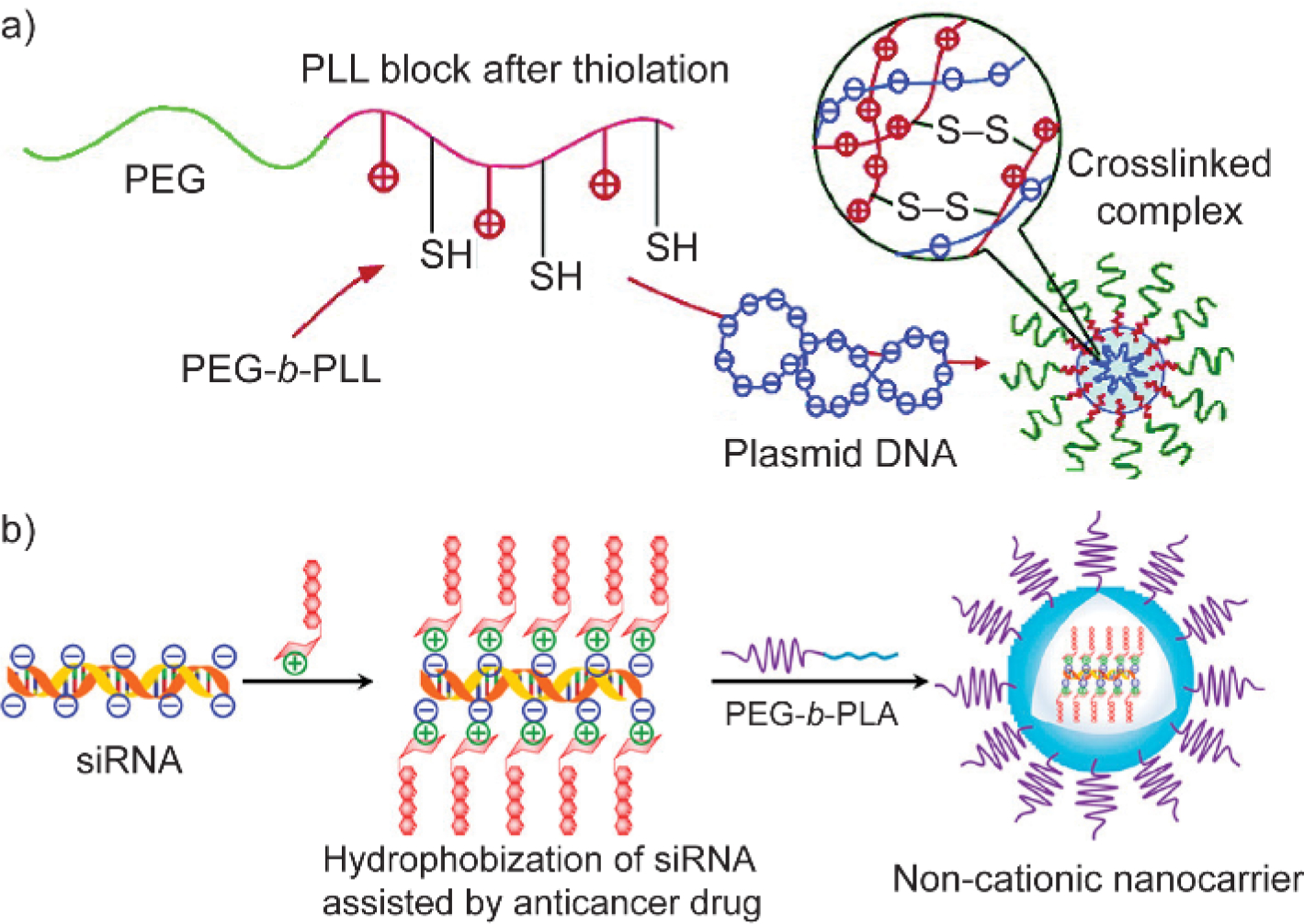
Schematic illustration of poly(ethylene glycol) shielding cationic moieties. (a) The poly(L-lysine) block was partially thiolated, initiating the complexation with plasmid DNA via electrostatic interaction as well as crosslinking the complex with disulfide linkage. PEG-b-PLL, poly(ethylene glycol)-poly(L-lysine) block copolymers. Adapted with permission.[21] Copyright 2004, American Chemical Society. (b) Hydrophobic anticancer drug was used to electrostatically complex with siRNA, followed by an encapsulation step using PEG-b-PLA block copolymers. PEG-b-PLA, poly(ethylene glycol)-polylactide block copolymers. Adapted with permission.[22] Copyright 2019, American Chemical Society.
Similarly, anionic polysaccharides can also shield the cationic moieties of delivery carriers. Involving anionic polysaccharides as a shielding layer on cationic complexes was achieved by either covalently conjugating polysaccharides with cationic materials, or directly incorporating polysaccharides on nucleic acid-containing complexes through electrostatic interaction. One unique advantage of polysaccharide coating is that certain type of polysaccharides has high binding affinity towards specific cell surface receptor,[23] e.g. hyaluronic acid (HA)-CD44 pair.[24] These negatively charged polysaccharides inherently bring cell surface targeting capability to the shell layer of nucleic acid-containing complexes. For example, Amiji and coworkers conjugated a series of amine derivatives on the carboxylate group of HA.[25] The conjugated amine moieties electrostatically interact with siRNA to initiate the assembly between HA derivatives and siRNA, which is not feasible for the anionic HA itself without chemical modifications. For tumors with similar vascular architecture, the siRNA-HA derivative assemblies demonstrated more efficient gene specific knockdown in CD44-overexpressing tumors than CD44-deficient tumors, confirming the contribution of HA backbone on the targeting of CD44 receptors. Other than synthesizing HA conjugates for nucleic acid binding, HA can also be involved to shield cationic moieties through electrostatic interaction. As an example, Cheng and coworkers first condensed pDNA with cationic polypeptide to form positively charged polyelectrolytes, followed by an HA-coating step on these polyelectrolytes (Figure 2).[26] The HA-coating reduced the cytotoxicity of the cationic polyelectrolytes and enhanced the transfection efficiency of pDNA towards CD44-positive cells. Thus, the design of delivery carriers for nucleic acids can benefit from anionic polysaccharides to achieve non-cationic feature and reduce the material toxicity. With suitable polysaccharide selection, using polysaccharide as the shield layer also improves the targeting capability of the delivery system.
Figure 2.
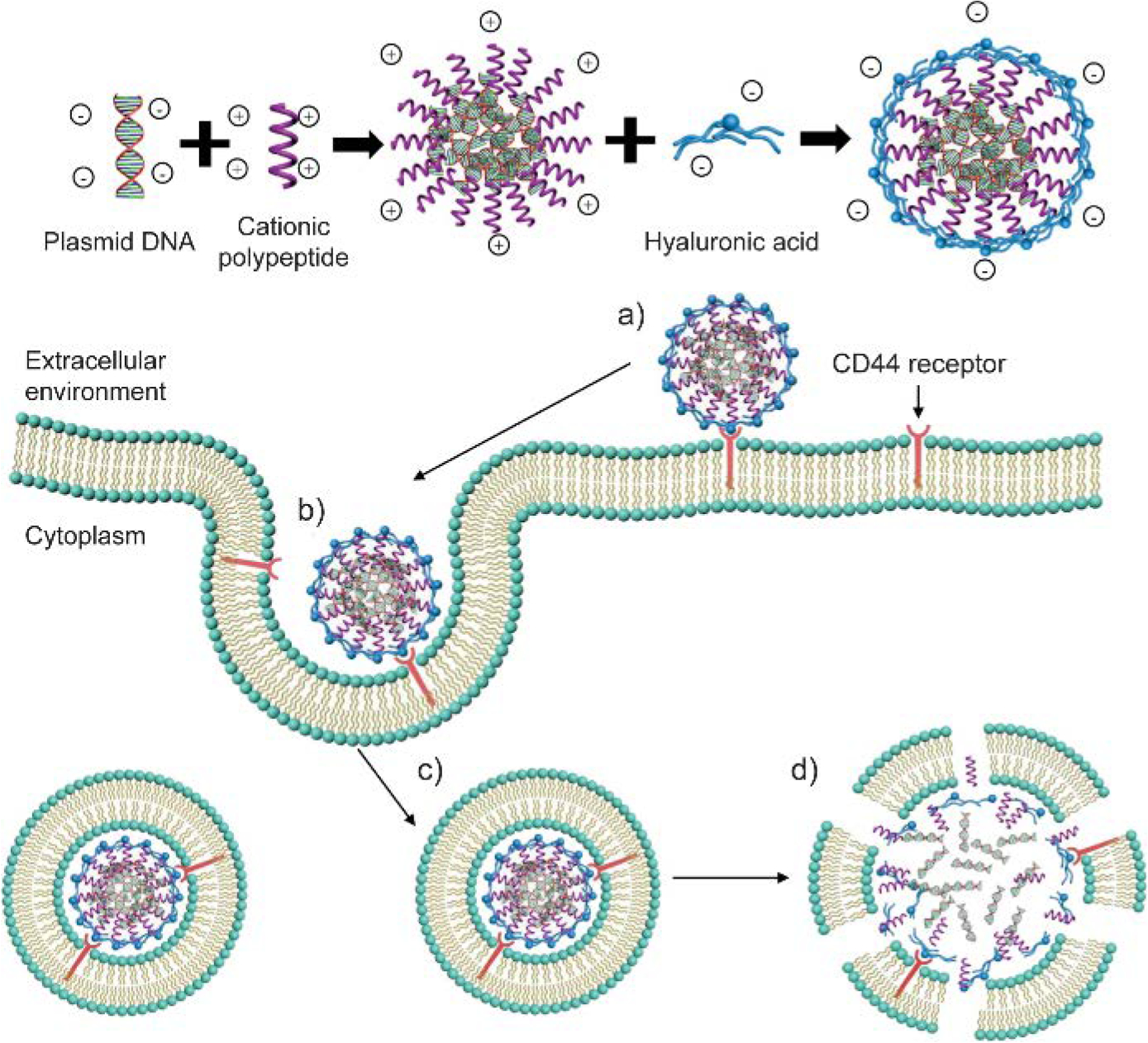
Schematic illustration of plasmid DNA-cationic polypeptide polyelectrolytes with hyaluronic acid (HA) coating. Cellular uptake process of HA-coated complexes: (a) Binding with cell surface CD44 receptor; (b~c) Endocytosis; (d) Disruption of endosomal membrane by cationic polypeptide and subsequent release of content. Adapted with permission.[26] Copyright 2016, American Chemical Society.
Nucleic acids themselves can shield cationic moieties through electrostatic interaction to reduce the cytotoxicity of delivery carries. In a gold nanoparticle (AuNP)-based siRNA delivery system, Ijiro and coworkers coated siRNA onto the surface of (16-mercaptohexadecyl)trimethylammonium bromide (MTAB)-stabilized cationic AuNPs.[27] After coating with siRNA, the surface charge of AuNPs was converted from positive to negative, ensuring high biocompatibility of the delivery system. With the help of an amphiphilic ligand anchored on the siRNA-coated AuNPs, these AuNPs efficiently entered HeLa cells and achieved gene specific knockdown. Gene specific knockdown mediated by siRNA is a commonly practiced assay to evaluate the efficiency of a nucleic acid delivery system. The siRNA of interest has two strands: a sense strand and an antisense strand. The antisense strand is designed to be complementary to the mRNA that is transcribed by the target gene. When siRNA enters the cytosol, with the help of Argonaute proteins, the target mRNA is recognized by the antisense strand and subsequently cleaved, causing the knockdown of the target gene expression. This is the RNA interference process as mentioned before.[28] RNAi is one of the most commonly used and successful technique for gene regulation. Recently, the first RNAi-based therapeutic has been approved by the US Food and Drug Administration (FDA) for hereditary ATTR amyloidosis treatment.[29]
2.2. Metal ion-induced coordination
Metal ions including Mg2+, Ca2+, and Zn2+ can be utilized to form a coordination network between nucleic acid and delivery vehicles. These metal ions simultaneously interact with the phosphate backbone of nucleic acids and the coordination site on the delivery vehicle, forming a nucleic acid-containing complex. Upon the formation of the tri-component complexes, the metal cations are expected to be embedded inside the complex, with either the nucleic acid or the delivery vehicles exposed on the outside. The variation of such metal cation-induced coordination design lies in the choice of coordination site on the delivery vehicle. In an recent example, the high affinity between Zinc(II)-dipicolylamine (DPA) and phosphate derivatives[30] was utilized to design polymeric vehicles for pDNA delivery (Figure 3a).[31] Zinc(II)-DPA derivative was decorated on the polymeric side chain and utilized to complex with pDNA. The metal coordination-induced complexes showed negligible cytotoxicity, while PEI at the same dosage resulted in more than 60% cell death. This non-cationic formulation achieved comparable transfection efficiency comparing to Lipofectamine 2000 and PEI. Note that since these non-cationic complexes do not interact as strongly as cationic materials to serum proteins, the non-cationic complexes achieved better transfection efficiency than Lipofectamine 2000 and PEI in high fetal bovine serum environment (> 10%). The interaction between Zinc(II)-DPA derivatives and nucleic acids has also been applied to develop non-cationic delivery materials for nucleic acids, including lipid-based[32] and hyaluronic acid-based[33] materials.
Figure 3.
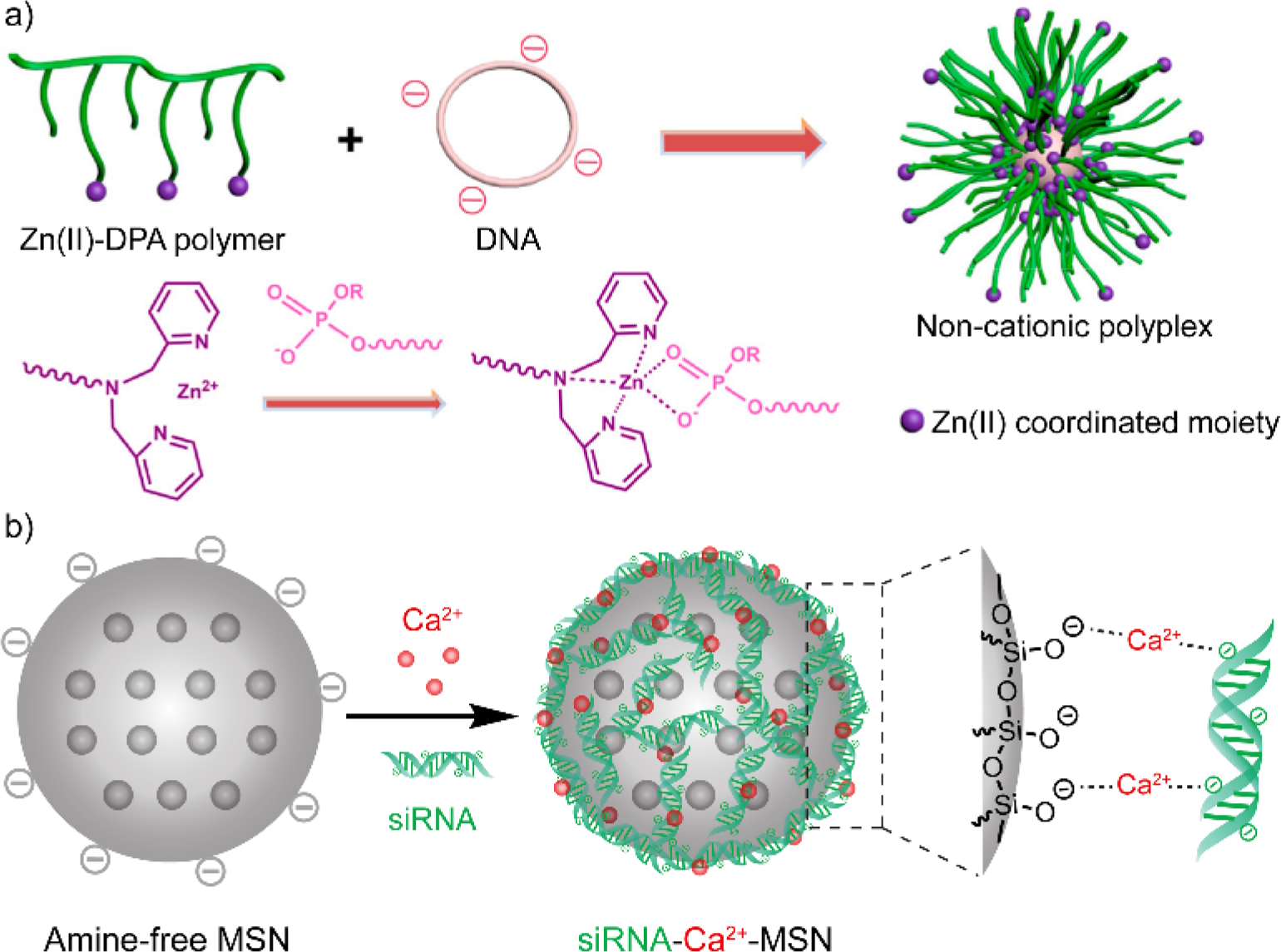
(a) Complexation between plasmid DNA and dipicolylamine-containing polymer through Zn(II) coordination. DPA, dipicolylamine. Adapted with permission.[31] Copyright 2018, American Chemical Society. (b) Complexation between siRNA and anionic mesoporous silica nanoparticle through their interaction with Ca2+ ion. MSN, mesoporous silica nanoparticle. Adapted with permission.[34] Copyright 2019, Elsevier Ltd.
Metal ion coordination also facilitates amine-free anionic materials for nucleic acid delivery. Calcium ion has been used as a bridge between the phosphate group on nucleic acids and negatively charged delivery vehicles,[35] forming non-cationic materials for nucleic acid delivery. For instance, silica nanoparticles (SiNPs) can be synthesized as an anionic form, displaying silicate groups on the surface. Using CaCl2 as the source of calcium ions, siRNA was coated on the surface of SiNPs through the interaction with the Ca2+ ions that immobilized on the SiNP surface (Figure 3b).[34] Before coating with siRNA, negatively charged SiNPs and Ca2+-immobilized SiNPs demonstrated higher biocompatibility than PEI-coated SiNPs and primary amine-coated SiNPs. The siRNA-Ca2+-SiNP complexes successfully delivered siRNA into cells and accomplished gene specific knockdown. Moreover, the mesoporous feature of these silica nanoparticles allows the loading of small molecule drugs inside the non-cationic complexes, enabling a co-delivery platform for siRNA and small molecules. Similarly, Burgess and coworkers reported that anionic liposomes based on 1,2-dioleoyl-sn-glycero-3-phospho-rac-(1-glycerol) (DOPG) were complexed with siRNA through their interactions with Ca2+ ions.[36] The non-cationic formulation achieved more efficient gene silencing than Lipofectamine 2000 at low lipid concentrations (< 0.5 μg·mL−1 lipid), as well as showing high cell viability (~100%) when the lipid concentration reaches 32 μg·mL−1. Meanwhile, the Lipofectamine 2000-based cationic formulation had an IC50 value of 22.9 μg·mL−1, denoting 50% cell viability at this concentration. The safe range of Ca2+ dosage was also confirmed to be within 4.8 mM, indicating the biocompatibility of such non-cationic formulations at a wide range of dosage.
2.3. Entrapping with crosslinked polymers
One ideal non-cationic material design for nucleic acid complexation is to eliminate the existence of cationic moieties in the final formulation. For example, Lopez-Berestein and coworkers successfully utilized neutral lipid-based liposomes for nucleic acid delivery.[37] These neutral lipids are zwitterionic phosphocholine derivatives, with a positively charged quaternary amine group and a negatively charged phosphate group displayed on the same molecule. Although the interaction between neutral lipids and nucleic acids were not clearly mentioned, it is reasonable to believe that quaternary amine group may have played a role in the interaction with the phosphate backbone of nucleic acids. Meanwhile, more options are available to result in platforms without cationic moieties in the final delivery formulation.
Crosslinked polymeric materials[38] present a covalently connected network to physically entrap nucleic acids, rather than complexing nucleic acids via electrostatic interaction. The versatile functionality on polymers allows a palette of methods to develop crosslinked polymers. Among several crosslinking methodologies,[39] disulfide crosslinking is a favorable option for intracellular therapeutic delivery.[40] Glutathione (GSH) is a cysteine-containing tripeptide that is highly involved in the intracellular redox regulation processes.[41] Because of the intracellular abundance (2~10 mM) of GSH comparing to the extracellular environment (2~20 μM),[42] cleavage of disulfide bonds within delivery vehicles can be refined within the cytosol, ensuring the intracellular release of cargos. For example, Park and coworkers developed thiol-functionalized HA derivative for siRNA delivery.[43] The non-cationic platform was carried out through a water-in-oil emulsion approach, where the HA-thiol derivatives and siRNA cargos were confined within the water droplet. The thiols conjugated on HA were crosslinked upon ultrasound treatment, forming disulfide crosslinks to trap siRNA cargos. As expected, the HA-based anionic delivery vehicles did not exhibit detectable cytotoxicity while PEI and PLL showed high cytotoxicity. Successful knockdown of transiently expressed green fluorescence protein (GFP) was achieved by the HA-based system. Note that the water-in-oil emulsion approach requires water-immiscible organic solvents and surfactants. These organic solvents and surfactants need to be carefully washed away to prevent their side effects on cell-based assays.
Crosslinked polymer for nucleic acid entrapment and delivery was also conducted completely in aqueous solutions, without involving organic solvents and surfactants. Very recently, Thayumanavan and coworkers designed a two-step approach to formulate non-cationic RNA-polymer complexes for RNA delivery (Figure 4).[44] The design starts with the complexation between cationic polymers and RNA to form RNA-polymer polyelectrolytes. The positive charge was contributed by the N-methylated pyridyl disulfide derivative on the polymer side chains. Next, dithiothreitol (DTT) was added into the polyelectrolytes to remove the cationic moieties, meanwhile crosslinking the polymers to lock the RNA molecules within the crosslinked complexes. Removing positive charge from the formulation has significantly reduced the cytotoxicity of the complex. The non-cationic RNA-polymer complexes showed gene specific knockdown in preimplantation mouse embryos, confirming the RNA interference capability of these non-cationic RNA-polymer complexes. Later, a post-functionalization approach was introduced to further develop this method. After the formation of RNA-polymer polyelectrolytes, DTT was added to partially crosslink the complex, followed by a post-PEGylation step using thiol-terminated PEG.[45] The post-functionalized RNA-containing complex was demonstrated to protect RNA from enzymatic degradation. By varying the DTT dosage, the density of disulfide bonds within the complex was controlled, allowing a tunable RNA release rate upon the GSH-triggered cleavage of disulfide bonds. Moreover, these non-cationic complexes delivered siRNA into human cervical cancer cells and specifically knocked down the targeted gene expression.
Figure 4.
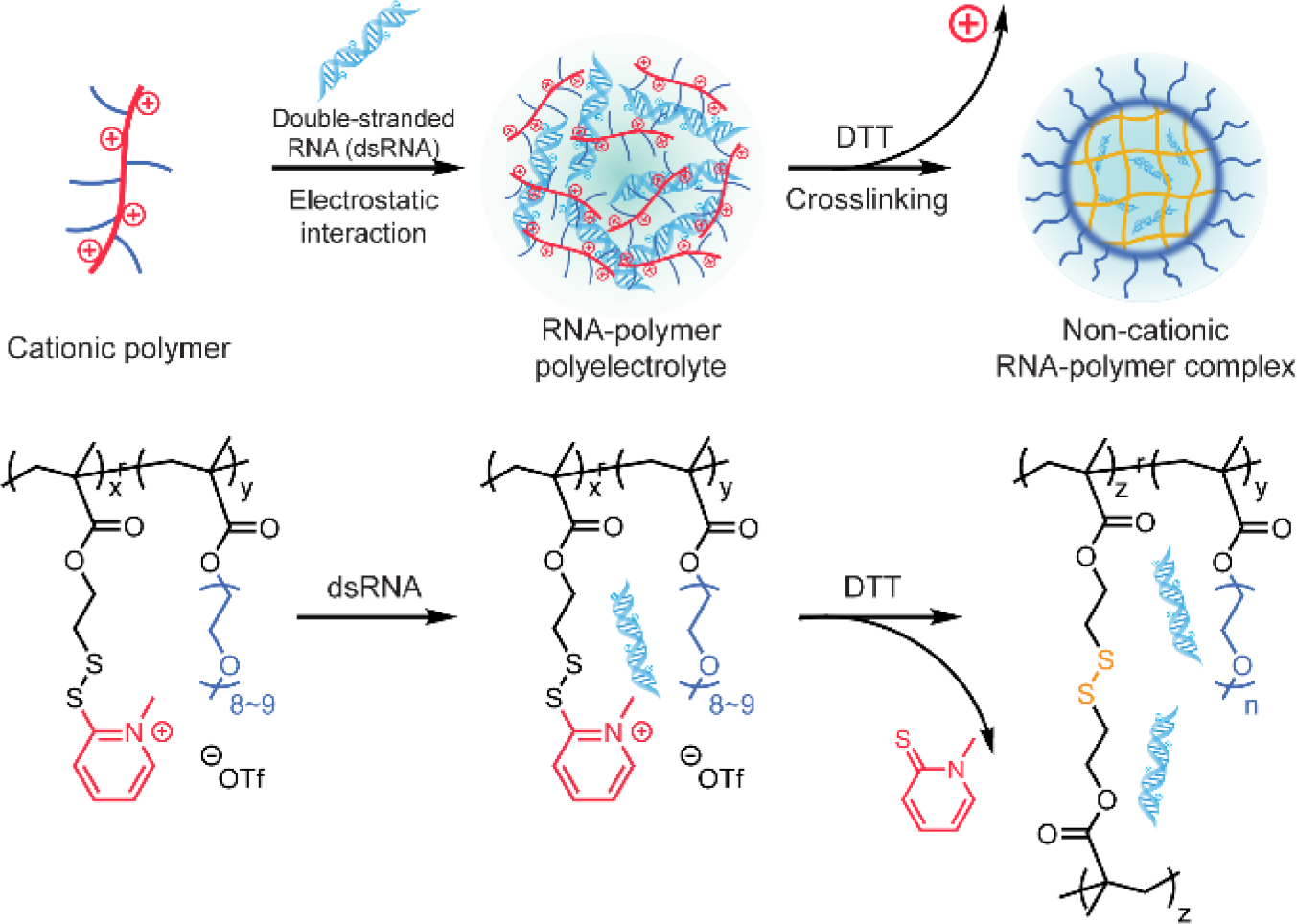
Scheme of non-cationic crosslinked polymer for RNA complexation. DTT, dithiothreitol. Adapted with permission.[44] Copyright 2019, American Chemical Society.
3. Conjugation with synthetic materials
Conjugating synthetic materials to nucleic acids requires structural modifications on nucleic acids as the first step.[46] The structural variation is typically designed on the 3’- or 5’- end of the nucleic acid strand, ensuring minimal interference on the overall activity of the nucleic acid.[47] Synthetic materials are conjugated with nucleic acids to facilitate their entry into cells. The choice of such synthetic materials includes small molecules, polymers, and inorganic nanoparticles (as summarized below). To obtain non-cationic delivery platforms, the conjugation approach does not require the involvement of electrostatic interaction, thus circumventing the need of cationic materials to bind with nucleic acids. However, if the biological process requires the participation of both exogenous nucleic acids and intracellular enzymes (e.g. the Argonaute protein during RNAi), the steric effect of conjugated materials must be carefully evaluated.
3.1. Small molecules
A majority of small molecule conjugation for nucleic acid delivery is based on lipophilic molecules. Lipid conjugation is one of the most effective and well-developed methods for nucleic acid delivery in non-cationic platforms.[48] Conjugation with lipids generally enhances the cell membrane permeability of nucleic acids.[49] Lipophilic molecules such as cholesterol,[50] fatty acid derivatives,[51] and α-tocopherol[52] have been successfully conjugated on siRNA, achieving RNA interference without the use of transfection reagent. The absence of transfection reagents reduces the cytotoxicity concern of delivery vehicles, while it also renders nucleic acids exposing to nucleases in biological fluids. Thus, the stability of nucleic acids needs to be improved against enzymatic degradation before lipid conjugation. One solution is to chemically modify the sugar, phosphate groups, or the entire backbone of nucleic acids, reducing their nuclease susceptibility. The detailed chemistry for such modifications has been summarized in previous review articles.[7a, 48, 53] Within these modifications, peptide nucleic acids (PNAs) are representative materials with the original sugar-phosphate backbone in nucleic acids replaced by N-2-aminoethylglycine repeating units, offering enhanced stability and scalability for nucleic acid-based materials.[54]
Nucleic acid modifications in combination with the lipid conjugation results in efficient non-cationic systems for nucleic acid delivery. For example, Stoffel and coworkers developed a library of lipid-conjugated siRNA for in vivo delivery (Figure 5a).[55] Before the lipid conjugation, the siRNA were chemically modified with a partial phosphorothioate backbone and 2’-O-methyl sugar modifications on both strands, enhancing their resistance to degradation by nuclease. The study showed that lipid-conjugation mediated efficient cellular uptake of siRNA without the requirement of cationic materials. Moreover, upon tuning the hydrophobic-hydrophilic balance of the conjugates, the incorporation of lipophilic molecules into nucleic acids allows the formation of micellar structures, broadening their applications in therapeutic delivery.[56]
Figure 5.
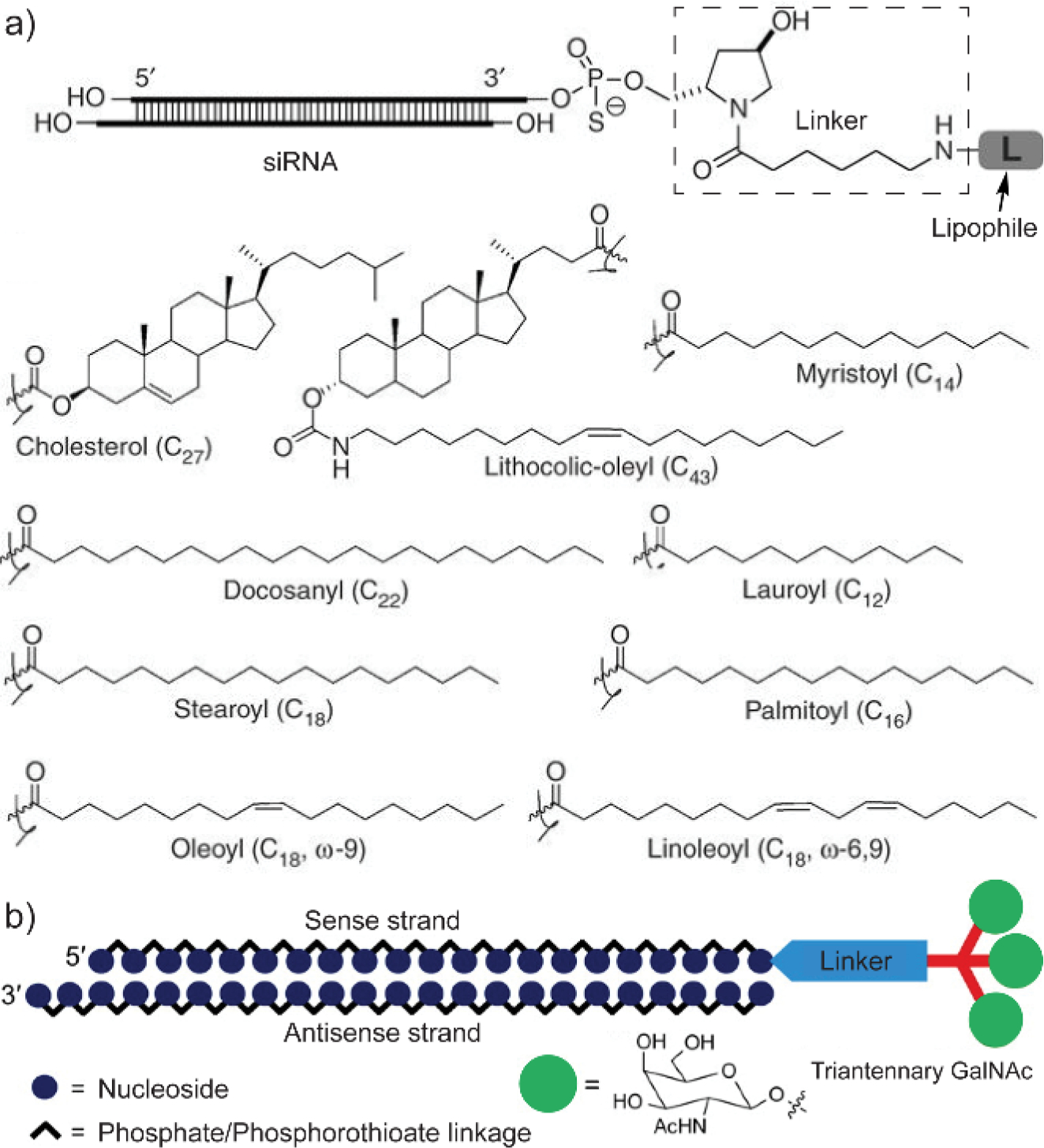
Small molecule modifications are generally designed on the 3’-end of the siRNA sense strand. (a) Molecular design of representative lipid-conjugated siRNAs. Adapted with permission.[55] Copyright 2007, Springer Nature. (b) Schematic illustration of triantennary N-acetylgalactosamine-conjugated siRNA. GalNAc, N-acetylgalactosamine. Adapted with permission.[57] Copyright 2014, American Chemical Society.
Apart from lipids, conjugation of water-soluble small molecules also helps the delivery of nucleic acids. One representative class of molecules is cell penetrating peptides (CPPs).[58] CPPs are typically positively charged due to the lysine and arginine residues, inherently having the similar cytotoxicity concerns with cationic delivery vehicles. Alternatively, conjugation of non-cationic small molecule ligands improves the cellular uptake of nucleic acids. Recently, Manoharan and coworkers reported the conjugation of an N-acetylgalactosamine (GalNAc) derivative with siRNA to improve their delivery into hepatocytes (Figure 5b).[57] GalNAc is a ligand that has high affinity towards the asialoglycoprotein receptor (ASGPR), which is a highly expressed receptor on hepatocytes.[59] A triantennary GalNAc ligand was synthesized and conjugated on the 3’-end of the siRNA sense strand. Note that the small molecule-conjugation is generally designed on the sense strand. Minimal modifications on the antisense strand allows the antisense strand to bind with Argonaute proteins without elevated steric interference, subsequently initiating the RNA interference process. The GalNAc-conjugated siRNA resulted in significantly enhanced cellular uptake in mouse hepatocytes than the unconjugated siRNA. Meanwhile, cellular uptake of the GalNAc derivative-conjugated siRNA was almost diminished in ASGPR-deficient cells, suggesting the effect of ligand-receptor binding on improving the cellular uptake. From the in vivo evaluation, the GalNAc-conjugated siRNA accumulation and gene specific knockdown was observed in mouse liver, further confirming the targeting capability that introduced by the conjugation.
3.2. Polymers
Conjugating nucleic acids with synthetic polymers improves the stability of nucleic acids by hindering nucleic acids from nuclease degradation. The structure of synthetic polymers needs to be rationally designed to improve the cellular uptake of nucleic acids. Meanwhile, intracellular cleavage of nucleic acids from the polymer conjugates is also important to initiate the upcoming biological process, avoiding the hinderance from polymers. In an example from Maynard and coworkers, PEG was synthesized with a pyridyl disulfide unit at the end of the polymer backbone, enabling their conjugation with thiol-modified siRNA through thiol-disulfide exchange reaction.[60] With the charge-neutral PEG conjugated on the siRNA, the overall charge of the delivery platform is maintained as non-cationic. Later, the PEG-siRNA conjugates demonstrated increased serum stability comparing to unmodified siRNA.[61] With significantly high biocompatibility presented, the PEG-siRNA conjugates were capable of gene specific silencing. After further formulating the non-cationic conjugates with a cationic peptide, a twofold increase in gene silencing efficiency was observed.
Likewise, block copolymers with both PEG and azide-functionalized block were conjugated with a dibenzocyclooctyne (DBCO)-functionalized DNA strand. The conjugation was initiated between the DBCO and azide group,[62] a well-known click reaction pair.[63] The resulted conjugates showed enhanced stability against enzymatic degradation and improved cellular uptake efficiency. However, the triazole linkage formed between DBCO and azide cannot be easily cleaved inside the cell. Thus, a disulfide-containing linker was developed to ensure the release of nucleic acids from the nucleic acid-polymer conjugates after the click reaction.[62b] As mentioned in the previous section, the intracellular GSH is expected to cleavage the disulfide linkage among the delivery vehicles. A largely improved gene specific knockdown efficiency was observed within siRNA-polymer conjugates that formed with the disulfide linkage, outperforming the uncleavable siRNA-polymer conjugates without disulfide linkages (Figure 6a).
Figure 6.
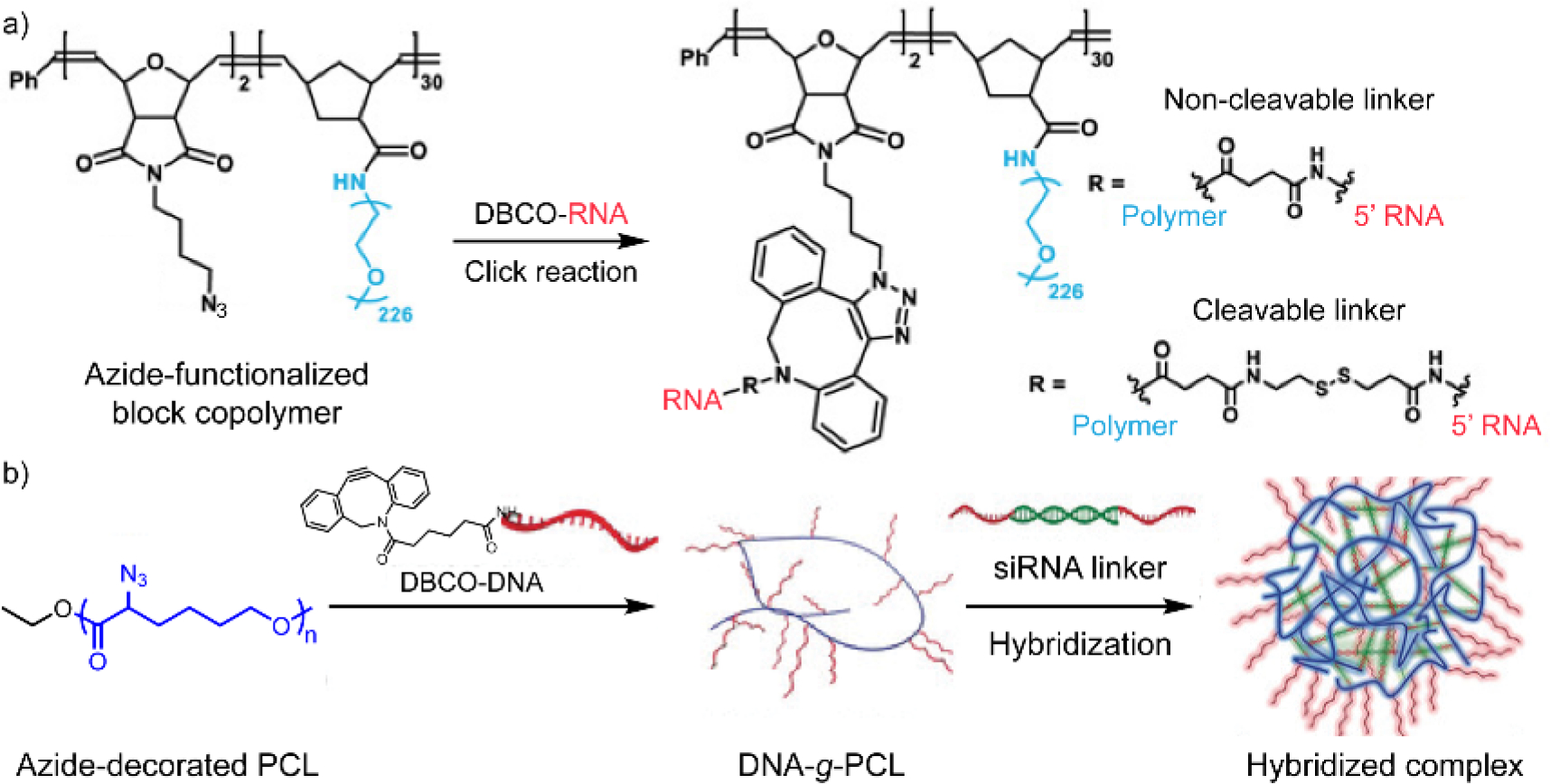
Polymer-conjugated nucleic acid via the click reaction between dibenzocyclooctyne (DBCO) and azide. (a) Conjugation between DBCO-functionalized RNA and azide-containing block copolymer. Adapted with permission.[62b] Copyright 2019, American Association for the Advancement of Science. (b) Formulation process of the hybridized complex between siRNA and DNA-grafted-polycaprolactone (PCL) (DNA-g-PCL). Before the hybridization step, DBCO-functionalized DNA was first conjugated with azide-decorated PCL. Adapted with permission.[64] Copyright 2018, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
DNA-polymer conjugate can also be used as a protective scaffold for siRNA delivery. In a recent report, a DBCO-terminated DNA strand was grafted on azide-decorated polycaprolactone (PCL) to form DNA-PCL conjugates (Figure 6b).[64] Next, the conjugates were complexed with siRNA via the nucleic acid hybridization between the DNA strand and the pre-designed single-stranded overhang at both end of the siRNA. The complexation embeds siRNA inside and provides a steric shield for siRNA against the degradation of ribonuclease. The gene silencing result and antitumor activities showed that Lipofectamine 2000 is more efficient than such non-cationic platform, suggesting that improvement is still needed for the material design to enhance the therapeutic efficacy.
3.3. Inorganic nanoparticles
Anchoring nucleic acids onto inorganic nanoparticles through metal-ligand interactions boots the stability of nucleic acid-particle conjugates.[65] Take gold nanoparticles (AuNPs) as an example, the Au-S interaction was analyzed to be a mixture of electrostatic (~65%) and covalent (~35%) character.[66] The partially covalent character of Au-S was utilized to construct AuNP-based conjugates with thiolated small molecules and biomacromolecules.[15b] One representative class of materials is the nucleic acid-AuNP conjugates (Figure 7).[67] In an example, Mirkin and coworkers started the material formulation from citrate-stabilized AuNPs, followed by a conjugation step with thiol-modified oligonucleotides.[68] The platform is ensured to be non-cationic because both the citrate-AuNPs and oligonucleotides are negatively charged. These conjugated nucleic acids were antisense oligonucleotides, a type of nucleic acid strand that is complementary to the target gene-transcribed mRNA. Once entering the cytosol, the antisense strand will bind with target mRNA and suppress the expression of the target gene, denoted as antisense therapy. Comparing to the unmodified oligonucleotide counterparts, the oligonucleotides conjugated on AuNPs demonstrated enhanced stability against enzymatic degradation, as well as higher binding affinity constants with the complementary sequence. Such conjugation on spherical nanoparticles lead to a spherically packed orientation of nucleic acids, decreasing the accessibility of nucleases. Cellular uptake efficiency of these nucleic acid-conjugated particles is correlated to the density of nucleic acids on each particle, as more efficient delivery was observed with higher density. The design of nucleic acid-AuNP conjugate was also successfully applied with other type of inorganic nanoparticles (e.g. iron oxide),[69] expanding the non-cationic platform toolkit for nucleic acid delivery.
Figure 7.
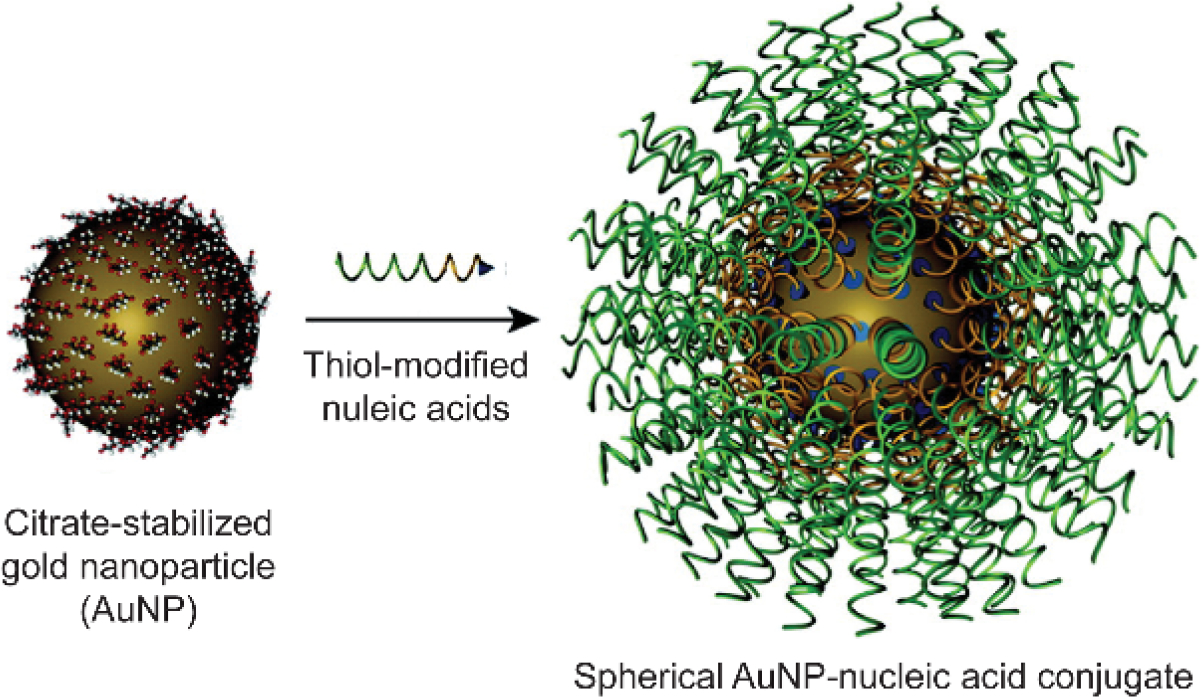
Synthesis of nucleic acid-gold nanoparticle conjugates. Adapted with permission.[67a] Copyright 2012, American Chemical Society.
4. Engineered nucleic acid-based structures
Engineered nucleic acid-based structures can be employed for nucleic acid delivery.[70] As these materials are completely constructed by nucleic acids, their overall charge is going to be guaranteed as anionic, inherently presenting less cytotoxicity than cationic materials.
4.1. Programmed nucleic acid structures
With specifically designed DNA/RNA sequences, based-pairing initiates the recognition and hybridization of single-stranded ends on two or multiple nucleic acid pieces, resulting in the self-assembly of nucleic acids.[71] Artificially programmed assemblies of nucleic acids create a large library of DNA/RNA nanostructures for biomedical applications.[72] Several of these nucleic acid architectures feature enhanced stability in presence of serum than linear nucleic acids.[73] Such stability advantage is presumably attributed to the densely packed morphology of engineered nucleic acid structures, similar to the nucleic acid-inorganic nanoparticle conjugates mentioned above.
One straightforward material design strategy is to incorporate nucleic acid cargos into nucleic acid-based delivery vehicles. The unique base-pairing interaction among nucleic acids allows the hybridization and complexation between vehicles and cargos. From an example from Anderson and coworkers, they designed six DNA single strands with complementary overhangs at the 3’ ends to self-assemble into a tetrahedron structure, with double-stranded DNA formed on each edge (Figure 8).[74] Once the DNA assembly forms, each edge has an overhang in the middle that is complementary to the overhang of cargo siRNA strands, allowing six siRNA molecules anchored on each DNA tetrahedron. The siRNA cargo was later modified to conjugate with a folic acid derivative, improving the cellular uptake and gene silencing efficiency of the complex. By varying the overhang sequence of siRNA, the number and location of hybridized siRNA on the DNA tetrahedron, consequently tuning the number and location of folic acid ligands. With such a precise control on ligand decoration, the design notably revealed the threshold number and optimal spatial orientation of targeting ligands for efficient nucleic acid delivery. The precise control on nucleic acid sequence enables a tunable control on the size and morphology of programmed nucleic acid structures, offering multiple design choices for nucleic acid-based delivery vehicle.
Figure 8.
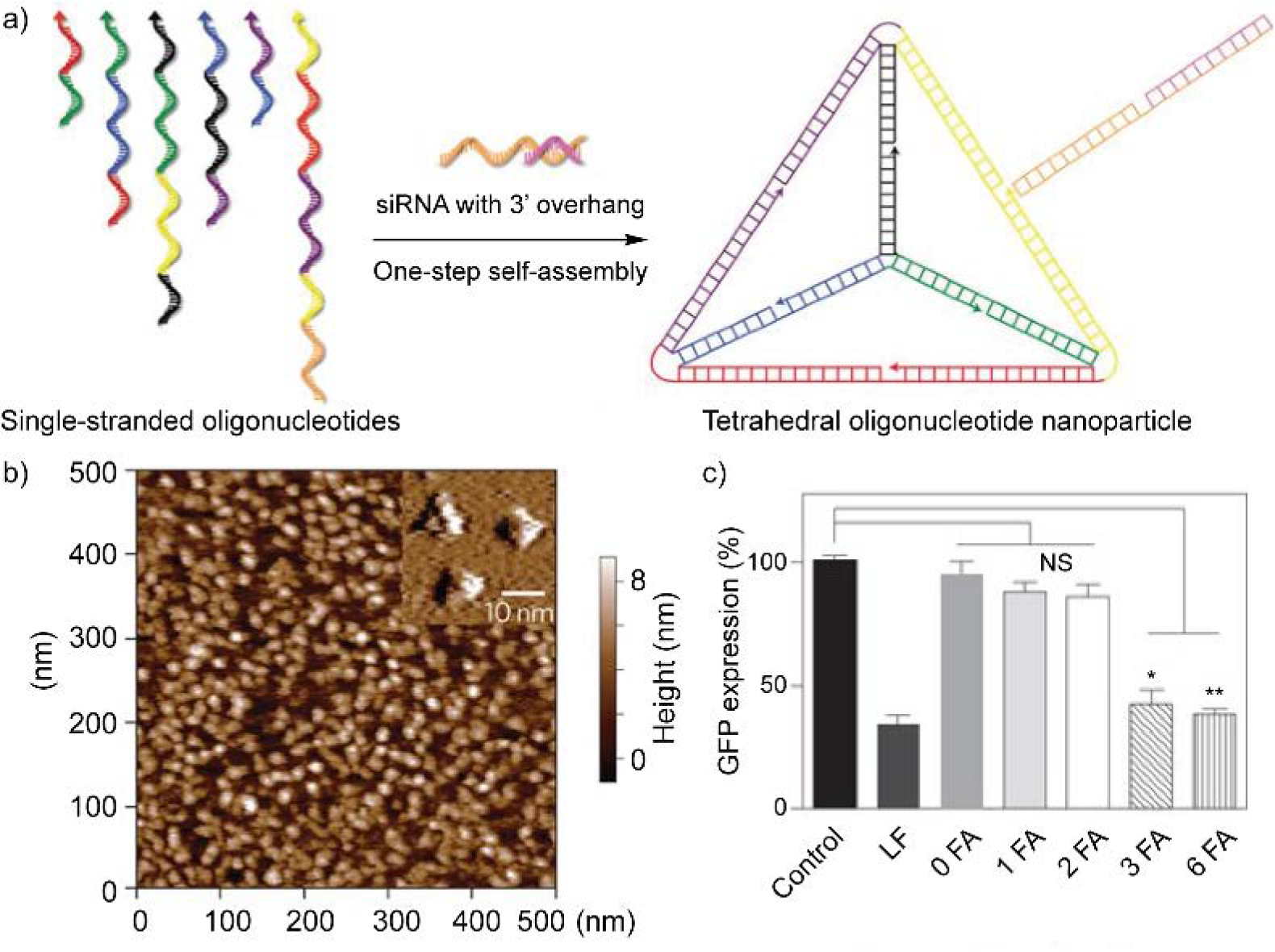
(a) One-step self-assembly of oligonucleotide nanoparticles. Six single-stranded DNA altogether self-assemble into tetrahedral structure. Arrowhead stands for the 5’-end of each DNA strand, with each color corresponding to one of the six edges of the tetrahedron. Meanwhile, the design allows site-specific hybridization of siRNA to the tetrahedron edge. (b) Atomic force microscopy image of the tetrahedral oligonucleotide nanoparticles. (c) Folic acid (FA) density on the oligonucleotide nanoparticle affects the GFP gene silencing efficiency of the particle. LF, Lipofectamine RNAiMAX formulated with siGFP (siRNA of GFP). 0, 1, 2, 3, or 6 FA represent oligonucleotide nanoparticles with same siGFP concentration and varied number of folic acid. *P < 0.003, **P < 0.001 compared with control (siGFP only). NS, not significant. Adapted with permission.[74] Copyright 2012, Springer Nature.
4.2. Rolling circle amplification
Rolling circle amplification (RCA) is an amplification process of nucleic acid based on circular DNA or RNA templates.[75] Inspired by the rolling circle replication processes in viruses and bacteria,[76] the RCA process produces long single-stranded nucleic acids with periodic sequence, where each repeat unit is complementary to the template sequence. The design of RCA process can also benefit from the strategies of programmed assembly, resulting in folded scaffolds for therapeutic delivery applications.[77] For example, initiated with the RCA process, Sleiman and coworkers developed a nucleic acid-based nanotube structure.[78] With an improved nuclease resistance, these nanotubes were internalized by cells more efficiently than regular double-stranded DNA.
Folding the long chain of RCA products creates substantial space for cargo encapsulation, including small molecule drugs and proteins.[79] Meanwhile, during the delivery process, the RCA product itself can act as a functional nucleic acid.[80] With a suitable cargo release mechanism designed, a synergistic therapeutic effect can be achieved by these cargos. For example, Gu and coworkers reported an RCA-based design for programmed delivery of nucleic acid and protein (Figure 9).[81] The choice of nucleic acid and protein were selected for cancer immunotherapy. In detail, the nucleic acid was produced by RCA process with CpG sequence and enzyme-cleavable region encoded into the DNA template. The CpG oligonucleotides within the nucleic acid act as potent immunostimulants by triggering Toll-like receptor 9-expressing cells, inducing enhanced anti-cancer activities in several reported cancer treatments.[82] The controlled release of CpG oligodeoxynucleotide (ODN) was designed to be triggered by a caged enzyme that is liberated at inflammatory conditions. Meanwhile, the folded nucleic acid functions as a delivery vehicle to load an antibody against programmed cell death protein 1 (anti-PD-1). The natural ligand of PD-1 (PD-L1/2) are highly expresses on multiple cancer cells.[83] These ligands interact with PD-1 on immune cells to prevent immune system from eliminating cancer cells. As a result, the anti-PD-1 antibodies are immune regulatory checkpoint inhibitors by recognizing PD-1 on immune cells, blocking its interaction with PD-L1/2 on cancer cells. Therefore, RCA product-based delivery of CpG oligonucleotides and anti-PD-1 antibodies boosts the immune response against cancer cells.
Figure 9.
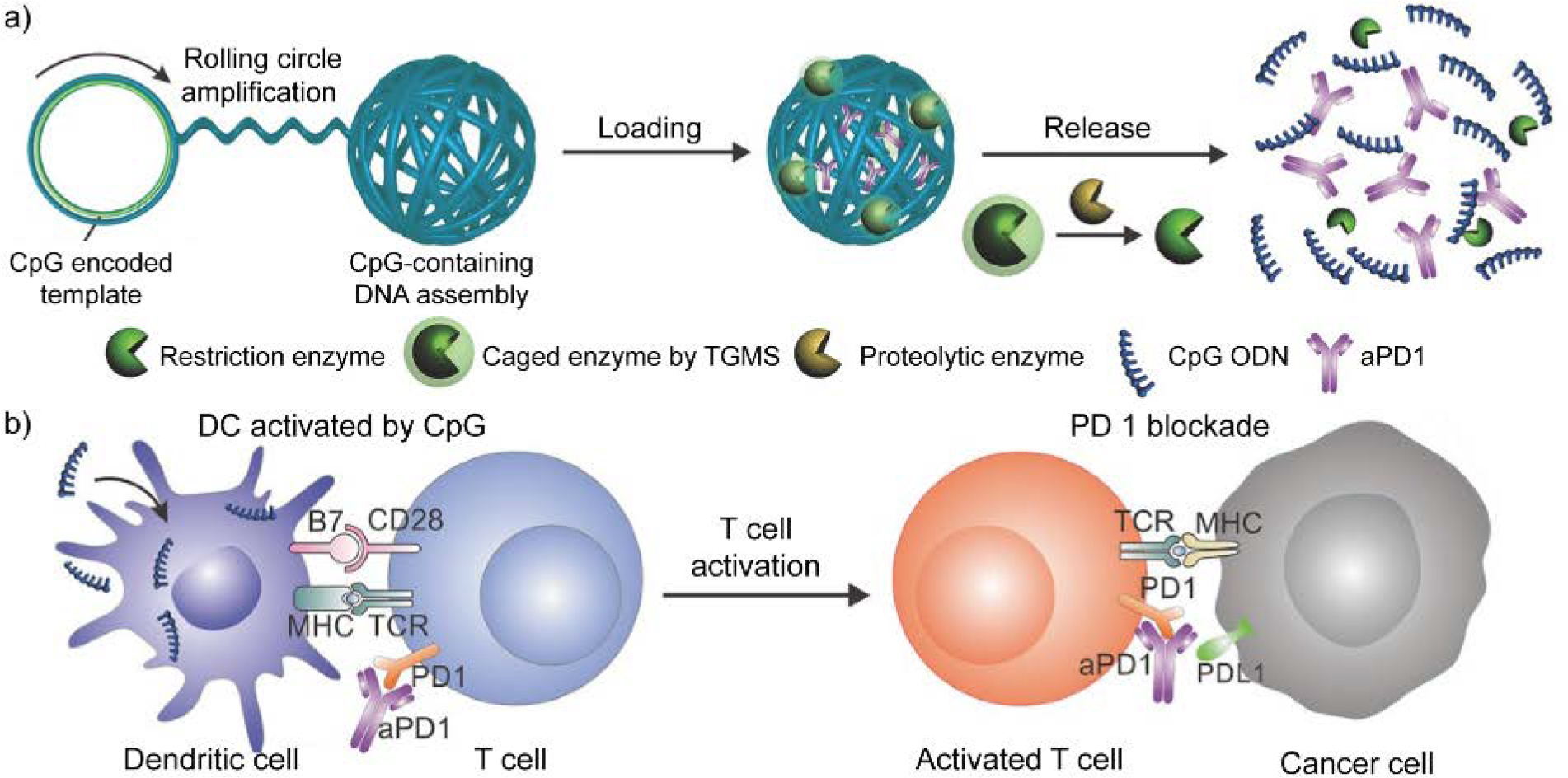
Schematic illustration of rolling circle amplification-assisted DNA assembly for CpG oligodeoxynucleotide (CpG ODN) and anti-PD-1 antibody (aPD1) delivery. (a) CpG-sequence containing DNA assembly was loaded with aPD1 and caged restriction enzyme. Under inflammation condition, proteolytic enzyme degrades triglycerol monostearate (TGMS) and releases the restriction enzyme, triggering the fragmentation of DNA assembly. As a result, CpG ODN and aPD1 are released at the inflammation site. (b) With aPD1 for PD 1 blockade, released CpG ODN activates dendritic cells (DCs) to drive T cell response, enhancing the immune response against cancer cells. Adapted with permission.[81] Copyright 2016, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.
4.3. Aptamer
Aptamers are single-stranded short nucleic acids or peptides with high affinity and specificity towards targets.[84] Common targets of aptamers widely range from small molecules to biomacromolecules. Nucleic acid-based aptamers are produced through a selection process in test-tube known as SELEX (systematic evolution of ligands by exponential enrichment).[85] Starting from a library of random nucleic acid sequences, SELEX recognizes and specifically amplifies the nucleic acid that has high affinity with the target molecule.
Incorporating aptamers into material design ideally will facilitate the delivery vehicles to recognize target of interest (e.g. cell surface receptor proteins), improving the specificity of nucleic acid delivery system. In an example, Sullenger and coworkers devised aptamer-siRNA chimeric structures to mediate RNA interference without the need of transfection reagent.[86] Within the chimeric nucleic acid structure (Figure 10), the aptamer was designed to specifically bind with an over-expressing cell surface receptor on prostate cancer cells, enabling cell type-specific siRNA delivery. Meanwhile, the siRNA region was designed to target at overexpressed survival genes, inducing cell apoptosis once being successfully involved in the RNAi pathway. The low immunogenicity of aptamers and siRNA adds up another advantage of the design for in vivo applications.[87] For example, such a chimeric design between aptamer and siRNA has also been used to develop liver fibrosis treatment.[88] Moreover, double-stranded RNA can also be engineered and self-assembled into organized structures with enhanced stability.[89] Other than utilizing transfection reagents, conjugating targeting ligands or recognition units for cell surface proteins may provide options to improve the delivery efficiency of engineered nucleic acid structures. The incorporation of small-molecule drugs[90] or other synthetic materials such as lipids and inorganic particles[91] is also an option to improve the therapeutic efficacy of aptamer-based delivery system.
Figure 10.
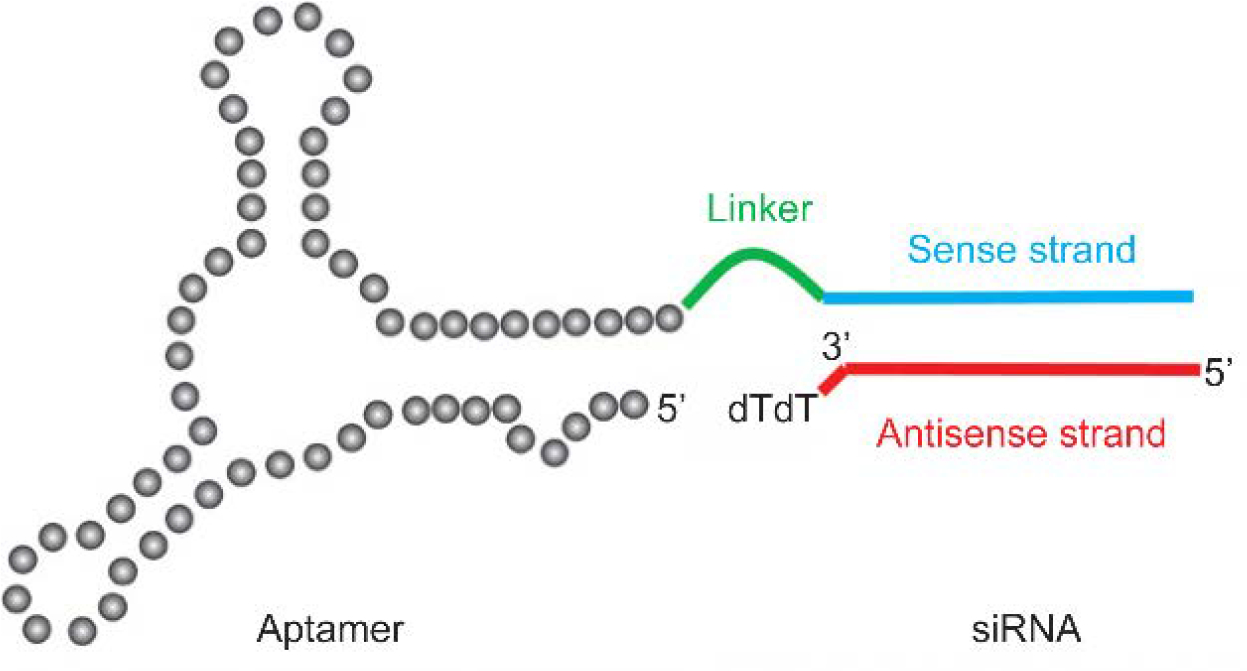
Schematic representation of aptamer-siRNA chimeric structure. Reproduced with permission.[92] Copyright 2011, American Association for the Advancement of Science.
5. Cellular uptake mechanism of non-cationic materials
Initiating the cellular internalization of non-cationic materials is not as straightforward as cationic materials. Non-cationic materials lack electrostatic interaction with the cell membrane as the driving force for their cellular internalization. Therefore, non-cationic delivery systems may be limited due to low cellular internalization. Understanding the cellular uptake mechanism will provide insights of how materials design affects the cellular uptake, and how to potentially improve the efficiency of non-cationic delivery systems. To elucidate the cellular uptake mechanism of new materials, the most common approach is the pharmacological approach. The pharmacological approach employs specific inhibitors for representative endocytic pathways.[93] Upon the treatment of inhibitor, the cellular uptake of delivery system is evaluated. Subsequently, the cellular uptake pathway is indicated based on the level of effect from different inhibitors. Next, it is ideal to consolidate the conclusion from the pharmacological approach with the genetic approach. The genetic approach refers to mutating or modulating the expression of a protein that is specifically involved in the pathway of interest, or a protein that is considered as the target of the pharmacological inhibitor.[94] Meanwhile, the protein should be selected with one that causes minimal interference on other pathways. Finally, referencing the results from two approaches will help rule out the results that caused by the off-target effect of inhibitors.[95]
Three major cellular uptake pathways are regularly investigated for materials less than 500 nm in size: macropinocytosis, clathrin-mediated endocytosis, and caveolae-mediated endocytosis.[96] Briefly, macropinocytosis describes an actin-driven non-selective process for cells to internalize extracellular fluid and its contents.[97] Clathrin-mediated endocytosis contains a key step in which clathrin proteins are coated on endocytic pits, with a series of proteins involved in the process.[98] As a clathrin-independent process, caveolae-mediated endocytosis is dependent on the cholesterol- and sphingolipid-rich fractions on cell membrane.[99] Since caveolae-mediated endocytosis shares similar signatures with lipid raft-mediate endocytosis, it can also be referred as caveolae/lipid raft-dependent endocytosis.[100] For materials with large size (≥ 500 nm), their engulfment by the cell membrane is defined as phagocytosis,[101] a process that generally happens in phagocytes such as neutrophils, monocytes and macrophages.[102] Commonly used pharmacological inhibitors for these endocytic pathways are summarized in previously published articles.[103] The cellular uptake evaluation of materials uptake upon the treatment of pharmacological inhibitors has been frequently employed to study the process of therapeutic materials.
Reflected from different systems (Table 1), the non-cationic nucleic acid delivery materials are majorly internalized by cells through clathrin-mediated and caveolae-mediated endocytosis, while macropinocytosis was not as significantly involved. Combining with the results from non-cationic nanomaterials in which nucleic acid is not involved,[104] the general resemblance is that the cholesterol within the cell membrane plays an important role in the cellular uptake of non-cationic materials. When cells were treated with methyl-β-cyclodextrin, a cholesterol-depletion reagent,[105] the cellular uptake of several non-cationic nucleic acid systems was effectively downregulated.
Table 1.
Summary of endocytic pathways in different non-cationic nucleic acid delivery systems.
| Materials | Macropinocytosis | Clathrin-mediated endocytosis | Caveolae-mediated endocytosis | Reference |
|---|---|---|---|---|
| Polyethylene glycol-coated siRNA | − | + | + | [22] |
| Hyaluronic acid-coated plasmid DNA | − | + | + | [26] |
| Polymer-Zn(II)-plasmid complex | + | − | + | [31] |
| Zn(II)-DNA complex | − | + | + | [106] |
| Anionic lipid-Ca(II)-siRNA complex | + | − | − | [107] |
| RNA-polymer complex | − | − | + | [45] |
| Polyethylene glycol-DNA conjugate | − | + | + | [108] |
| Self-assembled DNA nanostructure | − | + | − | [109] |
| Rolling circle amplification product (DNA) | + | + | − | [79] |
| Rolling circle amplification product (DNA) | − | + | + | [110] |
Since cholesterol has a relevant role in cell membrane fluidity and permeability,[111] one method to potentially improve the cellular uptake efficiency of non-cationic systems is to incorporate moieties with lipid affinity. For example, previous studies have reported therapeutic delivery systems with fusogenic feature.[112] While being internalized by cells, delivery vehicles with such feature will fuse with the cell membrane and efficiently deliver the cargos into the cytosol. From the material design perspective, the fusogenic feature can be tuned by using lipophilic molecules,[113] peptides,[114] or proteins (e.g. SNARE proteins,[115] Sendai virus accessory proteins[116]). Meanwhile, decorating delivery systems with known ligands (Table 2) towards cell surface receptors is also an effective method to improve the cellular uptake of non-cationic materials.
Table 2.
Cell surface protein receptors involved in the uptake of non-cationic nucleic acid delivery systems.
| Materials | Specific receptor(s) | Reference |
|---|---|---|
| Hyaluronic acid-siRNA complex | CD44 receptor | [25] |
| Hyaluronic acid-Zn(II)-siRNA complex | [33] | |
| Hyaluronic acid-crosslinked complex | [43] | |
| Lipid-siRNA conjugate | Lipoprotein receptors (including Scavenger receptor class B, type 1) |
[55] |
| N-Acetylgalactosamine-siRNA conjugate | Asialoglycoprotein receptor | [57] |
| Gold nanoparticle-DNA conjugate | Scavenger receptors | [117] |
| DNA tetrahedron-folic acid conjugate | Folate receptor | [74] |
Unlike the membrane fusion process, a majority of therapeutic delivery systems face the barrier of endosomal entrapment during their cellular internalization, limiting the delivery efficiency of therapeutics.[118] The fate of entrapped delivery vehicles and cargos is either degradation within endosomes/lysosomes or exocytosis. Therefore, sufficient endosomal escape will possibly reduce the required dosage for realizing the desired therapeutic effect of a delivery system. Strategies to initiate the endosomal escape of delivery systems are mainly focused on the disruption of endosomal membrane, releasing the cargos after endosomal rupture. Detailed strategies are summarized in previous review articles.[119] One representative hypothesis for endosomal escape is the proton sponge effect.[120] After entering the acidifying environment of endosomes/lysosomes, materials with amine groups are able to sequester endosomal protons and slow down the pH drop. As a result, increased protons are pumped into the endosomes by cells to reach the target pH, resulting in an increased influx of counterions (e.g. Cl−) into endosomes. The ion influx elevates the osmotic pressure within endosomes and consequently ruptures the endosomal membrane rupture, eventually leading to the endosomal escape of the delivery system. However, the hypothesis of proton sponge effect is debated in recent reports.[121] Currently, methods for efficient endosomal escape are still needed in therapeutic delivery systems, and such need is not limited to non-cationic material design.
6. Clinical relevance of non-cationic delivery carriers
So far, we have summarized multiple non-cationic materials design for nucleic acid delivery (Table 3). Several of these materials (not limited to non-cationic platforms) have been involved in preclinical and clinical trials,[124] including hyaluronic acids,[125] aptamers,[126] lipid-based,[127] and GalNAc-decorated nucleic acid therapeutics.[128] For non-cationic carriers, the pursuit for clinical trials is still in its early stage, specifically regarding nucleic acid delivery.[129] Readers are referred to the above references for detailed information.
Table 3.
Summary of non-cationic materials design for nucleic acids in the Progress Report.
| Category | Sub-category |
|---|---|
| Complexation with synthetic materials | Shielding cationic moieties of synthetic materials (polymer, inorganic nanoparticle) |
| Metal-ion induced coordination (polymer, lipid, inorganic nanoparticle) | |
| Entrapping nucleic acids with crosslinked polymer | |
| Conjugation with synthetic materials | Small molecule (Lipid-, sugar- derivatives) |
| Polymer | |
| Inorganic nanoparticle | |
| Engineered nucleic acid-based structures | Programmed nucleic acid structure |
| Rolling-circle amplification product | |
| Aptamer |
7. Conclusions and outlook
Non-cationic systems for nucleic acid delivery present materials with less cytotoxicity concerns than cationic materials, primarily due to the reduced interference of non-cationic materials on cell membrane integrity. Meanwhile, the absence of net positive charge is inherently compromising the material-cell interaction that contributed by electrostatic interaction, therefore reducing the cellular internalization of the delivery system. Considering that the majority of synthetic platforms for nucleic acid delivery are based on cationic materials, one possible solution is to formulate the delivery vehicle with a “cocktail” approach, incorporating non-cationic components into a cationic delivery platform.[130] Other than modulating the material toxicity, combination of components also provides variables in the efficacy, stability, and circulation time of the delivery system. Synthetic materials that are not commonly used for nucleic acid delivery may also contribute with their unique mechanical and biologically relevant properties.[131] From translational perspective, several aspects need to be explored to further understand the non-cationic delivery systems. First, apart from reduced cytotoxicity, the effect of non-cationic systems on immune systems is not well understood. Recent literatures have revealed the immune response against polyethylene glycol derivatives from animal studies,[132] suggesting that potential complications could arise from the immunological standpoint. Second, the efficiency of the delivery system can benefit from improved targeting capability. The incorporation of antibodies have demonstrated promising results to target the delivery of siRNA at diseased cells.[133] Last, in vivo evaluation of non-cationic formulations is certainly required regarding their pharmacokinetics, carcinogenesis, genotoxicity, and efficacy. While non-cationic synthetic materials for nucleic acid is a comparatively less explored area, in-depth understanding of their cellular uptake and improved delivery efficiency will advance their development, eventually providing prominent tools for nucleic acid delivery.
Figure 11.
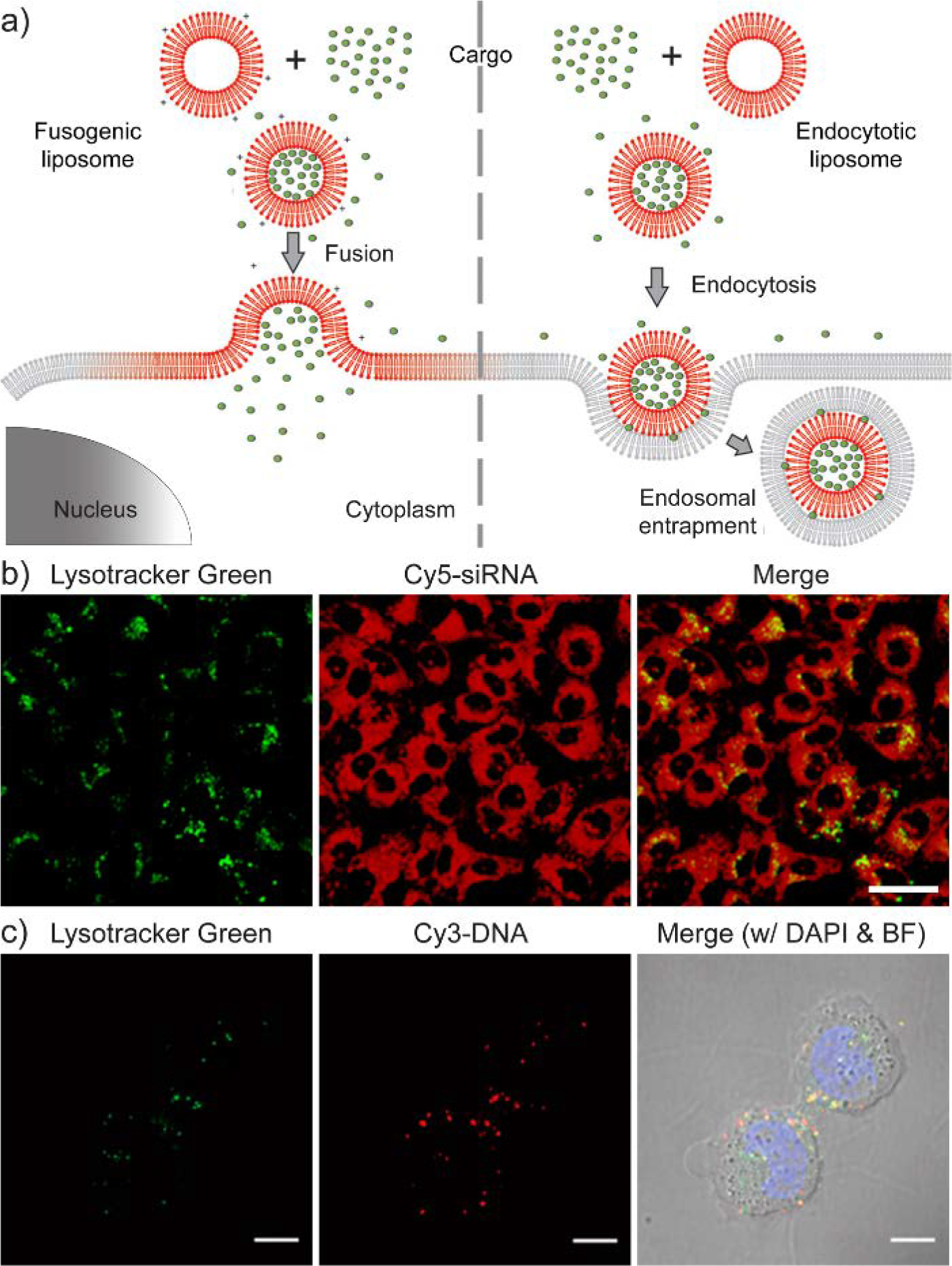
(a) Schematic comparison between membrane fusion and endocytosis using liposomes as the delivery vehicle. Adapted with permission.[122] Copyright 2011, American Chemical Society. (b) Confocal microscopy images of siRNA delivery via membrane fusion process. After the delivery, Cy5-labeled siRNAs are homogenously distributed in the cytosol of most cells. The scale bar represents 20 μm. Adapted with permission.[123] Copyright 2018, Elsevier Ltd. (c) Confocal microscopy images of the cellular internalization of DNA via endocytosis. When Cy3-labeled DNA overlaps with the Lysotracker Green (a fluorescent stain for cellular lysosomes), it is denoted in yellow and represents the endosomal entrapment of DNA. The scale bars represent 10 μm Adapted with permission.[110] Copyright 2015, American Chemical Society.
Acknowledgement
The authors acknowledge the support from the NIGMS of the National Institutes of Health (GM-128181) and the Army Research Office (W911NF-15-1-0568).
Contributor Information
Ziwen Jiang, Department of Chemistry, University of Massachusetts Amherst, Amherst, MA, 01003, USA; Department of Pharmaceutical Chemistry, University of California, San Francisco, CA, 94158, USA.
S. Thayumanavan, Department of Chemistry, University of Massachusetts Amherst, Amherst, MA, 01003, USA Molecular and Cellular Biology Program, Center for Bioactive Delivery at the Institute for Applied Life Sciences, University of Massachusetts Amherst, Amherst, MA 01003, USA.
References
- [1].Nguyen J, Szoka FC, Acc. Chem. Res 2012, 45, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Luo D, Saltzman WM, Nat. Biotechnol 2000, 18, 33; [DOI] [PubMed] [Google Scholar]; b) Jong YS, Jacob JS, Yip K-P, Gardner G, Seitelman E, Whitney M, Montgomery S, Mathiowitz E, J. Controlled Release 1997, 47, 123; [Google Scholar]; c) Abbas AO, Donovan MD, Salem AK, J. Pharm. Sci 2008, 97, 2448. [DOI] [PubMed] [Google Scholar]
- [3].a) Sahin U, Karikó K, Türeci Ö, Nat. Rev. Drug Discov 2014, 13, 759; [DOI] [PubMed] [Google Scholar]; b) Hajj KA, Whitehead KA, Nat. Rev. Mater 2017, 2, 17056; [Google Scholar]; c) Kowalski PS, Rudra A, Miao L, Anderson DG, Mol. Ther 2019, 27, 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC, Nature 1998, 391, 806; [DOI] [PubMed] [Google Scholar]; b) Tuschl T, Zamore PD, Lehmann R, Bartel DP, Sharp PA, Genes Dev. 1999, 13, 3191; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Scherer LJ, Rossi JJ, Nat. Biotechnol 2003, 21, 1457. [DOI] [PubMed] [Google Scholar]
- [5].a) Campolongo MJ, Tan SJ, Xu J, Luo D, Adv. Drug Del. Rev 2010, 62, 606; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kumar V, Palazzolo S, Bayda S, Corona G, Toffoli G, Rizzolio F, Theranostics 2016, 6, 710; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Angell C, Xie S, Zhang L, Chen Y, Small 2016, 12, 1117. [DOI] [PubMed] [Google Scholar]
- [6].a) Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD, Nat. Rev. Genet 2010, 11, 636; [DOI] [PubMed] [Google Scholar]; b) Wright Addison V., Nuñez James K., Doudna Jennifer A., Cell 2016, 164, 29; [DOI] [PubMed] [Google Scholar]; c) Hsu Patrick D., Lander Eric S., Zhang F, Cell 2014, 157, 1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Burnett JC, Rossi JJ, Chem. Biol 2012, 19, 60; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miyata K, Nishiyama N, Kataoka K, Chem. Soc. Rev 2012, 41, 2562; [DOI] [PubMed] [Google Scholar]; c) Wang J, Lu Z, Wientjes MG, Au JLS, AAPS J. 2010, 12, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Kay MA, Glorioso JC, Naldini L, Nat. Med 2001, 7, 33; [DOI] [PubMed] [Google Scholar]; b) Thomas CE, Ehrhardt A, Kay MA, Nat. Rev. Genet 2003, 4, 346; [DOI] [PubMed] [Google Scholar]; c) Robbins PD, Ghivizzani SC, Pharmacol. Ther 1998, 80, 35. [PubMed] [Google Scholar]
- [9].a) Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG, Nat. Rev. Genet 2014, 15, 541; [DOI] [PubMed] [Google Scholar]; b) Draghici B, Ilies MA, J. Med. Chem 2015, 58, 4091; [DOI] [PubMed] [Google Scholar]; c) Lostalé-Seijo I, Montenegro J, Nat. Rev. Chem 2018, 2, 258. [Google Scholar]
- [10].a) Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L, J. Virol 1998, 72, 8463; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L, Nat. Genet 2003, 33, 401. [DOI] [PubMed] [Google Scholar]
- [11].a) Medina-Kauwe LK, Adv. Drug Del. Rev 2003, 55, 1485; [DOI] [PubMed] [Google Scholar]; b) Mingozzi F, High KA, Nat. Rev. Genet 2011, 12, 341; [DOI] [PubMed] [Google Scholar]; c) Kotterman MA, Schaffer DV, Nat. Rev. Genet 2014, 15, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Grisham J, Nat. Biotechnol 2000, 18, 254; [DOI] [PubMed] [Google Scholar]; b) Nayak S, Herzog RW, Gene Ther. 2009, 17, 295; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Mingozzi F, High KA, Blood 2013, 122, 23; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Colella P, Ronzitti G, Mingozzi F, Mol. Ther. Methods Clin. Dev 2018, 8, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Miller AD, Angew. Chem. Int. Ed 1998, 37, 1768; [Google Scholar]; b) Li W, Szoka FC, Pharm. Res 2007, 24, 438; [DOI] [PubMed] [Google Scholar]; c) Akinc A, Zumbuehl A, Goldberg M, Leshchiner ES, Busini V, Hossain N, Bacallado SA, Nguyen DN, Fuller J, Alvarez R, Borodovsky A, Borland T, Constien R, de Fougerolles A, Dorkin JR, Narayanannair Jayaprakash K, Jayaraman M, John M, Koteliansky V, Manoharan M, Nechev L, Qin J, Racie T, Raitcheva D, Rajeev KG, Sah DWY, Soutschek J, Toudjarska I, Vornlocher H-P, Zimmermann TS, Langer R, Anderson DG, Nat. Biotechnol 2008, 26, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Merdan T, Kopecek J, Kissel T, Adv. Drug Del. Rev 2002, 54, 715; [DOI] [PubMed] [Google Scholar]; b) Pack DW, Hoffman AS, Pun S, Stayton PS, Nat. Rev. Drug Discov 2005, 4, 581; [DOI] [PubMed] [Google Scholar]; c) Roy R, Jerry DJ, Thayumanavan S, Biomacromolecules 2009, 10, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Xu ZP, Zeng QH, Lu GQ, Yu AB, Chem. Eng. Sci 2006, 61, 1027; [Google Scholar]; b) Ding Y, Jiang Z, Saha K, Kim CS, Kim ST, Landis RF, Rotello VM, Mol. Ther 2014, 22, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Bishop JR, Schuksz M, Esko JD, Nature 2007, 446, 1030; [DOI] [PubMed] [Google Scholar]; b) Cruz-Chu Eduardo R., Malafeev A, Pajarskas T, Pivkin Igor V., Koumoutsakos P, Biophys. J 2014, 106, 232; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen BD, Le WJ, Wang YL, Li ZQ, Wang D, Ren L, Lin L, Cui SB, Hu JJ, Hu YH, Yang PY, Ewing RC, Shi DL, Cui Z, Theranostics 2016, 6, 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].a) Fischer D, Li YX, Ahlemeyer B, Krieglstein J, Kissel T, Biomaterials 2003, 24, 1121; [DOI] [PubMed] [Google Scholar]; b) Goodman CM, McCusker CD, Yilmaz T, Rotello VM, Bioconjugate Chem. 2004, 15, 897; [DOI] [PubMed] [Google Scholar]; c) Lv H, Zhang S, Wang B, Cui S, Yan J, J. Controlled Release 2006, 114, 100; [DOI] [PubMed] [Google Scholar]; d) Hunter AC, Adv. Drug Del. Rev 2006, 58, 1523. [DOI] [PubMed] [Google Scholar]
- [18].a) Moghimi SM, Symonds P, Murray JC, Hunter AC, Debska G, Szewczyk A, Mol. Ther 2005, 11, 990; [DOI] [PubMed] [Google Scholar]; b) Roursgaard M, Knudsen KB, Northeved H, Persson M, Christensen T, Kumar PEK, Permin A, Andresen TL, Gjetting T, Lykkesfeldt J, Vesterdal LK, Loft S, Møller P, Toxicol. In Vitro 2016, 36, 164. [DOI] [PubMed] [Google Scholar]
- [19].a) Vatsal S, Oleh T, Olga BG, Mahesh LP, Ronak S, Min Z, Tamara M, Curr. Drug Disc. Technol 2013, 10, 8; [Google Scholar]; b) Knudsen KB, Northeved H, Kumar Ek P, Permin A, Gjetting T, Andresen TL, Larsen S, Wegener KM, Lykkesfeldt J, Jantzen K, Loft S, Møller P, Roursgaard M, Nanomed. Nanotechnol. Biol. Med 2015, 11, 467. [DOI] [PubMed] [Google Scholar]
- [20].a) Harris JM, Chess RB, Nat. Rev. Drug Discov 2003, 2, 214; [DOI] [PubMed] [Google Scholar]; b) Otsuka H, Nagasaki Y, Kataoka K, Adv. Drug Del. Rev 2003, 55, 403; [DOI] [PubMed] [Google Scholar]; c) Veronese FM, Pasut G, Drug Discov. Today 2005, 10, 1451; [DOI] [PubMed] [Google Scholar]; d) Knop K, Hoogenboom R, Fischer D, Schubert US, Angew. Chem. Int. Ed 2010, 49, 6288. [DOI] [PubMed] [Google Scholar]
- [21].Miyata K, Kakizawa Y, Nishiyama N, Harada A, Yamasaki Y, Koyama H, Kataoka K, J. Am. Chem. Soc 2004, 126, 2355. [DOI] [PubMed] [Google Scholar]
- [22].Xu C, Li D, Cao Z, Xiong M, Yang X, Wang J, Nano Lett 2019, 19, 2688. [DOI] [PubMed] [Google Scholar]
- [23].a) Laurent TC, Fraser JRE, FASEB J. 1992, 6, 2397; [PubMed] [Google Scholar]; b) Weis WI, Drickamer K, Annu. Rev. Biochem 1996, 65, 441; [DOI] [PubMed] [Google Scholar]; c) Ooi VEC, Liu F, Curr. Med. Chem 2000, 7, 715; [DOI] [PubMed] [Google Scholar]; d) Kwak JY, Mar. Drugs 2014, 12, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Naor D, Sionov RV, IshShalom D, in Adv. Cancer Res, Vol. 71 (Eds: VandeWoude GF, Klein G), Elsevier Academic Press Inc, San Diego: 1997, 241; [DOI] [PubMed] [Google Scholar]; b) Fraser JRE, Laurent TC, Laurent UBG, J. Intern. Med 1997, 242, 27. [DOI] [PubMed] [Google Scholar]
- [25].Ganesh S, Iyer AK, Morrissey DV, Amiji MM, Biomaterials 2013, 34, 3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yen J, Ying H, Wang H, Yin L, Uckun F, Cheng J, ACS Biomater. Sci. Eng 2016, 2, 326. [DOI] [PubMed] [Google Scholar]
- [27].Niikura K, Kobayashi K, Takeuchi C, Fujitani N, Takahara S, Ninomiya T, Hagiwara K, Mitomo H, Ito Y, Osada Y, Ijiro K, ACS Appl. Mater. Interfaces 2014, 6, 22146. [DOI] [PubMed] [Google Scholar]
- [28].a) Davidson BL, McCray PB, Nat. Rev. Genet 2011, 12, 329; [DOI] [PMC free article] [PubMed] [Google Scholar]; Bobbin ML, Rossi JJ, in Annu. Rev. Pharmacool. Toxicol, Vol. 56 (Ed: Insel PA), 2016, 103. [DOI] [PubMed] [Google Scholar]
- [29].Egli M, Manoharan M, Acc. Chem. Res 2019, 52, 1036. [DOI] [PubMed] [Google Scholar]
- [30].O’Neil EJ, Smith BD, Coord. Chem. Rev 2006, 250, 3068. [Google Scholar]
- [31].Liu S, Jia H, Yang J, Pan J, Liang H, Zeng L, Zhou H, Chen J, Guo T, ACS Macro Lett. 2018, 7, 868. [DOI] [PubMed] [Google Scholar]
- [32].Tajik-Ahmadabad B, Chollet L, White J, Separovic F, Polyzos A, Mol. Pharm 2019, 16, 978. [DOI] [PubMed] [Google Scholar]
- [33].Liu G, Choi KY, Bhirde A, Swierczewska M, Yin J, Lee SW, Park JH, Hong JI, Xie J, Niu G, Kiesewetter DO, Lee S, Chen X, Angew. Chem. Int. Ed 2012, 51, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Choi E, Lee J, Kwon IC, Lim D-K, Kim S, Biomaterials 2019, 209, 126. [DOI] [PubMed] [Google Scholar]
- [35].Somiya M, Yamaguchi K, Liu Q, Niimi T, Maturana AD, Iijima M, Yoshimoto N, Kuroda S. i., Int. J. Pharm 2015, 490, 316. [DOI] [PubMed] [Google Scholar]
- [36].Kapoor M, Burgess DJ, Int. J. Pharm 2012, 432, 80. [DOI] [PubMed] [Google Scholar]
- [37].a) Gutiérrez-Puente Y, Tari AM, Stephens C, Rosenblum M, Guerra RT, Lopez-Berestein G, J. Pharmacol. Exp. Ther 1999, 291, 865; [PubMed] [Google Scholar]; b) Landen CN, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK, Cancer Res. 2005, 65, 6910. [DOI] [PubMed] [Google Scholar]
- [38].a) Peppas NA, Hilt JZ, Khademhosseini A, Langer R, Adv. Mater 2006, 18, 1345; [Google Scholar]; b) Appel EA, del Barrio J, Loh XJ, Scherman OA, Chem. Soc. Rev 2012, 41, 6195. [DOI] [PubMed] [Google Scholar]
- [39].Chacko RT, Ventura J, Zhuang J, Thayumanavan S, Adv. Drug Del. Rev 2012, 64, 836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].a) Matsumoto S, Christie RJ, Nishiyama N, Miyata K, Ishii A, Oba M, Koyama H, Yamasaki Y, Kataoka K, Biomacromolecules 2009, 10, 119; [DOI] [PubMed] [Google Scholar]; b) Ryu J-H, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S, J. Am. Chem. Soc 2010, 132, 17227; [DOI] [PubMed] [Google Scholar]; c) Ryu J-H, Roy R, Ventura J, Thayumanavan S, Langmuir 2010, 26, 7086; [DOI] [PubMed] [Google Scholar]; d) Ryu J-H, Jiwpanich S, Chacko R, Bickerton S, Thayumanavan S, J. Am. Chem. Soc 2010, 132, 8246; [DOI] [PubMed] [Google Scholar]; e) Zhao M, Biswas A, Hu B, Joo K-I, Wang P, Gu Z, Tang Y, Biomaterials 2011, 32, 5223; [DOI] [PubMed] [Google Scholar]; f) Wang M, Zuris JA, Meng F, Rees H, Sun S, Deng P, Han Y, Gao X, Pouli D, Wu Q, Georgakoudi I, Liu DR, Xu Q, Proc. Natl. Acad. Sci. U.S.A 2016, 113, 2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].a) Pastore A, Federici G, Bertini E, Piemonte F, Clin. Chim. Acta 2003, 333, 19; [DOI] [PubMed] [Google Scholar]; b) Meister A, Anderson ME, Annu. Rev. Biochem 1983, 52, 711; [DOI] [PubMed] [Google Scholar]; c) Cheng R, Feng F, Meng F, Deng C, Feijen J, Zhong Z, J. Controlled Release 2011, 152, 2. [DOI] [PubMed] [Google Scholar]
- [42].a) Mura S, Nicolas J, Couvreur P, Nat. Mater 2013, 12, 991; [DOI] [PubMed] [Google Scholar]; b) Huo M, Yuan J, Tao L, Wei Y, Polym. Chem 2014, 5, 1519. [Google Scholar]
- [43].Lee H, Mok H, Lee S, Oh Y-K, Park TG, J. Controlled Release 2007, 119, 245. [DOI] [PubMed] [Google Scholar]
- [44].Jiang Z, Cui W, Prasad P, Touve MA, Gianneschi NC, Mager J, Thayumanavan S, Biomacromolecules 2019, 20, 435. [DOI] [PubMed] [Google Scholar]
- [45].Jiang Z, Cui W, Mager J, Thayumanavan S, Ind. Eng. Chem. Res 2019, 58, 6982. [Google Scholar]
- [46].a) Goodchild J, Bioconjugate Chem. 1990, 1, 165; [DOI] [PubMed] [Google Scholar]; b) Manoharan M, Antisense Nucleic Acid Drug Dev. 2002, 12, 103; [DOI] [PubMed] [Google Scholar]; c) Venkatesan N, Kim BH, Chem. Rev 2006, 106, 3712; [DOI] [PubMed] [Google Scholar]; d) Singh Y, Murat P, Defrancq E, Chem. Soc. Rev 2010, 39, 2054; [DOI] [PubMed] [Google Scholar]; e) Benizri S, Gissot A, Martin A, Vialet B, Grinstaff MW, Barthélémy P, Bioconjugate Chem. 2019, 30, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].a) Grijalvo S, Ocampo SM, Perales JC, Eritja R, Chem. Biodivers 2011, 8, 287; [DOI] [PubMed] [Google Scholar]; b) Grijalvo S, Ocampo SM, Perales JC, Eritja R, J. Org. Chem 2010, 75, 6806. [DOI] [PubMed] [Google Scholar]
- [48].Raouane M, Desmaële D, Urbinati G, Massaad-Massade L, Couvreur P, Bioconjugate Chem. 2012, 23, 1091. [DOI] [PubMed] [Google Scholar]
- [49].a) Winkler J, Ther. Deliv 2013, 4, 791; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Karaki S, Benizri S, Mejías R, Baylot V, Branger N, Nguyen T, Vialet B, Oumzil K, Barthélémy P, Rocchi P, J. Controlled Release 2017, 258, 1. [DOI] [PubMed] [Google Scholar]
- [50].Lorenz C, Hadwiger P, John M, Vornlocher H-P, Unverzagt C, Bioorg. Med. Chem. Lett 2004, 14, 4975. [DOI] [PubMed] [Google Scholar]
- [51].a) Raouane M, Desmaele D, Gilbert-Sirieix M, Gueutin C, Zouhiri F, Bourgaux C, Lepeltier E, Gref R, Ben Salah R, Clayman G, Massaad-Massade L, Couvreur P, J. Med. Chem 2011, 54, 4067; [DOI] [PubMed] [Google Scholar]; b) Musacchio T, Vaze O, D’Souza G, Torchilin VP, Bioconjugate Chem. 2010, 21, 1530. [DOI] [PubMed] [Google Scholar]
- [52].Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H, Yokota T, Mol. Ther 2008, 16, 734. [DOI] [PubMed] [Google Scholar]
- [53].Patwa A, Gissot A, Bestel I, Barthélémy P, Chem. Soc. Rev 2011, 40, 5844. [DOI] [PubMed] [Google Scholar]
- [54].a) Aldrian-Herrada G, Méry J, Brugidou J, Desarménien MG, Orcel H, Boissin-Agasse L, Rabié A, Nucleic Acids Res. 1998, 26, 4910; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jain A, Barve A, Zhao Z, Fetse JP, Liu H, Li Y, Cheng K, Adv. Ther 2019, 2, 1900046; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gupta A, Mishra A, Puri N, Biotechnol J. 2017, 259, 148; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jin W, Jain A, Liu H, Zhao Z, Cheng K, ACS Applied Bio Materials 2018, 1, 643; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gupta A, Bahal R, Gupta M, Glazer PM, Saltzman WM, J. Controlled Release 2016, 240, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, Zimmermann T, Koteliansky V, Manoharan M, Stoffel M, Nat. Biotechnol 2007, 25, 1149. [DOI] [PubMed] [Google Scholar]
- [56].a) Huo S, Li H, Boersma AJ, Herrmann A, Adv. Sci 2019, 6, 1900043; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu H, Zhu Z, Kang H, Wu Y, Sefan K, Tan W, Chem. Eur. J 2010, 16, 3791; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang Y, Wu C, Chen T, Sun H, Cansiz S, Zhang L, Cui C, Hou W, Wu Y, Wan S, Cai R, Liu Y, Sumerlin BS, Zhang X, Tan W, Chem. Sci 2016, 7, 6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nair JK, Willoughby JLS, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel’in AV, Milstein S, Taneja N, O’Shea J, Shaikh S, Zhang L, van der Sluis RJ, Jung ME, Akinc A, Hutabarat R, Kuchimanchi S, Fitzgerald K, Zimmermann T, van Berkel TJC, Maier MA, Rajeev KG, Manoharan M, J. Am. Chem. Soc 2014, 136, 16958. [DOI] [PubMed] [Google Scholar]
- [58].a) Lundin P, Johansson H, Guterstam P, Holm T, Hansen M, Langel Ü, El Andaloussi S, Bioconjugate Chem. 2008, 19, 2535; [DOI] [PubMed] [Google Scholar]; b) Said Hassane F, Saleh AF, Abes R, Gait MJ, Lebleu B, Cell. Mol. Life Sci 2010, 67, 715; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bolhassani A, Biochim. Biophys. Acta 2011, 1816, 232; [DOI] [PubMed] [Google Scholar]; d) Copolovici DM, Langel K, Eriste E, Langel Ü, ACS Nano 2014, 8, 1972. [DOI] [PubMed] [Google Scholar]
- [59].Huang X, Leroux J-C, Castagner B, Bioconjugate Chem. 2017, 28, 283. [DOI] [PubMed] [Google Scholar]
- [60].Heredia KL, Nguyen TH, Chang C-W, Bulmus V, Davis TP, Maynard HD, Chem. Commun 2008, 3245. [DOI] [PubMed] [Google Scholar]
- [61].Gunasekaran K, Nguyen TH, Maynard HD, Davis TP, Bulmus V, Macromol. Rapid Commun 2011, 32, 654. [DOI] [PubMed] [Google Scholar]
- [62].a) Lu X, Jia F, Tan X, Wang D, Cao X, Zheng J, Zhang K, J. Am. Chem. Soc 2016, 138, 9097; [DOI] [PubMed] [Google Scholar]; b) Wang D, Lin J, Jia F, Tan X, Wang Y, Sun X, Cao X, Che F, Lu H, Gao X, Shimkonis JC, Nyoni Z, Lu X, Zhang K, Sci. Adv 2019, 5, eaav9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].a) Sletten EM, Bertozzi CR, Acc. Chem. Res 2011, 44, 666; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Debets MF, van Berkel SS, Dommerholt J, Dirks AJ, Rutjes FPJT, van Delft FL, Acc. Chem. Res 2011, 44, 805. [DOI] [PubMed] [Google Scholar]
- [64].Ding F, Mou Q, Ma Y, Pan G, Guo Y, Tong G, Choi CHJ, Zhu X, Zhang C, Angew. Chem. Int. Ed 2018, 57, 3064. [DOI] [PubMed] [Google Scholar]
- [65].a) Seferos DS, Prigodich AE, Giljohann DA, Patel PC, Mirkin CA, Nano Lett. 2009, 9, 308; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu B, Huang Z, Liu J, Angew. Chem. Int. Ed 2018, 57, 9439. [DOI] [PubMed] [Google Scholar]
- [66].Pakiari AH, Jamshidi Z, J. Phys. Chem. A 2010, 114, 9212. [DOI] [PubMed] [Google Scholar]
- [67].a) Cutler JI, Auyeung E, Mirkin CA, J. Am. Chem. Soc 2012, 134, 1376; [DOI] [PubMed] [Google Scholar]; b) Zheng D, Giljohann DA, Chen DL, Massich MD, Wang X-Q, Iordanov H, Mirkin CA, Paller AS, Proc. Natl. Acad. Sci. U.S.A 2012, 109, 11975; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jensen SA, Day ES, Ko CH, Hurley LA, Luciano JP, Kouri FM, Merkel TJ, Luthi AJ, Patel PC, Cutler JI, Daniel WL, Scott AW, Rotz MW, Meade TJ, Giljohann DA, Mirkin CA, Stegh AH, Sci. Transl. Med 2013, 5, 209ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA, Science 2006, 312, 1027. [DOI] [PubMed] [Google Scholar]
- [69].a) Mitchell GP, Mirkin CA, Letsinger RL, J. Am. Chem. Soc 1999, 121, 8122; [Google Scholar]; b) Lee J-S, Lytton-Jean AKR, Hurst SJ, Mirkin CA, Nano Lett. 2007, 7, 2112; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cutler JI, Zheng D, Xu X, Giljohann DA, Mirkin CA, Nano Lett. 2010, 10, 1477; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Young KL, Scott AW, Hao L, Mirkin SE, Liu G, Mirkin CA, Nano Lett. 2012, 12, 3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].a) Pei H, Zuo X, Zhu D, Huang Q, Fan C, Acc. Chem. Res 2014, 47, 550; [DOI] [PubMed] [Google Scholar]; b) Grabow WW, Jaeger L, Acc. Chem. Res 2014, 47, 1871; [DOI] [PubMed] [Google Scholar]; c) Li J, Green AA, Yan H, Fan C, Nat. Chem 2017, 9, 1056; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Liu J, Wang Z, Zhao S, Ding B, Nano Res. 2018, 11, 5017. [Google Scholar]
- [71].a) Aldaye FA, Palmer AL, Sleiman HF, Science 2008, 321, 1795; [DOI] [PubMed] [Google Scholar]; b) Guo P, Nat. Nanotechnol 2010, 5, 833; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pinheiro AV, Han D, Shih WM, Yan H, Nat. Nanotechnol 2011, 6, 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].a) Wang F, Lu C-H, Willner I, Chem. Rev 2014, 114, 2881; [DOI] [PubMed] [Google Scholar]; b) Chen Y-J, Groves B, Muscat RA, Seelig G, Nat. Nanotechnol 2015, 10, 748; [DOI] [PubMed] [Google Scholar]; c) Bujold KE, Lacroix A, Sleiman HF, Chem 2018, 4, 495; [Google Scholar]; d) Hu Q, Li H, Wang L, Gu H, Fan C, Chem. Rev 2019, 119, 6459. [DOI] [PubMed] [Google Scholar]
- [73].a) Keum J-W, Bermudez H, Chem. Commun 2009, 7036; [DOI] [PubMed] [Google Scholar]; b) Mei Q, Wei X, Su F, Liu Y, Youngbull C, Johnson R, Lindsay S, Yan H, Meldrum D, Nano Lett. 2011, 11, 1477; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Conway JW, McLaughlin CK, Castor KJ, Sleiman H, Chem. Commun 2013, 49, 1172; [DOI] [PubMed] [Google Scholar]; d) Hahn J, Wickham SFJ, Shih WM, Perrault SD, ACS Nano 2014, 8, 8765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lee H, Lytton-Jean AKR, Chen Y, Love KT, Park AI, Karagiannis ED, Sehgal A, Querbes W, Zurenko CS, Jayaraman M, Peng CG, Charisse K, Borodovsky A, Manoharan M, Donahoe JS, Truelove J, Nahrendorf M, Langer R, Anderson DG, Nat. Nanotechnol 2012, 7, 389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].a) Zhao W, Ali MM, Brook MA, Li Y, Angew. Chem. Int. Ed 2008, 47, 6330; [DOI] [PubMed] [Google Scholar]; b) Ali MM, Li F, Zhang Z, Zhang K, Kang D-K, Ankrum JA, Le XC, Zhao W, Chem. Soc. Rev 2014, 43, 3324; [DOI] [PubMed] [Google Scholar]; c) Zhao Y, Chen F, Li Q, Wang L, Fan C, Chem. Rev 2015, 115, 12491. [DOI] [PubMed] [Google Scholar]
- [76].a) del Solar G, Giraldo R, Ruiz-Echevarría MJ, Espinosa M, Díaz-Orejas R, Microbiol. Mol. Biol. Rev 1998, 62, 434; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Khan SA, Microbiol. Mol. Biol. Rev 1997, 61, 442; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Flores R, Hernández C, Alba A. E. M. d., Daròs J-A, Serio FD, Annu. Rev. Phytopathol 2005, 43, 117. [DOI] [PubMed] [Google Scholar]
- [77].a) Hong CA, Jang B, Jeong EH, Jeong H, Lee H, Chem. Commun 2014, 50, 13049; [DOI] [PubMed] [Google Scholar]; b) Sun W, Lu Y, Gu Z, Ther. Deliv 2015, 6, 765. [DOI] [PubMed] [Google Scholar]
- [78].Hamblin GD, Carneiro KMM, Fakhoury JF, Bujold KE, Sleiman HF, J. Am. Chem. Soc 2012, 134, 2888. [DOI] [PubMed] [Google Scholar]
- [79].Sun W, Jiang T, Lu Y, Reiff M, Mo R, Gu Z, J. Am. Chem. Soc 2014, 136, 14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ouyang X, Li J, Liu H, Zhao B, Yan J, Ma Y, Xiao S, Song S, Huang Q, Chao J, Fan C, Small 2013, 9, 3082. [DOI] [PubMed] [Google Scholar]
- [81].Wang C, Sun W, Wright G, Wang AZ, Gu Z, Adv. Mater 2016, 28, 8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].a) Krieg AM, Oncogene 2008, 27, 161; [DOI] [PubMed] [Google Scholar]; b) Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, Rajapaksa R, Green MR, Torchia J, Brody J, Luong R, Rosenblum MD, Steinman L, Levitsky HI, Tse V, Levy R, J. Clin. Invest 2013, 123, 2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].a) Pardoll DM, Nat. Rev. Cancer 2012, 12, 252; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ribas A, New Engl. J. Med 2015, 373, 1490; [DOI] [PubMed] [Google Scholar]; c) Zou W, Wolchok JD, Chen L, Sci. Transl. Med 2016, 8, 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].a) Keefe AD, Pai S, Ellington A, Nat. Rev. Drug Discov 2010, 9, 537; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bunka DHJ, Stockley PG, Nat. Rev. Microbiol 2006, 4, 588; [DOI] [PubMed] [Google Scholar]; c) Nimjee SM, Rusconi CP, Sullenger BA, Annu. Rev. Med 2005, 56, 555. [DOI] [PubMed] [Google Scholar]
- [85].a) Fang X, Tan W, Acc. Chem. Res 2010, 43, 48; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stoltenburg R, Reinemann C, Strehlitz B, Biomol. Eng 2007, 24, 381. [DOI] [PubMed] [Google Scholar]
- [86].McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH, Nat. Biotechnol 2006, 24, 1005. [DOI] [PubMed] [Google Scholar]
- [87].Dassie JP, Liu X.-y., Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara Ii JO, Giangrande PH, Nat. Biotechnol 2009, 27, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chen Z, Liu H, Jain A, Zhang L, Liu C, Cheng K, Theranostics 2017, 7, 2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Lee JB, Hong J, Bonner DK, Poon Z, Hammond PT, Nat. Mater 2012, 11, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Jeong H, Lee SH, Hwang Y, Yoo H, Jung H, Kim SH, Mok H, Macromol. Biosci 2017, 17, 1600343. [DOI] [PubMed] [Google Scholar]
- [91].Kim MW, Jeong HY, Kang SJ, Choi MJ, You YM, Im CS, Lee TS, Song IH, Lee CG, Rhee K-J, Lee YK, Park YS, Sci. Rep 2017, 7, 9474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Neff CP, Zhou J, Remling L, Kuruvilla J, Zhang J, Li H, Smith DD, Swiderski P, Rossi JJ, Akkina R, Sci. Transl. Med 2011, 3, 66ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Ivanov AI, in Exocytosis and Endocytosis, (Ed: Ivanov AI), Humana Press, Totowa, NJ: 2008, 15. [Google Scholar]
- [94].Iversen T-G, Skotland T, Sandvig K, Nano Today 2011, 6, 176. [Google Scholar]
- [95].Jiang Z, He H, Liu H, Thayumanavan S, Biomacromolecules 2019, 20, 4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].a) Conner SD, Schmid SL, Nature 2003, 422, 37; [DOI] [PubMed] [Google Scholar]; b) Doherty GJ, McMahon HT, Annu. Rev. Biochem 2009, 78, 857. [DOI] [PubMed] [Google Scholar]
- [97].a) Kerr MC, Teasdale RD, Traffic 2009, 10, 364; [DOI] [PubMed] [Google Scholar]; b) Bloomfield G, Kay RR, J. Cell Sci 2016, 129, 2697. [DOI] [PubMed] [Google Scholar]
- [98].a) Mousavi SA, Malerød L, Berg T, Kjeken R, Biochem. J 2004, 377, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kaksonen M, Roux A, Nat. Rev. Mol. Cell Biol 2018, 19, 313; [DOI] [PubMed] [Google Scholar]; c) Mettlen M, Chen P-H, Srinivasan S, Danuser G, Schmid SL, Annu. Rev. Biochem 2018, 87, 871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].a) Pelkmans L, Helenius A, Traffic 2002, 3, 311; [DOI] [PubMed] [Google Scholar]; b) Mayor S, Pagano RE, Nat. Rev. Mol. Cell Biol 2007, 8, 603. [DOI] [PubMed] [Google Scholar]
- [100].a) Galbiati F, Razani B, Lisanti MP, Cell 2001, 106, 403; [DOI] [PubMed] [Google Scholar]; b) Parton RG, Richards AA, Traffic 2003, 4, 724; [DOI] [PubMed] [Google Scholar]; c) Nabi IR, Le PU, J. Cell Biol 2003, 161, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].a) Tabata Y, Ikada Y, Biomaterials 1988, 9, 356; [DOI] [PubMed] [Google Scholar]; b) Champion JA, Walker A, Mitragotri S, Pharm. Res 2008, 25, 1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].a) and AA, Underhill DM, Annu. Rev. Immunol 1999, 17, 593; [DOI] [PubMed] [Google Scholar]; b) Stuart LM, Ezekowitz RAB, Immunity 2005, 22, 539. [DOI] [PubMed] [Google Scholar]
- [103].a) Dutta D, Donaldson JG, Cell. Logist 2012, 2, 203; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang LW, Monteiro-Riviere NA, Toxicol. Sci 2009, 110, 138. [DOI] [PubMed] [Google Scholar]
- [104].a) He C, Hu Y, Yin L, Tang C, Yin C, Biomaterials 2010, 31, 3657; [DOI] [PubMed] [Google Scholar]; b) Jiang Y, Huo S, Mizuhara T, Das R, Lee Y-W, Hou S, Moyano DF, Duncan B, Liang X-J, Rotello VM, ACS Nano 2015, 9, 9986; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wong NKY, Shenoi RA, Abbina S, Chafeeva I, Kizhakkedathu JN, Khan MK, Biomacromolecules 2017, 18, 2427; [DOI] [PubMed] [Google Scholar]; d) Ho LWC, Yung W-Y, Sy KHS, Li HY, Choi CKK, Leung KC-F, Lee TWY, Choi CHJ, ACS Nano 2017, 11, 6085. [DOI] [PubMed] [Google Scholar]
- [105].Zidovetzki R, Levitan I, Biochim. Biophys. Acta 2007, 1768, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Lim KS, Lee DY, Valencia GM, Won Y-W, Bull DA, Adv. Funct. Mater 2015, 25, 5445. [Google Scholar]
- [107].Kapoor M, Burgess DJ, Pharm. Res 2013, 30, 1161. [DOI] [PubMed] [Google Scholar]
- [108].Cao X, Lu X, Wang D, Jia F, Tan X, Corley M, Chen X, Zhang K, Small 2017, 13, 1701432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ren K, Liu Y, Wu J, Zhang Y, Zhu J, Yang M, Ju H, Nat. Commun 2016, 7, 13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen G, Liu D, He C, Gannett TR, Lin W, Weizmann Y, J. Am. Chem. Soc 2015, 137, 3844. [DOI] [PubMed] [Google Scholar]
- [111].a) Barenholz Y, Prog. Lipid Res 2002, 41, 1; [DOI] [PubMed] [Google Scholar]; b) Edidin M, Annu. Rev. Biophys. Biomol. Struct 2003, 32, 257; [DOI] [PubMed] [Google Scholar]; c) Korade Z, Kenworthy AK, Neuropharmacology 2008, 55, 1265; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mahammad S, Parmryd I, in Methods in Membrane Lipids, (Ed: Owen DM), Springer New York, New York, NY: 2015, 91. [Google Scholar]
- [112].a) Kunisawa J, Nakagawa S, Mayumi T, Adv. Drug Del. Rev 2001, 52, 177; [DOI] [PubMed] [Google Scholar]; b) Jiang Y, Tang R, Duncan B, Jiang Z, Yan B, Mout R, Rotello VM, Angew. Chem. Int. Ed 2015, 54, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].a) Umeda M, Nojima S, Inoue K, Biochem J. 1985, 97, 1301; [DOI] [PubMed] [Google Scholar]; b) Hu Q, Shew CR, Bally MB, Madden TD, Biochim. Biophys. Acta 2001, 1514, 1; [DOI] [PubMed] [Google Scholar]; c) Kolašinac R, Kleusch C, Braun T, Merkel R, Csiszár A, Int. J. Mol. Sci 2018, 19, 346; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dutta K, Bochicchio D, Ribbe AE, Alfandari D, Mager J, Pavan GM, Thayumanavan S, ACS Appl. Mater. Interfaces 2019, 11, 24971. [DOI] [PubMed] [Google Scholar]
- [114].a) Oliveira S, van Rooy I, Kranenburg O, Storm G, Schiffelers RM, Int. J. Pharm 2007, 331, 211; [DOI] [PubMed] [Google Scholar]; b) Tai W, Gao X, Adv. Drug Del. Rev 2017, 110–111, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Bonifacino JS, Glick BS, Cell 2004, 116, 153. [DOI] [PubMed] [Google Scholar]
- [116].a) Verma SK, Mani P, Sharma NR, Krishnan A, Kumar VV, Reddy BS, Chaudhuri A, Roy RP, Sarkar DP, J. Biol. Chem 2005, 280, 35399; [DOI] [PubMed] [Google Scholar]; b) Yoshikawa T, Okada N, Nakagawa S, Future Lipidol. 2006, 1, 735. [Google Scholar]
- [117].Patel PC, Giljohann DA, Daniel WL, Zheng D, Prigodich AE, Mirkin CA, Bioconjugate Chem. 2010, 21, 2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Pei D, Buyanova M, Bioconjugate Chem. 2019, 30, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].a) El-Sayed A, Futaki S, Harashima H, AAPS J 2009, 11, 13; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Varkouhi AK, Scholte M, Storm G, Haisma HJ, J. Controlled Release 2011, 151, 220; [DOI] [PubMed] [Google Scholar]; c) Smith SA, Selby LI, Johnston APR, Such GK, Bioconjugate Chem. 2019, 30, 263; [DOI] [PubMed] [Google Scholar]; d) Brock DJ, Kondow-McConaghy HM, Hager EC, Pellois J-P, Bioconjugate Chem. 2019, 30, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].a) Behr J-P, Chimia 1997, 51, 34; [Google Scholar]; b) Yang S, May S, J. Chem. Phys 2008, 129, 185105; [DOI] [PubMed] [Google Scholar]; c) Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M, Nat. Mater 2009, 8, 543. [DOI] [PubMed] [Google Scholar]
- [121].a) Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL, Mol. Ther 2013, 21, 149; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vermeulen LMP, De Smedt SC, Remaut K, Braeckmans K, Eur. J. Pharm. Biopharm 2018, 129, 184. [DOI] [PubMed] [Google Scholar]
- [122].Kube S, Hersch N, Naumovska E, Gensch T, Hendriks J, Franzen A, Landvogt L, Siebrasse J-P, Kubitscheck U, Hoffmann B, Merkel R, Csiszár A, Langmuir 2017, 33, 1051. [DOI] [PubMed] [Google Scholar]
- [123].Tai W, Gao X, Biomaterials 2018, 178, 720. [DOI] [PubMed] [Google Scholar]
- [124].Shi J, Kantoff PW, Wooster R, Farokhzad OC, Nat. Rev. Cancer 2017, 17, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Gupta RC, Lall R, Srivastava A, Sinha A, Frontiers in Veterinary Science 2019, 6, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].a) Kaur H, Bruno JG, Kumar A, Sharma TK, Theranostics 2018, 8, 4016; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ponce AT, Hong KL, Biomedicines 2019, 7, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Barba AA, Bochicchio S, Dalmoro A, Lamberti G, Pharmaceutics 2019, 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Huang Y, Mol. Ther. Nucleic Acids 2017, 6, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Choi CKK, Zhang L, Choi CHJ, Nat. Biomed. Eng 2018, 2, 275. [DOI] [PubMed] [Google Scholar]
- [130].a) Jayaraman M, Ansell SM, Mui BL, Tam YK, Chen J, Du X, Butler D, Eltepu L, Matsuda S, Narayanannair JK, Rajeev KG, Hafez IM, Akinc A, Maier MA, Tracy MA, Cullis PR, Madden TD, Manoharan M, Hope MJ, Angew. Chem. Int. Ed 2012, 51, 8529; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Semple SC, Akinc A, Chen JX, Sandhu AP, Mui BL, Cho CK, Sah DWY, Stebbing D, Crosley EJ, Yaworski E, Hafez IM, Dorkin JR, Qin J, Lam K, Rajeev KG, Wong KF, Jeffs LB, Nechev L, Eisenhardt ML, Jayaraman M, Kazem M, Maier MA, Srinivasulu M, Weinstein MJ, Chen QM, Alvarez R, Barros SA, De S, Klimuk SK, Borland T, Kosovrasti V, Cantley WL, Tam YK, Manoharan M, Ciufolini MA, Tracy MA, de Fougerolles A, MacLachlan I, Cullis PR, Madden TD, Hope MJ, Nat. Biotechnol 2010, 28, 172; [DOI] [PubMed] [Google Scholar]; c) Cheng X, Lee RJ, Adv. Drug Del. Rev 2016, 99, 129. [DOI] [PubMed] [Google Scholar]
- [131].a) Wang H, Feng Z, Xu B, Theranostics 2019, 9, 3213; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lu N, Wang L, Lv M, Tang Z, Fan C, Nano Res. 2019, 12, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].a) Schellekens H, Hennink WE, Brinks V, Pharm. Res 2013, 30, 1729; [DOI] [PubMed] [Google Scholar]; b) Yang Q, Lai SK, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2015, 7, 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].a) Song E, Zhu P, Lee S-K, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J, Nat. Biotechnol 2005, 23, 709; [DOI] [PubMed] [Google Scholar]; b) Zhang K, Hao L, Hurst SJ, Mirkin CA, J. Am. Chem. Soc 2012, 134, 16488. [DOI] [PMC free article] [PubMed] [Google Scholar]