

Abstract
Cyclic nucleotide‐gated channel β1 (CNGB1) encodes the 240‐kDa β subunit of the rod photoreceptor cyclic nucleotide‐gated ion channel. Disease‐causing sequence variants in CNGB1 lead to autosomal recessive rod‐cone dystrophy/retinitis pigmentosa (RP). We herein present a comprehensive review and analysis of all previously reported CNGB1 sequence variants, and add 22 novel variants, thereby enlarging the spectrum to 84 variants in total, including 24 missense variants (two of which may also affect splicing), 21 nonsense, 19 splicing defects (7 at noncanonical positions), 10 small deletions, 1 small insertion, 1 small insertion–deletion, 7 small duplications, and 1 gross deletion. According to the American College of Medical Genetics and Genomics classification criteria, 59 variants were considered pathogenic or likely pathogenic and 25 were variants of uncertain significance. In addition, we provide further phenotypic data from 34 CNGB1‐related RP cases, which, overall, are in line with previous findings suggesting that this form of RP has long‐term retention of useful central vision despite the early onset of night blindness, which is valuable for patient counseling, but also has implications for it being considered a priority target for gene therapy trials.
Keywords: CNGB1, cyclic nucleotide‐gated channel, genotype‐phenotype correlation, inherited retinal disease, retinitis pigmentosa, rod‐cone dystrophy
1. BACKGROUND
Inherited retinal diseases (IRDs) are a group of clinically and genetically heterogeneous disorders with an overall estimated prevalence that ranges between 1/3500 and 1/5000 worldwide. They are characterized by progressive photoreceptor degeneration (Hartong et al., 2006) with variable age of onset and degree of severity of vision loss. Up to now, more than 300 genes have been associated with IRDs (source: https://sph.uth.edu/retnet/) following all modes of inheritance, including Mendelian, mitochondrial, and rarely, digenism. Most of the proteins encoded by these genes are involved in cellular pathways that are crucial for photoreceptor or retinal pigment epithelium (RPE) homeostasis and functions (Wright et al., 2010). Retinitis pigmentosa (RP; MIM# 268000) or rod‐cone dystrophy (RCD) is the most common form of IRD. It affects approximately 1/4000 individuals and is characterized by rod dysfunction, followed by cone impairment (Hartong et al., 2006). Typical symptoms are night blindness, progressive visual field constriction and reduced visual acuity in later stages of the disease, resulting in some individuals being classified as “legally blind.” Over 80 different genes are associated with nonsyndromic RP (source: https://sph.uth.edu/retnet/). There is no current treatment for RP, however, there are several clinical trials underway (phase I–III), including various strategies (e.g., gene augmentation therapy, gene editing, antisense oligonucleotides, and others) showing promising results (Bainbridge et al., 2008, 2015; Cideciyan et al., 2013, 2019; MacLaren et al., 2014; Maguire et al., 2008, 2019; Schimmer & Breazzano, 2015).
Variants in CNGB1 (MIM# 600724, Cyclic nucleotide‐gated channel β1) are associated with autosomal recessive RP 45 (RP45, MIM# 613767; Bareil et al., 2001), accounting for approximately 1%–4% of autosomal recessive RP cases (Ge et al., 2015; Maeda et al., 2008; Xu et al., 2014). Patients affected by CNGB1‐related RP usually experience the onset of night blindness during childhood. However, the constriction of the visual field becomes symptomatic much later and the visual acuity is usually well‐preserved during adulthood. Fundus abnormalities are typical of RP, with retinal vessel attenuation and mid‐peripheral “bone spicule” pigmentary clumping. Electroretinography shows a generalized dysfunction of the photoreceptors, predominantly affecting the rod system, with relative preservation of macular function in young adults (Hull et al., 2017).
CNGB1 (MIM# 600724) encodes the 240‐kDa β subunit (CNGB1a) of the rod photoreceptor cyclic nucleotide‐gated (CNG) ion channel, containing a long cytosolic N‐terminus (glutamic acid‐rich protein [GARP]) domain and a channel domain, which includes a cyclic nucleotide‐binding domain (CNBD) (Ko¨rschen et al., 1995). This full‐length CNGB1 is exclusively expressed in the retina, predominantly in the rod photoreceptors. However, shorter transcripts that encode the CNGB1b protein lacking the GARP domain are expressed in sensory neurons of the olfactory epithelium and form together with CNGA2 and CNGA4 subunits the native olfactory CNG channel (Kaupp & Seifert, 2002). In line with this, CNGB1 variants within the channel‐encoding part of the sequence result in peculiar olfactory symptoms. In particular, CNGB1‐mutant patients might experience an impaired sense of smell because the splice variant of the gene, CNGB1b, is expressed in the olfactory epithelium. The rod CNG channel consists of four subunits: one CNGB1a and three cyclic nucleotide‐gated channel α1 (CNGA1) (Kaupp & Seifert, 2002; Shuart et al., 2011). More specifically, CNGB1a subunits are important to ensure the correct localization of the channel in the plasma membrane of the outer segment of photoreceptors and help to modulate the channel activity (Biel et al., 2009). Under scotopic conditions, the CNBD of the CNG channels is occupied by cyclic GMP (cGMP), resulting in transient channel opening and a depolarizing inward current in rods. Upon light‐induced initiation of the phototransduction cascade, the intracellular cGMP concentration decreases through activation of the phosphodiesterase PDE6, which cleaves cGMP. This leads to the closure of the channels and hyperpolarization of the rods (Biel et al., 2009).
A mouse model of CNGB1‐retinopathy lacking exon 26 (Cngb1‐X26) is characterized by a slow progressive retinal degeneration with cell apoptosis and retinal gliosis (Huttl, 2005; Zhang et al., 2009). In this model, rods are the first to degenerate producing early (around 1 month of age) undetectable scotopic responses on the electroretinogram (ERG). Cone degeneration usually follows after 6 months of age and eventually these mice become blind after one year. In the inner retina, some morphological alterations were described in rod bipolar cells and horizontal cells (e.g., sprouting extensions, retraction of processes, and misplacement of cell bodies). More recently, Winkler et al. (2013) discovered a canine model of progressive retinal degeneration connected with a spontaneous variant in Cngb1, which closely resembled not only the Cngb1‐X26 mouse model but also the human RP45 phenotype. These studies using naturally occurring or generated animal models can provide further insights into the function of the protein and are important for preclinical studies in the development of therapeutic trials. For example, CNGB1 was at first considered a modulatory subunit of the CNG channels, and the retinal phenotype in Cngb1‐deficient mice and dogs was expected to be absent or milder than the one actually expressed. For this reason, it has been hypothesized that CNGB1 could be more than a modulatory subunit but it turned out to be required for assembly and targeting of native CNG channels to the outer segment, ensuring that only properly formed heteromeric channels reach the outer segment membrane. In fact, in Cngb1‐deficient mice only traces of the CNGA1 subunit are present at the membrane of outer segments of rods, confirming that homomeric CNGA1 channels are not efficiently trafficked and/or transported in the absence of CNGB1. The absence (or downregulation) of the CNG channels thus compromises rod function and eventually leads to the death of the photoreceptors through pathways that are still unclear. One possibility is the “equivalent light” hypothesis, which assumes that the absence of CNG channels is equivalent to the permanent closure of channels (as under a continuous bright light stimulation). Continuous exposure of experimental animals to light may result in photoreceptor degeneration through yet undefined mechanisms (Biel et al., 2009). Both CNGB1‐deficient mouse and canine models were used to attempt the rescue of the phenotype through gene augmentation therapy. This resulted in a robust, sustained restoration of rod function and retinal structural preservation in both models (Koch et al., 2012; Petersen‐Jones et al., 2018).
These preclinical successes raised the hope for applying CNGB1 gene augmentation therapy to humans in the near future. However, before clinical trials, large cohort studies and natural history studies are warranted to ascertain whether there are suitable biomarkers of progression of retinal disease that can be used to assess the efficacy of therapy within a practical time frame. At the same time, the review and classification of the previously reported CNGB1 variants in IRD cases could serve as a guide for the enrollment of patients in such trials.
2. MATERIAL AND METHODS
2.1. Literature search
A literature search was performed on Pubmed (updated on June 1st, 2020) to collect all reported variants on CNGB1 (NM_001135639) in association with IRDs. Additional databases were queried, such as The Human Gene Mutation Database (HGMD® Professional 2017.4, last queried on September 1st, 2020), Leiden Open Variation Database (LOVD V.3.0, last queried on September 1st, 2020), and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, last queried on September 1st, 2020). For each of the reported variants, we collected information on phenotype/genotype correlation, familial segregation, and documented minor allele frequency (MAF) as well as assessed functional impact through in silico pathogenic predictions tools (i.e., Mutation Taster [http://www.mutationtaster.org/ (Schwarz et al., 2014)], Sorting Intolerant From Tolerant SIFT [https://sift.bii.a-star.edu.sg/ (Sim et al., 2012)], Polymorphism Phenotyping v2 Polyphen‐2 [http://genetics.bwh.harvard.edu/pph2/ (Adzhubei et al., 2010)], Splice Site Prediction by Neural Network, NNSPLICE, [https://www.fruitfly.org/seq_tools/splice.html (Reese et al., 1997)]; Human Splicing Finder v.2.4.1, HSF, [http://www.umd.be/HSF/ (Desmet et al., 2009)] and evolutionary conservation (HomoloGene, available at https://www.ncbi.nlm.nih.gov/homologene and UCSC Genome Browser, available at https://genome.ucsc.edu/index.html). Guidelines from the American College of Medical Genetics and Genomics (ACMG) were used for variant classification (Richards et al., 2015).
2.2. Novel CNGB1 variants
In addition to the comprehensive literature search, we analyzed a multicenter large cohort of unrelated subjects with IRDs to find RP45 patients harboring known and novel CNGB1 variants to prepare patients for future clinical trials. Each patient underwent a full ophthalmic examination by one of the investigators. Written informed consent was obtained from each subject. The study protocol adhered to the tenets of the Declaration of Helsinki and was approved by the local ethics committees. The affected index cases recruited at the Centre Hospitalier National d'Ophtalmologie des Quinze‐Vingts in Paris were genetically screened through targeted next generation sequencing (NGS) at the Institut de la Vision as previously described (Audo et al., 2012; Boulanger‐Scemama et al., 2015). The affected index cases recruited at the University Eye Hospital in Tübingen, Germany were genetically screened through targeted (NGS) at the Wissinger Lab, Institute for Ophthalmic Research in Tübingen as previously described (Glöckle et al., 2014; Weisschuh et al., 2016, 2018). The affected index cases recruited at the Department of Ophthalmology, Columbia University in New York, USA and in the Chang Gung Memorial Hospital in Taoyuan, Taiwan were genetically screened through whole exome sequencing as previously described (Tsang et al., 2014). The affected index case recruited at Moorfields Eye Hospital, London, UK was genetically tested through NGS at the Genomics Diagnostic Laboratory (Manchester Centre for Genomic Medicine, Manchester, UK). The affected index cases recruited at the Lille University Hospital, France, were genetically tested by NGS of a panel of 156 IRD genes in the genetics diagnostic laboratory (Department of Biochemistry and Molecular Biology, Lille, France). The data analysis was performed by a standard bioinformatics pipeline. The raw reads were filtered and aligned with the hg19 reference sequence from UCSC Genome Browser (https://genome.ucsc.edu/index.html) database (Kent et al., 2002). Variants were annotated using ANNOVAR (http://annovar.openbioinformatics.org/en/latest/) (Wang et al., 2010). The functional impact was assessed using in silico prediction tools described in the previous paragraph. Only rare variants with an MAF below or equal to 0.005 in the genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org/) were considered. All variants of interest were confirmed by Sanger sequencing, and segregation analyses were performed on the other family members when available.
2.3. Clinical investigation
A retrospective analysis was performed on the charts of the patients and their exams. Herein we reported the data from the last, most complete visit performed. When available, age of onset, best‐corrected visual acuity (BCVA), color vision, full‐field ERG (ff‐ERG), kinetic visual field, fundus examination, optical coherence tomography (OCT), and short‐wavelength fundus autofluorescence (SW‐FAF) data were collected.
2.4. CNGB1 VARIANTS
A total of 62 genetic changes in CNGB1 were found in the literature underlying IRD. In addition, we identified 22 novel changes in our cohort (Figure 1 and Table S1). Altogether, the sequence variant spectrum includes 24 missense variants (28%), 2 of which may also affect splicing, 21 nonsense (25%), 19 splicing defects (23%; 7 at noncanonical positions), and 20 changes in protein length and/or frameshift (24%; 10 small deletions, 1 small insertion, 1 small insertion–deletion, 7 small duplications, and 1 gross deletion) (Figure 2 and Table 1).
Figure 1.
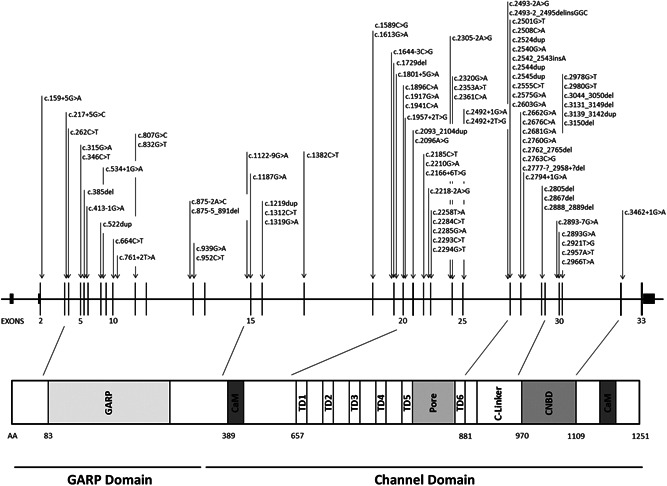
CNGB1 variants underlying inherited retinal dystrophies. Nucleotide positions and translation correspond to CCDS45495.1 and NP_001288.3, respectively
Figure 2.
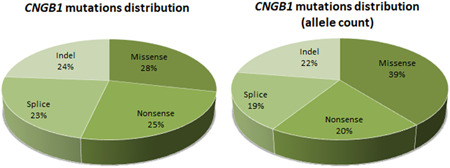
Pie charts showing the distribution of the CNGB1 variants underlying inherited retinal diseases according to their types. On the left, the absolute distribution is shown. On the right, the distribution according to the allele count performed through the revision of the literature and the analysis of the cohort in this study is shown
Table 1.
List of CNGB1 variants associated with inherited retinal dystrophies
| Genomic start position (hg19) | Exon/intron | cDNA (NM_001297.5) | Protein change (NP_001288.3) | ACMG classification (criteria) | Phenotype | References | Frequency | ||
|---|---|---|---|---|---|---|---|---|---|
| Index Cases | Allele count | Hom Cases | |||||||
| 58001027 | IVS2 | c.159+5G>A | p.? | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 1 | 0 |
| 57998386 | IVS3 | c.217+5G>C | p.? | Uncertain significance (PM2, PP3) | RCD | Oishi et al. (2014); This study | 3 | 4 | 1 |
| 57998062 | 4 | c.262C>T | p.(Gln88*) |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Ellingford et al. (2016); Afshar et al. (2019); Carss et al. (2017) | 4 | 4 | 0 |
| 57996944 | 5 | c.315G>A | p.(Trp105*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | This study | 1 | 2 | 1 |
| 57996913 | 5 | c.346C>T | p.(Gln116*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Afshar et al. (2019); This study | 2 | 2 | 0 |
| 57996773 | 6 | c.385del | p.(Leu129Trpfs*148) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Xiang et al. (2018) | 1 | 2 | 1 |
| 57996515 | IVS6 | c.413‐1G>A | p.[=;Cys139Alafs*138]a |
Pathogenic (PVS1,PM2, PS3, PP3, PP5) |
RCD | Azam et al. (2011); Afshar et al. (2019); Charbel Issa et al. (2018) | 6 | 11 | 5 |
| 57994756 | 8 | c.522dup | p.(Lys175Glnfs*4) |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Lingao et al. (2016) | 3 | 3 | 0 |
| 57994743 | IVS8 | c.534+1G>A | p.? |
Pathogenic (PVS1, PM2, PP3) |
RCD | Afshar et al. (2019) | 1 | 1 | 0 |
| 57993889 | 10 | c.664C>T | p.(Gln222*) |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Afshar et al. (2019); Carss et al. (2017) | 2 | 2 | 0 |
| 57993790 | IVS10 | c.761+2T>A | p.? |
Pathogenic (PVS1, PM2,PP3) |
RCD | Hull et al. (2017) | 1 | 2 | 1 |
| 57992344 | 11 | c.807G>C | p.(Gln269His) |
Likely pathogenic (PM1, PM2, PM3, PP3) |
Isolated rod dysfunction | Afshar et al. (2019); Ba‐Abbad et al. (2019) | 2 | 2 | 0 |
| 57992319 | 11 | c.832G>T | p.(Glu278*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | This study | 1 | 1 | 0 |
| 57984446 | IVS12 | c.875‐2A>C | p.? |
Pathogenic (PVS1, PM2,PP3) |
RCD | Maeda et al. (2018) | 1 | 2 | 1 |
| 57984428 | IVS12 | c.875‐5_891del | p.? |
Pathogenic (PVS1, PM2, PP3) |
RCD | Bernardis et al. (2016) | 2 | 3 | 1 |
| 57984380 | 13 | c.939G>A | p.(Trp313*) |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Fradin et al. (2016); This study | 2 | 4 | 2 |
| 57984367 | 13 | c.952C>T | p.(Gln318*) |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Afshar et al. (2019); Hull et al. (2017) | 2 | 2 | 0 |
| 57974234 | IVS14 | c.1122‐9G>A | p.[=; Glu374Glufs*7]b |
Pathogenic (PS3, PM2, PM3, PP1, PP3) |
RCD | Petersen‐Jones et al. (2018) | 1 | 1 | 0 |
| 57974160 | 15 | c.1187G>A | p.(Arg396Gln) | Uncertain significance (BP4, PM2) | RCD | Comander et al. (2017) | 1 | 1 | 0 |
| 57973487 | 16 | c.1219dup | p.(Glu407Glyfs*12) |
Pathogenic (PVS1, PM2,PP3) |
RCD | Charbel Issa et al. (2018); This study | 2 | 2 | 1 |
| 57973394 | 16 | c.1312C>T | p.(Gln438*) |
Pathogenic (PVS1, PM1, PM2) |
RCD | Charbel Issa et al. (2018) | 1 | 2 | 1 |
| 57973387 | 16 | c.1319G>A | p.(Trp440*) |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | This study | 1 | 1 | 0 |
| 57965773 | 17 | c.1382C>T | p.(Thr461Met) |
Uncertain significance (BS1, PP3) |
RCD | Ellingford et al. (2016) | 1 | 1 | 0 |
| 57957231 | 18 | c.1589C>G | p.(Pro530Arg) | Uncertain significance (PP1, PP3, BS1) | RCD | Fu et al. (2013) | 1 | 2 | 1 |
| 57957207 | 18 | c.1613G>A | p.(Trp538*) |
Pathogenic (PVS1, PM2,PP3) |
RCD | This study | 1 | 2 | 1 |
| 57954451 | IVS18 | c.1644‐3C>G | p.? | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 2 | 1 |
| 57954363 | 19 | c.1729del | p.(Glu577Serfs*6) |
Pathogenic (PVS1, PM1, PM2) |
RCD | Afshar et al. (2019) | 1 | 2 | 1 |
| 57954286 | IVS19 | c.1801+5G>A | p.? | Uncertain significance (PM2, PP3) | RCD | Comander et al. (2017) | 1 | 1 | 0 |
| 57953064 | 20 | c.1896C>A | p.(Cys632*) |
Pathogenic (PVS1, PM2, PP1) |
RCD | Nishiguchi et al. (2013); Petersen‐Jones et al. (2018) | 2 | 2 | 0 |
| 57953043 | 20 | c.1917G>A | p.(Trp639*) |
Pathogenic (PVS1, PM2, PM3) |
RCD | Banerjee et al. (2017) | 1 | 1 | 0 |
| 57953019 | 20 | c.1941C>A | p.(Ser647Arg) | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 1 | 0 |
| 57953001 | IVS20 | c.1957+2T>G | p.? |
Pathogenic (PVS1,PM2, PP3) |
RCD | This study | 1 | 1 | 0 |
| 57951242 | 21 | c.2096A>G | p.(Asp699Gly) | Uncertain significance (PM2, PP3) | RCD | Ellingford et al. (2016) | 1 | 1 | 0 |
| 57951234 | 21 | c.2093_2104dup | p.(Cys698_Ile701dup) | Likely pathogenic (PM2, PM4, PP1, PP3) | RCD | Alshamrani et al. (2020) | 1 | 1 | 0 |
| 57951166 | IVS21 | c.2166+6T>G | p.? | Uncertain significance (PM3,PP3) | RCD | This study | 1 | 1 | 0 |
| 57950065 | 22 | c.2185C>T | p.(Arg729*) |
Pathogenic (PVS1, PM2,PP3) |
RCD | Carss et al. (2017); Hull et al. (2017) | 3 | 5 | 2 |
| 57950040 | 22 | c.2210G>A | p.(Arg737His) | Uncertain significance (PM2, PP3) | RCD | Charbel Issa et al. (2018) | 1 | 1 | 0 |
| 57949241 | IVS22 | c.2218‐2A>G | p.? |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Petersen‐Jones et al. (2018) | 1 | 1 | 0 |
| 57949199 | 23 | c.2258T>A | p.(Leu753*) |
Pathogenic (PVS1, PM1, PM2) |
Isolated rod dysfunction | Afshar et al. (2019); Ba‐Abbad et al. (2019) | 2 | 2 | 0 |
| 57949173 | 23 | c.2284C>T | p.(Arg762Cys) |
Likely pathogenic (PM2, PM3, PP1, PP3, PP5) |
RCD | Azam et al. (2011); Bocquet et al. (2013); Beryozkin et al. (2015); Consugar et al. (2015); Bernardis et al. (2016); Charbel Issa et al. (2018); Petersen‐Jones et al. (2018); This study | 12 | 21 | 9 |
| 57949172 | 23 | c.2285G>A | p.(Arg762His) | Uncertain significance (PM2, PP1, PP3) | RCD | Afshar et al. (2019); Carss et al. (2017) | 2 | 4 | 2 |
| 57949164 | 23 | c.2293C>T | p.(Arg765Cys) | Uncertain significance (PM2, PP1, PP3) | RCD | Schorderet et al. (2014); Habibi et al. (2016) | 2 | 4 | 2 |
| 57949163 | 23 | c.2294G>T | p.(Arg765Leu) | Uncertain significance (PM2, PP3) | RCD | Patel et al. (2016) | 1 | 2 | 1 |
| 57946900 | IVS23 | c.2305‐2A>G | p.? |
Pathogenic (PVS1, PM2, PP1) |
RCD | This study | 2 | 3 | 1 |
| 57912979 | 24 | c.2320G>A | p.(Glu774Lys) | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 1 | 0 |
| 57946850 | 24 | c.2353A>T | p.(Lys785*) |
Pathogenic (PVS1, PM2, PP1) |
RCD | This study | 1 | 2 | 1 |
| 57946842 | 24 | c.2361C>A | p.(Tyr787*) |
Pathogenic (PVS1,PM2, PM3,PP3) |
RCD | Xu et al. (2014); Banerjee et al. (2017) | 2 | 2 | 0 |
| 57945656 | IVS25 | c.2492+1G>A | p.? |
Pathogenic (PVS1, PM2, PP3) |
RCD | Charbel Issa et al. (2018) | 2 | 3 | 1 |
| 57911751 | IVS25 | c.2492+2T>G | p.? |
Pathogenic (PVS1, PM2, PP3) |
RCD | This study | 1 | 1 | 0 |
| 57938777 | IVS25 | c.2493‐2_2495delinsGGC | p.? |
Pathogenic (PVS1, PM2, PP1,PP3) |
RCD | Maranhao et al. (2015) | 1 | 2 | 1 |
| 57938781 | IVS25 | c.2493‐2A>G | p.? |
Pathogenic (PVS1, PM2, PP1, PP3) |
RCD | Maria et al. (2015) | 1 | 1 | 0 |
| 57938771 | 26 | c.2501G>T | p.(Arg834Leu) | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 1 | 0 |
| 57938764 | 26 | c.2508C>A | p.(Tyr836*) |
Pathogenic (PVS1, PM2,PP1,PP3) |
RCD | Consugar et al. (2015); Petersen‐Jones et al. (2018) | 2 | 2 | 0 |
| 57938748 | 26 | c.2524dup | p.(Thr842Asnfs*10) |
Likely pathogenic (PVS1, PM2) |
RCD | Oishi et al. (2014) | 1 | 2 | 1 |
| 57938732 | 26 | c.2540G>A | p.(Gly847Glu) | Uncertain significance (PM2, PP3) | RCD | Afshar et al. (2019) | 1 | 1 | 0 |
| 57938730 | 26 | c.2542_2543insA | p.(Gly848Glufs*4) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Charbel Issa et al. (2018) | 1 | 2 | 1 |
| 57938728 | 26 | c.2544dup | p.(Leu849Alafs*3) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Kondo et al. (2004); Consugar et al. (2015); Ge et al. (2015; Ellingford et al. (2016); Afshar et al. (2019); Hull et al. (2017) | 8 | 10 | 2 |
| 57938727 | 26 | c.2545dup | p.(Leu849Profs*3) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Petersen‐Jones et al. (2018) | 1 | 1 | 0 |
| 57938717 | 26 | c.2555C>T | p.(Pro852Leu) | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 2 | 1 |
| 57937858 | 26 | c.2575G>A | p.(Val859Ile) | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 1 | 0 |
| 57938669 | 26 | c.2603G>A | p.(Gly868Asp) | Uncertain significance (PM2, PP3) | RCD | Alshamrani et al. (2020); This study | 2 | 3 | 1 |
| 57938697 | 26 | c.2662G>A | p.(Ala888Thr) | Uncertain significance (PM2, PP3) | RCD | This study | 1 | 1 | 0 |
| 57937844 | 27 | c.2676C>A | p.(Tyr892*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Afshar et al. (2019); Carss et al. (2017) | 2 | 2 | 0 |
| 57937839 | 27 | c.2681G>A | p.(Arg894His) | Uncertain significance (PP1, PP3, BS1) | RCD | Ellingford et al. (2016) | 1 | 1 | 0 |
| 57937760 | 27 | c.2760G>A | p.(Trp920*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Beryozkin et al. (2015) | 1 | 2 | 1 |
| 57937755 | 27 | c.2762_2765del | p.(Tyr921Cysfs*15) |
Pathogenic (PVS1, PM2, PP3) |
RCD | de Castro‐Miró et al. (2016); This study | 2 | 4 | 2 |
| 57937757 | 27 | c.2763C>G | p.(Tyr921*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Charbel Issa et al. (2018) | 1 | 1 | 0 |
| 57935256 | 28‐29 | c.2777‐?_2958+?del | p.? |
Pathogenic (PVS1, PM2, PP3) |
RCD | Afshar et al. (2019) | 1 | 2 | 1 |
| 57937725 | IVS27 | c.2794+1G>A | p.? |
Pathogenic (PVS1, PM2, PP3) |
RCD | This study | 1 | 1 | 0 |
| 57935519 | 28 | c.2805del | p.(Glu935Aspfs*2) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Ge et al. (2015); This study | 2 | 2 | 0 |
| 57935457 | 28 | c.2867del | p.(Ile956Thrfs*15) |
Pathogenic (PVS1, PM2, PP3) |
RCD | This study | 2 | 2 | 0 |
| 57935435 | 28 | c.2888_2889del | p.(Phe963Serfs*4) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Xu et al. (2014) | 1 | 1 | 0 |
| 57935346 | IVS28 | c.2893‐7G>A | p.? | Uncertain significance (PM2, PM3, PP3) | RCD | This study | 3 | 5 | 2 |
| 57935339 | 29 | c.2893G>A | p.(Gly965Ser;?) |
Likely pathogenic (PM1, PM2, PP1, PP3) |
RCD | Ellingford et al. (2016) | 1 | 1 | 0 |
| 57935311 | 29 | c.2921T>G | p.(Met974Arg) | Uncertain significance (PM1, PM2, PP3) | RCD | Dan et al. (2020); This study | 2 | 3 | 1 |
| 57935275 | 29 | c.2957A>T | p.(Asn986Ile) |
Likely pathogenic (PM1, PM2, PP3, PP5) |
RCD | Simpson et al. (2011); Abu‐Safieh et al. (2013); Bernardis et al. (2016); Ellingford et al. (2016); Patel et al. (2016); Carss et al. (2017); Charbel Issa et al. (2018); Hull et al. (2017); Pérez‐Carro et al. (2018); Afshar et al. (2019); Fuster‐García et al. (2019); This study | 24 | 34 | 10 |
| 57935266 | 29 | c.2966T>A | p.(Val989Glu) |
Likely pathogenic (PM1, PM2, PP1, PP3) |
RCD | This study | 1 | 2 | 1 |
| 57931817 | 30 | c.2978G>T | p.(Gly993Val) |
Likely pathogenic (PM1, PM2, PP1, PP3) |
RCD | Bareil et al. (2001) | 1 | 2 | 1 |
| 57931815 | 30 | c.2980G>T | p.(Glu994*) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Afshar et al. (2019); Carss et al. (2017) | 2 | 2 | 0 |
| 57931745 | 30 | c.3044_3050del | p.(Gly1015Valfs*4) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Charbel Issa et al. (2018) | 1 | 1 | 0 |
| 57931394 | 30 | c.3131_3149del | p.(Ala1044Glyfs*13) |
Pathogenic (PVS1, PM2, PP3) |
RCD | This study | 1 | 1 | 0 |
| 57931401 | 31 | c.3139_3142dup | p.(Ala1048Glyfs*13) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Carss et al. (2017); Charbel Issa et al. (2018) | 3 | 3 | 0 |
| 57931393 | 31 | c.3150del | p.(Phe1051Leufs*12) |
Pathogenic (PVS1, PM2, PP3) |
RCD | Nishiguchi et al. (2013); Ge et al. (2015); Petersen‐Jones et al. (2018); This study | 5 | 7 | 2 |
| 57921758 | IVS32 | c.3462+1G>A | p.[=;Arg1081Argfs*68]c |
Pathogenic (PVS1, PM2, PP3) |
RCD | Kondo et al. (2004) | 1 | 2 | 1 |
| Total | 170 | 236 | 67 | ||||||
Note: ACMG criteria for this study: PVS1: null variant (nonsense, frameshift, canonical ±1 or 2 splice sites or initiation codon); PS3: well‐established in vitro and/or in vivo functional studies supportive of damaging effect on the gene or gene product; PM1: located on CNBD domain; PM2: frequency on gnomAD <0.5% and no homozygous cases (if not: BS1); PM3: variant detected in trans with a pathogenic variant; PM4: protein length changes as a result of in‐frame deletions/insertions in a nonrepeat region or stop‐loss variants; PP1: cosegregation with disease verified; PP3: at least 1 predictive algorithm suggest pathogenicity (for splice variants, score ≤−10%), if not: BP4; PP5: at least three previous publications report the variant as pathogenic; BP3: in‐frame deletions/insertions in a repetitive region without a known function; BP7: a synonymous variant for which splicing prediction algorithms predict no impact to the splice sequence nor the creation of a new splice site and the nucleotide is not highly conserved.
Abbreviations: ACMG, American College of Medical Genetics and Genomics; Hom, homozygous; RCD, rod‐cone dystrophy.
Effect validated by means of minigene assay by (Saqib et al., 2015).
Effect validated by means of in vitro assay by (Petersen‐Jones et al., 2018)
Effect validated by means of in vitro assay by (Becirovic et al., 2010).
The distribution of the different types of variants slightly changes when counting the alleles across all the reported cohorts; in fact, there is a higher prevalence of missense variants (39%) and a lower prevalence of nonsense (20%), splicing defects (19%), and indels (22%) (Figure 2 and Table 1). This is mostly related to the fact that the two missense variants c.2284C>T p.(Arg762Cys) (allele count: 21/236) and c.2957A>T p.(Asn986Ile) (allele count: 34/236) account together for the 23% of the investigated alleles. These variants were both found in different populations from different continents (Table 1).
According to the ACMG criteria, 59 variants were considered pathogenic or likely pathogenic and 25 variants were classified of uncertain significance (VUS; Table 1). All reported variants span the entire gene and therefore impact all functional domains (Figure 1). It is worth noticing that most of the variants occurring in the GARP domain of the protein are nonsense, frameshift, or affect splicing. Very few are the exceptions: c.1187G>A p.(Arg396Gln), c.1382C>T p.(Thr461Met), and c.807G>C p.(Gln269His). Although its frequency among the general population is as high as ~0.9% with 44 homozygous cases in gnomAD, variant c.1382C>T p.(Thr461Met) was considered VUS as Polyphen2 predicted it as “probably damaging.” On the contrary, variant c.1187G>A p.(Arg396Gln) was classified as VUS since its frequency among the general population is as low as 0.0046% but it was not predicted to be pathogenic by any of the applied algorithms, probably because of the low PhyloP (conservation) and Grantham (difference between amino acids) scores. Variant c.807G>C p.(Gln269His) is absent in the general population and is predicted as pathogenic by PolyPhen2 and SIFT. However, the associated phenotype was not the classical RP45, but an isolated rod dysfunction with only subtle peripheral pigmentary changes on fundus examination (Ba‐Abbad et al., 2019). In this case, the authors hypothesized a role of c.807G>C p.(Gln269His) in the sensitivity of the rod CNG channels to cGMP modulation: while the structural integrity of the channel might still allow the flow of Ca2+ ions to an extent that achieves nontoxic intracellular concentration, the variant might somehow affect the closure in response to light, abolishing the rod response. Overall, the interpretation of the missense variants on the GARP domain is still controversial as the precise function of the domain itself has not been established. However, its high conservation among vertebrates might suggest an important function. Of note, soluble GARP proteins are translated from the first 11–16 exons (Ardell et al., 2000) as separate cytosolic proteins that might take part in other cellular functions and interact with unknown partners (Ko¨rschen et al., 1995). Expression and proper outer segment localization of soluble GARP proteins is not affected in Cngb1‐X26 mice and dogs (Huttl, 2005; Winkler et al., 2013). To study the effect of GARP deletion another mouse model with a genetic modification in exon 1 of Cngb1 (Cngb1‐X1 mice) was generated (DeRamus et al., 2017; Zhang et al., 2009). While Cngb1‐X1 mice showed similar functional defects as Cngb1‐X26 mice, they in addition showed a dramatically compromised rod outer segment morphology suggesting that soluble and channel‐attached GARP proteins are essential for rod disk morphogenesis and outer segment integrity (Zhang et al., 2009). Other studies suggested a functional role of GARP in the control of rod CNG channel activity by interacting with the CNBD (Michalakis et al., 2011). GARP was shown to act as a gatekeeper, which directly binds to the CNBD to dampen channel activity, thus, lowering background channel noise and increasing the fidelity of the phototransduction cascade. This inhibitory effect of GARP can be relieved by binding of cGMP to the CNBD of both rod CNG channel subunits (CNGA1 and CNGB1). The clinical relevance of this function of GARP became evident when analyzing CNGB1 variants, which structurally impair the CNBD. For instance, c.2978G>T leads to a Gly to Val substitution within the CNBD and was shown to abolish channel function by impairing the cGMP‐dependent release of this autoinhibitory effect of GARP on the channel (Michalakis et al., 2011). Further biochemical analyses are required to clarify the impact of the GARP domain and its mutant forms on the overall activity of the CNG channels.
Most of the CNGB1 missense variants associated with the RP45 phenotype have been located in the channel domain, with two “hot spots” at the level of the transmembrane domains and at the level of the CNBD, both crucial for the protein structure and function. In our cohort, we identified 32 families (34 patients) carrying biallelic CNGB1 variants, 23 of which carried at least one novel variant (Table 2). Overall, we found 35 distinct variants, including 12 missense, 8 nonsense, 9 splice defects, 5 small deletions, and 1 small duplication. Recurrent variants were the known c.2957A>T (six families: two European, three North African, and one unknown origin) and the novel c.2893‐7G>A p.(?) (3 families: two European, one Turkish origin).
Table 2.
Clinical and genetic characteristics of patients carrying novel likely disease‐associated CNGB1 variants included in the study
| ID | Geographic origin | CNGB1 mutations (NM_001297.5) | Cosegregation | Symptoms at onset | Age at diagnosis (years) | Age at visit (years) | BCVA (Snellen) | ff‐ERG | Fundus | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OD | OS | OD | OS | |||||||||
| F5240 | European | c.2575G>A,p.(Val859Ile) | c.2662G>A p.(Ala888Thr) | Unaffected mother (CIC09167) hetc.2575G>A; unaffected father(CIC09168) hetc.2662G>A | Night blindness during infancy | 12 | 14 | 20/50 | 20/50 | NA | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| CIC09166 | ||||||||||||
| OPH2286 | European | c.1917G>A p.(Trp639*) | c.2492+2T>G p.(?) | NA | Night blindness during early infancy | 16 | 18 | 20/32 | 20/32 | Undetectable scotopic responses, residual photopic responses | Waxy disk and narrow vessels. Few bone spicules along the retinal vessels in mid‐periphery | |
| SRP541 | European | c.2867del p.(Ile956Thrfs*15) | c.2957A>T p.(Asn986Ile) | NA | Night blindness during infancy | 29 | 29 | 20/25 | 20/20 | NA | Waxy disk, narrow vessels, central atrophy, few mid‐peripheral bone spicules | |
| ARRP278 | European(?) | c.413‐1G>A p.(?) | c.413‐1G>A p.(?) | NA | Visual field constriction | 27 | 30 | 20/32 | 20/25 | Undetectable | Waxy disk, narrow vessels, mid‐peripheral bone spicules | |
| TW8999826# | Asian |
c.217+5G>C, p.(?) |
c.2903T>G, p.(Met968Arg) |
NA | Night blindness since the age of 18 | NA | 30 | 20/20 | 20/20 | Undetectable | Waxy disk, narrow vessels, mid‐peripheral bone spicules | |
| TW20024045# | Asian |
c.217+5G>C, p.(?) |
c.2903T>G, p.(Met968Arg) |
NA | Night blindness childhood | NA | 31 | 20/20 | 20/20 | Undetectable scotopic responses, residual photopic responses | Waxy disk, narrow vessels, mid‐peripheral bone spicules | |
| SRP995 | European | c.2867del p.(Ile956Thrfs*15) |
c.2893‐7G>A p.(?) |
NA | Night blindness during infancy | 27 | 31 | 20/25 | 20/20 | Undetectable scotopic responses, residual photopic responses | Waxy disk, narrow vessels, mid‐peripheral bone spicules | |
| MRN:6822243 | Hispanic/Caucasian |
c.159+5G>A p.? |
c.1941C>A p.(Ser647Arg) | NA | Night blindness during infancy, photophobia, decreased peripheral vision | 31 | 34 | 20/32 | 20/32 | Scotopic rod specific ERG b‐wave amplitudes were extinguished in both eyes | Bony spicules located on the outer equator and there is slightly enlarged cupping in the left eye | |
| OPH3784 | North Africa | c.2320G>A p.(Glu774Lys) | c.2957A>T p.(Asn986Ile) | NA | Night blindness during early infancy | 35 | 36 | 20/20 | 20/25 | Undetectable scotopic responses, residual photopic responses | Normal optic nerve head. Some vessel narrowing. Few bone spicules in mid peripheral. Hypopigmented retina | |
| ARRP400 | Western Asia? | c.2555C>T p.(Pro852Leu) | c.2555C>T p.(Pro852Leu) | NA | Night blindness during infancy | 34 | 36 | 20/20 | 20/20 | Some residual scotopic responses, reduced photopic responses | Waxy disk, narrow vessels, ERM, few mid‐peripheral bone spicules | |
|
F4300 |
North Africa | c.2957A>T p.(Asn986Ile) | c.2957A>T p.(Asn986Ile) | NA | 39 | 20/25 | 20/32 | NA | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |||
| SRP266 | Unknown |
c.2794+1G>A p.(?) |
c.2957A>T p.(Asn986Ile) | NA | Night blindness at the age of 20 | 20 | 41 | 20/20 | 20/20 | Some residual scotopic responses, reduced photopic responses | Mid‐peripheral bone spicules and atrophy | |
| ARRP349 | Western Asia | c.2603G>A p.(Gly868Asp) | c.2603G>A p.(Gly868Asp) | NA | Night blindness during infancy | 10 | 41 | 20/80 | 20/100 | Undetectable | Waxy disk, narrow vessels, mid‐peripheral bone spicules | |
| F141 CIC00189 | European | c.832G>T p.(Glu278*) | c.2805del p.(Glu935Aspfs*2) | NA | Night blindness during infancy | 32 | 43 | 20/320 | 20/250 | Undetectable | Waxy optic disc with peripapillary atrophy, vessels narrowing, peripheral atrophy with bone spicules and central atrophy with RPE mottling | |
| MEH28189 | European | c.346C>T p.(Gln116*) | c.3139_3142dup p.(Ala1048Glyfs*13) | NA | Night blindness from age 7 | 27 | 44 | 20/30 | 20/40 | NA | Peripheral bone spicule pigmentation and outer retinal atrophy | |
| F1107 CIC01530 | European |
c.1644‐3C>G p.? |
c.1644‐3C>G p.? |
Unaffected father (CIC03328) hetc.1644‐3C>G; unaffected mother (CIC03508) hetc.1644‐3C>G | Night blindness during infancy | 19 | 45 | 20/63 | 20/32 | Undetectable | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
|
F3791 |
North Africa | c.2957A>T p.(Asn986Ile) | c.2957A>T p.(Asn986Ile) | NA | Night blindness during infancy | 39 | 45 | 20/20 | 20/20 | Undetectable scotopic responses, residual photopic responses | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| ARRP386 | Western Asia(?) |
c.2893‐7G>A p.(?) |
c.2893‐7G>A p.(?) |
NA | Night blindness during infancy | 23 | 49 | 20/20 | 20/20 | Undetectable | Waxy disk, narrow vessels, ERM, mid‐peripheral bone spicules | |
| ARRP398 | Western Asia? |
c.2893‐7G>A p.(?) |
c.2893‐7G>A p.(?) |
NA | Night blindness since the age of 15–16 | 19 | 49 | 20/20 | 20/20 | Undetectable | Waxy disk, narrow vessels, ERM, mid‐peripheral bone spicules | |
| OPH1710 | European | c.2185C>T p.(Arg729*) | c.2957A>T p.(Asn986Ile) | NA | Night blindness during teen‐age | NA | 50 | 20/25 | 20/20 | Undetectable scotopic responses, residual photopic responses | Normal optic nerve head. Some vessel narrowing. Few bone spicules in nasal and inferior retina. Hypopigmented retina | |
| MRN:8759303 | European |
c.3131_3149del, p.(Ala1044Glyfs*13) |
c.2166+6T>G, p.(?) |
NA | Night blindness and peripheral vision loss since childhood | NA | 50 | 20/100 | 20/32 | NA | Waxy disk, narrow vessels, mid‐peripheral bone spicules, macular hole | Waxy disk, narrow vessels, mid‐peripheral bone spicules, ERM |
| F463 CIC00691* | European | c.1319G>A p.(Trp440*) |
c.2305‐2A>G p.? |
Unaffected sister (CIC00732) het c.1319G>A; unaffected sister (CIC00692) het c.2305‐2A>G |
NA | NA | 51 | 20/125 | 20/50 | Undetectable | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| F463 CIC02695* | European | c.1319G>A p.(Trp440*) |
c.2305‐2A>G p.? |
Unaffected sister (CIC00732) het c.1319G>A; unaffected sister (CIC00692) het c.2305‐2A>G |
NA | NA | 51 | 20/50 | 20/20 | Undetectable | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| F2070 CIC04317 | European | c.1219dup p.Glu407Glyfs*12 |
c.1957+2T>G p.? |
NA | Night blindness during infancy | 45 | 51 | 20/20 | 20/32 | Undetectable scotopic responses, residual photopic responses | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
|
F5462 |
North Africa | c.2284C>Tp.(Arg762Cys) | c.2284C>Tp.(Arg762Cys) | NA | Night blindness during infancy | NA | 52 | 20/125 | 20/800 | Undetectable | Waxy optic disc, vessels narrowing, peripheral and central atrophy; bone spicules in the periphery | |
| F4053 CIC07355 | European |
c.2305‐2A>G p.? |
c.2305‐2A>G p.? |
NA | Night blindness during infancy | 45 | 55 | 20/25 | 20/20 | Undetectable scotopic responses, residual photopic responses | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| F5830 CIC10130 | European | c.939G>A p.(Trp313*) | c.939G>A p.(Trp313*) | NA | Night blindness during infancy | 22 | 56 | 20/32 | 20/32 | Undetectable scotopic responses, residual photopic responses | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| ARRP396 | Western Asia(?) | c.315G>A p.(Trp105*) | c.315G>A p.(Trp105*) | NA | Night blindness during infancy | NA | 58 | 20/40 | 20/63 | Undetectable | Pallor of the optic disk, narrow vessels, diffuse RPE atrophy with sparing of the macula, peripheral bone spicules | |
|
F3038 |
North Africa | c.2762_2765del p.(Tyr921Cysfs*15) | c.2762_2765del p.(Tyr921Cysfs*15) | NA | 64 | Hands Motion | 20/160 | Undetectable | Waxy optic disc, vessels narrowing, peripheral and central atrophy; bone spicules in the periphery | |||
| MRN:1203567687 | Unknown | c.2353A>T, p.(Lys785*) | c.2353A>T, p.(Lys785*) | NA | Night blindness during infancy | 58 | 72 | No light perception | No light perception | NA | Diffuse chorioretinal atrophy with peripheral pigmentation | |
| MEH16550 | Unknown | c.1613G>A, p.(Trp538*) | c.1613G>A, p.(Trp538*) | NA | Night blindness during infancy | NA | 73 | 20/30 | No light perception | NA | Waxy disk, narrow vessels, mid‐peripheral bone spicules | Impossible to perform |
|
F4517 |
European | c.3150del p.(Phe1051Leufs*12) | c.217+5G>A p? | NA | Night blindness during infancy | 20 | 77 | 20/100 | 20/125 | Undetectable | Waxy optic disc, vessels narrowing, peripheral atrophy with bone spicules | |
| SRP480 | Unknown | c.2957A>T p.(Asn986Ile) | c.2501G>T p.(Arg834Leu) | NA | ||||||||
| SRP629 | Unknown | c.2966T>A p.(Val989Glu) | c.2966T>A p.(Val989Glu) | NA | ||||||||
Note: * and #: Patients are siblings.
Abbreviations: BCVA, best‐corrected visual acuity; ERM, epiretinal membrane; ff‐ERG, full‐field electroretinogram; het, heterozygous; NA, not available; OD, right eye; OS, left eye; RPE, retinal pigment epithelium.
Among the cohort, we identified 22 novel variants of which, according to the ACMG classification criteria, 12 were pathogenic or likely pathogenic while 10 were VUS (Table 1). The latter included six missense variants and four noncanonical splice variants. All missense variants were predicted as “deleterious” by at least two prediction algorithms; they exhibit very low frequency in the general population and the corresponding amino acids are highly conserved across different species (Tables S1 and S2). Furthermore, five of them are located in the channel domain, at the level of the mutational “hot spot” previously mentioned: variants c.2320G>A p.(Glu774Lys), c.2501G>T p.(Arg834Leu), c.2555C>T p.(Pro852Leu), c.2575G>A p.(Val859Ile), and c.2662G>A p.(Ala888Thr) are in the transmembrane regions of the protein and very close to the channel pore. Variant c.1941C>A p.(Ser647Arg) is located between the calmodulin‐binding site and the transmembrane domains. A change in polarity given from the substitution of the neutral serine with an arginine (positively charged) might have an important impact on the activity and/or the structure of this region. However, further investigations are warranted to confirm the pathogenicity of these variants. The four noncanonical splice variants classified as VUS were c.159+5G>A, c.1644‐3C>G, c.2166+6T>G, and c.2893‐7G>A. All four variants have very low frequency in the general population, they were predicted as pathogenic by in silico analysis and the corresponding nucleotides were highly conserved among species (Tables S2–S4). Variants c.159+5G>A and c.2166+6T>G are predicted to shift the closest donor sites from their physiologic positions to c.159+7G and c.2166+12G, respectively. On the other hand, variants c.1644‐3C>G and c.2893‐7G>A are predicted to shift the closest physiologic acceptor sites in positions c.1644‐2A and c.2893‐5G, respectively. While this is highly suggestive for a pathogenic effect of these variants, further functional tests are needed for confirmation.
3. PREVALENCE DATA
The screening of the cohorts of IRDs from the different centers involved in this study revealed that biallelic variants of CNGB1 would account for a prevalence ranging between 0.42% and 0.78%. In particular, a prevalence of 0.78% was found at the Centre Hospitalier National d'Ophtalmologie des Quinze‐Vingts in Paris, France (12 index subjects out of 1533 screened); a prevalence of 0.42% was found at the University Eye Hospital in Tübingen, Germany (9 index subjects out of 2156 screened); a prevalence of 0.46% was found at the Lille University Hospital in France (3 index subjects out of 649 screened); finally a prevalence of 0.63% was found at Moorfields Eye Hospital, London, UK (20 index subjects out of 3195 screened). These numbers are comparable with previous studies that reported a prevalence ranging from ∼0.4% to 1% among IRDs (Carss et al., 2017; Consugar et al., 2015; Habibi et al., 2016; Haer‐Wigman et al., 2017; Oishi et al., 2014; Pontikos et al., 2020).
4. CLINICAL CHARACTERISTICS OF PATIENTS CARRYING CNGB1 VARIANTS
There are limited published data on the CNGB1 retinal phenotype, including all imaging modalities and functional assessment (Ba‐Abbad et al., 2019; Bareil et al., 2001; Fradin et al., 2016; Hull et al., 2017). The CNGB1‐related IRD phenotype is usually a young adult onset disease with slow progression and preserved visual acuity through late adulthood (Hull et al., 2017). However, most of the patients report the occurrence of night vision disturbances since childhood or infancy. As previously mentioned, only one case of CNGB1‐related isolated rod dysfunction (as evaluated on ff‐ERGs) has been described (Ba‐Abbad et al., 2019). This patient had slightly prolonged adaptation to dim lighting conditions and no retinal alterations except for a mild mid‐peripheral retinal pallor. To the best of our knowledge, our cohort is currently the largest cohort with CNGB1‐related RP ever reported (Tables 2 and S5).
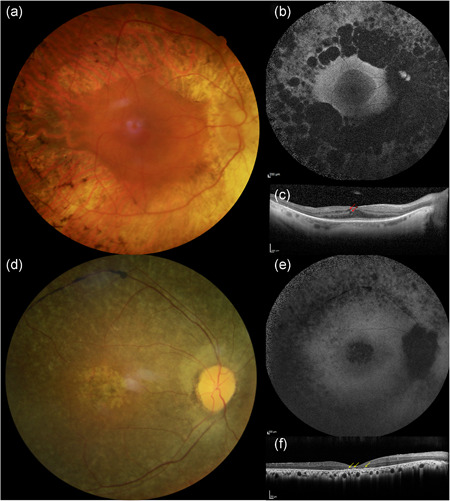
The 34 patients included had a mean age at the time of examination of 45.16 ± 14.67 years (median: 45 years; range 14–77 years; available data for 32 patients [n]). Age of diagnosis ranged between 10 and 58 (mean: 28.09 ± 11.81 years, median: 27; n: 21). All subjects experienced the onset of symptoms (primarily night blindness) during infancy or childhood. Visual acuity ranged from no light perception to 20/20 Snellen; however, most of the patients retained a visual acuity ≥20/40 Snellen in at least one eye (24/32 patients, 75%). Color vision was abnormal in at least one eye of 9/17 patients (52.94%), with 17 eyes showing dyschromatopsia, of which 9 with tritan (5 patients), 6 deutan (3 patient), and 2 tetartan (1 patient) defects. Visual fields were constricted in all patients, with a majority reduced to the 40 central degrees (vertical and horizontal diameters) or below in both eyes (V4 or III4 isopters; 13/20 patients, 65%). All patients had undetectable scotopic responses on ff‐ERGs. However, some residual cone responses were detectable in some of them (11 patients, 44%, n: 25), even in adult age. All subjects presented a classic form of RP with waxy optic disc pallor, attenuated retinal vessels, and peripheral bone spicules at fundus examination (Figure 2). The SW‐FAF showed the presence of a preserved central area of autofluorescence surrounded by a ring of hyperautofluorescence that clearly demarked the advancement of the peripheral outer retinal atrophy (Figures 2 and 3). This sign may represent an abnormal perifoveal accumulation of lipofuscin in the RPE due to increased outer segment dysgenesis (Lima et al., 2009; Robson et al., 2006) or a window defect caused by the atrophic alterations of the photoreceptors above a still intact RPE (Boulanger‐Scemama et al., 2019; Khateb, Nassisi, et al., 2019). In a recent study employing quantitative fundus autofluorescence, it was shown that SW‐AF in the ring reflects an actual increase in AF intensity within the ring relative to corresponding areas in the healthy retina. This finding is indicative of increased fluorophore production in impaired photoreceptor cells within the ring (Schuerch et al., 2017). This sign is usually associated with a preserved central vision (Khateb, Mohand‐Saïd, et al., 2019; Khateb, Nassisi, et al., 2019; Lima et al., 2012; Robson et al., 2003). Abnormal parafoveal rings of increased SW‐AF have been reported in approximately 59% of patients with RP (Murakami et al., 2008) although different genetic causes may have different prevalence (e.g., ~63% PDE6A, ~79% PDE6B, ~68% MYO7A (Khateb, Mohand‐Saïd, et al., 2019; Khateb, Nassisi, et al., 2019)). In our cohort, only 6/29 subjects did not show this para‐foveal feature (20.68%). All these cases showed an advanced atrophic involvement of all the posterior poles and very poor BCVA. Only in one case (F141‐CIC00189), there was evidence of central hypoautofluorescence with foveal involvement (BCVA: 20/320 and 20/250 Snellen in the right and left eye, respectively), even though the visual field was still well preserved (Figure 3). Consistently, this patient had central outer retinal thinning/atrophy on OCT unlike those with the normal foveal autofluorescence (Figure 3). A total of 10/29 patients (34.48%) had an epiretinal membrane (ERM) evident on OCT, although in most of the cases, it did not involve the center. Previous studies reported a prevalence of ERM that ranged between 0.6% and 64% in RP, depending on the OCT used and above all, not considering the genetic causes (Hagiwara et al., 2011; Khateb, Mohand‐Saïd, et al., 2019; Liew et al., 2019; Testa et al., 2014; Triolo et al., 2013). In our cohort, the prevalence of ERM is slightly higher than the 20% (2/10) previously reported by Hull et al. (2017) in CNGB1‐related RP. Other common OCT features were intraretinal hyperreflective foci (IHF) and intraretinal cysts (Figure 3). IHF may represent outer segment debris, RPE cell migration and/or phagocytes (Ho et al., 2011; Kuroda et al., 2014; Nagasaka et al., 2018). However, they are not specific to CNGB1‐related RP (Chen et al., 2016; Piri et al., 2015; Romano et al., 2019), neither of IRDs (Bolz et al., 2009; Christenbury et al., 2013; Matet et al., 2018; Nassisi et al., 2018; Ogino et al., 2012). In RCD, Kuroda et al. (2014) suggested that these hyper‐reflective dots could be associated with disease progression. However, the genetic analysis was not available in this study, therefore, the occurrence of IHF in correlation with specific gene defects in IRD remains to be established. Finally, intraretinal cysts were present bilaterally in 5/29 cases (17.24%), all of them without foveal involvement. This data is inferior to the previously reported prevalence of cystoid macular edema in RCD, which ranged among 26.9% and 58.6% (Adackapara et al., 2008; Hajali et al., 2008; Liew et al., 2019) but slightly higher than the 10% (1/10) reported in CNGB1 patients (Hull et al., 2017). As the shorter isoform CNGB1b transcribed from the CNGB1 locus is involved in the olfactory signal transduction, recent reports highlighted the presence of an impaired sense of smell in patients carrying biallelic CNGB1 variants, in particular in the channel domain (as the GARP domain is not expressed in the olfactory cells) (Afshar et al., 2019; Charbel Issa et al., 2018). Given the retrospective nature of our study, we did not have any data regarding the sense of smell in our cohort, neither subjective nor objective. Overall, our study is consistent with the previous findings that suggest that CNGB1‐related RP has a good prognosis for central vision despite the early onset of night blindness. Even though the present cohort is the largest CNGB1‐related retinopathy cohort reported so far in the literature, we were not able to draw any specific conclusion on phenotype/genotype correlations, most likely due to the diversity of the sequence variant spectrum in a yet limited number of patients. However, coherently with previous reports, we did not see any direct correlation between different variants and phenotypic severity. For example, patients ARRP400 and F3791‐CIC06919, who harbor missense variants, had well preserved central vision and visual field into their third and fourth decades; however, SRP995, who also carries missense variants, showed an early onset disease with a significant reduction of BCVA and visual field. Conversely, among the four patients with relatively preserved visual fields (≥120° of field) in their 30s and 40s, there were two subjects (F141‐CIC00189 and SRP995) with null variants on both alleles.
Figure 3.
Clinical phenotype of a CNGB1‐related retinitis pigmentosa patient (F3791‐CIC06919). Fundus photographs of the right (a) and left (d) eyes show a waxy optic disc, narrow vessels and peripheral bone spicules. On short‐wavelength fundus autofluorescence (b, e), the central area of preserved tissue is surrounded by a ring of increased autofluorescence that demarks the limits of the peripheral atrophy. On optical coherence tomography (c, e) all retinal layers look centrally well preserved, while peripherally a thinning of the outer layers is evident
Figure 4.
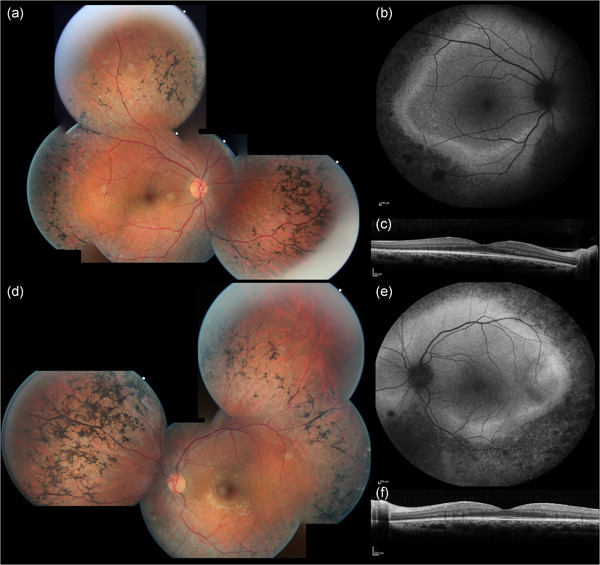
Clinical phenotype of two CNGB1‐related retinitis pigmentosa patients. Patient F1107‐CIC01530 (a) shows widespread peripheral RPE changes with bone spicules on fundus photographs (top row). These changes (black on the SW‐FAF [middle row]) surround an area with three different types of autofluorescence: a central normal autofluorescence (area of preserved tissue), a thin ring of intense hyperautofluorescence, and an outer ring of relatively preserved autofluorescence (outer retinal atrophy). These three areas are easy to recognize on OCT (bottom row) where there is also evidence of small intraretinal cysts (red arrows) and a small ERM. Patient F141‐CIC00189 (b) shows a central area of RPE and outer retina atrophy (top row), also evident on the SW‐FAF, which is surrounded by a preserved area of autofluorescence. In this patient, the demarcation between preserved and diseased tissue is not well demarked. OCT (bottom row) confirms the central atrophy and shows also the presence of intraretinal hyperreflective foci (yellow arrows) as well as an adherent ERM. ERM, epiretinal membrane; OCT, optical coherence tomography; RPE, retinal pigment epithelium; SW‐FAF, short‐wavelength fundus autofluorescence
5. CONCLUSIONS AND FUTURE DIRECTIONS
Our work provides a comprehensive overview of the sequence variant spectrum of CNGB1‐linked RP in the largest cohort reported to date, including 22 novel identified variants and sets the basis for the recruitment of patients in future therapeutic trials of gene augmentation or cell therapies. Our study also provides a complete phenotypic characterization of a relatively large cohort of subjects affected by this form of RP. However, natural history studies with longitudinal follow‐up are warranted to better define prognostic factors and window of intervention.
WEB RESOURCES
RETNET: https://sph.uth.edu/retnet/
HGMD® Professional 2017.4: http://www.hgmd.cf.ac.uk/ac/index.php
Leiden Open Variation Database (LOVD V.3.0): https://www.lovd.nl/
ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
Mutation Taster: http://www.mutationtaster.org/
Sorting Intolerant From Tolerant SIFT: https://sift.bii.a-star.edu.sg/
Polymorphism Phenotyping v2 Polyphen‐2: http://genetics.bwh.harvard.edu/pph2/
Splice Site Prediction by Neural Network, NNSPLICE: https://www.fruitfly.org/seq_tools/splice.html
Human Splicing Finder v.2.4.1, HSF: http://www.umd.be/HSF/
HomoloGene: https://www.ncbi.nlm.nih.gov/homologene
UCSC Genome Browser: https://genome.ucsc.edu/index.html).
ANNOVAR: http://annovar.openbioinformatics.org/en/latest/
Genome Aggregation Database (gnomAD): https://gnomad.broadinstitute.org/
UniProt Align tool: https://www.uniprot.org/align/.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors thank the families in the study and the clinical staff from the different centers for collecting phenotypic data. DNA samples from the Parisian group were obtained from the NeuroSensCol DNA bank, for research in neuroscience (PI: JA Sahel, co‐PI I Audo, partner with CHNO des Quinze‐Vingts, Inserm and CNRS, certified NFS96‐900). National Institute of Health (NIH)‐National Eye Institute (NEI), R24EY027285; LABEX LIFESENSES (reference ANR‐10‐LABX‐65) supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d'Avenir program (ANR‐11‐IDEX‐0004‐0); IHU FOReSIGHT (ANR‐18‐IAHU‐0001) supported by French state funds managed by the Agence Nationale de la Recherche within the Investissements d'Avenir program; Foundation Fighting Blindness center grant (C‐CMM‐0907‐0428‐INSERM04) and fellowship award (MN) (CD‐CL‐0619‐0759‐INSERM). Supported by a grant from the National Institute for Health Research Biomedical Research Centre at Moorfields Eye Hospital National Health Service Foundation Trust and UCL Institute of Ophthalmology; OAM is funded by Wellcome Trust grant 206619/Z/17/Z; Research to Prevent Blindness supported some of this work at the University of Florida.
Nassisi, M. , Smirnov, V. , Hernandez, C. S. , Mohand‐Saïd, S. , Condroyer, C. , Antonio, A. , Kühlewein, L. , Kempf, M. , Kohl, S. , Wissinger, B. , Nasser, F. , Ragi, S. D. , Wang, N. , Sparrow, J. R. , Greenstein, V. , Michalakis, S. , Mahroo, O. , Ba‐Abbad, R. , Michaelides, M. , … Audo, I. (2021). CNGB1‐related rod‐cone dystrophy: A mutation review and update. Human Mutation, 42, 641–666. 10.1002/humu.24205
Christina Zeitz and Isabelle Audo contributed equally to this study.
Contributor Information
Christina Zeitz, Email: christina.zeitz@inserm.fr.
Isabelle Audo, Email: isabelle.audo@inserm.fr.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study will be openly available in LOVD at https://www.lovd.nl/ after the acceptance of the study.
REFERENCES
- Abu‐Safieh, L. , Alrashed, M. , Anazi, S. , Alkuraya, H. , Khan, A. O. , Al‐Owain, M. , Al‐Zahrani, J. , Al‐Abdi, L. , Hashem, M. , Al‐Tarimi, S. , Sebai, M. ‐A. , Shamia, A. , Ray‐Zack, M. D. , Nassan, M. , Al‐Hassnan, Z. N. , Rahbeeni, Z. , Waheeb, S. , Alkharashi, A. , Abboud, E. , & Alkuraya, F. S. (2013). Autozygome‐guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Research, 23, 236–247. 10.1101/gr.144105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adackapara, C. A. , Sunness, J. S. , Dibernardo, C. W. , Melia, B. M. , & Dagnelie, G. (2008). Prevalence of cystoid macular edema and stability in oct retinal thickness in eyes with retinitis pigmentosa during a 48‐week lutein trial. Retina, 28(1), 103–110. 10.1097/IAE.0b013e31809862aa [DOI] [PubMed] [Google Scholar]
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar, F. , Arno, G. , Ba‐Abbad, R. , Esposti, S. D. , Michaelides, M. , Webster, A. R. , & Mahroo, O. A. (2019). Awareness of olfactory impairment in a cohort of patients with CNGB1‐associated retinitis pigmentosa. Eye, 34(4), 783–784. 10.1038/s41433-019-0609-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alshamrani, A. A. , Raddadi, O. , Schatz, P. , Lenzner, S. , Neuhaus, C. , Azzam, E. , & Abdelkader, E. (2020). Severe retinitis pigmentosa phenotype associated with novel CNGB1 variants. American Journal of Ophthalmology Case Reports, 19, 100780. 10.1016/j.ajoc.2020.100780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardell, M. D. , Bedsole, D. L. , Schoborg, R. V. , & Pittler, S. J. (2000). Genomic organization of the human rod photoreceptor cGMP‐gated cation channel beta‐subunit gene. Gene, 245(2), 311–318. 10.1016/s0378-1119(00)00023-8 [DOI] [PubMed] [Google Scholar]
- Audo, I. , Bujakowska, K. M. , Léveillard, T. , Mohand‐Saïd, S. , Lancelot, M.‐E. , Germain, A. , Antonio, A. , Michiels, C. , Saraiva, J. P. , Letexier, M. , Sahel, J. A. , Bhattacharya, S. S. , & Zeitz, C. (2012). Development and application of a next‐generation‐sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet Journal of Rare Diseases, 7, 8. 10.1186/1750-1172-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam, M. , Collin, R. W. J. , Malik, A. , Khan, M. I. , Shah, S. T. A. , Shah, A. A. , Hussain, A. , Sadeque, A. , Arimadyo, K. , Ajmal, M. , Azam, A. , Qureshi, N. , Bokhari, H. , Strom, T. M. , Cremers, F. P. M. , & Qamar, R. (2011). Identification of novel mutations in Pakistani families with autosomal recessive retinitis pigmentosa. Archives of Ophthalmology, 129, 1377–1378. 10.1001/archophthalmol.2011.290 [DOI] [PubMed] [Google Scholar]
- Ba‐Abbad, R. , Holder, G. E. , Robson, A. G. , Neveu, M. M. , Waseem, N. , Arno, G. , & Webster, A. R. (2019). Isolated rod dysfunction associated with a novel genotype of CNGB1. American Journal of Ophthalmology Case Reports, 14, 83–86. 10.1016/j.ajoc.2019.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge, J. W. B. , Mehat, M. S. , Sundaram, V. , Robbie, S. J. , Barker, S. E. , Ripamonti, C. , Georgiadis, A. , Mowat, F. M. , Beattie, S. G. , Gardner, P. J. , Feathers, K. L. , Luong, V. A. , Yzer, S. , Balaggan, K. , Viswanathan, A. , de Ravel, T. J. L. , Casteels, I. , Holder, G. E. , Tyler, N. , … Ali, R. R. (2015). Long‐term effect of gene therapy on Leber's congenital amaurosis. The New England Journal of Medicine, 372(20), 1887–1897. 10.1056/NEJMoa1414221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge, J. W. B. , Smith, A. J. , Barker, S. S. , Robbie, S. , Henderson, R. , Balaggan, K. , Viswanathan, A. , Holder, G. E. , Stockman, A. , Tyler, N. , Petersen‐Jones, S. , Bhattacharya, S. S. , Thrasher, A. J. , Fitzke, F. W. , Carter, B. J. , Rubin, G. S. , Moore, A. T. , & Ali, R. R. (2008). Effect of gene therapy on visual function in Leber's congenital amaurosis. The New England Journal of Medicine, 358(21), 2231–2239. 10.1056/NEJMoa0802268 [DOI] [PubMed] [Google Scholar]
- Banerjee, S. , Yao, J. , Zhang, X. , Niu, J. , & Chen, Z. (2017). Next generation sequencing identified novel heterozygous nonsense mutation in CNGB1 gene associated with retinitis pigmentosa in a Chinese patient. Oncotarget, 8, 88345–88350. 10.18632/oncotarget.21728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareil, C. , Hamel, C. P. , Delague, V. , Arnaud, B. , Demaille, J. , & Claustres, M. (2001). Segregation of a mutation in CNGB1 encoding the beta‐subunit of the rod cGMP‐gated channel in a family with autosomal recessive retinitis pigmentosa. Human Genetics, 108(4), 328–334. 10.1007/s004390100496 [DOI] [PubMed] [Google Scholar]
- Becirovic, E. , Nakova, K. , Hammelmann, V. , Hennel, R. , Biel, M. , & Michalakis, S. (2010). The retinitis pigmentosa mutation c.3444+1G>A in CNGB1 results in skipping of exon 32. PLoS ONE, 5, e8969. 10.1371/journal.pone.0008969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardis, I. , Chiesi, L. , Tenedini, E. , Artuso, L. , Percesepe, A. , Artusi, V. , Simone, M. L. , Manfredini, R. , Camparini, M. , Rinaldi, C. , Ciardella, A. , Graziano, C. , Balducci, N. , Tranchina, A. , Cavallini, G. M. , Pietrangelo, A. , Marigo, V. , & Tagliafico, E. (2016). Unravelling the complexity of inherited retinal dystrophies molecular testing: Added value of targeted next‐generation sequencing. BioMed Research International, 2016, 6341870. 10.1155/2016/6341870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel, M. , Wahl‐Schott, C. , Michalakis, S. , & Zong, X. (2009). Hyperpolarization‐activated cation channels: From genes to function. Physiological Reviews, 89(3), 847–885. 10.1152/physrev.00029.2008 [DOI] [PubMed] [Google Scholar]
- Bolz, M. , Schmidt‐Erfurth, U. , Deak, G. , Mylonas, G. , Kriechbaum, K. , & Scholda, C. , Diabetic Retinopathy Research Group Vienna . (2009). Optical coherence tomographic hyperreflective foci: A morphologic sign of lipid extravasation in diabetic macular edema. Ophthalmology, 116(5), 914–920. 10.1016/j.ophtha.2008.12.039 [DOI] [PubMed] [Google Scholar]
- Boulanger‐Scemama, E. , Mohand‐Saïd, S. , El Shamieh, S. , Démontant, V. , Condroyer, C. , Antonio, A. , Michiels, C. , Boyard, F. , Saraiva, J. P. , Letexier, M. , Sahel, J. A. , Zeitz, C. , & Audo, I. (2019). Phenotype analysis of retinal dystrophies in light of the underlying genetic defects: Application to cone and cone‐rod dystrophies. International Journal of Molecular Sciences, 20(19), 4854. 10.3390/ijms20194854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger‐Scemama, E. , El Shamieh, S. , Démontant, V. , Condroyer, C. , Antonio, A. , Michiels, C. , Boyard, F. , Saraiva, J. P. , Letexier, M. , Souied, E. , Mohand‐Saïd, S. , Sahel, J. A. , Zeitz, C. , & Audo, I. (2015). Next‐generation sequencing applied to a large French cone and cone‐rod dystrophy cohort: Mutation spectrum and new genotype‐phenotype correlation. Orphanet Journal of Rare Diseases, 10, 85. 10.1186/s13023-015-0300-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carss, K. J. , Arno, G. , Erwood, M. , Stephens, J. , Sanchis‐Juan, A. , Hull, S. , Megy, K. , Grozeva, D. , Dewhurst, E. , Malka, S. , Plagnol, V. , Penkett, C. , Stirrups, K. , Rizzo, R. , Wright, G. , Josifova, D. , Bitner‐Glindzicz, M. , Scott, R. H. , Clement, E. , … Huissoon, A. (2017). Comprehensive rare variant analysis via whole‐genome sequencing to determine the molecular pathology of inherited retinal disease. American Journal of Human Genetics, 100(1), 75–90. 10.1016/j.ajhg.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro‐Miró, M. , Tonda, R. , Escudero‐Ferruz, P. , Andrés, R. , Mayor‐Lorenzo, A. , Castro, J. , Ciccioli, M. , Hidalgo, D. A. , Rodríguez‐Ezcurra, J. J. , Farrando, J. , Pérez‐Santonja, J. J. , Cormand, B. , Marfany, G. , & Gonzàlez‐Duarte, R. (2016). Novel candidate genes and a wide spectrum of structural and point mutations responsible for inherited retinal dystrophies revealed by exome sequencing. PLoS ONE, 11, e0168966. 10.1371/journal.pone.0168966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbel Issa, P. , Reuter, P. , Kühlewein, L. , Birtel, J. , Gliem, M. , Tropitzsch, A. , Whitcroft, K. L. , Bolz, H. J. , Ishihara, K. , MacLaren, R. E. , Downes, S. M. , Oishi, A. , Zrenner, E. , Kohl, S. , & Hummel, T. (2018). Olfactory dysfunction in patients with CNGB1‐associated retinitis pigmentosa. JAMA Ophthalmology, 136(7), 761–769. 10.1001/jamaophthalmol.2018.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, K. C. , Jung, J. J. , Curcio, C. A. , Balaratnasingam, C. , Gallego‐Pinazo, R. , Dolz‐Marco, R. , Freund, K. B. , & Yannuzzi, L. A. (2016). Intraretinal hyperreflective foci in acquired vitelliform lesions of the macula: Clinical and histologic study. American Journal of Ophthalmology, 164, 89–98. 10.1016/j.ajo.2016.02.002 [DOI] [PubMed] [Google Scholar]
- Christenbury, J. G. , Folgar, F. A. , O'Connell, R. V. , Chiu, S. J. , Farsiu, S. , & Toth, C. A. , Age‐related Eye Disease Study 2 Ancillary Spectral Domain Optical Coherence Tomography Study Group . (2013). Progression of intermediate age‐related macular degeneration with proliferation and inner retinal migration of hyperreflective foci. Ophthalmology, 120(5), 1038–1045. 10.1016/j.ophtha.2012.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan, A. V. , Jacobson, S. G. , Beltran, W. A. , Sumaroka, A. , Swider, M. , Iwabe, S. , Roman, A. J. , Olivares, M. B. , Schwartz, S. B. , Komaromy, A. M. , Hauswirth, W. W. , & Aguirre, G. D. (2013). Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proceedings of the National Academy of Sciences of the United States of America, 110(6), E517–E525. 10.1073/pnas.1218933110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan, A. V. , Jacobson, S. G. , Drack, A. V. , Ho, A. C. , Charng, J. , Garafalo, A. V. , Roman, A. J. , Sumaroka, A. , Han, I. C. , Hochstedler, M. D. , Pfeifer, W. L. , Sohn, E. H. , Taiel, M. , Schwartz, M. R. , Biasutto, P. , Wit, W. , Cheetham, M. E. , Adamson, P. , Rodman, D. M. , … Russell, S. R. (2019). Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nature Medicine, 25(2), 225–228. 10.1038/s41591-018-0295-0 [DOI] [PubMed] [Google Scholar]
- Comander, J. , Weigel‐DiFranco, C. , Maher, M. , Place, E. , Wan, A. , Harper, S. , Sandberg, M. A. , Navarro‐Gomez, D. , & Pierce, E. A. (2017). The genetic basis of pericentral retinitis pigmentosa‐A form of mild retinitis pigmentosa. Genes (Basel), 8. 10.3390/genes8100256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consugar, M. B. , Navarro‐Gomez, D. , Place, E. M. , Bujakowska, K. M. , Sousa, M. E. , Fonseca‐Kelly, Z. D. , Taub, D. G. , Janessian, M. , Wang, D. Y. , Au, E. D. , Sims, K. B. , Sweetser, D. A. , Fulton, A. B. , Liu, Q. , Wiggs, J. L. , Gai, X. , & Pierce, E. A. (2015). Panel‐based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in Medicine, 17(4), 253–261. 10.1038/gim.2014.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRamus, M. L. , Stacks, D. A. , Zhang, Y. , Huisingh, C. E. , McGwin, G. , & Pittler, S. J. (2017). GARP2 accelerates retinal degeneration in rod cGMP‐gated cation channel β‐subunit knockout mice. Scientific Reports, 7, 42545. 10.1038/srep42545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet, F.‐O. , Hamroun, D. , Lalande, M. , Collod‐Béroud, G. , Claustres, M. , & Béroud, C. (2009). Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Research, 37(9), e67. 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellingford, J. M. , Barton, S. , Bhaskar, S. , O'Sullivan, J. , Williams, S. G. , Lamb, J. A. , Panda, B. , Sergouniotis, P. I. , Gillespie, R. L. , Daiger, S. P. , Hall, G. , Gale, T. , Lloyd, I. C. , Bishop, P. N. , Ramsden, S. C. , & Black, G. C. M. (2016). Molecular findings from 537 individuals with inherited retinal disease. Journal of Medical Genetics, 53, 761–767. 10.1136/jmedgenet-2016-103837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradin, M. , Colin, E. , Hannouche‐Bared, D. , Audo, I. , Sahel, J. A. , Biskup, S. , Carré, W. , Ziegler, A. , Wilhelm, C. , Guichet, A. , Odent, S. , & Bonneau, D. (2016). Run of homozygosity analysis reveals a novel nonsense variant of the CNGB1 gene involved in retinitis pigmentosa 45. Ophthalmic Genetics, 37(3), 357–359. 10.3109/13816810.2015.1087578 [DOI] [PubMed] [Google Scholar]
- Fu, Q. , Wang, F. , Wang, H. , Xu, F. , Zaneveld, J. E. , Ren, H. , Keser, V. , Lopez, I. , Tuan, H. ‐F. , Salvo, J. S. , Wang, X. , Zhao, L. , Wang, K. , Li, Y. , Koenekoop, R. K. , Chen, R. , & Sui, R. (2013). Next‐generation sequencing‐based molecular diagnosis of a chinese patient cohort with autosomal recessive retinitis pigmentosa. Investigative Ophthalmology & Visual Science, 54, 4158–4166. 10.1167/iovs.13-11672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, Z. , Bowles, K. , Goetz, K. , Scholl, H. P. N. , Wang, F. , Wang, X. , Xu, S. , Wang, K. , Wang, H. , & Chen, R. (2015). NGS‐based Molecular diagnosis of 105 eyeGENE(®) probands with retinitis pigmentosa. Scientific Reports, 5, 18287. 10.1038/srep18287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glöckle, N. , Kohl, S. , Mohr, J. , Scheurenbrand, T. , Sprecher, A. , Weisschuh, N. , Bernd, A. , Rudolph, G. , Schubach, M. , Poloschek, C. , Zrenner, E. , Biskup, S. , Berger, W. , Wissinger, B. , & Neidhardt, J. (2014). Panel‐based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. European Journal of Human Genetics, 22(1), 99–104. 10.1038/ejhg.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi, I. , Chebil, A. , Falfoul, Y. , Allaman‐Pillet, N. , Kort, F. , Schorderet, D. F. , & El Matri, L. (2016). Identifying mutations in Tunisian families with retinal dystrophy. Scientific Reports, 6, 37455. 10.1038/srep37455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haer‐Wigman, L. , van Zelst‐Stams, W. A. , Pfundt, R. , van den Born, L. I. , Klaver, C. C. , Verheij, J. B. , Hoyng, C. B. , Breuning, M. H. , Boon, C. J. , Kievit, A. J. , Verhoeven, V. J. , Pott, J. W. , Sallevelt, S. C. , van Hagen, J. M. , Plomp, A. S. , Kroes, H. Y. , Lelieveld, S. H. , Hehir‐Kwa, J. Y. , Castelein, S. , … Yntema, H. G. (2017). Diagnostic exome sequencing in 266 Dutch patients with visual impairment. European Journal of Human Genetics, 25(5), 591–599. 10.1038/ejhg.2017.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara, A. , Yamamoto, S. , Ogata, K. , Sugawara, T. , Hiramatsu, A. , Shibata, M. , & Mitamura, Y. (2011). Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmologica, 89(2), e122–e125. 10.1111/j.1755-3768.2010.01866.x [DOI] [PubMed] [Google Scholar]
- Hajali, M. , Fishman, G. A. , & Anderson, R. J. (2008). The prevalence of cystoid macular oedema in retinitis pigmentosa patients determined by optical coherence tomography. The British Journal of Ophthalmology, 92(8), 1065–1068. 10.1136/bjo.2008.138560 [DOI] [PubMed] [Google Scholar]
- Hartong, D. T. , Berson, E. L. , & Dryja, T. P. (2006). Retinitis pigmentosa. Lancet, 368(9549), 1795–1809. 10.1016/S0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- Ho, J. , Witkin, A. J. , Liu, J. , Chen, Y. , Fujimoto, J. G. , Schuman, J. S. , & Duker, J. S. (2011). Documentation of intraretinal retinal pigment epithelium migration via high‐speed ultrahigh‐resolution optical coherence tomography. Ophthalmology, 118(4), 687–693. 10.1016/j.ophtha.2010.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull, S. , Attanasio, M. , Arno, G. , Carss, K. , Robson, A. G. , Thompson, D. A. , Plagnol, V. , Michaelides, M. , Holder, G. E. , Henderson, R. H. , Raymond, F. L. , Moore, A. T. , & Webster, A. R. (2017). Clinical characterization of CNGB1‐related autosomal recessive retinitis pigmentosa. JAMA Ophthalmology, 135(2), 137–144. 10.1001/jamaophthalmol.2016.5213 [DOI] [PubMed] [Google Scholar]
- Huttl, S. (2005). Impaired channel targeting and retinal degeneration in mice lacking the cyclic nucleotide‐gated channel subunit CNGB1. The Journal of Neuroscience, 25(1), 130–138. 10.1523/JNEUROSCI.3764-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupp, U. B. , & Seifert, R. (2002). Cyclic nucleotide‐gated ion channels. Physiological Reviews, 82(3), 769–824. 10.1152/physrev.00008.2002 [DOI] [PubMed] [Google Scholar]
- Kent, W. J. , Sugnet, C. W. , Furey, T. S. , Roskin, K. M. , Pringle, T. H. , Zahler, A. M. , & Haussler, D. (2002). The human genome browser at UCSC. Genome Research, 12(6), 996–1006. 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateb, S. , Mohand‐Saïd, S. , Nassisi, M. , Bonnet, C. , Roux, A.‐F. , Andrieu, C. , & Audo, I. (2019). Phenotypic characteristics of rod‐cone dystrophy associated with myo7a mutations in a large french cohort. Retina, 40(8), 1603–1615. 10.1097/IAE.0000000000002636 [DOI] [PubMed] [Google Scholar]
- Khateb, S. , Nassisi, M. , Bujakowska, K. M. , Méjécase, C. , Condroyer, C. , Antonio, A. , Foussard, M. , Démontant, V. , Mohand‐Saïd, S. , Sahel, J. A. , Zeitz, C. , & Audo, I. (2019). Longitudinal clinical follow‐up and genetic spectrum of patients with rod‐cone dystrophy associated with mutations in PDE6A and PDE6B. JAMA Ophthalmology, 137(6), 669–679. 10.1001/jamaophthalmol.2018.6367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch, S. , Sothilingam, V. , Garcia Garrido, M. , Tanimoto, N. , Becirovic, E. , Koch, F. , Seide, C. , Beck, S. C. , Seeliger, M. W. , Biel, M. , Muhlfriedel, R. , & Michalakis, S. (2012). Gene therapy restores vision and delays degeneration in the CNGB1(‐/‐) mouse model of retinitis pigmentosa. Human Molecular Genetics, 21(20), 4486–4496. 10.1093/hmg/dds290 [DOI] [PubMed] [Google Scholar]
- Kondo, H. , Qin, M. , Mizota, A. , Kondo, M. , Hayashi, H. , Hayashi, K. , Oshima, K. , Tahira, T. , & Hayashi, K. (2004). A homozygosity‐based search for mutations in patients with autosomal recessive retinitis pigmentosa, using microsatellite markers. Investigative Ophthalmology & Visual Science, 45, 4433–4439. 10.1167/iovs.04-0544 [DOI] [PubMed] [Google Scholar]
- Ko¨rschen, H. G. , Illing, M. , Seifer, R. , Sesti, F. , Williams, A. , Gotzes, S. , Colville, C. , Muller, F. , Dose´, A. , Godde, M. , Molday, L. , Kaupp, U. B. , & Molday, R. S. (1995). A 240 kDa protein represents the complete beta subunit of the cyclic nucleotide‐gated channel from rod photoreceptor. Neuron, 15(3), 627–636. 10.1016/0896-6273(95)90151-5 [DOI] [PubMed] [Google Scholar]
- Kuroda, M. , Hirami, Y. , Hata, M. , Mandai, M. , Takahashi, M. , & Kurimoto, Y. (2014). Intraretinal hyperreflective foci on spectral‐domain optical coherence tomographic images of patients with retinitis pigmentosa. Clinical Ophthalmology, 8, 435–440. 10.2147/OPTH.S58164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew, G. , Strong, S. , Bradley, P. , Severn, P. , Moore, A. T. , Webster, A. R. , Mitchell, P. , Kifley, A. , & Michaelides, M. (2019). Prevalence of cystoid macular oedema, epiretinal membrane and cataract in retinitis pigmentosa. The British Journal of Ophthalmology, 103(8), 1163–1166. 10.1136/bjophthalmol-2018-311964 [DOI] [PubMed] [Google Scholar]
- Lima, L. H. , Burke, T. , Greenstein, V. C. , Chou, C. L. , Cella, W. , Yannuzzi, L. A. , & Tsang, S. H. (2012). Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. American Journal of Ophthalmology, 153(4), 718–727.e2. 10.1016/j.ajo.2011.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima, L. H. , Cella, W. , Greenstein, V. C. , Wang, N.‐K. , Busuioc, M. , Smith, R. T. , YANNUZZI, L. A. , & TSANG, S. H. (2009). Structural assessment of hyperautofluorescent ring in patients with retinitis pigmentosa. Retina, 29(7), 1025–1031. 10.1097/IAE.0b013e3181ac2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingao, M. D. , Ganesh, A. , Karthikeyan, A. S. , Al Zuhaibi, S. , Al‐Hosni, A. , Al Khayat, A. , Capasso, J. , Trumler, A. A. , Stroh, E. , Al Shekaili, H. , Cater, J. R. , & Levin, A. V. (2016). Macular cystoid spaces in patients with retinal dystrophy. Ophthalmic Genetics, 37, 377–383. 10.3109/13816810.2015.1101775 [DOI] [PubMed] [Google Scholar]
- MacLaren, R. E. , Groppe, M. , Barnard, A. R. , Cottriall, C. L. , Tolmachova, T. , Seymour, L. , Clark, K. R. , During, M. J. , Cremers, F. P. M. , Black, G. C. M. , Lotery, A. J. , Downes, S. M. , Webster, A. R. , & Seabra, M. C. (2014). Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet, 383(9923), 1129–1137. 10.1016/S0140-6736(13)62117-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, A. , Maeda, T. , Golczak, M. , & Palczewski, K. (2008). Retinopathy in mice induced by disrupted all‐trans‐retinal clearance. The Journal of Biological Chemistry, 283(39), 26684–26693. 10.1074/jbc.M804505200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, A. , Yoshida, A. , Kawai, K. , Arai, Y. , Akiba, R. , Inaba, A. , Takagi, S. , Fujiki, R. , Hirami, Y. , Kurimoto, Y. , Ohara, O. , & Takahashi, M. (2018). Development of a molecular diagnostic test for retinitis pigmentosa in the japanese population. Japanese Journal of Ophthalmology, 62, 451–457. 10.1007/s10384-018-0601-x [DOI] [PubMed] [Google Scholar]
- Maguire, A. M. , Russell, S. , Wellman, J. A. , Chung, D. C. , Yu, Z.‐F. , Tillman, A. , Wittes, J. , Pappas, J. , Elci, O. , Marshall, K. A. , McCague, S. , Reichert, H. , Davis, M. , Simonelli, F. , Leroy, B. P. , Wright, J. F. , High, K. A. , & Bennett, J. (2019). Efficacy, safety, and durability of voretigene neparvovec‐rzyl in RPE65 mutation‐associated inherited retinal dystrophy: Results of phase 1 and 3 trials. Ophthalmology, 126(9), 1273–1285. 10.1016/j.ophtha.2019.06.017 [DOI] [PubMed] [Google Scholar]
- Maguire, A. M. , Simonelli, F. , Pierce, E. A. , Pugh, E. N. , Mingozzi, F. , Bennicelli, J. , Banfi, S. , Marshall, K. A. , Testa, F. , Surace, E. M. , Rossi, S. , Lyubarsky, A. , Arruda, V. R. , Konkle, B. , Stone, E. , Sun, J. , Jacobs, J. , Dell'Osso, L. , Hertle, R. , … Bennett, J. (2008). Safety and efficacy of gene transfer for Leber's congenital amaurosis. The New England Journal of Medicine, 358(21), 2240–2248. 10.1056/NEJMoa0802315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranhao, B. , Biswas, P. , Gottsch, A. D. H. , Navani, M. , Naeem, M. A. , Suk, J. , Chu, J. , Khan, S. N. , Poleman, R. , Akram, J. , Riazuddin, S. , Lee, P. , Riazuddin, S. A. , Hejtmancik, J. F. , & Ayyagari, R. (2015). Investigating the molecular basis of retinal degeneration in a familial cohort of pakistani decent by exome sequencing. PLOS ONE, 10, e0136561. 10.1371/journal.pone.0136561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria, M. , Ajmal, M. , Azam, M. , Waheed, N. K. , Siddiqui, S. N. , Mustafa, B. , Ayub, H. , Ali, L. , Ahmad, S. , Micheal, S. , Hussain, A. , Shah, S. T. A. , Ali, S. H. B. , Ahmed, W. , Khan, Y. M. , Hollander, A. I. d , Haer‐Wigman, L. , Collin, R. W. J. , Khan, M. I. , … Cremers, F. P. M. (2015). Homozygosity mapping and targeted sanger sequencing reveal genetic defects underlying inherited retinal disease in families from pakistan. PLOS ONE, 10, e0119806. 10.1371/journal.pone.0119806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matet, A. , Daruich, A. , Zola, M. , & Behar‐Cohen, F. (2018). Risk factors for recurrences of central serous chorioretinopathy. Retina, 38(7), 1403–1414. 10.1097/IAE.0000000000001729 [DOI] [PubMed] [Google Scholar]
- Michalakis, S. , Zong, X. , Becirovic, E. , Hammelmann, V. , Wein, T. , Wanner, K. T. , & Biel, M. (2011). The glutamic acid‐rich protein is a gating inhibitor of cyclic nucleotide‐gated channels. The Journal of Neuroscience, 31(1), 133–141. 10.1523/JNEUROSCI.4735-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, T. , Akimoto, M. , Ooto, S. , Suzuki, T. , Ikeda, H. , Kawagoe, N. , Takahashi, M. , & Yoshimura, N. (2008). Association between abnormal autofluorescence and photoreceptor disorganization in retinitis pigmentosa. American Journal of Ophthalmology, 145(4), 687–694. 10.1016/j.ajo.2007.11.018 [DOI] [PubMed] [Google Scholar]
- Nagasaka, Y. , Ito, Y. , Ueno, S. , & Terasaki, H. (2018). Number of hyperreflective foci in the outer retina correlates with inflammation and photoreceptor degeneration in retinitis pigmentosa. Ophthalmology Retina, 2(7), 726–734. 10.1016/j.oret.2017.07.020 [DOI] [PubMed] [Google Scholar]
- Nassisi, M. , Fan, W. , Shi, Y. , Lei, J. , Borrelli, E. , Ip, M. , & Sadda, S. R. (2018). Quantity of intraretinal hyperreflective foci in patients with intermediate age‐related macular degeneration correlates with 1‐year progression. Investigative Ophthalmology & Visual Science, 59(8), 3431–3439. 10.1167/iovs.18-24143 [DOI] [PubMed] [Google Scholar]
- Nishiguchi, K. M. , Tearle, R. G. , Liu, Y. P. , Oh, E. C. , Miyake, N. , Benaglio, P. , Harper, S. , Koskiniemi‐Kuendig, H. , Venturini, G. , Sharon, D. , Koenekoop, R. K. , Nakamura, M. , Kondo, M. , Ueno, S. , Yasuma, T. R. , Beckmann, J. S. , Ikegawa, S. , Matsumoto, N. , Terasaki, H. , & Rivoltaa, C. (2013). Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proceedings of the National Academy of Sciences of the United States of America, 110, 16139–16144. 10.1073/pnas.1308243110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino, K. , Murakami, T. , Tsujikawa, A. , Miyamoto, K. , Sakamoto, A. , Ota, M. , & Yoshimura, N. (2012). Characteristics of optical coherence tomographic hyperreflective foci in retinal vein occlusion. Retina, 32(1), 77–85. 10.1097/IAE.0b013e318217ffc7 [DOI] [PubMed] [Google Scholar]
- Oishi, M. , Oishi, A. , Gotoh, N. , Ogino, K. , Higasa, K. , Iida, K. , Makiyama, Y. , Morooka, S. , Matsuda, F. , & Yoshimura, N. (2014). Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and Usher syndrome patients by next‐generation sequencing. Investigative Ophthalmology & Visual Science, 55(11), 7369–7375. 10.1167/iovs.14-15458 [DOI] [PubMed] [Google Scholar]
- Patel, N. , Aldahmesh, M. A. , Alkuraya, H. , Anazi, S. , Alsharif, H. , Khan, A. O. , Sunker, A. , Al‐Mohsen, S. , Abboud, E. B. , Nowilaty, S. R. , Alowain, M. , Al‐Zaidan, H. , Al‐Saud, B. , Alasmari, A. , Abdel‐Salam, G. M. H. , Abouelhoda, M. , Abdulwahab, F. M. , Ibrahim, N. , Naim, E. , & Alkuraya, F. S. (2016). Expanding the clinical, allelic, and locus heterogeneity of retinal dystrophies. Genetics in Medicine, 18, 554–562. 10.1038/gim.2015.127 [DOI] [PubMed] [Google Scholar]
- Petersen‐Jones, S. M. , Occelli, L. M. , Winkler, P. A. , Lee, W. , Sparrow, J. R. , Tsukikawa, M. , Boye, S. L. , Chiodo, V. , Capasso, J. E. , Becirovic, E. , Schön, C. , Seeliger, M. W. , Levin, A. V. , Michalakis, S. , Hauswirth, W. W. , & Tsang, S. H. (2018). Patients and animal models of CNGβ1‐deficient retinitis pigmentosa support gene augmentation approach. The Journal of Clinical Investigation, 128(1), 190–206. 10.1172/JCI95161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piri, N. , Nesmith, B. L. W. , & Schaal, S. (2015). Choroidal hyperreflective foci in Stargardt disease shown by spectral‐domain optical coherence tomography imaging: Correlation with disease severity. JAMA Ophthalmology, 133(4), 398–405. 10.1001/jamaophthalmol.2014.5604 [DOI] [PubMed] [Google Scholar]
- Pontikos, N. , Arno, G. , Jurkute, N. , Schiff, E. , Ba‐Abbad, R. , Malka, S. , Gimenez, A. , Georgiou, M. , Wright, G. , Armengol, M. , Knight, H. , Katz, M. , Moosajee, M. , Yu‐Wai‐Man, P. , Moore, A. T. , Michaelides, M. , Webster, A. R. , & Mahroo, O. A. (2020). Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology, 127, 1384–1394. 10.1016/j.ophtha.2020.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, M. G. , Eeckman, F. H. , Kulp, D. , & Haussler, D. (1997). Improved splice site detection in Genie. Journal of Computational Biology, 4(3), 311–323. 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. , ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson, A. G. , El‐Amir, A. , Bailey, C. , Egan, C. A. , Fitzke, F. W. , Webster, A. R. , Bird, A. C. , & Holder, G. E. (2003). Pattern ERG correlates of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Investigative Ophthalmology & Visual Science, 44(8), 3544–3550. 10.1167/iovs.02-1278 [DOI] [PubMed] [Google Scholar]
- Robson, A. G. , Saihan, Z. , Jenkins, S. A. , Fitzke, F. W. , Bird, A. C. , Webster, A. R. , & Holder, G. E. (2006). Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. The British Journal of Ophthalmology, 90(4), 472–479. 10.1136/bjo.2005.082487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, F. , Arrigo, A. , MacLaren, R. E. , Charbel Issa, P. , Birtel, J. , Bandello, F. , & Battaglia Parodi, M. (2019). Hyperreflective foci as a pathogenetic biomarker in choroideremia. Retina, 40(8), 1634–1640. 10.1097/IAE.0000000000002645 [DOI] [PubMed] [Google Scholar]
- Saqib, M. A. N. , Nikopoulos, K. , Ullah, E. , Sher Khan, F. , Iqbal, J. , Bibi, R. , Jarral, A. , Sajid, S. , Nishiguchi, K. M. , Venturini, G. , Ansar, M. , & Rivolta, C. (2015). Homozygosity mapping reveals novel and known mutations in Pakistani families with inherited retinal dystrophies. Scientific Reports, 5, 9965. 10.1038/srep09965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimmer, J. , & Breazzano, S. (2015). Investor outlook: Significance of the positive LCA2 gene therapy phase III results. Human Gene Therapy. Clinical Development, 26(4), 208–210. 10.1089/humc.2015.29004.sch [DOI] [PubMed] [Google Scholar]
- Schorderet, D. F. , Bernasconi, M. , Tiab, L. , Favez, T. , & Escher, P. (2014). IROme, a new high‐throughput molecular tool for the diagnosis of inherited retinal dystrophies‐a price comparison with Sanger sequencing. Advances in Experimental Medicine and Biology, 801, 171–176. 10.1007/978-1-4614-3209-8_22 [DOI] [PubMed] [Google Scholar]
- Schuerch, K. , Woods, R. L. , Lee, W. , Duncker, T. , Delori, F. C. , Allikmets, R. , Tsang, S. H. , & Sparrow, J. R. (2017). Quantifying fundus autofluorescence in patients with retinitis pigmentosa. Investigative Ophthalmology & Visual Science, 58(3), 1843–1855. 10.1167/iovs.16-21302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Shuart, N. G. , Haitin, Y. , Camp, S. S. , Black, K. D. , & Zagotta, W. N. (2011). Molecular mechanism for 3:1 subunit stoichiometry of rod cyclic nucleotide‐gated ion channels. Nature Communications, 2, 457. 10.1038/ncomms1466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim, N.‐L. , Kumar, P. , Hu, J. , Henikoff, S. , Schneider, G. , & Ng, P. C. (2012). SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Research, 40(W1), W452–W457. 10.1093/nar/gks539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, D. A. , Clark, G. R. , Alexander, S. , Silvestri, G. , & Willoughby, C. E. (2011). Molecular diagnosis for heterogeneous genetic diseases with targeted high‐throughput DNA sequencing applied to retinitis pigmentosa. Journal of Medical Genetics, 48, 145–151. 10.1136/jmg.2010.083568 [DOI] [PubMed] [Google Scholar]
- Testa, F. , Rossi, S. , Colucci, R. , Gallo, B. , Di Iorio, V. , della Corte, M. , Azzolini, C. , Melillo, P. , & Simonelli, F. (2014). Macular abnormalities in Italian patients with retinitis pigmentosa. The British Journal of Ophthalmology, 98(7), 946–950. 10.1136/bjophthalmol-2013-304082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triolo, G. , Pierro, L. , Parodi, M. B. , De Benedetto, U. , Gagliardi, M. , Manitto, M. P. , & Bandello, F. (2013). Spectral domain optical coherence tomography findings in patients with retinitis pigmentosa. Ophthalmic Research, 50(3), 160–164. 10.1159/000351681 [DOI] [PubMed] [Google Scholar]
- Tsang, S. H. , Burke, T. , Oll, M. , Yzer, S. , Lee, W. , Xie, Y. A. , & Allikmets, R. (2014). Whole exome sequencing identifies CRB1 defect in an unusual maculopathy phenotype. Ophthalmology, 121(9), 1773–1782. 10.1016/j.ophtha.2014.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), e164–e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisschuh, N. , Feldhaus, B. , Khan, M. I. , Cremers, F. P. M. , Kohl, S. , Wissinger, B. , & Zobor, D. (2018). Molecular and clinical analysis of 27 German patients with Leber congenital amaurosis. PLOS One, 13(12), e0205380. 10.1371/journal.pone.0205380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisschuh, N. , Mayer, A. K. , Strom, T. M. , Kohl, S. , Glöckle, N. , Schubach, M. , Andreasson, S. , Bernd, A. , Birch, D. G. , Hamel, C. P. , Heckenlively, J. R. , Jacobson, S. G. , Kamme, C. , Kellner, U. , Kunstmann, E. , Maffei, P. , Reiff, C. M. , Rohrschneider, K. , Rosenberg, T. , … Wissinger, B. (2016). Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLOS ONE, 11(1), e0145951. 10.1371/journal.pone.0145951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler, P. A. , Ekenstedt, K. J. , Occelli, L. M. , Frattaroli, A. V. , Bartoe, J. T. , Venta, P. J. , & Petersen‐Jones, S. M. (2013). A large animal model for CNGB1 autosomal recessive retinitis pigmentosa. PLOS ONE, 8(8), e72229. 10.1371/journal.pone.0072229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, A. F. , Chakarova, C. F. , Abd El‐Aziz, M. M. , & Bhattacharya, S. S. (2010). Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait. Nature Reviews Genetics, 11(4), 273–284. 10.1038/nrg2717 [DOI] [PubMed] [Google Scholar]
- Xiang, Q. , Guo, Y. , Cao, Y. , Xiong, W. , Deng, X. , Xu, H. , Li, Y. , Du, D. , & Deng, H. (2018). Identification of a CNGB1 frameshift mutation in a han chinese family with retinitis pigmentosa. Optometry and Vision Science, 95, 1155–1161. 10.1097/OPX.0000000000001305 [DOI] [PubMed] [Google Scholar]
- Xu, Y. , Guan, L. , Shen, T. , Zhang, J. , Xiao, X. , Jiang, H. , Li, S. , Yang, J. , Jia, X. , Yin, Y. , Guo, X. , Wang, J. , & Zhang, Q. (2014). Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Human Genetics, 133(10), 1255–1271. 10.1007/s00439-014-1460-2 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Molday, L. L. , Molday, R. S. , Sarfare, S. S. , Woodruff, M. L. , Fain, G. L. , Kraft, T. W. , & Pittler, S. J. (2009). Knockout of GARPs and the β‐subunit of the rod cGMP‐gated channel disrupts disk morphogenesis and rod outer segment structural integrity. Journal of Cell Science, 122(Pt 8), 1192–1200. 10.1242/jcs.042531 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The data that support the findings of this study will be openly available in LOVD at https://www.lovd.nl/ after the acceptance of the study.