

Abstract
PROTACs (proteolysis targeting chimeras) are an emerging class of promising therapeutic modalities that degrade intracellular protein targets by hijacking the cellular ubiquitin–proteasome system. However, potential toxicity of PROTACs in normal cells due to the off-tissue on-target degradation effect limits their clinical applications. Precise control of a PROTAC’s on-target degradation activity in a tissue-selective manner could minimize potential toxicity/side-effects. To this end, we developed a cancer cell selective delivery strategy for PROTACs by conjugating a folate group to a ligand of the VHL E3 ubiquitin ligase, to achieve targeted degradation of proteins of interest (POIs) in cancer cells versus noncancerous normal cells. We show that our folate-PROTACs, including BRD PROTAC (folate-ARV-771), MEK PROTAC (folate-MS432), and ALK PROTAC (folate-MS99), are capable of degrading BRDs, MEKs, and ALK, respectively, in a folate receptor-dependent manner in cancer cells. This design provides a generalizable platform for PROTACs to achieve selective degradation of POIs in cancer cells.
Graphical Abstract
INTRODUCTION
By hijacking an endogenous E3 ubiquitin ligase and the ubiquitin–proteasome system (UPS), the proteolysis targeting chimera (PROTAC) technology could potentially be applied to target any intracellular proteins for degradation, including those so-called undruggable targets such as transcriptional factors and scaffold proteins.1-3 Compared to small-molecule inhibitors, PROTACs are potentially more powerful therapeutic modalities, as they do not rely on occupancy-driven pharmacology, in part due to the catalytic nature of PROTACs in degrading their protein targets.4 However, potential off-tissue effect(s), i.e., diffused distribution of PROTAC molecules in nontarget normal tissues/organs after being systemically administered, may lead to unwanted toxicity issues and complications in the clinic.5,6 Recently, we and others have independently reported light-controllable PROTACs, either by incorporating photocage groups, such as nitroveratryloxycarbonyl (NVOC),7-9 4,5-dimethoxy-2-nitrobenzyl (DMNB), and [7-(diethylamino)coumarin-4-yl]methyl (DEACM),10 onto the pomalidomide or von Hippel–Lindau (VHL) ligand moieties in PROTACs or by installing a photoswitchable azobenzene in the linker region.11-13 These light-controllable PROTACs provide an avenue to achieve spatiotemporal regulation of the catalytic activity of the PROTAC molecules, but there is also limitations of these methods, as they could be used only in limited cancer types with light accessibility.14,15
To achieve the targeted-delivery goal, several antibody-based PROTACs have also been developed recently to degrade either membrane proteins or intracellular protein targets, via targeting cell membrane-anchored receptors, such as HER2 and ERα.16-22 However, a major disadvantage of antibody-based PROTACs is due to their relatively high molecular weight and instability during systemic administration, which limits their effective application in the clinic. Furthermore, besides using membrane-anchored antigen(s) as cellular clues for antibody–drug conjugates such as antibody-based PROTACs, several small molecule ligand–receptor pairs have also been used in targeted delivery of drugs, such as vitamin B12 and the transcobalamin receptor,23 transferrin and the transferrin receptor,24 and folate and the folate receptor.25
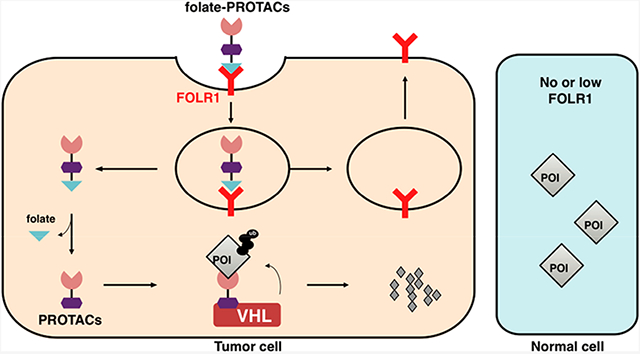
Folate receptor α (FOLR1) is the most well-defined target for drug delivery into cancer cells, because FOLR1 is highly expressed in many cancer types, such as ovarian cancer, lung cancer, and breast cancer, while normal tissues or cells have very low or no FOLR1 expression.25 Besides FOLR1, other receptors, such as FOLR2 and FOLR3, are also capable of transporting folate into cells, while their affinity to folate is relatively lower than FOLR1.25 As such, a FOLR1-targeting strategy has already been used for decades in both tumor imaging26 and cancer-targeting drug delivery,27 and several FOLR1-targeting drugs are in phase II/III clinical trials.25 Moreover, FOLR2 is also used as a target for drug delivery.28 These prompted us to use the folate-conjugating strategy for the specific delivery of PROTACs into cancer cells to achieve controllable targeted degradation of a protein of interest (POI), thus eliminating potential unwanted toxicity to normal tissues. To this end, we designed folate-PROTACs, which are preferentially transported into cancer cells with high FOLR1 expression. After entry into cancer cells, folate-PROTACs are catalyzed by intracellular hydrolases29 to release the folate moiety, and then the uncaged PROTAC recruits endogenous VHL E3 ubiquitin ligase to ubiquitinate the POI for subsequent degradation by the 26S proteasome (Figure 1A).
Figure 1.

FOLR1 transports folate-PROTAC into cancer cells for targeted degradation of a protein of interest (POI). (A) Schematic representation of the folate-PROTAC strategy. Following the FOLR1-mediated entrance into cancer cells, the folate group (light blue triangle) is cleaved by endogenous hydrolase, releasing the active PROTAC to degrade the POI. (B) Schematic illustration of the activation of folate-ARV-771 by endogenous hydrolase. (C, D) Western blot analysis of BRD protein levels from HeLa or HFF-1 cells treated with the indicated doses of ARV-771, folate-ARV-771, or folate-ARV771N for 12 h. (E, F) Cell viability of HeLa or HFF-1 cells after treatment with ARV-771, folate-ARV-771, or folate-ARV-771N for 72 h.
RESULTS AND DISCUSSION
Design and Synthesis of Folate-ARV-771.
A typical PROTAC molecule consists of three functional parts: a warhead to recruit the POI, a ligand to recruit the E3 ubiquitin ligase, and a linker between these two moieties.3 To ensure that the folate-PROTAC design is generally applicable, we chose to install a folate group onto the E3 ubiquitin ligase ligand. For a VHL-based PROTAC, the hydroxyl group in the VHL ligand is critical for the recruitment of VHL E3 ubiquitin ligase,30,31 and inversion of the stereochemistry from R to S or installation of a bulky caging group on the hydroxyl moiety abolishes PROTAC activity,10,32 primarily due to the loss of VHL-binding ability. To this end, we designed a lead compound, folate-PROTAC (folate-ARV-771), by incorporating folate via an ester bond29 onto the hydroxyl group of a well-studied VHL-based bromodomain (BRD) degrader, ARV-771.5 Through this strategy, we achieve two goals: (1) the folate moiety aids targeted enrichment of PROTACs into cancer cells; and (2) similar to the other caging strategy,9,10 the caged folate-PROTAC compound is inert to begin with and can be activated after being uncaged via cleavage by endogenous hydrolases in cells (Figure 1A and B).
Folate-ARV-771 was synthesized through a Cu-catalyzed azide–alkyne cycloaddition (CuAAC) reaction to conjugate alkyne-modified folic acid and azide-modified ARV-771 ester (Scheme S1). In addition, a negative control of folate-PROTAC, folate-ARV-771N, was designed and synthesized by replacing the ester bond with a noncleavable amide bond (Figure 1B and Scheme S2); thus folate-ARV-771N is resistant to cleavage and remains inactive even after entering cancer cells. Furthermore, two stereochemical negative controls, namely, ARV-7665 and folate-ARV-766, were also synthesized (Scheme S3) with the reversed configuration at the hydroxyproline in the VHL ligand moiety, which were incapable of binding with VHL E3 ubiquitin liase and degrading BRDs.5 Then, the stability of folate-ARV-771 was measured after incubating either in phosphate-buffered saline (PBS), in cell culture media, or in media plus 10% fetal bovine serum (FBS) at 37 °C. These results indicated that folate-ARV-771 was relatively stable in physiological conditions that are used to culture cells in our experimental setting (Figure S1).
Folate-ARV-771 Preferentially Degrades BRDs in Cancer Cells.
In order to determine the specific role of folate-PROTAC in degrading POIs in cancer cells versus normal cells, we took advantage of three cancer cell lines with high FOLR1 expression, including HeLa cells,33 OVCAR-8 ovary cancer cells,25 and T47D breast cancer (BRCA) cells,34 as well as three noncancerous normal cell lines with low FOLR1 expression, including human fibroblast cells (HFF-1), human normal kidney epithelia cells (HK2), and mouse fibroblast cells (3T3) (Figure S2A). Notably, in HeLa cancer cells, folate-ARV-771 degraded BRD4 as efficiently as ARV-771, while the noncleavable negative control, folate-ARV-771N, and the stereochemical negative control ARV-766 and folate-ARV-766 were incapable of degrading BRDs even at 100 nM (Figures 1C, S2B, and S2C). Similarly, folate-ARV-771 had comparable efficiency with ARV-771 in degrading BRDs in both OVCAR-8 (Figure S2D and E) and T47D cancer cells (Figure S2F), while folate-ARV-771N did not (Figure S2D-F). In contrast, folate-ARV-771 was less efficient than ARV-771 in degrading BRDs in HFF-1 (Figure 1D), HK2 (Figure S2G), and 3T3 (Figure S2H) noncancerous normal cells.
In keeping with these results, folate-ARV-771 had a comparable cell killing IC50 with ARV-771 in three FOLR1-expressing cancer cell lines, including HeLa cells (246 nM vs 183 nM, Figure 1E), OVCAR8 cells (297 nM vs 215 nM, Figure S3A), and T47D cells (18 nM vs 13 nM, Figure S3B). In contrast, folate-ARV-771 was much less efficient than ARV-771 in noncancerous normal cell lines, including HFF-1 cells (>10 μM vs 1.1 μM, Figure 1F), HK2 cells (2.1 μM vs 166 nM, Figure S3C) and 3T3 cells (1.4 μM vs 210 nM, Figure S3D). Furthermore, we also compared the expression of BRD proteins among those cell lines (Figure S2A) and found that the difference of sensitivity to ARV-771 or folate-ARV-771 may not be due to the different expression levels of protein targets (Figures 1 and S2). Taken together, these results indicated that folate-ARV-771 is specifically enriched and degrades POI in cancer cells versus noncancerous normal cells.
Folate-ARV-771 Degrades BRD4 in VHL- and FOLR1-Dependent Manners.
To show that the degradation of POI by folate-PROTAC depends on VHL E3 ubiquitin ligase, we further treated HeLa cells with free VHL ligand (VH-032) together with folate-ARV-771. We found that cotreatment of VH-032 effectively blocked the degradation of BRDs by folate-ARV-771 (Figure S4A) and increased the IC50 of folate-ARV-771 (Figure S4B). Moreover, deletion of endogenous VHL completely abolished the effect of folate-ARV-771 on both degradation of BRDs and inhibition of cell proliferation (Figure S4C and D), further supporting the dependence of folate-ARV-771 on endogenous VHL E3 ubiquitin ligase. In addition, the proteasome inhibitor MG132 and the Cullin neddylation inhibitor MLN4924,35 which represses the activation of Cullin RING ubiquitin ligases (CRLs), blocked the effect of folate-ARV-771 in degrading BRDs in cancer cells (Figure S4E-G). These results demonstrate that folate conjugation will not introduce nonspecific function to original PROTACs, and their effects on degradation are proteasome dependent.
The entry of a folate-conjugate into cells largely depends on its receptor FOLR1 on the cancer cell membrane,25 and FOLR1-mediated drug entry can be antagonized by free folic acid.26 To this end, HeLa cells were pretreated with free folic acid and then challenged with either ARV-771 or folate-ARV-771. In keeping with previous findings,25,26 we found that free folic acid antagonized the ability of folate-ARV-771 in degrading BRD4, but not ARV-771 (Figure 2A). As such, pretreatment with free folic acid significantly increased the IC50 of folate-ARV-771 from 365 nM to 1.5 μM (Figure 2B). Furthermore, depletion of endogenous FOLR1 also eliminated the effect of folate-ARV-771 in degrading BRD4 in HeLa and T47D cancer cells (Figures 2C and S5A). Notably, excessive free folic acid or VHL ligand could not completely abolish the cytotoxicity of folate-ARV-771, even when the degradation event was efficiently blocked (Figures 2A,B and S4A,B), suggesting that PROTAC derived from a BET inhibitor likely retains its original inhibitory function on BRDs, which should be noted during its clinic usage. After binding with FOLR1, folate-conjugates take advantage of the endocytosis process to enter cells.25 To further examine the role of endocytosis in this process, we pretreated HeLa cells with an endocytosis inhibitor, MβCD, 36 and found that MβCD efficiently blocked the effect of folate-ARV-771 in degrading BRD4 (Figure S5B).
Figure 2.

Folate-ARV-771 degrades BRD4 in a FOLR1-dependent manner. (A) Western blot analysis of BRD4 level in HeLa cells after treatment with the indicated doses of free folic acid with either 30 nM folate-ARV-771 or ARV-771 for 12 h. (B) Cell viability of HeLa cells treated with folate-ARV-771 with or without free folic acid for 72 h. (C) HeLa cells with or without knockdown of endogenous FOLR1 were treated with folate-ARV-771 for 2 h, and then the levels of BRD4 were determined by Western blot. (D) Western blot analysis of basal BRDs and FOLR1/2 levels in a panel of BRCA cell lines and the noncancerous normal breast epithelial MCF10A cells. (E) Western blot analysis of BRD4 level in BRCA cell lines with either high FOLR1 expression (ZR-75-1, SK-BR-3, and AU565) or low FOLR1 expression levels (BT549 and MDA-MB-231) or noncancerous normal breast epithelial MCF10A cells after treatment with folate-ARV-771 for 12 h.
To further determine the critical role of FOLR1 for dictating the activity of folate-PROTAC, we measured the expression of FOLR1 in a panel of BRCA cell lines and a noncancerous breast epithelial cell line, MCF10A, and found that ZR-75-1, SK-BR-3, and AU565 cells have high FOLR1 expression, while BT549, MDA-MB-231, and MCF10A cells have relatively lower or no FOLR1 expression (Figure 2D). Then, we measured the effects of folate-ARV-771 in these cell lines with distinct FOLR1 expression levels. Notably, folate-ARV-771 degraded BRD4, as efficiently as ARV-771, in those BRCA cells with relatively high FOLR1 expression (Figure 2E). In contrast, in either normal breast epithelial MCF10A cells or FOLR1-low cells (BT549 and MDA-MB-231), folate-ARV-771 was much less efficient in degrading BRD4 compared with ARV-771 (Figure 2E). Notably, folate-ARV-771 could still degrade BRD4 with low efficiency in cells lines with low FOLR1 expression, such as BT549 and MCF10A (Figure 2E), and this effect could possibly be in part due to the existence of other folate receptors, such as FOLR2 (Figure 2D). On the other hand, the effect of folate-ARV-771 could be antagonized by free folic acid (Figure 2A) or abolished by genetic depletion of FOLR1 (Figures 2C and S5A), indicating that FOLR1 might be the major transporter for folate-ARV-771. Given that folate-PROTAC is relatively stable in cell culture media supplied with serum (Figure S1), and the degradation of POI occurs as early as 2 h after folate-PROTAC treatment (Figure S2), this finding largely excludes the possibility of an uncaging process before the entry of folate-PROTAC into cells. Taken together, these results indicated that folate-ARV-771 degrades BRDs most likely in a FOLR1-dependent manner in cancer cells.
Folate-MS432 Degrades MEK1/2 in a FOLR1-Dependent Manner.
To further examine whether the folate-mediated caging strategy can also be applied to other VHL-based PROTACs, we fUrther designed and synthesized two other folate-PROTACs, folate-MS432 (Figure 3A) and folate-MS99 (Figure 4A), based on our previously reported MEK1/2 degrader (MS432)37 and a close analogue of a reported ALK degrader (MS99),38 respectively. Folate-MS432 and its negative control folate-MS432N (Figure 3A) were synthesized through a similar strategy with folate-ARV-771 and folate-ARV-771N (Schemes S4 and S5). We examined the effect of folate-MS432 in HeLa cells and found that folate-MS432 degraded MEK1 and MEK2 as efficiently as MS432, while folate-MS432N was incapable of degrading either MEK1 or MEK2 even at 1 μM (Figure S6A).
Figure 3.

Folate-MS432 degrades MEK1/2 in a FOLR1-dependent manner. (A) Schematic illustration of the activation of folate-MS432 by endogenous hydrolase. (B) Western blot analysis of MEK1 and MEK2 levels in HT-29 cells treated with indicated doses of MS432, folate-MS432, or folate-MS432N for 12 h. (C, D) Western blot analysis of MEK1 and MEK2 levels in HT-29 cells after the cotreatment with free folic acid or VHL ligand (VH-032) and 100 nM folate-MS432 for 12 h. (E) Western blot analysis of MEK1 and MEK2 levels in HT-29 cells after the cotreatment with 10 μM MG132 or 1 μM MLN4924 and 100 nM folate-MS432 for 12 h. (F) Cell viability of HT-29 cells after treatment with MS432, folate-MS432, or folate-MS432N for 72 h.
Figure 4.

Folate-MS99 degrades ALK fusion proteins in a FOLR1-dependent manner. (A) Schematic illustration of the activation of folate-MS99 by endogenous hydrolase. (B) Western blot analysis of the NPM-ALK fusion protein level from SU-DHL-1 cells treated with indicated doses of MS99, folate-MS99, or folate-MS99N for 12 h. (C) Western blot analysis of NPM-ALK fusion protein from HT-29 cells after the cotreatment with free folic acid and 0.3 μM folate-MS99 for 12 h. (D) Western blot analysis of NPM-ALK fusion protein level in SU-DHL-1 cells after the cotreatment with 10 μM MG132 or 1 μM MLN4924 and 0.3 μM folate-MS99 for 12 h. (E) Cell viability of SU-DHL-1 cells after treatment with MS99, folate-MS99, or folate-MS99N for 72 h.
Since MEK/ERK signaling is critical for the survival of BRAF mutant cancer cells, we further evaluated the effect of folate-MS432 in BRAF-V600E mutant harboring cells, including a colorectal cancer cell line, HT29,39 and a melanoma cell line, SK-MEL-28.39 As expected, folate-MS432 degraded MEK1 and MEK2 in both HT29 and SK-MEL-28 cells in dose- and time-dependent manners, while folate-MS432N could not do so (Figures 3B and S6B-E). Moreover, pretreatment of folic acid blocked the effect of folate-MS432 in degrading MEK1 and MEK2 in both HT-29 (Figure 3C) and SK-MEL-28 cells (Figure S6F). Furthermore, cotreatment with the VHL ligand, VH-032 (Figure 3D), the proteasome inhibitor MG132, or the Cullin neddylation inhibitor MLN4924 (Figures 3E and S6G) blocked the degradation of MEK1/2 in HT-29 cells, indicating that folate-MS432 degrades MEK1/2 in VHL E3 ubiquitin ligase-and proteasome-dependent manners. However, VH-032 was relatively less effective in repressing the effect of folate-MS432 than MG132. Finally, we measured the cell viability of both HT-29 and SK-MEL-28 cells after treatment with either MS432, folate-MS432, or folate-MS432N. In HT-29 cells, the IC50 of MS432 and folate-MS432 was 176 and 436 nM, respectively (Figure 3F). In SK-MEL-28 cells, the IC50 of MS432 and folate-MS432 was 32 nM and 390 nM, respectively (Figure S6H). These results together indicated that folate-MS432 degrades MEK1/2 largely in a FOLR1-dependent manner.
Folate-MS99 Degrades ALK Fusion Proteins in a FOLR1-Dependent Manner.
Anaplastic lymphoma kinase (ALK) fusion proteins are the drivers in several types of cancer, including the EML4-ALK fusion in non-small-cell lung cancer (NSCLC)40 and NPM-ALK fusion in leukemia41 (Figure S7A and B). These fusion proteins are constitutively active and confer resistance to the ALK inhibitor in the clinic,42,43 while ALK degraders are expected to overcome such drug resistance.38,44 To this end, a new folate-conjugated VHL-based degrader, folate-MS99, and its negative control, folate-MS99N, were synthesized (Figure 4A and Schemes S6-S8). We first determined the efficiency of folate-MS99 in degrading ALK fusion proteins in SU-DHL-1 cells, an anaplastic large cell lymphoma (ALCL) cell line with NPM-ALK fusion.44 Both MS99 and folate-MS99 degraded NPM-ALK fusion protein efficiently in SU-DHL-1 cells, while folate-MS99N was incapable of degrading NPM-ALK even at 3 μM (Figures 4B and S7C). Furthermore, folate-MS99 efficiently degraded the EML4-ALK fusion proteins in two NSCLC cell lines, NCI-H2228 and NCI-H3122 cells (Figure S7D and E). More importantly, pretreatment with free folic acid antagonized the effect of folate-MS99 in degrading NPM-ALK fusion protein in SU-DHL-1 cells (Figure 4C) and EML4-ALK fusion protein in both NSCLC cells (Figure S7F and G), suggesting the key role of FOLR1 in mediating the effect of folate-MS99. Furthermore, the proteasome inhibitor MG132 and the Cullin neddylation inhibitor MLN4924 largely abolished the effect of folate-MS99 in degrading ALK fusion proteins in both SU-DHL-1 cells (Figure 4D) and NSCLC cells (Figure S7H and I). We further measured the cell viability of SU-DHL-1 cells after treatment with MS99, folate-MS99, or folate-MS99N. The IC50 values of MS99 and folate-MS99 were 91 nM and 200 nM, respectively (Figure 4E). These results indicated that folate-MS99 is efficient in degrading ALK fusion proteins likely in a FOLR1-dependent manner.
CONCLUSION
Taken together, we provide a FOLR1-targeting delivery strategy for PROTACs to selectively degrade POIs in cancer cells versus noncancerous normal cells and have validated three lead folate-PROTACs (folate-ARV-771, folate-MS432, and folate-MS99) that effectively degraded BRDs and MEK1/2 and ALK fusion proteins, respectively, in a FOLR1-dependent manner in cancer cells. Our results also indicate that, besides FOLR1, other receptors/transporters such as FOLR2 might also help the specific recruitment of folate-caged PROTACs to enter cells. Furthermore, passive diffusion of folate-caged PROTACs to transduce into cells through the cell membrane could also be possible,45 although it might not be the major route due to the hydrophilic nature of the charged folate molecule.46 Moreover, the conjugation of the folate group leads to an increase of the molecular weight of PROTACs to over 1000 Da, which might compromise the oral bioavailability and pharmacokinetics of folate-PROTACs. Thus, additional indepth studies are warranted to optimize the stability of folate-caged PROTACs and to evaluate their efficiency in cancer-specific delivery of PROTACs in vivo. Taken together, our results clearly demonstrate that this approach is generalizable and could be applied to all VHL-recruiting PROTACs, thereby providing a targeting strategy to selectively degrade POIs in cancer cells and to minimize potential toxicity/side-effects in normal tissues/cells, thus enhancing therapeutic windows of PROTACs.
EXPERIMENTAL METHODS
General Chemistry Methods.
Common reagents or materials were purchased from commercial sources and used without further purification. Ultraperformance liquid chromatography (UPLC) spectra for compounds were acquired using a Waters Acquity I-Class UPLC system with a PDA detector. Chromatography was performed on a 2.1 Å, 30 mm ACQUITY UPLC BEH C18 1.7 μm column with water containing 3% acetonitrile and 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.8 mL/min. The gradient program was as follows: 1–99% B (1–1.5 min) and 99–1% B (1.5–2.5 min). High-performance liquid chromatography (HPLC) spectra were acquired using an Agilent 1200 Series system with a DAD detector for all the intermediates and final products below. Chromatography was performed on a 2.1 × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. NMR spectra were acquired on a Bruker DRX-600 spectrometer with 600 MHz for proton (1H NMR) and 151 MHz for carbon (13C NMR); chemical shifts are reported in (δ). Preparative HPLC was performed on Agilent Prep 1200 series with the UV detector set to 220 or 254 nm. Samples were injected onto a Phenomenex Luna 250 × 30 mm, 5 μm, C18 column at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% acetonitrile in H2O (with 0.1% TFA) (B) to 100% acetonitrile (A). HPLC was used to establish the purity of target compounds. All final compounds had >96% purity using the HPLC methods described above. ARV-771,5 MS432,37 and VH-03247 were synthesized according to the published procedures.
Stability Assay of Folate-ARV-771.
The stability assay of folate-ARV-771 (100 μM) was performed using HPLC after incubating either in PBS or in cell culture media (DMEM) alone, or DMEM plus 10% FBS at 37 °C for 2, 4, 8, or 16 h. To precipitate proteins in FBS, the mixture was diluted with an equal volume of acetonitrile and centrifuged, followed by HPLC analysis for the top clear solution.
Cell Culture.
Human embryonic kidney 293T (HEK293T), human fibroblast (HFF1), mouse 3T3 fibroblast, human kidney epithelial cells HK2, HeLa, OVCAR8, MDA-MB-231, and SK-MEL-28 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 100 units/mL of penicillin, and 100 μg/mL streptomycin. T47D, BT549, ZR-75-1, SK-BR-3, AU565, SU-DHL1, NCI-H2228, NCI-H3122, and HT-29 cells were cultured in RPMI1640 containing 10% FBS, 100 units/mL of penicillin, and 100 μg/mL streptomycin. MCF10A cells were cultured in MEGM media (CC-3150, Lonza) plus 100 ng/mL cholera toxin. The panels of breast cancer and epithelial cells were cultured as previously described.48,49 The usage of SU-DHL1, NCI-H2228, and NCI-H3122 cells for evaluation of ALK degrader and the usage of HT29 and SK-MEK-28 cells for evaluation of MEK degrader is based on our previous reports.8 The shRNA for FOLR1 was purchased from Sigma. The lentivirus of shFOLR1 and sgVHL was generated in HEK293T cells for the infection of HeLa and T47D cells as previously described.48,49 Cells were infected with lentivirus, selected with puromycin for 72 h, followed by further PROTAC treatment.
For all PROTAC treatment, cells were incubated with chemicals for 12 h unless otherwise indicated. For the competition assay, cells were incubated with the indicated dose of folic acid (F8758, Sigma), VHL ligand (VHL-032), or MβD (21633, Cayman) together with respective PROTACs for 12 h. For proteasome or CRL inhibition assays, cells were treated with 10 μM MG132 (BML-Pl102, ENZO Life Sciences) or 1 μM MLN4924 (S7109, SelleckChem) together with respective PROTACs for 12 h.
Antibodies.
Anti-BRD3 (11859-1-AP) and FOLR1 (23355-1-AP) antibodies were purchased from Proteintech. Anti-BRD4 (A301-985A-M) antibody was purchased from Bethyl Laboratories. Anti-ALK (3633), MEK1 (2352), and MEK2 (9147) antibodies were purchased from Cell Signaling Technologies. FOLR2 (PA5-45768) antibodies were purchased from ThermoFisher. Monoclonal anti-Vinculin antibody (V-4505), peroxidase-conjugated anti-mouse secondary antibody (A-4416), and peroxidase-conjugated anti-rabbit secondary antibody (A-4914) were purchased from Sigma. All antibodies were used at a 1:1000 dilution in 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.1% Tween-20 (TBST) buffer for Western blots.
Immunoblot (IB) Assay.
Cells were lysed in EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) supplemented with protease inhibitors (Pierce) and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II, Calbiochem). The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. The lysates (30–60 μg protein) were then resolved by 10% or 7% (used for blotting BRD4 and BRD3) SDS-PAGE at 130 V for 80–100 min and immunoblotted with the indicated antibodies at 4 °C overnight, washed four times with TBST, incubated with secondary antibody in 5% nonfat milk for 1 h at room temperature, and then washed four times with TBST.
CCK-8 Cell Proliferation Assay.
Cell viability was analyzed as previously described.8 Briefly, cells in 96-well plate were treated with the indicated doses of respective PROTACs for 72 h and then incubated with 10 μL/well of CCK-8 (K1018, APExBIO) solution at 37 °C for 1–2 h, followed by the measurement of optical density at 450 nm. Cells treated with vehicle or puromycin were used as the negative and the positive control, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH grants R35CA253027 (W.W.), R01CA218600 (J.J.), R01CA230854 (J.J.), R01GM122749 (J.J.), R01CA260666 (J.J.), and P30CA196521 (J.J.). This work utilized the AVANCE NEO 600 MHz NMR spectrometer system that was upgraded with funding from a National Institutes of Health SIG grant 1S10OD025132-01A1. We thank Wei lab members for critical reading of the manuscript and members of the Wei and Jin laboratories for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c00451.
Compound synthesis and experimental details, including Schemes S1-S8 and Figures S1-S7 (PDF)
The authors declare the following competing financial interest(s): W.W. is co-founder and stockholder of the Rekindle Therapeutics. J.J. is an equity shareholder and consultant of Cullgen, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, and Cullgen, Inc. All other authors declare no competing interests.
Contributor Information
Jing Liu, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
He Chen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
Yi Liu, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
Yudao Shen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
Fanye Meng, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
H. ϋmit Kaniskan, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
Wenyi Wei, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
REFERENCES
- (1).Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (15), 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discovery 2019, 18 (12), 949–963. [DOI] [PubMed] [Google Scholar]
- (3).Schneekloth JS Jr.; Fonseca FN; Koldobskiy M; Mandal A; Deshaies R; Sakamoto K; Crews CM Chemical genetic control of protein levels: selective in vivo targeted degradation. J. Am. Chem. Soc 2004, 126 (12), 3748–3754. [DOI] [PubMed] [Google Scholar]
- (4).Burslem GM; Smith BE; Lai AC; Jaime-Figueroa S; McQuaid DC; Bondeson DP; Toure M; Dong H; Qian Y; Wang J; Crew AP; Hines J; Crews CM The Advantages of Targeted Protein Degradation Over Inhibition: An RTK Case Study. Cell. Chem. Biol 2018, 25 (1), 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AM; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (26), 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Moreau K; Coen M; Zhang AX; Pachl F; Castaldi MP; Dahl G; Boyd H; Scott C; Newham P Proteolysis-targeting chimeras in drug development: A safety perspective. Br. J. Pharmacol 2020, 177 (8), 1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Xue G; Wang K; Zhou D; Zhong H; Pan Z Light-induced protein degradation with photo-caged PROTACs. J. Am. Chem. Soc 2019, 141 (46), 18370–18374. [DOI] [PubMed] [Google Scholar]
- (8).Liu J; Chen H; Ma L; He Z; Wang D; Liu Y; Lin Q; Zhang T; Gray N; Kaniskan H. ϋ.; Jin J; Wei W Light-induced control of protein destruction by opto-PROTAC. Sci. Adv 2020, 6 (8), eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kounde CS; Shchepinova MM; Saunders CN; Muelbaier M; Rackham MD; Harling JD; Tate EW A caged E3 ligase ligand for PROTAC-mediated protein degradation with light. Chem. Commun 2020, 56 (41), 5532–5535. [DOI] [PubMed] [Google Scholar]
- (10).Naro Y; Darrah K; Deiters A Optical Control of Small Molecule-Induced Protein Degradation. J. Am. Chem. Soc 2020, 142 (5), 2193–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Reynders M; Matsuura BS; Berouti M; Simoneschi D; Marzio A; Pagano M; Trauner D PHOTACs enable optical control of protein degradation. Sci. Adv 2020, 6 (8), eaay5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jin YH; Lu MC; Wang Y; Shan WX; Wang XY; You QD; Jiang ZY Azo-PROTAC: Novel Light-Controlled Small-Molecule Tool for Protein Knockdown. J. Med. Chem 2020, 63 (9), 4644–4654. [DOI] [PubMed] [Google Scholar]
- (13).Pfaff P; Samarasinghe KT; Crews CM; Carreira EM Reversible Spatiotemporal Control of Induced Protein Degradation by Bistable photoPROTACs. ACS Cent. Sci 2019, 5 (10), 1682–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Zhao W; Zhao Y; Wang Q; Liu T; Sun J; Zhang R Remote Light-Responsive Nanocarriers for Controlled Drug Delivery: Advances and Perspectives. Small 2019, 15 (45), e1903060. [DOI] [PubMed] [Google Scholar]
- (15).Karisma VW; Wu W; Lei M; Liu H; Nisar MF; Lloyd MD; Pourzand C; Zhong JL UVA-Triggered Drug Release and Photo-Protection of Skin. Front. Cell Dev. Biol 2021, 9, 598717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cotton AD; Nguyen DP; Gramespacher JA; Seiple IB; Wells JA Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc 2021, 143 (2), 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pillow TH; Adhikari P; Blake RA; Chen J; Del Rosario G; Deshmukh G; Figueroa I; Gascoigne KE; Kamath AV; Kaufman S; Kleinheinz T; Kozak KR; Latifi B; Leipold DD; Sing Li C; Li R; Mulvihill MM; O’Donohue A; Rowntree RK; Sadowsky JD; Wai J; Wang X; Wu C; Xu Z; Yao H; Yu SF; Zhang D; Zang R; Zhang H; Zhou H; Zhu X; Dragovich PS Antibody Conjugation of a Chimeric BET Degrader Enables in vivo Activity. ChemMedChem 2020, 15 (1), 17–25. [DOI] [PubMed] [Google Scholar]
- (18).Banik SM; Pedram K; Wisnovsky S; Ahn G; Riley NM; Bertozzi CR Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584 (7820), 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Maneiro MA; Forte N; Shchepinova MM; Kounde CS; Chudasama V; Baker JR; Tate EW Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol 2020, 15 (6), 1306–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Dragovich PS; Adhikari P; Blake RA; Blaquiere N; Chen J; Cheng YX; den Besten W; Han J; Hartman SJ; He J; He M; Rei Ingalla E; Kamath AV; Kleinheinz T; Lai T; Leipold DD; Li CS; Liu Q; Lu J; Lu Y; Meng F; Meng L; Ng C; Peng K; Lewis Phillips G; Pillow TH; Rowntree RK; Sadowsky JD; Sampath D; Staben L; Staben ST; Wai J; Wan K; Wang X; Wei B; Wertz IE; Xin J; Xu K; Yao H; Zang R; Zhang D; Zhou H; Zhao Y Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERalpha). Bioorg. Med. Chem. Lett 2020, 30 (4), 126907. [DOI] [PubMed] [Google Scholar]
- (21).Dragovich PS; Pillow TH; Blake RA; Sadowsky JD; Adaligil E; Adhikari P; Bhakta S; Blaquiere N; Chen J; Dela Cruz-Chuh J; Gascoigne KE; Hartman SJ; He M; Kaufman S; Kleinheinz T; Kozak KR; Liu L; Liu Q; Lu Y; Meng F; Mulvihill MM; O’Donohue A; Rowntree RK; Staben LR; Staben ST; Wai J; Wang J; Wei B; Wilson C; Xin J; Xu Z; Yao H; Zhang D; Zhang H; Zhou H; Zhu X Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem 2021, 64 (5), 2534–2575. [DOI] [PubMed] [Google Scholar]
- (22).Dragovich PS; Pillow TH; Blake RA; Sadowsky JD; Adaligil E; Adhikari P; Chen J; Corr N; Dela Cruz-Chuh J; Del Rosario G; Fullerton A; Hartman SJ; Jiang F; Kaufman S; Kleinheinz T; Kozak KR; Liu L; Lu Y; Mulvihill MM; Murray JM; O’Donohue A; Rowntree RK; Sawyer WS; Staben LR; Wai J; Wang J; Wei B; Wei W; Xu Z; Yao H; Yu SF; Zhang D; Zhang H; Zhang S; Zhao Y; Zhou H; Zhu X Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 2: Improvement of In Vitro Antiproliferation Activity and In Vivo Antitumor Efficacy. J. Med. Chem 2021, 64 (5), 2576–2607. [DOI] [PubMed] [Google Scholar]
- (23).Clardy SM; Allis DG; Fairchild TJ; Doyle RP Vitamin B12 in drug delivery: breaking through the barriers to a B12 bioconjugate pharmaceutical. Expert Opin. Drug Delivery 2011, 8 (1), 127–140. [DOI] [PubMed] [Google Scholar]
- (24).Daniels TR; Bernabeu E; Rodriguez JA; Patel S; Kozman M; Chiappetta DA; Holler E; Ljubimova JY; Helguera G; Penichet ML The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta, Gen. Subj 2012, 1820 (3), 291–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Scaranti M; Cojocaru E; Banerjee S; Banerji U Exploiting the folate receptor alpha in oncology. Nat. Rev. Clin. Oncol 2020, 17 (6), 349–359. [DOI] [PubMed] [Google Scholar]
- (26).Numasawa K; Hanaoka K; Saito N; Yamaguchi Y; Ikeno T; Echizen H; Yasunaga M; Komatsu T; Ueno T; Miura M; Nagano T; Urano Y A Fluorescent Probe for Rapid, High-Contrast Visualization of Folate-Receptor-Expressing Tumors In Vivo. Angew. Chem., Int. Ed 2020, 59 (15), 6015–6020. [DOI] [PubMed] [Google Scholar]
- (27).Yang Z; Lee JH; Jeon HM; Han JH; Park N; He Y; Lee H; Hong KS; Kang C; Kim JS Folate-based near-infrared fluorescent theranostic gemcitabine delivery. J. Am. Chem. Soc 2013, 135 (31), 11657–11662. [DOI] [PubMed] [Google Scholar]
- (28).Chandrupatla D; Molthoff CFM; Lammertsma AA; van der Laken CJ; Jansen G The folate receptor beta as a macrophage-mediated imaging and therapeutic target in rheumatoid arthritis. Drug Delivery Transl. Res 2019, 9 (1), 366–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mahato R; Tai W; Cheng K Prodrugs for improving tumor targetability and efficiency. Adv. Drug Delivery Rev 2011, 63 (8), 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Min JH; Yang H; Ivan M; Gertler F; Kaelin WG Jr.; Pavletich NP Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 2002, 296 (5574), 1886–1889. [DOI] [PubMed] [Google Scholar]
- (31).Hon WC; Wilson MI; Harlos K; Claridge TD; Schofield CJ; Pugh CW; Maxwell PH; Ratcliffe PJ; Stuart DI; Jones EY Structural basis for the recognition of hydroxyproline in HIF-1 alpha by pVHL. Nature 2002, 417 (6892), 975–978. [DOI] [PubMed] [Google Scholar]
- (32).Buckley DL; Van Molle I; Gareiss PC; Tae HS; Michel J; Noblin DJ; Jorgensen WL; Ciulli A; Crews CM Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J. Am. Chem. Soc 2012, 134 (10), 4465–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Salim L; Islam G; Desaulniers JP Targeted delivery and enhanced gene-silencing activity of centrally modified folic acid-siRNA conjugates. Nucleic Acids Res. 2020, 48 (1), 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Cheung A; Opzoomer J; Ilieva KM; Gazinska P; Hoffmann RM; Mirza H; Marlow R; Francesch-Domenech E; Fittall M; Dominguez Rodriguez D; Clifford A; Badder L; Patel N; Mele S; Pellizzari G; Bax HJ; Crescioli S; Petranyi G; Larcombe-Young D; Josephs DH; Canevari S; Figini M; Pinder S; Nestle FO; Gillett C; Spicer JF; Grigoriadis A; Tutt ANJ; Karagiannis SN Anti-Folate Receptor Alpha-Directed Antibody Therapies Restrict the Growth of Triple-negative Breast Cancer. Clin. Cancer Res 2018, 24 (20), 5098–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Soucy TA; Smith PG; Milhollen MA; Berger AJ; Gavin JM; Adhikari S; Brownell JE; Burke KE; Cardin DP; Critchley S An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458 (7239), 732–736. [DOI] [PubMed] [Google Scholar]
- (36).Subtil A; Gaidarov I; Kobylarz K; Lampson MA; Keen JH; McGraw TE Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. U. S. A 1999, 96 (12), 6775–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wei J; Hu J; Wang L; Xie L; Jin MS; Chen X; Liu J; Jin J Discovery of a First-in-Class Mitogen-Activated Protein Kinase Kinase 1/2 Degrader. J. Med. Chem 2019, 62 (23), 10897–10911. [DOI] [PubMed] [Google Scholar]
- (38).Kang CH; Lee DH; Lee CO; Du Ha J; Park CH; Hwang JY Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem. Biophys. Res. Commun 2018, 505 (2), 542–547. [DOI] [PubMed] [Google Scholar]
- (39).Davies H; Bignell GR; Cox C; Stephens P; Edkins S; Clegg S; Teague J; Woffendin H; Garnett MJ; Bottomley W; Davis N; Dicks E; Ewing R; Floyd Y; Gray K; Hall S; Hawes R; Hughes J; Kosmidou V; Menzies A; Mould C; Parker A; Stevens C; Watt S; Hooper S; Wilson R; Jayatilake H; Gusterson BA; Cooper C; Shipley J; Hargrave D; Pritchard-Jones K; Maitland N; Chenevix-Trench G; Riggins GJ; Bigner DD; Palmieri G; Cossu A; Flanagan A; Nicholson A; Ho JW; Leung SY; Yuen ST; Weber BL; Seigler HF; Darrow TL; Paterson H; Marais R; Marshall CJ; Wooster R; Stratton MR; Futreal PA Mutations of the BRAF gene in human cancer. Nature 2002, 417 (6892), 949–954. [DOI] [PubMed] [Google Scholar]
- (40).Soda M; Choi YL; Enomoto M; Takada S; Yamashita Y; Ishikawa S; Fujiwara S; Watanabe H; Kurashina K; Hatanaka H; Bando M; Ohno S; Ishikawa Y; Aburatani H; Niki T; Sohara Y; Sugiyama Y; Mano H Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448 (7153), 561–566. [DOI] [PubMed] [Google Scholar]
- (41).Morris SW; Kirstein MN; Valentine MB; Dittmer KG; Shapiro DN; Saltman DL; Look AT Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263 (5151), 1281–1284. [DOI] [PubMed] [Google Scholar]
- (42).Choi YL; Soda M; Yamashita Y; Ueno T; Takashima J; Nakajima T; Yatabe Y; Takeuchi K; Hamada T; Haruta H; Ishikawa Y; Kimura H; Mitsudomi T; Tanio Y; Mano H EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N. Engl. J. Med 2010, 363 (18), 1734–1739. [DOI] [PubMed] [Google Scholar]
- (43).Lin JJ; Zhu VW; Yoda S; Yeap BY; Schrock AB; Dagogo-Jack I; Jessop NA; Jiang GY; Le LP; Gowen K; Stephens PJ; Ross JS; Ali SM; Miller VA; Johnson ML; Lovly CM; Hata AN; Gainor JF; Iafrate AJ; Shaw AT; Ou SI Impact of EML4-ALK Variant on Resistance Mechanisms and Clinical Outcomes in ALK-Positive Lung Cancer. J. Clin. Oncol 2018, 36 (12), 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang C; Han XR; Yang X; Jiang B; Liu J; Xiong Y; Jin J Proteolysis Targeting Chimeras (PROTACs) of Anaplastic Lymphoma Kinase (ALK). Eur. J. Med. Chem 2018, 151, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Sugano K; Kansy M; Artursson P; Avdeef A; Bendels S; Di L; Ecker GF; Faller B; Fischer H; Gerebtzoff G Coexistence of passive and carrier-mediated processes in drug transport. Nat. Rev. Drug Discovery 2010, 9 (8), 597–614. [DOI] [PubMed] [Google Scholar]
- (46).Zhao R; Matherly LH; Goldman ID Membrane transporters and folate homeostasis: intestinal absorption and transport into systemic compartments and tissues. Expert Rev. Mol. Med 2009, 28 (11), e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Galdeano C; Gadd MS; Soares P; Scaffidi S; Van Molle I; Birced I; Hewitt S; Dias DM; Ciulli A Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem 2014, 57 (20), 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Zhang J; Bu X; Wang H; Zhu Y; Geng Y; Nihira NT; Tan Y; Ci Y; Wu F; Dai X; Guo J; Huang YH; Fan C; Ren S; Sun Y; Freeman GJ; Sicinski P; Wei W Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553 (7686), 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gao Y; Nihira NT; Bu X; Chu C; Zhang J; Kolodziejczyk A; Fan Y; Chan NT; Ma L; Liu J; Wang D; Dai X; Liu H; Ono M; Nakanishi A; Inuzuka H; North BJ; Huang YH; Sharma S; Geng Y; Xu W; Liu XS; Li L; Miki Y; Sicinski P; Freeman GJ; Wei W Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat. Cell Biol 2020, 22 (9), 1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.