

Abstract
Epithelial–mesenchymal transition (EMT) is an important biological process through which epithelial cells undergo phenotypic transitions to mesenchymal cells by losing cell–cell adhesion and gaining migratory properties that cells use in embryogenesis, wound healing, and cancer metastasis. An important research topic is to identify the underlying gene regulatory networks (GRNs) governing the decision making of EMT and develop predictive models based on the GRNs. The advent of recent genomic technology, such as single-cell RNA sequencing, has opened new opportunities to improve our understanding about the dynamical controls of EMT. In this article, we review three major types of computational and mathematical approaches and methods for inferring and modeling GRNs driving EMT. We emphasize (1) the bottom-up approaches, where GRNs are constructed through literature search; (2) the top-down approaches, where GRNs are derived from genome-wide sequencing data; (3) the combined top-down and bottom-up approaches, where EMT GRNs are constructed and simulated by integrating bioinformatics and mathematical modeling. We discuss the methodologies and applications of each approach and the available resources for these studies.
Keywords: bottom-up approach, epithelial-mesenchymal transition, gene regulatory network, network construction, network modeling, top-down approach
1. INTRODUCTION
Epithelial–mesenchymal transition (EMT) is an important cellular process, during which epithelial cells (E) convert to mesenchymal cells (M) by changing their morphology from cobblestone shape to spindle shape, losing tight cell–cell adhesion, and gaining motility and invasiveness [1, 2]. EMT and its reverse process, mesenchymal–epithelial transition (MET), have been shown to play a crucial role in multiple biological phenomena, such as embryonic development, wound healing, and cancer metastasis [3]. It is worth noting that recent studies have identified a spectrum of hybrid EMT states, featuring the coexistence of both E and M traits [4, 5]. In a hybrid state, cells retain cell–cell adhesion and meanwhile become motile, thus allowing collective cell migration, a phenomenon related to cancer invasiveness [6].
To understand the properties of the EMT-related state transitions, many experimental and computational studies have been undertaken to elucidate the gene regulatory mechanisms driving EMT. In particular, substantial efforts have been made with computational systems biology approaches to model EMT gene regulatory networks (GRNs). A high-quality GRN model can enhance our understanding of the molecular drivers of EMT, the relationship between various EMT states, and the coupling of EMT with other biological processes. GRN models also allow us to generate new predictions, such as the outcomes of gene knockdown, which lead to testable hypotheses for new experimental studies. So far, the existing network modeling studies can be categorized into three types: (1) the bottom-up approach, where GRNs are derived from the analysis and synthesis of literature data, followed by mathematical modeling for network dynamics simulations; (2) the top-down approach, where GRNs are derived from genomics data, such as gene expression, by bioinformatics methods featuring statistical analysis; (3) a more recent methodology that integrates both the bottom-up and top-down approaches, typically involving both bioinformatics and network simulations (Figure 1). Here, we will explain and review these types of computational and theoretical studies on EMT GRN modeling. For each approach, we will discuss the methodology and its applications and the currently available resources for its studies.
FIGURE 1.
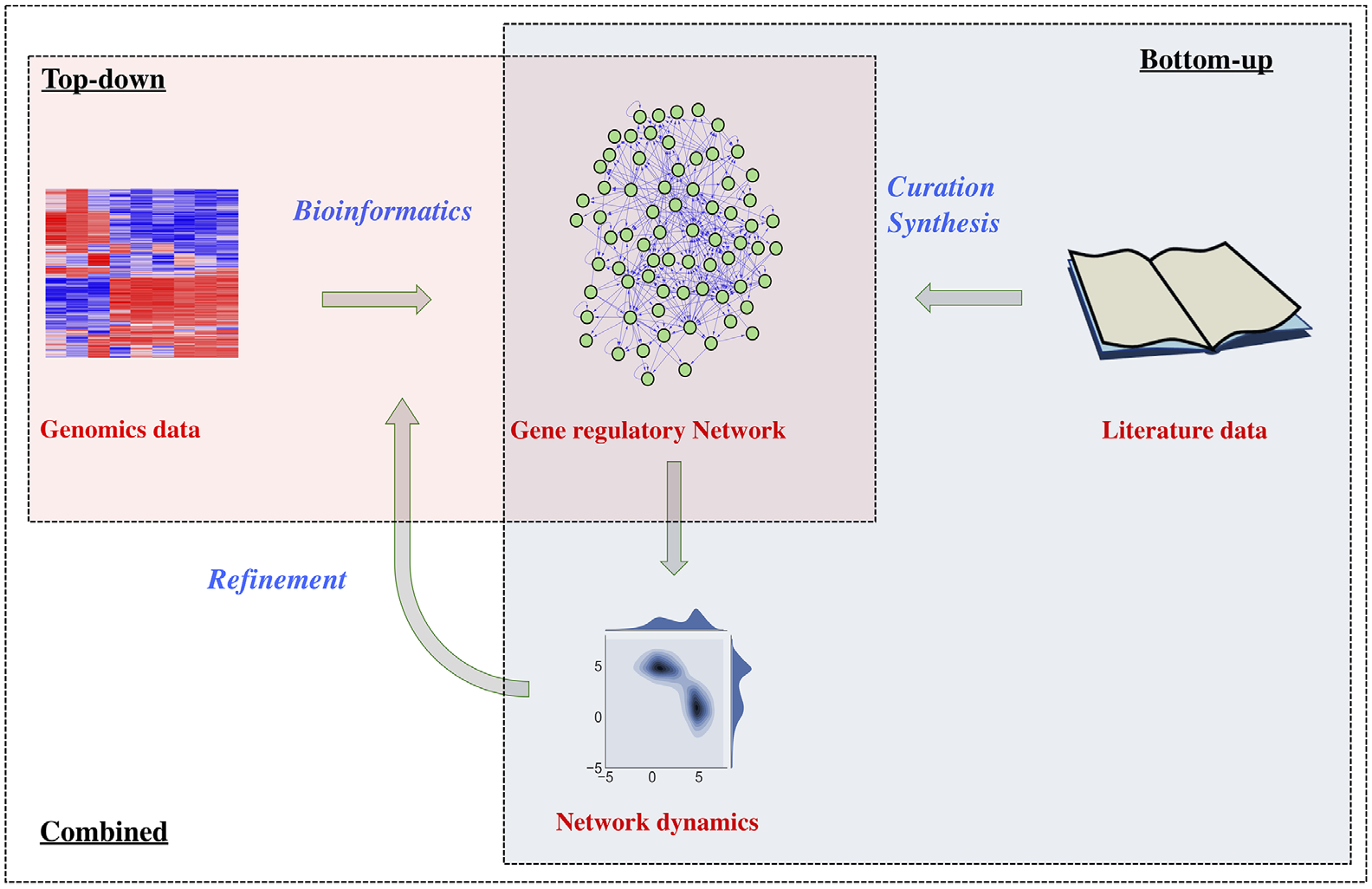
Graphical summary of the three broad approaches discussed in this review. (A) Bottom-up approaches (blue shaded box): manual literature review or database search is used to construct a GRN, which is then modeled with mathematical modeling to elucidate the network gene expression dynamics and the property of network control. GRNs are usually constructed based on simulation outcomes. (B) Top-down approaches (red shaded box): algorithmic and statistical tools are used to infer GRNs from various omics assays of a dataset of interest. GRNs are usually constructed based on statistical test. (C) Integrated top-down and bottom-up approaches (outermost box): these studies combine mathematical modeling, with an emphasis on the system dynamics, with high-throughput bioinformatics assays to construct GRNs that agree with both specific experimental observations and general understanding of EMT dynamics
2. THE BOTTOM-UP APPROACH
The most common and popular approach for modeling EMT GRNs relies on an extensive literature search for biological evidence of gene regulatory interactions, from which researchers assemble a gene network. Mathematical modeling is then applied to the constructed GRNs to evaluate their gene expression dynamics. A good GRN model can not only capture the essential dynamical behavior of a biological system, but also provides new testable predictions for experimental validation, shedding new insights and permitting a deeper understanding of the system. Due to extensive previous studies on EMT [7, 8], abundant biological evidence for gene regulatory interactions during EMT is available, particularly in the area of cancer research [9, 10] and developmental biology [11]. These experimental studies have led to some successful modeling efforts on EMT GRNs [12–14], where literature-based GRNs were simulated to elucidate the heterogeneity of EMT states and the control mechanism of the cellular state transitions between them. These simulation studies have generated new predictions, which can then be tested experimentally.
GRN models in the bottom-up approach can be of three categories: those that focus on a core gene regulatory circuit of EMT master regulators, those that model a large GRN of detailed gene regulators and/or upstream signaling pathways, and those that investigate the coupling of EMT circuit with circuits of other biological processes. In the following, we will describe the research efforts in those directions.
2.1. Small EMT circuits
In a typical study using the bottom-up approach, one synthesizes the literature data to construct a small circuit model, from which one elucidates its regulatory mechanism. Some early modeling studies on EMT GRNs focused on core gene regulatory circuits, consisting of the EMT master regulators: two microRNA families miR34 and miR200 and two transcription factor families ZEB and SNAIL [15, 16]. These models incorporated signaling nodes such as transforming growth factor beta (TGF-β) to drive the circuits and some targeted genes such as CDH1 and VIM as circuit readout. Because of the essential role of microRNAs in the translational regulation of key transcription factors [17], new mathematical formalisms were introduced [15, 18] to model microRNA-mediated translational inhibition and mRNA degradation. A typical way to model a GRN is to first write down the chemical rate equations (typically ordinary differential equations) and then apply nonlinear dynamics methods, such as nullcline and bifurcation, to identify the possible stable steady states of the GRN. These ordinary differential equation (ODE)-based modeling studies predicted not only the epithelial (E) and mesenchymal (M) states, but also a hybrid state (E/M) with features of both epithelial and mesenchymal phenotypes. The predicted hybrid E/M state was later identified experimentally [6], and its important role was found in tumorigenesis [19, 20]. Core EMT circuit models have also been carefully evaluated [21] and validated experimentally, for example, by flow cytometry measurement of E-cadherin and vimentin in TGF-β1-induced EMT of MCF10A cell line [22]. Furthermore, energy landscape analysis has been applied to the core EMT circuit, from which access to a hybrid state was shown to depend on the extracellular environment [23].
These circuit models have further been extended to incorporate additional genes, such as OVOL2, GRHL2, Np63α, NFATc, and NRF2, with a research focus on their role in stabilizing/destabilizing the hybrid state in cancer metastasis [18, 24–28]. Recently, Celia-Terrassa et al. characterized two distinct types of EMT dynamics (hysteretic and nonhysteretic) through their ODE/PDE-based modeling of a small EMT circuit (TGF-β, Zeb1/2, miR-200, and E-cadherin) and identified their association with metastasis and clinical outcomes using mouse models [29].
Overall, these studies of the EMT circuits demonstrate the usefulness of investigating small circuit models, typically constructed based on expert knowledge in the EMT literature. Mathematical modeling of these small EMT circuits sheds light on a mechanistic understanding of EMT. However, in some cases certain regulators of interest may not be captured by a small EMT circuit, therefore researchers are also interested in constructing and modeling larger EMT GRNs.
2.2. Large EMT networks
Construction of large EMT GRNs relies on more extensive literature search, typically incorporating (1) more detailed gene components, including factors from the same gene families, (2) signaling pathways upstream to EMT master regulators, and (3) in some cases, readout nodes representing the downstream EMT-related processes.
In particular, Steinway et al. synthesized existing literature data and constructed a 70-node EMT network representing the conserved regulation of EMT [30]. They gathered interactions primarily from hepatocellular carcinoma (HCC) EMT and secondarily from other tissue types, which produced an EMT GRN incorporating different molecular processes involving growth factors, signal transduction pathways, and transcription regulators. They simulated the GRN using a Boolean network model to understand the signaling abnormalities in the HCC progression [30] and their implication of combinatorial therapy by gene perturbation [31]. Here, Boolean network models describe the node status (gene expression or activity of a biological process) with two discrete values (i.e., 0 and 1) and simulate the network dynamics by updating the node status using Boolean functions [32]. Font-Clos et al. extended the GRN to a 72-node network and performed Boolean network modeling to construct a topographic map [33]. They studied the phenotypic stability of the topographic landscape using Ising model, where they identified a series of metastable hybrid EMT states, a prediction that is supported by RNA-seq data from both lung adenocarcinoma and embryonic differentiation. In a recent study, Silveira et al. constructed an 18-node literature-based EMT GRN to simulate EMT using Boolean network modeling [34]. In addition, some researchers augmented the literature-based approach by incorporating bioinformatics methods to construct larger EMT networks [35, 36] (details in Section 6).
Besides, Huang et al. [37] extended a core EMT circuit [15, 16] to a 22-node GRN by incorporating EMT factors from Ingenuity Pathway Analysis [38] and additional literature data [30, 31, 39, 40]. Instead of using Boolean network modeling as in many other large network studies, they devised a modeling method named random circuit perturbation (RACIPE), an ODE-based modeling method to generate an ensemble of kinetic models corresponding to a fixed GRN topology. Because RACIPE allows to model the time dynamics of continuous gene expression levels, it can better capture the intermediate levels of gene expression and is more effective to characterize hybrid states of a GRN, as supported by a recent study that compared RACIPE and Boolean simulations for various EMT GRNs of different sizes [41]. Also, with the RACIPE framework, Kohar and Lu showed that stochasticity in gene regulation and cell-to-cell variability can stabilize these hybrid EMT states [42].
In summary, by carefully integrating an extensive collection of literature data, researchers have developed large size EMT GRNs, from which the dynamic features of cellular state transitions can be identified. However, EMT is not a standalone process, but tightly associated with other biological processes, including, but not limited to, intercellular communication by Notch signaling pathway, cell motility, metabolism, cell proliferation, stem cell differentiation, and immunity.
2.3. EMT circuits coupled with other processes
Many efforts have been made to understand the role of Notch signaling pathway in regulating EMT-induced cell motility during normal development and cancer metastasis [43–45]. These led to modeling the coupling between the Notch–Delta signaling pathway and the EMT core circuit, which provided a mechanistic understanding of how the hybrid E/M state induces and maintains the metastatic cellular clusters via intercellular communication [46, 47]. Cohen et al. developed a 30-node Boolean network to study synergistic combination of Notch overexpression and p53 deletion in cancer metastasis [39].
Furthermore, several studies modeled the coupling between metabolic pathways, EMT, and metastasis. Yu et al. constructed a coarse-grained 4-node network model of two metabolic pathways glycolysis and oxidative phosphorylation (OXPHOS) to study the interplay between the two pathways and the gene regulation of metabolic plasticity [48], with the implication of their roles in cancer metastasis. Jia et al. further extended the network with detailed interactions among regulatory genes and metabolites; their modeling predictions of metabolic plasticity were experimentally validated using several cancer cell lines [49]. Subsequently, Kang et al. [50] modeled, with a landscape approach, a 16-node metabolism-EMT-metastasis network that integrates metabolism circuit [48], EMT core circuit [15, 51, 52], and metastasis circuit [53]. Some recent mathematical models focused on mechanical interactions to understand the gene regulation of cells losing cellular cohesion during EMT [54–56].
In summary, researchers employed the bottom-up approaches to construct EMT GRNs of different sizes, whose mathematical modeling elucidated the regulatory mechanism of EMT and its coupling with other pathways. Despite its success in modeling EMT GRN, the bottom-up approach is typically limited by the following factors: (1) literature synthesis can be quite tedious and time consuming; (2) because of the involvement of significant manual curation, it is not straightforward to reproduce literature-based GRNs; (3) there may not be sufficient literature data to investigate the EMT process in a particular biological context. Additional information on the bottom-up approach can also be found in some recent reviews [14, 57–59].
3. THE TOP-DOWN APPROACH
Another approach to model EMT GRNs is a top-down approach of constructing networks from bioinformatics algorithms using genome-wide sequencing data, such as transcriptomics data (bulk and single-cell RNA-seq) and epigenomics data (Assay for Transposase-Accessible Chromatin [ATAC-seq], chromatin immunoprecipitation [ChIP-seq]). These genome-wide data can be utilized to unbiasedly infer transcription factor (TF)-target relations based on statistical association (such as correlation, mutual information and regression) and their occurrence in experimental and literature databases (such as TF-target databases and TF binding motif database) [60–64]. One advantage of these top-down approaches is that they help tailor the GRN to the dataset of interest by emphasizing interactions reflected in the (epi)genomics data [65–68]. Compared to bottom-up approaches, top-down approaches also streamline the network construction process, making GRN modeling analysis more efficient and reproducible. On the other hand, the top-down approaches usually lead to large GRNs, therefore the network construction is more liable to overfitting and is more adversely affected by sparsity and noise in the data [69]. Moreover, most bioinformatics-based GRNs are not evaluated according to their ability to capture network dynamics. Below, we will summarize the basic components of top-down methodologies and describe some of their recent applications to EMT.
3.1. Bioinformatics algorithms for GRN construction
The increasing availability of multiple omics studies represents an opportunity to develop a new, more cohesive model of EMT regulation. Indeed, a rich resource of transcriptomics and epigenomics data are publicly available on the study of EMT GRNs, as summarized in Table 1 [5, 65, 67, 68, 70–92]. Many bioinformatics algorithms have been developed to construct GRNs from these resources [60–64, 69]. In recent years, scRNA-seq data have become particularly popular for GRN construction, mainly because of rapid advances in genomic technology and computational methodologies. In the study of EMT, single-cell transcriptomics can be especially important for the discovery of cell phenotypic heterogeneity and the dynamical transitions between cellular states [5, 36, 93]. Thus, network construction using scRNA-seq is more likely to generate networks capturing these features of EMT.
TABLE 1.
Published EMT transcriptomics and epigenomics datasets
| Assay type | Assay name | Experiment | Description | Reference |
|---|---|---|---|---|
| Transcriptomic | Microarray | GSE121372 | Human HPMCs treated with TGF-b1 | Han et al.,2019 [73] |
| GSE88762 | EMT in mouse tumor-initiating cells | Latil et al., 2017 [76] | ||
| GSE87877 | EMT in mouse tumor-initiating cells | Latil et al., 2017 [76] | ||
| GSE53923 | Ovol2 in EMT in mouse terminal end buds | Watanabe et al., 2014 [89] | ||
| GSE53175 | EMT in a breast cancer primary culture | Minafra et al., 2014 [79] | ||
| GSE39368 | Molecular subtypes of head and neck cancer | Walter et al., 2013 [86] | ||
| GSE42373 | TGF-β/TNF-α-treated A549 spheroids | Wamsley et al., 2015 [87] | ||
| GSE17708 | Time course of A549 cells treated with TGF-β | Sartor et al., 2010 [82] | ||
| GSE17538 | Four experiments, colon cancer in humans and mice | Smith et al., 2010 [83] | ||
| GSE9691 | E-cadherin loss in human epithelial cells | Onder et al., 2008 [80]; Taube et al., 2010 [84] | ||
| RNA-seq | GSE145850 | MCF10A cells treated with TGF-β | Johnson et al., 2020 [75] | |
| GSE124843 | Perturbing TGF-β and ZEB1 in MCF10A | Watanabe et al., 2019 [90] | ||
| GSE110585 | Hybrid EMT states in mouse tumor tissues | Pastushenko et al., 2018 [81] | ||
| GSE70741 | hESC differentiation into hepatoctyes | Li et al., 2017 [77] | ||
| GSE88989 | EMT in mouse tumor-initiating cells | Latil et al., 2017 [76] | ||
| GSE59987 | Hypoxia-induced EMT in human cancer cells | Tsai et al., 2014 [85]; Wang et al., 2020 [88] | ||
| scRNA-seq | GSE147405 | Time course scRNA-seq in human cancer cell lines | Cook et al., 2020 [65] | |
| GSE134432 | scRNA-seq and ATAC-seq of melanoma tissues | Wouters et al., 2020 [92] | ||
| GSE135893 | EMT in pulmonary fibrosis and healthy lungs | Habermann et al., 2020 [72] | ||
| GSE114687 | EMT in MCF10A and HuMEC cells | McFaline-Figueroa et al., 2019 [78] | ||
| GSE137749 | Two triple knockout SCLC mouse models | Wooten et al., 2019 [91] | ||
| GSE110357 | Hybrid EMT states in mouse tumor tissues | Pastushenko et al., 2018 [81] | ||
| GSE114397 | TGF-β-induced EMT in HMLE cells | van Dijk et al., 2018 [71] | ||
| GSE100037 | Mouse bone marrow lymphoid progenitors | Herman et al., 2018 [74] | ||
| GSE87038 | EMT in mouse organogenesis | Dong et al., 2018 [5] | ||
| GSE103322 | Head and neck cancer | Puram et al., 2017 [67] | ||
| Epigenomic | ChIP-seq | GSE80218 | Hypoxia-regulated EMT in FADU cell line | Wang et al., 2020 [88] |
| GSE61198 | EMT in normal and cancerous mouse stem cells | Ye et al., 2015 [68] | ||
| GSM1303689 | Ovol2 ChIP-seq in mouse terminal end buds | Watanabe et al., 2014 [89] | ||
| GSE42374 | TGF-β/TNF-α-treated A549 spheroids | Cieslik et al., 2013 [70] | ||
| ATAC-seq | GSE145851 | TGF-β-induced EMT in MCF10A cells | Johnson et al., 2020 [75] | |
| GSE134432 | scRNA-seq and ATAC-seq of melanoma tissues | Wouters et al., 2020 [92] | ||
| GSE114397 | TGF-β-induced EMT in HMLE cells | van Dijk et al., 2018 [71] | ||
| GSE110584 | Hybrid EMT states in mouse tumor tissues | Pastushenko et al., 2018 [81] | ||
| GSE70474 | EMT in mouse tumor-initiating cells | Latil et al., 2017 [76] | ||
| hMeDIP-seq | GSE59989 | Hypoxia-induced EMT in human cancer cells | Tsai et al., 2014 [85] |
Although different GRN construction methods have their own approaches, they typically deploy common steps of bioinformatic analyses as part of their algorithms. In the following, we will take scRNA-seq data as an example to illustrate these bioinformatic techniques. First, the raw sequencing data need to be aligned to a reference genome and converted to gene expression counts [94]. Second, the count data are normalized by gene length and library size and log-transformed [95]. The gene expression data must also be processed to correct batch effects and/or remove cells/genes with low counts [96]. Third, having preprocessed the data, one can perform certain downstream analyses such as (1) visualizing the transcriptomic landscape via dimensional reduction [97] (principal component analysis [PCA] [98], t-stochastic neighbor embedding [t-SNE] [99], uniform manifold approximation and projection [UMAP]) [100]; (2) identifying distinct cellular phenotypes by gene expression clustering [101] (k-means, hierarchical clustering, etc.); (3) identifying important genes, pathways, or gene ontology (GO) terms that are distinct between the cellular phenotypes using differential expression analysis [102] (limma [103], DESeq2) [104] and gene-set-based enrichment analysis (GSEA [105, 106], GSA [107], GSVA) [108]; (4) inferring pseudo-time [109] in the case that time series data are unavailable.
Finally, there is a growing suite of software packages designed to analyze single-cell sequencing data, some of which have provided functionality for GRN construction. For example, a package termed single-cell regulatory network inference and clustering (SCENIC) [60] works by identifying highly correlated modules of genes and cross-referencing these with TF binding motifs from the cisTarget database [110]. Another method, Dynamic Regulatory Events Miner (DREM) [111, 112], can be used to construct dynamic GRNs from time series data by identifying timepoints where coexpressed genes diverge, using GO terms to annotate the biological mechanisms behind each split. Other tools like Cicero [113] are used to construct GRNs from chromatin accessibility data instead of RNA-seq by identifying regulatory elements coaccessible with gene promoters [63, 114]. Recently, Pratapa et al. [69] developed a framework entitled BEELINE to evaluate the quality of network construction algorithms using scRNA-seq data based on criteria including accuracy, scalability, and the level of detail they output. The authors benchmarked 12 network construction algorithms with several simulated and experimental datasets with known network topologies and identified PIDC [115], GENIE3 [116], and GrnBoost2 [117] as having the best overall performance. They also found that inaccurate pseudo-time labels can be detrimental, and that many methods infer edges where only an indirect relationship exists, creating unintended feedforward structures. In summary, computational methods for GRN construction are growing in number and sophistication. Although sparsity and noise remain challenging obstacles, these tools provide an accessible framework to infer regulatory links from transcriptomics and epigenomic data.
3.2. EMT GRN Construction
An example that encapsulates the top-down approach to EMT modeling is a 2016 work from Chang et al. [118] where the authors uncovered synergistic behavior of three EMT regulators: ETS2, HNF4A, and JUNB. The authors first performed RNA-seq on TGF-β treated A549 cells over a period of 96 h, identifying three distinct, sequentially activated groups of genes, which they associate to E, hybrid, and M phenotypes. GO terms confirmed these findings, as the gene sets enriched in the hybrid and M cells were increasingly related to cell motility and adhesion. Interestingly, however, certain canonical EMT markers like SNAI1/2, TWIST1/2, and ZEB1/2 did not appear to be key regulators in this dataset. Hypothesizing that important transcription factors (TFs) may have been as yet unknown, the authors performed binding motif enrichment for putative EMT TFs based on the time series data. They then examined ChIP-seq data, finding additional evidence that the candidate TFs indeed bind to the locations of hundreds of differentially expressed genes in the experiment. Finally, the authors applied DREM to the time series data to pinpoint temporal changes in regulation. Major splitting points were identified at the 6-h and 48-h timepoints and included regulatory changes among the previously indicated TFs, possibly reflecting transitions from E to hybrid and hybrid to M states, respectively. The approach adopted by Chang et al. permitted a thorough and contextual analysis of EMT in A549 cells, despite the apparent lack of activity among many canonical EMT factors. By examining EMT on the basis of multi-gene signatures and quantified trends in gene expression, top-down approaches thus stand to improve the accuracy and applicability of EMT GRNs.
Top-down approaches can also reveal context specific (i.e., dependent on tissue type, time, input signal, etc.) EMT regulatory mechanisms, by applying inference tools to transcriptomic data or epigenetic sequencing like ATAC-seq [65, 67, 68]. Cook and Vanderhyden recently examined four cancer cell lines undergoing EMT induced via three different signaling conditions, using time series measurements to observe distinct trajectories and patterns of TF activity according to the context of the EMT [65]. Only a small number of the genes that responded to the three signals were shared across all conditions, demonstrating how much context can influence the EMT regulatory network. In another study, Wouters et al. [92] constructed GRNs based on SOX10 KD-induced EMT in melanoma at various timepoints by taking the consensus results of SCENIC over 100 runs, supplementing SCENIC’s use of TF binding motifs with ATAC-seq chromatin accessibility information. The authors found that much of the data could be explained by a spectrum of melanocytic, intermediate, and mesenchymal-like phenotypes, noting that the consensus GRN for intermediate states was a stable mixture of regulations from both extreme phenotypes. The authors leveraged software tools and public repositories to map the EMT trajectory in melanoma with a high degree of detail. Although algorithmic GRN inference has far to go, in the case of EMT many of these tools have proven capable of recapitulating known findings and identifying new and/or cell type-specific regulatory interactions.
Top-down, bioinformatic-based approaches to model EMT have proven useful in characterizing the transcriptomic landscape of EMT and even in algorithmically constructing GRNs. This approach permits a thorough analysis of the phenotypic space, with single-cell sequencing providing the necessary granularity to construct GRNs that accurately reflect the observed distribution of cell states. Additionally, computational tools can make GRN construction more efficient, scalable, and reproducible. However, despite many available tools and datasets, constructing highly accurate EMT GRNs from bioinformatics results alone has proven challenging. Feature measurements are often noisy, impacting the accuracy of downstream analyses. Additionally, it remains challenging to distinguish, directly from the data, different types of regulation (e.g., methylation, transcriptional, translational) and identify key regulators. As a result, automatically constructed networks are prone to contain redundant structures or spurious links between genes that may be in shared modules, but do not directly interact [64, 69]. Although top-down analyses are especially useful for examining phenotypic heterogeneity, algorithmically constructed GRNs can benefit greatly from additional validation or optimization. Mathematical network modeling is thus a natural progression from bioinformatics approaches; inferred GRNs can be integrated into dynamical models and interactions iteratively refined by examining their dynamical properties in comparison to experimentally observed behaviors.
4. COMBINED TOP-DOWN AND BOTTOM-UP APPROACH
To overcome the limitations of both the bottom-up and top-down approaches, some recent studies seek to combine mathematical modeling with bioinformatic network construction. This approach offers a number of potential advantages as follows. First, EMT transition paths and key regulators can depend on the system in which they occur, so networks pulled together from general databases and literature search may not be relevant to a particular system of interest [65, 78, 119]. In these scenarios, bioinformatic analysis on associated transcriptomics and epigenetic data can contribute to incorporate context-specific regulatory relationships [35, 66, 91]. Second, the combined approach can improve the quality of the GRNs constructed by bioinformatics methods, as mathematical modeling can evaluate whether the GRNs can capture the gene expression dynamics of the biological process. Ideally, this approach combines the features of simplicity and predictivity from the bottom-up approach and the features of reproducibility and robustness to literature bias/errors from the top-down approach. Examples of studies with combined approaches and their corresponding methodology are summarized in Table 2 [35, 36, 42, 66, 91, 120–123].
TABLE 2.
Combined-approach study methodologies
| Reference | Subject | Modeling | Bioinformatics | Integration |
|---|---|---|---|---|
| Examples of GRN modeling of EMT | ||||
| Khan et al., 2017 [35] | E2F-mediated EMT in cancer | Boolean network simulations and in silico perturbations | E2F family interactions curated from TRANSFAC, STRING, HPRD, MiRTarBase; >98% validated by domain experts | GRNs for breast and bladder cancer constructed by ranking global network motifs by (1) topological properties, (2) agreement with gene expression in target datasets, (3) agreement with KEGG cancer pathways |
| Udyavar et al., 2017 [36]; Wooten et al., 2019 [91] | EMT in SCLC | Developed BooleaBayes, a Boolean network modeling framework that can also estimate probabilities | Clustering, weighted gene coexpression network analysis (WGCNA), and GRN inference with ARACNE filtered with TF-target databases, literature review | Boolean network modeling to predict multiple SCLC subtypes and subtype-specific master regulators |
| Kohar and Lu, 2018 [42] | EMT in SCC | Ensemble ODE-based simulations with RACIPE and stochastic noise | Incorporated GRNs from a previous study on Epcam+ and Epcam− cells using RNA-seq and ATAC-seq | Combination of manually curated core EMT network with SCC-specific networks from previous genome-wide study |
| Ramirez et al., 2020 [66] | EMT in cancer | Ensemble ODE-based simulations with RACIPE | SCENIC used to infer GRNs for each dataset and identify conserved and context-specific interactions | Iterative GRN construction and SCENIC parameter optimization by comparing simulated and experimental data |
| Sha et al., 2020 [123] | EMT in cancer and embryogenesis |
Stochastic ODE-based multiscale simulation of a core EMT circuit | QuanTC is developed, which identifies clusters, marker genes, and transition genes from scRNA-seq data | QuanTC applied to multiple EMT datasets to validate the behaviors predicted by the model |
| Examples of GRN modeling of other processes | ||||
| Moignard et al., 2015 [120] | Mouse hematopoiesis | Boolean network modeling | Single-cell quantitative reverse transcription polymerase chain reaction (qRT-PCR) on ~40 genes; Developed single-cell network synthesis (SCNS) toolkit to construct Boolean networks from discretized expression data | Using SCNS, a GRN was constructed to identify key regulators, which were later validated experimentally |
| Dunn et al., 2014 [122]; Dunn et al., 2019 [121] | mESCs | Abstract Boolean network (ABN) modeling—ensemble Boolean networks based on experimental constraints | Initial coexpression network from microarray and RNA-seq data, qRT-PCR and clonal assays with siRNA to test model predictions | Iteratively refined a meta-model of multiple Boolean networks by experimentally validating model predictions |
One approach that combines top-down and bottom-up methodologies is to first construct a large network, then identify subnetworks that describe EMT in different contexts. Khan et al. [35] constructed an 879-node, 2278-edge network for the E2F TF family based on extensive manual review of published literature, characterizing its role in processes including EMT, cell cycle, DNA repair, and apoptosis. This large network, while comprehensive, would be unwieldy to investigate in the context of specific tumor types. Therefore, the authors identified subnetworks that described EMT in breast and bladder cancer by identifying the most important network structures in each type. Motifs were ranked on multiple metrics including involvement in cancer pathways, fold-change between invasive and noninvasive specimens, and topological properties. They conducted Boolean simulations on 41- and 35-node subnetworks for bladder and breast cancer respectively, finding unique combinatorial EMT-inducing signals, each associated with more aggressive tumors of their respective tissue type in The Cancer Genome Atlas (TCGA) cohort data [35]. By ranking key network motifs according to multiple factors including transcriptomics data and topological properties, Khan et al. facilitate the construction of GRNs that are highly representative of specific biological conditions.
Udyavar et al. [36] describes another integrated study examining subtypes of small cell lung cancer (SCLC). Using ARACNE, a large network was generated and subsequently filtered by cross-referencing with multiple binding site and databases including ENCODE [124], TRANSFAC [125], EnrichR [126], and PubMed [127]. Subsequent Boolean simulations of this GRN predicted the expected NE and ML subtypes, but failed to capture a hybrid phenotype present in tumor samples. In a follow-up work by Wooten et al., a Boolean modeling approach called BooleaBayes was developed that infers the probability that each node is ON or OFF based on gene expression patterns of similar states, allowing more nuanced relationships than traditional Boolean modeling. Conducting in silico perturbations with BooleaBayes revealed likely stabilizers and destabilizers of each SCLC subtype, suggesting targets for therapies aimed at driving SCLC tumors from an aggressive subtype to a more tractable one [91]. These studies together illustrate the complementary nature of top-down with bottom-up methods: the initial top-down GRN alone, while in agreement with experimental data, failed to accurately recapitulate the observed phenotypic landscape in a simple mathematical model. Integrating systematic validation against literature and binding motifs improved the model’s predictive capabilities, with a more sophisticated mathematical model finally bringing the simulated results into close agreement with observed data.
Another strategy for combining top-down and bottom-up methods is to begin from a well-supported, manually curated core topology and augment it with a context-specific set of interactions such that modeling can approximate the observed bioinformatic data. Kohar et al., integrated a literature-based GRN with networks from squamous cell carcinoma and modeled it with RACIPE. The GRN simulations accurately depict the E, M, and hybrid states observed in the gene expression data, with further improvements in accuracy when gene expression noise was implemented in the modeling [42]. The integration of SCC-specific topologies and well-established EMT motifs improved the agreement between the steady states predicted by RACIPE and those observed in the data. Furthermore, some efforts have been made to systematically generate the context specific interaction set while preserving the fundamental behavior of the core circuit. Ramirez et al. combined a core EMT topology with new interactions found by applying SCENIC to time series scRNA-seq data comparing EMT in four cell lines as induced by three different signaling conditions [65]. Considering each experimental condition separately, Ramirez et al. constructed, simulated, and refined context-specific GRNs by testing an ensemble of network construction parameters and finding the optimal GRN for each case. The primary criteria for inclusion in the network were (1) a correlation between regulator and target gene in the expression data for the relevant cell line, and (2) proximity to the core topology, as interactions were added incrementally moving outward from the core (both upstream and downstream). Although the resulting GRNs varied between experimental conditions, they included several highly conserved genes, suggesting that EMT may be governed by a small set of master regulators with flexible roles [66]. An iterative, optimization-based approach to network construction is expected to greatly improve the accuracy of EMT modeling studies.
This third category of studies, wherein networks are constructed using both broadly supported evidence from the literature and context-specific interactions from bioinformatics, then subsequently simulated with mathematical models, represents an evolution in quality and reproducibility in EMT modeling research. Integrated studies can not only identify genes of interest or infer individual regulatory links but can make testable predictions about complex dynamical behaviors and master regulators, facilitating the discovery of clinical tools targeting EMT. On the other hand, integrated methodologies are early in development, with few established best practices or formalized workflows, and some critical limitations. One obstacle is the breadth of background knowledge required to properly integrate bioinformatics with more traditional modeling approaches. Moreover, combined approaches tend to involve larger GRNs, which can be both more difficult to validate experimentally and more computationally expensive to model.
5. DISCUSSION AND PERSPECTIVES
One of the major challenges in biology is to understand the gene regulatory mechanisms that determine the decision making of cellular state transitions. In this paper, we reviewed three different types of computational systems biology approaches for modeling EMT-associated GRNs. The first approach relies on literature data for network construction. Being the gold-standard methodology in the field of systems biology, the literature-based method utilizes network interactions derived from dedicated experimental studies in biochemistry, cell biology and genetics, most of them having high accuracy. Thus, the literature-based approach results in high-quality GRNs to recapitulate existing biology. However, it may not work well in the case where biology literature is incomplete and/or inconsistent (e.g., in the studies of cancer biology) [128]. It is also tedious, time consuming, and error-prone to construct a large GRN. Note that, although most literature-based GRN modeling provides a list of experimental evidences for GRN regulatory interactions, little is usually given to describe how GRNs were constructed step by step, making most of the literature synthesis steps irreproducible. The literature-based approach also does not work well to study GRNs specific to a particular experimental condition, disease type, and subjects of certain genetic background.
The second approach constructs GRNs using bioinformatics analysis on genomics data from a specific experiment. Being a mainstream approach in current genomics and computational biology studies, it utilizes statistical analysis on gene expression data (e.g., bulk RNA-seq, scRNA-seq) and/or epigenetics data (such as ATAC-seq, Hi-C) to identify potential gene regulatory interactions. In some studies, literature data were also integrated, but in the format of a database containing curated gene regulatory interactions, biochemical/metabolic reactions, or from in silico prediction based on transcription factor binding sites. This approach addresses certain issues from the former approach—in particular, it allows modeling for a specific biological context and potentially identifying novel interactions. Because of the top-down approach, it usually results in GRNs of larger size. However, it has been shown that the current network construction methods are still insufficient to construct high-quality GRNs [69]. One of the issues is network redundancy. As many regulators and interactions between them are redundant in a biological system to achieve robustness, it is hard to reverse engineer the correct interactions back directly from data such as gene expression. Moreover, although bioinformatics is an ideal tool to identify regulators and biological pathways, it is seldom evaluated whether a GRN constructed through bioinformatics can operate as a dynamic biological system. This becomes a critical problem, particularly in the studies of cellular state transition like EMT, as network dynamics is an essential component of the biological process.
The third approach combines both the bottom-up and top-down approaches to construct GRNs. Conceptually, this is a better way to address the issues of the previous two approaches. By incorporating genomics data and literature databases together with mathematical modeling, one can model context specific GRNs that capture the dynamical behavior of cellular state transitions. We have seen recent studies on EMT GRN modeling with such a strategy, yet it remains a quite challenging task owing to the following reasons. First, systems biology modeling and bioinformatics belong to two very distinct research disciplines, making it difficult for researchers to grasp sufficient knowledge to be experienced in both research fields. Second, building a high-quality GRN model remains difficult with the combined approach. It is not uncommon that important regulators and/or signaling pathways, which are well known in the literature, cannot be identified from the genome-wide data directly. Thus, it is important to have better databases containing high-quality regulatory interactions and signaling pathways. More sophisticated computational algorithms are also needed to accurately identify context specific regulatory interactions, for example, by integrating a variety of types of genomics data and biological evidence. Third, as another central component of this approach, a powerful mathematical modeling algorithm is needed to capture the dynamics of a large GRN in an unbiased and efficient way. In particular, the ensemble-based approach in some recent studies seems to be a promising technique [37, 42, 66]. Last but not least, experimental validation is crucial for better GRN modeling. As the constructed GRNs can be especially large, it is important to devise validations, such as high-throughput gene perturbation, that allow to evaluate the quality of a large system. In summary, we foresee that the combined top-down and bottom-up approach, although still in its infancy, could be a powerful tool in the future GRN modeling studies on EMT and also other biological cellular state transitions.
Funding information
Northeastern University Startup Grant; University of Maine/Northeastern University Seed Grant; National Institutes of Health, Grant/Award Number: R35GM128717
REFERENCES
- 1.Nieto MA et al. , EMT: 2016, Cell 166 (2016), no. 1, 21–45. 10.1016/j.cell.2016.06.028 [DOI] [PubMed] [Google Scholar]
- 2.Nisticò P, Bissell MJ, and Radisky DC, Epithelial-mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metallo-proteinases, Cold Spring Harb. Perspect. Biol 4 (2012), no. 2, a011908. 10.1101/cshperspect.a011908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiery JP et al. , Epithelial-mesenchymal transitions in development and disease, Cell 139 (2009), no. 5, 871–890. 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 4.Bartoschek M et al. , Spatially and functionally distinct sub-classes of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun 9 (2018), no. 1. 10.1038/s41467-018-07582-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong J et al. , Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 19 (2018), no. 1. 10.1186/s13059-018-1416-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jolly MK et al. , Implications of the hybrid epithelial/mesenchymal phenotype in metastasis, Front. Oncol 5 (2015), 10.3389/fonc.2015.00155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dongre A and Weinberg RA, New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer, Nat. Rev. Mol. Cell Biol 20 (2019), no. 2, 69–84. 10.1038/s41580-018-0080-4 [DOI] [PubMed] [Google Scholar]
- 8.Kalluri R and Weinberg RA, The basics of epithelial-mesenchymal transition, J. Clin. Invest 119 (2009), no. 6, 1420–1428. 10.1172/JCI39104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu H et al. , The biological and clinical importance of epithelial-mesenchymal transition in circulating tumor cells, J. Cancer Res. Clin. Oncol 141 (2015), no. 2, 189–201. 10.1007/s00432-014-1752-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park S-M et al. , The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2, Genes Dev. 22 (2008), no. 7, 894–907. 10.1101/gad.1640608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim DH et al. , Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: A comprehensive overview, J. Clin. Med 7 (2017), no. 1. 10.3390/jcm7010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devaraj V and Bose B, The mathematics of phenotypic state transition: paths and potential, J. Indian Inst. Sci 100 (2020), no. 3, 451–464. 10.1007/s41745-020-00173-6 [DOI] [Google Scholar]
- 13.Jia D et al. , Epithelial-mesenchymal transition in cancer. In: Phenotypic switching (Levine H, Jolly MK, Kulkarni P, and Nanjundiah V, eds.), Academic Press, Cambridge, MA, 553–568, 2020. 10.1016/B978-0-12-817996-3.00018-9 [DOI] [Google Scholar]
- 14.Xing J and Tian X-J, Investigating epithelial-to-mesenchymal transition with integrated computational and experimental approaches, Phys. Biol 16 (2019), no. 3, 031001. 10.1088/1478-3975/ab0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu M et al. , MicroRNA-based regulation of epithelial–hybrid–mesenchymal fate determination, Proc. Natl. Acad. Sci 110 (2013), no. 45, 18144–18149. 10.1073/pnas.1318192110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian X-J, Zhang H, and Xing J, Coupled reversible and irreversible bistable switches underlying TGFβ-induced epithelial to mesenchymal transition, Biophys. J 105 (2013), no. 4, 1079–1089. 10.1016/j.bpj.2013.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burk U et al. , A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9 (2008), no. 6, 582–589. 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hong T et al. , An Ovol2-Zeb1 mutual inhibitory circuit governs bidirectional and multi-step transition between epithelial and mesenchymal states, PLOS Comput. Biol 11 (2015), no. 11, e1004569. 10.1371/journal.pcbi.1004569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao T-T and Yang M-H, Hybrid epithelial/mesenchymal state in cancer metastasis: Clinical significance and regulatory mechanisms, Cells 9 (2020), no. 3, 10.3390/cells9030623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y and Weinberg RA, Epithelial-to-mesenchymal transition in cancer: complexity and opportunities, Front. Med 12 (2018), no. 4, 361–373. 10.1007/s11684-018-0656-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia D et al. , Distinguishing mechanisms underlying EMT tristability. Cancer Convergence. 1 (2017), no. 1. 10.1186/s41236-017-0005-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J et al. , TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci. Signal 7 (2014), no. 345, ra91–ra91. 10.1126/scisignal.2005304. [DOI] [PubMed] [Google Scholar]
- 23.Li C, Hong T, and Nie Q, Quantifying the landscape and kinetic paths for epithelial–mesenchymal transition from a core circuit, Phys. Chem. Chem. Phys 18 (2016), no. 27, 17949–17956. 10.1039/C6CP03174A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bocci F et al. , NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol 11 (2019), no. 6, 251–263. 10.1093/intbio/zyz021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia D et al. , OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget. 6 (2015), no. 17, 15436–15448. 10.18632/oncotarget.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jolly MK et al. , Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget. 7 (2016), no. 19, 27067–27084. 10.18632/oncotarget.8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jolly MK et al. , Inflammatory breast cancer: a model for investigating cluster-based dissemination, npj Breast Cancer. 3 (2017), no. 1. 10.1038/s41523-017-0023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subbalakshmi AR et al. , NFATc Acts as a Non-Canonical Phenotypic Stability Factor for a Hybrid Epithelial/Mesenchymal Phenotype. Front. Oncol 10 (2020), 10.3389/fonc.2020.553342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Celià-Terrassa T et al. , Hysteresis control of epithelial-mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun 9 (2018), no. 1. 10.1038/s41467-018-07538-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinway SN et al. , Network Modeling of TGFß Signaling in Hepatocellular Carcinoma Epithelial-to-Mesenchymal Transition Reveals Joint Sonic Hedgehog and Wnt Pathway Activation. Cancer Res. 74 (2014), no. 21, 5963–5977. 10.1158/0008-5472.can-14-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinway SN et al. , Combinatorial interventions inhibit TGFβ-driven epithelial-to-mesenchymal transition and support hybrid cellular phenotypes, Npj Syst. Biol. Appl 1 (2015), 15014. 10.1038/npjsba.2015.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saadatpour A and Albert R, Boolean modeling of biological regulatory networks: A methodology tutorial, Methods 62 (2013), no. 1, 3–12. 10.1016/j.ymeth.2012.10.012 [DOI] [PubMed] [Google Scholar]
- 33.Font-Clos F, Zapperi S, and La Porta CAM, Topography of epithelial–mesenchymal plasticity, Proc. Natl. Acad. Sci 115 (2018), no. 23, 5902–5907. 10.1073/pnas.1722609115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silveira DA, Gupta S, Mombach JCM, Systems biology approach suggests new miRNAs as phenotypic stability factors in the epithelial–mesenchymal transition, J. R. Soc. Interface 17 (2020), no. 171, 20200693. 10.1098/rsif.2020.0693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan FM et al. , Unraveling a tumor type-specific regulatory core underlying E2F1-mediated epithelial-mesenchymal transition to predict receptor protein signatures. Nat. Commun 8 (2017), no. 1. 10.1038/s41467-017-00268-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Udyavar AR et al. , Novel Hybrid Phenotype Revealed in Small Cell Lung Cancer by a Transcription Factor Network Model That Can Explain Tumor Heterogeneity. Cancer Res. 77 (2017), no. 5, 1063–1074. 10.1158/0008-5472.can-16-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang B et al. , Interrogating the topological robustness of gene regulatory circuits by randomization, PLoS Comput. Biol 13 (2017), no. 3, e1005456. 10.1371/journal.pcbi.1005456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krämer A et al. , Causal analysis approaches in ingenuity pathway analysis, Bioinformatics 30 (2014), no. 4, 523–530. 10.1093/bioinformatics/btt703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cohen DPA et al. , Mathematical modelling of molecular pathways enabling tumour cell invasion and migration, PLoS Comput. Biol 11 (2015), no. 11, e1004571. 10.1371/journal.pcbi.1004571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamouille S, Xu J, and Derynck R, Molecular mechanisms of epithelial–mesenchymal transition, Nat. Rev. Mol. Cell Biol 15 (2014), no. 3, 178–196. 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hari K et al. , Identifying inhibitors of epithelial–mesenchymal plasticity using a network topology-based approach, npj. Systems Biology and Applications. 6 (2020), no. 1. 10.1038/s41540-020-0132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohar V and Lu M, Role of noise and parametric variation in the dynamics of gene regulatory circuits, npj Syst. Biol. Appl 4 (2018), no. 1, 40. 10.1038/s41540-018-0076-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campbell K and Casanova J, A common framework for EMT and collective cell migration, Development 143 (2016), no. 23, 4291–4300. 10.1242/dev.139071 [DOI] [PubMed] [Google Scholar]
- 44.Campbell K et al. , Collective cell migration and metastases induced by an epithelial-to-mesenchymal transition in Drosophila intestinal tumors. Nat. Commun 10 (2019), no. 1. 10.1038/s41467-019-10269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fuss B et al. , Cell movements controlled by the Notch signalling cascade during foregut development in Drosophila, Development 131 (2004), no. 7, 1587–1595. 10.1242/dev.01057 [DOI] [PubMed] [Google Scholar]
- 46.Boareto M et al. , Notch-Jagged signalling can give rise to clusters of cells exhibiting a hybrid epithelial/mesenchymal phenotype. J. R. Soc., Interface 13 (2016), no. 118, 20151106 10.1098/rsif.2015.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bocci F et al. , Numb prevents a complete epithelial–mesenchymal transition by modulating Notch signalling. J. R. Soc., Interface 14 (2017), no. 136, 20170512 10.1098/rsif.2017.0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu L et al. , Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 77 (2017), no. 7, 1564–1574. 10.1158/0008-5472.can-16-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jia D et al. , Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci 116 (2019), no. 9, 3909–3918. 10.1073/pnas.1816391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kang X, Wang J, and Li C, Exposing the underlying relationship of cancer metastasis to metabolism and epithelial-mesenchymal transitions, iScience 21 (2019), 754–772. 10.1016/j.isci.2019.10.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jolly Mohit K et al. , Coupling the modules of EMT and stemness: A tunable ‘stemness window’ model. Oncotarget. 6 (2015), no. 28, 25161–25174. 10.18632/oncotarget.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li C and Wang J. Quantifying the landscape for development and cancer from a core cancer stem cell circuit, Cancer Res. 75 (2015), no. 13, 2607–2618. 10.1158/0008-5472.CAN-15-0079 [DOI] [PubMed] [Google Scholar]
- 53.Lee J et al. , Network of mutually repressive metastasis regulators can promote cell heterogeneity and metastatic transitions. Proc. Natl. Acad. Sci 111 (2014), no. 3, E364–E373. 10.1073/pnas.1304840111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar S, Das A, and Sen S, Extracellular matrix density promotes EMT by weakening cell–cell adhesions, Mol. Biosyst 10 (2014), no. 4, 838–850. 10.1039/C3MB70431A [DOI] [PubMed] [Google Scholar]
- 55.Murphy RJ et al. , The role of mechanical interactions in EMT. Phys. Biol 18 (2021), no. 4, 046001. 10.1088/1478-3975/abf425. [DOI] [PubMed] [Google Scholar]
- 56.Ramis-Conde I et al. , Modeling the influence of the E-cadherin-β-catenin pathway in cancer cell invasion: A multiscale approach, Biophys. J 95 (2008), no. 1, 155–165. 10.1529/biophysj.107.114678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bocci F et al. , Deciphering the dynamics of epithelial-mesenchymal transition and cancer stem cells in tumor progression, Curr. Stem Cell Rep 5 (2019), no. 1, 11–21. 10.1007/s40778-019-0150-3 [DOI] [Google Scholar]
- 58.Burger GA, Danen EHJ, and Beltman JB, Deciphering epithelial–mesenchymal transition regulatory networks in cancer through computational approaches, Front. Oncol 7 (2017). 10.3389/fonc.2017.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cortesi M et al. , Computational models to explore the complexity of the epithelial to mesenchymal transition in cancer, WIREs Syst. Biol. Med 12 (2020), no. 6, e1488. 10.1002/wsbm.1488 [DOI] [PubMed] [Google Scholar]
- 60.Aibar S et al. , Aerts Stein SCENIC: single-cell regulatory network inference and clustering. Nat. Methods 14 (2017), no. 11, 1083–1086. 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deshpande A et al. , Network inference with granger causality ensembles on single-cell transcriptomic data, BioRkiv (2019). 10.1101/534834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hirotaka M et al. , SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics. 33 (2017), no. 15, 2314–2321. 10.1093/bioinformatics/btx194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pranzatelli TJF, Michael DG, and Chiorini JA, ATAC2GRN: Optimized ATAC-seq and DNase1-seq pipelines for rapid and accurate genome regulatory network inference, BMC Genomics 19 (2018), no. 1, 563. 10.1186/s12864-018-4943-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsumoto MA et al. , ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinformatics. 7 (2006), no. S1. 10.1186/1471-2105-7-s1-s7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook DP, and Vanderhyden BC, Context specificity of the EMT transcriptional response, Nat. Commun 11 (2020), no. 1, 2142. 10.1038/s41467-020-16066-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramirez D, Kohar V, and Lu M, Toward modeling context-specific EMT regulatory networks using temporal single cell RNA-seq data, Front. Mol. Biosci 7 (2020), 54. 10.3389/fmolb.2020.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puram SV et al. , Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 171 (2017), no. 7, 1611–1624.e24. 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye X et al. , Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 525 (2015), no. 7568, 256–260. 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pratapa A et al. , Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data, Nat. Methods 17 (2020), no. 2, 147–154. 10.1038/s41592-019-0690-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cieslik M et al. , Epigenetic coordination of signaling pathways during the epithelial-mesenchymal transition. Epigenetics Chromatin. 6 (2013), no. 1, 28. 10.1186/1756-8935-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Dijk D et al. , Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 174 (2018), no. 3, 716–729.e27. 10.1016/j.cell.2018.05.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Habermann AC et al. , Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv 6 (2020), no. 28, eaba1972. 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Han SM et al. , Network-based integrated analysis of omics data reveal novel players of TGF-ß1-induced EMT in human peritoneal mesothelial cells. Sci. Rep 9 (2019), no. 1. 10.1038/s41598-018-37101-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Herman JS and Sagar GD, FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data, Nat. Methods 15 (2018), no. 5, 379–386. 10.1038/nmeth.4662 [DOI] [PubMed] [Google Scholar]
- 75.Johnson KS et al. , Gene expression and chromatin accessibility during progressive EMT and MET linked to dynamic CTCF engagement, BioRkiv (2020), 10.1101/2020.05.11.089110 [DOI] [Google Scholar]
- 76.Latil M et al. , Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell. 20 (2017), no. 2, 191–204.e5. 10.1016/j.stem.2016.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Q et al. , A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat. Commun 8 (2017), no. 1. 10.1038/ncomms15166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McFaline-Figueroa JL et al. , A pooled single-cell genetic screen identifies regulatory checkpoints in the continuum of the epithelial-to-mesenchymal transition, Nat. Genet 51 (2019), no. 9, 1389–1398. 10.1038/s41588-019-0489-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Minafra L et al. , Gene expression profiling of epithelial–mesenchymal transition in primary breast cancer cell culture, Anticancer. Res 34 (2014), no. 5, 2173–2183. [PubMed] [Google Scholar]
- 80.Onder TT et al. , Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways, Cancer Res. 68 (2008), no. 10, 3645–3654. 10.1158/0008-5472.CAN-07-2938 [DOI] [PubMed] [Google Scholar]
- 81.Pastushenko I et al. , Identification of the tumour transition states occurring during EMT. Nature. 556 (2018), no. 7702, 463–468. 10.1038/s41586-018-0040-3. [DOI] [PubMed] [Google Scholar]
- 82.Sartor MA et al. , ConceptGen: a gene set enrichment and gene set relation mapping tool. Bioinformatics. 26 (2010), no. 4, 456–463. 10.1093/bioinformatics/btp683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith JJ et al. , Experimentally Derived Metastasis Gene Expression Profile Predicts Recurrence and Death in Patients With Colon Cancer. Gastroenterology. 138 (2010), no. 3, 958–968. 10.1053/j.gastro.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Taube JH et al. , Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudinlow and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci 107 (2010), no. 35, 15449–15454. 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsai Y-P et al. , TET1 regulates hypoxia-induced epithelial-mesenchymal transition by acting as a co-activator. Genome Biol. 15 (2014), no. 12. 10.1186/s13059-014-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walter V et al. , Molecular Subtypes in Head and Neck Cancer Exhibit Distinct Patterns of Chromosomal Gain and Loss of Canonical Cancer Genes. PLoS One. 8 (2013), no. 2, e56823. 10.1371/journal.pone.0056823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wamsley JJ et al. , Activin Upregulation by NF-?B Is Required to Maintain Mesenchymal Features of Cancer Stem–like Cells in Non–Small Cell Lung Cancer. Cancer Res. 75 (2015), no. 2, 426–435. 10.1158/0008-5472.can-13-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang J-Q et al. , Identification of new hypoxia-regulated epithelial-mesenchymal transition marker genes labeled by H3K4 acetylation, Genes, Chromosomes and Cancer. 59 (2020), no. 2, 73–83. 10.1002/gcc.22802. [DOI] [PubMed] [Google Scholar]
- 89.Watanabe K et al. , Mammary Morphogenesis and Regeneration Require the Inhibition of EMT at Terminal End Buds by Ovol2 Transcriptional Repressor. Dev. Cell 29 (2014), no. 1, 59–74. 10.1016/j.devcel.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watanabe K et al. , Combinatorial perturbation analysis reveals divergent regulations of mesenchymal genes during epithelial-to-mesenchymal transition, npj Syst. Biol. Appl 5 (2019), no. 1, 21. 10.1038/s41540-019-0097-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wooten DJ et al. , Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. PLoS Comput. Biol 15 (2019), no. 10, e1007343. 10.1371/journal.pcbi.1007343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wouters J et al. , Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat. Cell Biol 22 (2020), no. 8. 986–998. 10.1038/s41556-020-0547-3. [DOI] [PubMed] [Google Scholar]
- 93.Tripathi S et al. , A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells, PLoS Comput. Biol 16 (2020), no. 2, e1007619. 10.1371/journal.pcbi.1007619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baruzzo G et al. , Simulation-based comprehensive benchmarking of RNA-seq aligners, Nat Methods 14 (2017), no. 2, 135–139. 10.1038/nmeth.4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lytal N, Ran D, and An L, Normalization methods on single-cell RNA-seq data: An empirical survey, Front. Genet 11 (2020), 41. 10.3389/fgene.2020.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Vieth B et al. , A systematic evaluation of single cell RNA-seq analysis pipelines, Nat. Commun 10 (2019), no. 1, 4667. 10.1038/s41467-019-12266-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sun S et al. , Accuracy, robustness and scalability of dimensionality reduction methods for single-cell RNA-seq analysis, Genome Biol. 20 (2019), no. 1, 269. 10.1186/s13059-019-1898-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jolliffe IT, and Cadima J, Principal component analysis: A review and recent developments, Philos. Trans. R. Soc. Math. Phys. Eng. Sci 374 (2016), no. 2065, 20150202. 10.1098/rsta.2015.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Van der Maaten L, and Hinton G, Visualizing Data using t-SNE, J. Mach. Learn. Res 9 (2008), 2579–2605. [Google Scholar]
- 100.McInnes L, Healy J, and Melville J, UMAP: Uniform manifold approximation and projection for dimension reduction. ArXiv180203426 Cs Stat. Published online September 17, 2020, available at http://arxiv.org/abs/1802.03426. Accessed April 5, 2021.
- 101.Duò A, Robinson MD, and Soneson C, A systematic performance evaluation of clustering methods for single-cell RNA-seq data, F1000Research 7 (2020), 1141. 10.12688/f1000research.15666.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang T et al. , Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data, BMC Bioinformatics 20 (2019), no. 1, 40. 10.1186/s12859-019-2599-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ritchie Matthew E et al. , Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43 (2015), no. 7, e47–e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Love MI, Huber W, and Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome Biol. 15 (2014), no. 12, 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Subramanian A et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci 102 (2005), no. 43, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mootha VK et al. , PGC-1a-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34 (2003), no. 3, 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 107.Efron B and Tibshirani R, On testing the significance of sets of genes, Ann. Appl. Stat 1 (2007), no. 1. 10.1214/07-AOAS101 [DOI] [Google Scholar]
- 108.Hänzelmann S, Castelo R, and Guinney J, GSVA: Gene set variation analysis for microarray and RNA-seq data, BMC Bioinformatics 14 (2013), no. 1, 7. 10.1186/1471-2105-14-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Saelens W et al. , A comparison of single-cell trajectory inference methods, Nat. Biotechnol 37 (2019), no. 5, 547–554. 10.1038/s41587-019-0071-9 [DOI] [PubMed] [Google Scholar]
- 110.Herrmann C et al. , i-cisTarget: An integrative genomics method for the prediction of regulatory features and cis-regulatory modules, Nucleic Acids Res. 40 (2012), no. 15, e114–e114. 10.1093/nar/gks543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schulz MH et al. , DREM 2.0: Improved reconstruction of dynamic regulatory networks from time-series expression data, BMC Syst Biol. 6 (2012), no. 1, 104. 10.1186/1752-0509-6-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ernst J et al. , Reconstructing dynamic regulatory maps, Mol.Syst. Biol 3 (2007), no. 1, 74. 10.1038/msb4100115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pliner Hannah A et al. , Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71 (2018), no. 5, 858–871.e8. 10.1016/j.molcel.2018.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miraldi ER et al. , Leveraging chromatin accessibility for transcriptional regulatory network inference in T Helper 17 Cells. Genome Res. 29 (2019), no. 3, 449–463. 10.1101/gr.238253.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chan TE, Stumpf MPH, and Babtie AC, Gene regulatory network inference from single-cell data using multivariate information measures, Cell Syst. 5 (2017), no. 3, 251–267.e3. 10.1016/j.cels.2017.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huynh-Thu VA et al. , Inferring regulatory networks from expression data using tree-based methods, PLoS ONE 5 (2010), no. 9, e12776. 10.1371/journal.pone.0012776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Moerman T, et al. , GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics. 35 (2019), no. 12, 2159–2161. 10.1093/bioinformatics/bty916. [DOI] [PubMed] [Google Scholar]
- 118.Chang H et al. , Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 44 (2016), no. 6, 2514–2527. 10.1093/nar/gkw126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Karacosta LG et al. , Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nat. Commun 10 (2019), no. 1. 10.1038/s41467-019-13441-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moignard V et al. , Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat. Biotechnol 33 (2015), no. 3, 269–276. 10.1038/nbt.3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dunn S et al. , A common molecular logic determines embryonic stem cell self-renewal and reprogramming, EMBO J. 38 (2019), no. 1, 10.15252/embj.2018100003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dunn S-J et al. , Defining an essential transcription factor program for naive pluripotency, Science 344 (2014), no. 6188, 1156–1160. 10.1126/science.1248882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sha Y et al. , Inference and multiscale model of epithelial-to-mesenchymal transition via single-cell transcriptomic data, Nucleic Acids Res. 48 (2020), no. 17, 9505–9520. 10.1093/nar/gkaa725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Davis CA et al. , The Encyclopedia of DNA elements (ENCODE): data portal update, Nucleic Acids Res. 46, (2018), no. D1, D794–D801, 10.1093/nar/gkx1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Matys V et al. , TRANSFAC ® : transcriptional regulation, from patterns to profiles, Nucleic Acids Res. 31, (2003), no. 1, 374–378, 10.1093/nar/gkg108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuleshov MV et al. , Enrichr: a comprehensive gene set enrichment analysis web server 2016 update, Nucleic Acids Res. 44, (2016), no. W1, W90–W97, 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.National Center for Biotechnology Information (NCBI)[Internet]. Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information; (1988). Available from: https://www.ncbi.nlm.nih.gov/ [Google Scholar]
- 128.Wen H et al. , On the low reproducibility of cancer studies, Natl. Sci. Rev 5 (2018), no. 5, 619–624. 10.1093/nsr/nwy021 [DOI] [PMC free article] [PubMed] [Google Scholar]