

Abstract
Aims
Cardiac involvement in Fabry disease (FD) occurs prior to left ventricular hypertrophy (LVH) and is characterized by low myocardial native T1 with sphingolipid storage reflected by cardiovascular magnetic resonance (CMR) and electrocardiogram (ECG) changes. We hypothesize that a pre-storage myocardial phenotype might occur even earlier, prior to T1 lowering.
Methods and results
FD patients and age-, sex-, and heart rate-matched healthy controls underwent same-day ECG with advanced analysis and multiparametric CMR [cines, global longitudinal strain (GLS), T1 and T2 mapping, stress perfusion (myocardial blood flow, MBF), and late gadolinium enhancement (LGE)]. One hundred and fourteen Fabry patients (46 ± 13 years, 61% female) and 76 controls (49 ± 15 years, 50% female) were included. In pre-LVH FD (n = 72, 63%), a low T1 (n = 32/72, 44%) was associated with a constellation of ECG and functional abnormalities compared to normal T1 FD patients and controls. However, pre-LVH FD with normal T1 (n = 40/72, 56%) also had abnormalities compared to controls: reduced GLS (−18 ± 2 vs. −20 ± 2%, P < 0.001), microvascular changes (lower MBF 2.5 ± 0.7 vs. 3.0 ± 0.8 mL/g/min, P = 0.028), subtle T2 elevation (50 ± 4 vs. 48 ± 2 ms, P = 0.027), and limited LGE (%LGE 0.3 ± 1.1 vs. 0%, P = 0.004). ECG abnormalities included shorter P-wave duration (88 ± 12 vs. 94 ± 15 ms, P = 0.010) and T-wave peak time (Tonset – Tpeak; 104 ± 28 vs. 115 ± 20 ms, P = 0.015), resulting in a more symmetric T wave with lower T-wave time ratio (Tonset – Tpeak)/(Tpeak – Tend) (1.5 ± 0.4 vs. 1.8 ± 0.4, P < 0.001) compared to controls.
Conclusion
FD has a measurable myocardial phenotype pre-LVH and pre-detectable myocyte storage with microvascular dysfunction, subtly impaired GLS and altered atrial depolarization and ventricular repolarization intervals.
Keywords: Fabry disease, cardiovascular magnetic resonance, electrocardiogram, microvascular dysfunction, global longitudinal strain
Introduction
Fabry disease (OMIM 301500; FD) is a rare X-linked lysosomal storage disorder caused by mutations in the α-galactosidase A gene (GLA). The consequence is progressive sphingolipid accumulation1 affecting multiple organs. Since the availability of renal replacement therapy, the main cause of death has been cardiac, through heart failure or arrhythmia.2,3 Therapy is available for FD (enzyme replacement and oral chaperone therapies) but the effect is incomplete likely due to late initiation. The presence of overt left ventricular hypertrophy (LVH) and myocardial fibrosis has been shown to negatively affect treatment outcome, suggesting the importance of early initiation of treatment.4 Whilst treating all from diagnosis is not an option due to the therapeutic and financial burden,5 the optimal timing of intervention is not known. Cardiac response to treatment is typically assessed by measuring the left ventricular (LV) mass, but this method does not quantify myocardial biology or earlier stages when the benefit could be exploited. Better description of myocardial disease stages and processes might lead to a better understanding of the earliest commitment to irreversible disease and opportunity to commence treatment.4
Cardiovascular magnetic resonance (CMR) is a key tool for studying cardiac manifestations of FD. Early descriptions were of LVH and late gadolinium enhancement (LGE) in the basal inferolateral wall, thought initially to reflect only fibrosis.6–8 Later, multiparametric CMR has been able to measure sphingolipid infiltration (T1 mapping, low native T1)9 and apparent oedema (T2 mapping, high native T2) with blood troponin elevation suggesting inflammation.10,11 A pre-LVH phase of cardiac FD was subsequently defined by a low T1 phenotype, occurring in up to 60% of FD patients; slightly elevated LV ejection fraction and electrocardiographic (ECG) changes were also observed pre-LVH.12,13
However, as sphingolipid storage starts before birth14 and T1 mapping presumably has a detection threshold, a ‘silent’ sphingolipid accumulation stage before T1 lowers must also occur. To explore this, new techniques would be needed. By CMR, two pathways and their measurement techniques show promise. First, global longitudinal strain (GLS), which is robustly measured and changes earlier in many diseases including FD.15 Second, myocardial blood flow (MBF) measured by stress perfusion mapping, which reflects smooth muscle/endothelial changes that occur early in FD.16
A third approach has also been suggested. The ‘traditional’ 12-lead ECG is informative in FD, with some of the first reports of abnormal ECG in FD being published in the 1970s.17 Since then, several ECG features have been described in FD: atrioventricular (AV) block, short and long PQ interval, changes in QRS width and repolarization abnormalities, associated with arrhythmias and disease progression.18–20 Some features, particularly with advanced ECG analysis, occur early, pre-LVH and include accelerated atrial and ventricular depolarization/conductivity (shortening of P-wave duration and QRS width),21 with sphingolipid storage as a potential cause.19
Accordingly, we hypothesized that an even earlier phase of cardiac FD (pre-LVH, pre-low-T1) might be identifiable using a combination of CMR perfusion mapping, GLS assessment, and advanced 12-lead surface ECG analysis.
Methods
Study population
Fabry patients were recruited from the Lysosomal Storage Disorders Unit at Royal Free Hospital (RFH) London between 2015 and 2019, as part of the prospective Fabry400 study (NCT03199001). We included all consecutive FD patients with a confirmed GLA mutation (Supplementary data online, Table S1), multiparametric CMR and same day ECG. Patients <18 years old, with standard contraindications to CMR or with known pregnancy were excluded.
In addition, we included age-, sex-, and heart rate-matched healthy controls who also underwent parametric CMR and same day ECG. The healthy control group were volunteers free of any history or symptoms of cardiovascular disease or other comorbidities, and who were not taking any medications.
The study conformed to the principles of the Helsinki Declaration and ethical approval was obtained for both study groups. Written informed consent was obtained from all participants.
Clinical data and blood biomarkers
Clinical data collected included enzyme replacement and oral chaperone therapies status, GLA variant (Supplementary data online, Table S1), body surface area (BSA), and systolic and diastolic peripheral blood pressures. All FD patients had blood collected just before the scan and analysed for high-sensitivity troponin T (hsTnT, normal <15 ng/L) and NT-proBNP (brain natriuretic peptide, normal <47 pmol/L).
ECG analysis
Twelve-lead surface ECG was acquired at rest (Welch Allyn CP 200 Electrocardiograph) and independently analysed by two experts in a consensus reading (N.J. and M.N.), blinded to both clinical status (patient or healthy control) and CMR. Measurements were taken manually from the tracings at a sweep of 25 mm/s and standard criteria and normal values for ECG findings were applied (Supplementary data online, Methods).
P wave and PQ interval
P-wave indices were recorded in lead II. Atrial dimensions are known to affect the P-wave morphology and duration, and thus the PQ interval, which is why a ‘corrected’ PQ interval was derived by measuring the PQ interval minus P-wave duration in lead II (Pend – Q), better reflecting AV conduction.22
QRS complex
QRS axis (degrees) and the presence of left bundle branch block or right bundle branch block (RBBB), incomplete RBBB or intraventricular conduction delay (100–120 ms) were recorded. ECG LVH criteria were assessed using Sokolow–Lyon index [S-wave voltage in V1 + R-wave voltage in V5 or V6 (whichever larger) >35 mm], Cornell index [(R-wave voltage in aVL + S-wave voltage in V3) × QRS duration ≥2440 mm·ms for males (adding 8 mm for females)], and the ratio between T- and R-wave amplitudes in V5 (T-wave amplitude measured in the concordant part with R wave). R-wave amplitude in V1 was also recorded (a prominent R in V1 can be a marker of septal LVH or right ventricular hypertrophy). Presence of fractionated QRS (fQRS, a marker of intraventricular conduction delay) was checked for each patient and the number of leads with fQRS was noted.
T wave
The time interval between the onset and the peak of T wave (Tonset – Tpeak) and from the peak until the end of the T wave (Tpeak – Tend, an index of transmural dispersion of ventricular repolarization23) were recorded in II or V5; in the presence of negative or biphasic T wave, the peak was measured from the nadir of the T wave and leads with T waves with less than 1.5 mm in amplitude were excluded from the analysis. A measure of T-wave skewness/symmetry was calculated as the ratio between Tonset – Tpeak and Tpeak – Tend. Pathological repolarization was defined as a discordant ST/T as compared to the main QRS axis.
We also noted the presence of atrial fibrillation and atrial and/or ventricular pacing. None of the patients had higher than first degree AV block. ECG intervals of interest are summarized in Figure 1.
Figure 1.
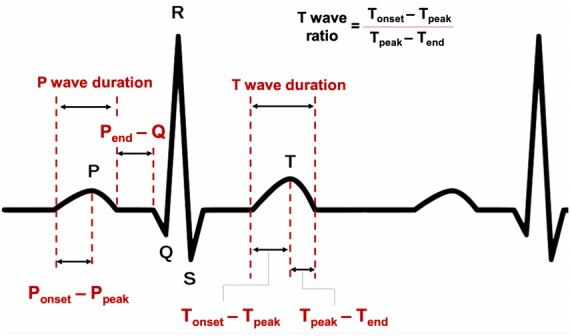
Schematic illustration of ECG intervals of interest.
CMR acquisition
All participants underwent CMR at 1.5 Tesla (Avanto or Aera, Siemens Healthcare, Erlangen, Germany) in one of three centres: the Heart Hospital, Barts Heart Centre, or Chenies Mews Imaging Centre. Standard cine imaging for ventricular volume analysis was performed.3 Native T1 mapping was performed on four-chamber, three-chamber views, and three (basal, mid, and apical) LV short-axis (SAX) slices using a modified Look-Locker inversion recovery sequence (MOLLI, 5b[3s]3b). Native T2 mapping was acquired on the same slices using a steady-state free-precession sequence in 99 Fabry patients and 26 controls. Adenosine stress perfusion mapping was performed in 45 Fabry patients (32 were LVH negative) and 27 controls16 (most common reason for not being performed was patient preference). Stress perfusion images were obtained in the basal, mid, and apical LV SAX slices. LGE images were acquired (not performed in eight patients due to contrast contraindication) following a bolus of 0.1 mmol/kg gadolinium contrast agent (Gadoterate meglumine, Dotarem, Guerbet S.A., France) using a phase sensitive inversion recovery sequence. Post-contrast T1 mapping was performed 15 min after gadolinium administration on the same location as native T1 for extracellular volume fraction (ECV) quantification. The T1 Mapping and ECV Standardization Program (T1MES) phantom was scanned to quality control for T1 and T2 mapping stability across all sites; the results have been published elsewhere.3,12,16
CMR analysis
All images were analysed using CVI42 software (Circle Cardiovascular Imaging Inc. v.5.9.4, Calgary, Canada). Measurements were performed by two expert readers (C.L. and M.A.F.). LV volumes, ejection fraction and mass (papillary muscles included as part of the LV mass) were measured using a semiautomated threshold-based technique and indexed. LVH was defined as a maximum wall thickness (MWT) >12 mm and/or an increased LV mass indexed to the body surface area with reference to age- and gender-adjusted CMR nomograms.24
Pixel-by-pixel colour maps were displayed for T1, T2 and stress MBF mappings using custom 12-bit lookup tables. Regions of interest with 20% offset were drawn for T1 in the basal and mid septum and for T2 in the basal inferolateral (BIFL) wall. The lowest septal T1 value was considered for ‘low T1’ recognition. Normal T1 and T2 ranges for each centre are detailed in the Supplementary data online, Methods. For ECV and MBF, endo- and epicardial contours were manually drawn and the right ventricular insertion points identified. The borders were offset by 20% and a global mean value (% and mL/g/min, respectively) across all segments was recorded. Global longitudinal 2D strain (GLS) values were obtained using feature tracking analysis.15 LV LGE quantification (percentage of total LV mass) was performed in the SAX LGE slices using manually drawn endo- and epicardial borders and a cut-off of 5 SDs above the mean signal in remote myocardium (with minimal manual adjustment when needed).
Statistical analysis
Statistical analysis was performed using SPSS (version 24.0, IBM Corp., Armonk, NY, USA). Discrete variables are presented as absolute frequencies with percentages; continuous as mean ± standard deviation if normally distributed, otherwise as median and interquartile ranges. Data were checked for normal distribution using Kolmogorov–Smirnov test and visual Q–Q plots assessment. Comparisons between groups were performed using Students’ t-test or Mann–Whitney U test as appropriate. Categorical variables were compared using Fisher’s exact test. Correlations between continuous variables were assessed using Pearson’s correlation (r). Reproducibility analysis for CMR is detailed in the Supplementary data online, Methods and Table S2. Two-sided P-values <0.05 were considered significant.
Logistic regression analysis was performed to assess the determinants of very early cardiac involvement in FD (pre-hypertrophy and normal T1 mapping) vs. healthy controls; independent variables were selected based on their relevance in the baseline tables. Significant factors in univariable analysis (P < 0.05) were selected for the multivariable model and inputted using an ‘enter’ method. Receiver-operator characteristics (ROC) analysis was performed to test the performance of different CMR and ECG parameters to detect very early cardiac involvement (Supplementary data online, Methods).
Results
One hundred and fourteen Fabry patients (age 46 ± 13 years, 61% female) and 76 healthy controls (age 49 ± 15 years, 50% female, P = 0.153 for age and P = 0.180 for sex vs. FD) were recruited. Average heart rate was similar between FD and healthy controls (61 ± 12 vs. 64 ± 11 bpm, P = 0.178). Forty-two (37%) Fabry patients had LVH. Among the 72 Fabry patients with no LVH (‘pre-LVH’) (63%), 32 had low T1 (44%) and 40 had normal T1 (56%) (see Tables 1 and 2 and Supplementary data online, Tables S3–S5).
Table 1.
Clinical and cardiovascular magnetic resonance findings in healthy controls and Fabry patients without LVH
| Healthy controls (n = 76) | Fabry disease without LVH |
P-value normal T1 vs. healthy controls | P-value normal T1 vs. low T1 | ||
|---|---|---|---|---|---|
| Normal T1 (n = 40) | Low T1 (n = 32) | ||||
| Age (years) | 49 ± 15 | 40 ± 13 | 43 ± 11 | 0.002 | 0.332 |
| Male, n (%) | 38 (50) | 5 (13) | 8 (25) | <0.001 | 0.222 |
| SBP (mmHg) | 122 ± 13 | 110 ± 11 | 115 ± 11 | <0.001 | 0.155 |
| DBP (mmHg) | 76 ± 9 | 71 ± 8 | 75 ± 5 | 0.010 | 0.082 |
| BSA (m2) | 1.8 ± 0.2 | 1.8 ± 0.2 | 1.8 ± 0.2 | 0.448 | 0.753 |
| Cardiac variant, n (%) | NA | 13 (33) | 10 (31) | 1.000 | |
| ERT/OCT, n (%) | NA | 13 (33) | 12 (38) | 0.804 | |
| hs-TnT (ng/L) | NA | 1 (1–5) | 3 (1–6) | 0.693 | |
| NT-proBNP (pmol/L) | NA | 8 (1–14) | 6 (1–12) | 0.630 | |
| CMR | |||||
| LV EDVI (mL/m2) | 72 ± 11 | 74 ± 11 | 72 ± 12 | 0.437 | 0.564 |
| LVEF (%) | 67 ± 4 | 70 ± 7 | 73 ± 6 | 0.018 | 0.097 |
| LVMI (g/m2) | 65 ± 13 | 59 ± 10 | 67 ± 14 | 0.008 | 0.011 |
| MWT (mm) | 9 (7–10) | 8 (7–9) | 10 (9–11) | 0.527 | 0.001 |
| Septal T1 (ms) | 1029 ± 38 | 1000 ± 28 | 913 ± 35 | <0.001 | <0.001 |
| BIFL T2 (ms) | 48 ± 2 | 50 ± 4 | 46 ± 2 | 0.027 | 0.021 |
| Global ECV | 24 ± 3 | 26 ± 2 | 25 ± 2 | 0.029 | 0.243 |
| LGE, n (%) | 0 | 5/37 (14) | 8/31 (26) | 0.003 | 0.230 |
| LV LGE (%)a | 0 | 0.3 ± 1.1 | 0.7 ± 1.4 | 0.004 | 0.156 |
| GLS (%) | −20.3 ± 2.3 | −18.3 ± 2.1 | −18.7 ± 2.5 | <0.001 | 0.457 |
| Stress MBF (mL/g per min) | 3.0 ± 0.8 | 2.5 ± 0.7 | 2.5 ± 0.5 | 0.028 | 0.961 |
BIFL, basal inferolateral wall; BNP, brain natriuretic peptide; BSA, body surface area; CMR, cardiovascular magnetic resonance; DBP, diastolic blood pressure; ECV, extracellular volume fraction; EDVI, end-diastolic volume index; ERT, enzyme replacement therapy; GLS, global longitudinal strain; hs-TnT, high-sensitivity troponin T; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; LVMI, left ventricular mass index; MBF, myocardial blood flow; MWT, maximum wall thickness; NA, not available/not applicable; OCT, oral chaperone therapy; SBP, systolic blood pressure.
Non-normally distributed variable.
Table 2.
Electrocardiographic findings in healthy controls and Fabry patients without LVH
| Healthy controls (n = 76) | Fabry disease without LVH |
P-value normal T1 vs. healthy controls | P-value normal T1 vs. low T1 | ||
|---|---|---|---|---|---|
| Normal T1 (n = 40) | Low T1 (n = 32) | ||||
| HR (bpm) | 64 ± 11 | 63 ± 11 | 61 ± 11 | 0.691 | 0.450 |
| PQ (ms) | 163 ± 22 | 152 ± 27 | 151 ± 32 | 0.017 | 0.943 |
| Short PQ, n (%) | 0 | 3 (8) | 5 (16) | 0.039 | 0.453 |
| Long PQ, n (%) | 2 (3) | 1 (3) | 2 (6) | 1.000 | 0.581 |
| P wave (ms) | 94 ± 15 | 88 ± 12 | 90 ± 13 | 0.010 | 0.451 |
| P onset – Ppeak (ms) | 48 ± 13 | 45 ± 14 | 48 ± 14 | 0.235 | 0.286 |
| P end – Q (ms) | 69 ± 19 | 64 ± 24 | 62 ± 30 | 0.252 | 0.675 |
| QRS width (ms) | 83 ± 11 | 85 ± 12 | 90 ± 11 | 0.317 | 0.064 |
| Maximum Q-wave amp. (mm) | 0.8 (0.5–1) | 1 (1–2) | 2 (1–2) | 0.117 | <0.001 |
| SLI (mm) | 19 (16–27) | 17 (13–23) | 22 (16–28) | 0.040 | 0.031 |
| Cornell index (mm·ms) | 990 (600–1310) | 578 (433–984) | 911 (590–1330) | 0.006 | 0.042 |
| T/R amp. ratio in V5 | 0.3 (0.2–0.4) | 0.3 (0.2–0.4) | 0.2 (0.1–0.3) | 0.014 | 0.093 |
| R peak time V5 | 37 ± 7 | 39 ± 5 | 42 ± 6 | 0.062 | 0.006 |
| R amp. in V1 (mm) | 2 (1–3) | 2 (1–3) | 3 (2–4) | 0.488 | 0.003 |
| fQRS, n (%) | 16 (21) | 7 (18) | 14 (44) | 0.807 | 0.020 |
| T onset – Tpeak (ms) | 115 ± 20 | 104 ± 28 | 102 ± 25 | 0.015 | 0.838 |
| T peak – Tend (ms) | 67 ± 12 | 72 ± 14 | 65 ± 17 | 0.053 | 0.073 |
| (Tonset – Tpeak)/(Tpeak – Tend) | 1.8 ± 0.4 | 1.5 ± 0.4 | 1.6 ± 0.3 | <0.001 | 0.199 |
| T-wave amp. (mm) | 4 (3–6) | 3 (2–4) | 3 (1–4) | <0.001 | 0.923 |
| Pathological repolarization, n (%) | 2 (3) | 4 (10) | 8 (25) | 0.180 | 0.117 |
Additional electrocardiographic features as detailed in Supplementary data online, Table S5.
fQRS, fractionated QRS; HR, heart rate; LVH, left ventricular hypertrophy; SLI, Sokolow–Lyon index.
Fabry disease with LVH
Fabry patients with LVH (overt disease), had lower MBF, GLS and T1, and higher T2 and %LGE than those without LVH (Supplementary data online, Results and Tables S3 and S4). ECG changes were also pronounced with LVH and included longer P wave, QRS and QT times, negative T wave/pathological repolarization, and LVH voltage criteria (Supplementary data online, Results and Tables S3 and S4).
Early Fabry disease without LVH: with and without detectable storage (low vs. normal T1)
Pre-LVH Fabry patients with low T1 (vs. normal T1) had higher LV mass index (67 ± 14 vs 59 ± 10g/m2, P = 0.011) and MWT [, 10 (9–11) vs. 8 (7–9)mm, P = 0.001]. Both hsTnT and NT-proBNP were normal for both groups, Table 1. Low T1 patients had higher maximum Q-wave amplitude [2 (1–2) vs. 1 (1–2) mm, P < 0.001], R-wave amplitude in V1 [3 (2–4) vs. 2 (1–3) mm, P = 0.003], greater Sokolow–Lyon [22 (16–28) vs. 17 (13–23) mm, P = 0.031] and Cornell indexes [911 (590–1330) vs. 578 (433–984) mm·ms, P = 0.042], longer R-wave peak times in V5 (42 ± 6 vs. 39 ± 5 ms, P = 0.006), and a higher prevalence of fQRS (44 vs. 18%, P = 0.020) (see Table 2, Figure 2 and Supplementary data online, Table S5). Of note, GLS was significantly lower in these low T1 patients compared to healthy controls (−18.7 ± 2.5 vs. −20.3 ± 2.3%, P = 0.003).
Figure 2.

Boxplots of maximum Q-wave amplitude (A), R-wave amplitude in V1 (B), Cornell index (C), Sokolow–Lyon index (D), and R-wave duration in V5 (E) according to T1 status in pre-hypertrophic Fabry patients. SL, Sokolow–Lyon.
Very early FD: pre-LVH and pre-storage (normal T1) vs. controls
FD with no LVH and a normal T1 still had a lower T1 compared to controls (1000 ± 28 vs. 1029 ± 38 ms, P < 0.001, Table 1). These FD patients had lower Sokolow–Lyon [17 (13–23) vs. 19 (16–27) mm, P = 0.040] and Cornell index [578 (433–984) vs. 990 (600–1310) mm·ms, P = 0.006] (Tables 1 and 2). GLS was lower (−18.3 ± 2.1 vs. −20.3 ± 2.3%, P < 0.001) as was stress MBF (2.5 ± 0.7 vs. 3.0 ± 0.8 mL/g/min, P = 0.028). There was slightly higher BIFL T2 (50 ± 4 vs. 48 ± 2 ms, P = 0.027), ECV (26 ± 2 vs. 24 ± 3%, P = 0.029), and %LGE (0.3 ± 1.1 vs. 0%, P = 0.004) than controls. PQ was shorter (152 ± 27 vs. 163 ± 22ms, P = 0.017), mostly due to shorter P-wave duration (88 ± 12 vs. 94 ± 15 ms, P = 0.010) with lower T-wave amplitudes [3 (2–4) vs. 4 (3–6) mm, P < 0.001] and shorter Tonset – Tpeak (104 ± 28 vs. 115 ± 20 ms, P = 0.015) but longer Tpeak – Tend (72 ± 14 vs. 67 ± 12 ms, P = 0.053), resulting in significantly lower (Tonset – Tpeak)/(Tpeak – Tend) ratio (1.5 ± 0.4 vs. 1.8 ± 0.4, P < 0.001) than controls (Table 2 and Supplementary data online, Table S5).
Very early FD: observation robustness
To assess the robustness of these changes, biomarkers of interest (GLS, MBF, T2, ECV, %LV LGE, PQ interval, P-wave duration, Tonset – Tpeak, Tpeak – Tend, [Tonset – Tpeak]/[Tpeak – Tend] and T-wave amplitude) were selected for regression analysis (Table 3). GLS [2.9, 95% confidence interval (1.2–7.2), P = 0.026], P-wave duration [1.2 (1.0–1.5), P = 0.029] and (Tonset – Tpeak)/(Tpeak – Tend) ratio [976 (2.2–425219), P = 0.026] predict very early cardiac FD involvement (pre-LVH normal T1) in multivariable regression analysis. The three aforementioned variables were computed in ROC curve analysis, either isolated or in combination (Figure 3, Supplementary data online, Results). The best discriminative ability however was a ROC curve that used the logit value of all three variables combined (see Supplementary data online, Results)—area under the curve (AUC) 0.87 (0.79–0.95, P < 0.001), significantly superior to other curves’ AUC (P < 0.05 for all). A selection of findings across subgroups is summarized in Figure 4.
Table 3.
Uni- and multivariable regression analysis of the determinants of very early cardiac involvement in Fabry disease (pre-LVH and normal T1 mapping)
| Dependent variable | Variables in model | P-value | Multivariable Exp(B) (95% CI) | P-value |
|---|---|---|---|---|
| Normal T1, No LVH FD (vs. controls) | GLS, per 1% decrease | <0.001 | 2.9 (1.2–7.2) | 0.026 |
| Global stress MBF, per 1 mL/g/min decrease | 0.035 | 2.1 (0.4–9.7) | 0.353 | |
| %LV LGE, per 1% increase | 0.996 | |||
| BIFL T2, per 1 ms increase | 0.038 | 1.0 (0.6–1.7) | 0.985 | |
| ECV, per 1% increase | 0.037 | 0.5 (0.2–1.4) | 0.181 | |
| PQ interval, per 1 ms decrease | 0.020 a | |||
| P-wave duration, per 1 ms decrease | 0.020 | 1.2 (1.0–1.5) | 0.029 | |
| T onset – Tpeak, per 1 point decrease | 0.021 b | |||
| T peak – Tend, per 1 point increase | 0.058 | |||
| (Tonset – Tpeak)/(Tpeak – Tend), per 1 point decrease | 0.001 | 976 (2.2–425219) | 0.026 | |
| T-wave amplitude, per 1 mm decrease | 0.001 | 1.4 (0.7–2.6) | 0.363 |
BIFL, basal inferolateral; CI, confidence interval; ECV, extracellular volume fraction; FD, Fabry disease; GLS, global longitudinal strain; LGE, late gadolinium enhancement; LV, left ventricular; LVH, LV hypertrophy; MBF, myocardial blood flow.
P-wave duration was included instead.
T-wave time ratio was included instead.
Figure 3.

Receiver-operator characteristic curves and corresponding AUCs for detection of cardiac involvement in pre-hypertrophic normal T1 Fabry disease. AUC, area under the curve; CI, confidence interval; GLS, global longitudinal strain.
Figure 4.

Multiparametric cardiovascular magnetic resonance and electrocardiographic assessment in patients with FD and healthy controls. Left to right—steady-state free precession cines, native T1 mapping, stress MBF mapping, GLS, P-wave duration, and T-wave ratio. (A) Healthy control, no LVH, normal T1, MBF, GLS, P-wave time, and T-wave ratio. (B) FD with normal T1 and without LVH; MBF and GLS are mildly reduced, P wave is short and T-wave ratio reduced. (C) FD with low T1 and without LVH, low MBF and GLS, P-wave duration, and T-wave ratio are no different from control. (D) FD with LVH; T1 is low, MBF and GLS are significantly impaired, P wave is long and T-wave ratio increased.
Discussion
In recent years, a pre-hypertrophic phase of FD with ECG abnormalities and sphingolipid storage detected by T1 mapping has been described. Here, we sought an even earlier phase of cardiac FD pre-LVH and pre-detectable storage by using advanced ECG analysis and two CMR methods, GLS measurement and quantitative perfusion mapping. In both overt and pre-LVH disease with storage, we found the expected changes in all parameters. For pre-LVH, pre-detectable storage there was an identifiable phenotype with lower ECG conventional voltages than healthy volunteers, and a number of other more robust features: reduced MBF and GLS, PQ shortening (mainly from a shorter P-wave duration), and a shorter Tonset – Tpeak time (with a shorter (Tonset – Tpeak)/(Tpeak – Tend) ratio also). Prior staging of Fabry cardiomyopathy3 included a pre-LVH stage (accumulation/storage) and two LVH stages (hypertrophy and inflammation; fibrosis and impairment). Here we define a silent (pre-clinical) pre-LVH phase, with two stages: accumulation/storage and an even earlier stage microvascular/pre-detectable storage (Figure 5).
Figure 5.

Proposed stages of cardiac involvement in Fabry disease. A new pre-storage stage is proposed.
The early accumulation phase of FD
Sphingolipid storage has been shown to start before birth14 and some cells including endothelial cells may be more susceptible to sphingolipid storage than myocytes, hence an early degree of microvascular dysfunction would be plausible. Similarly, a shorter P-wave duration pre-storage likely reflects a phenomenon of accelerated intra-atrial conduction with sphingolipids19 which has not yet been counterbalanced by extracellular expansion and/or atrial electroanatomical remodelling processes. At this very early stage, T-wave amplitude was lower than in healthy controls, Tpeak – Tend intervals longer (indicating ventricular repolarization dispersion, a marker for ventricular arrhythmias23) and Tonset – Tpeak times shorter, both resulting in a lower (Tonset – Tpeak)/(Tpeak – Tend) ratio and thus more symmetric T waves. Of note, a normal T wave is asymmetric with a steeper downslope (second half, Tpeak – Tend) than its upslope (first half, Tonset – Tpeak) and is a result of transmural endo-epicardial action potential gradients and their duration, endocardial ones being physiologically slightly longer.25 Any alteration of the myocardial microarchitecture and its perfusion instantaneously entails changes in resting gradients (increased), maximal potentials (decreased), and action potential duration (endocardial shortening).23 To this effect, the observed T wave changes make sense and are in line with our concomitant CMR findings.
But what about GLS and the reduced MBF? To interpret these, whole organism changes should be considered. Before overt cardiac diseases, FD patients manifest small fibre changes (acroparesthesia, GI disturbance, alterations in sweating, and other autonomic abnormalities), but they also show vascular changes and other features that are not so easily quantified—these could include microvascular disease, here expressed by a very early reduced MBF. Whilst slightly reduced GLS might be a primary cardiac phenomenon, it may reflect altered myocardial coupling to the systemic vasculature due to systemic endothelial and smooth muscle changes. Both of these hypotheses are testable and further work is needed.
The effects of sphingolipid storage and progressive remodelling
In the later disease stages, our results corroborate previous findings that sphingolipid storage impairs GLS,15 MBF16 and may result in myocardial oedema, fibrosis and LV impairment3 (Figure 5). Here, we have now described for the first time novel ECG features of cardiac involvement in FD that occur even before native T1 mapping lowering and overt LVH.
With low T1, electrocardiographic indexes of hypertrophy become more pronounced, and ventricular depolarization is delayed (longer QRS and R-wave peak times more fQRS) and so does atrial depolarization (longer P-wave duration). These observations may seem counterintuitive, since one would expect an increase in the diameter of conducting cells with storage eventually leading to faster conduction velocities19 and thus shorter depolarization times. However, P-wave duration, for instance, follows an interesting ‘biphasic’ pattern with disease progression. It first gets shorter pre-detectable storage, reflecting accelerated intra-atrial conduction, but with progressive storage also comes extracellular expansion and left atrial remodelling, progressively slowing down intra-atrial conduction, first ‘pseudonormalizing’ P-wave duration, and finally prolonging it. This sort of pattern hinders ECG interpretation in FD patients who have a ‘normal’ P-wave duration and may be misclassified as not having cardiac involvement, but low T1 is able to rule in cardiac involvement in such patients.
Limitations
This is a cross-sectional single time-point analysis. Longitudinal changes (e.g. how P-wave might behave overtime) are therefore hypothesized rather than observed. The Fabry population with pacemaker is not represented in this study as some CMR parameters (e.g. T1, T2 mapping) would likely be affected by the metallic artefact and introduce error in the analysis. Tissue characterization assumptions (microvascular disease, storage, oedema, and fibrosis) are based on CMR rather than biopsy and may have more than one biological explanation—for example stress blood flow reduction could be capillary rarefaction or insensitivity to adenosine instead of alteration in endothelial/smooth muscle behaviour. However, we believe that some histological findings would only be present in a later stage and are likely not sensitive enough to detect some very early changes such as cardiac electrophysiological involvement. A more comprehensive blood biomarker analysis (e.g. proteomics) could also have a role in understanding pathophysiology and detection of early forms of cardiac involvement in FD (see Figure 5, bottom), but further studies are needed.
Conclusions
There is a pre-LVH, pre-detectable storage phase of cardiac involvement in FD characterized by subtle abnormalities of microvascular dysfunction, impaired LV mechanics and altered atrial depolarization and ventricular repolarization intervals. Further studies are required to understand whether this observation provides a window for optimal therapeutic intervention.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary data
Supplementary data are available at European Heart Journal - Cardiovascular Imaging online.
Conflict of interest: U.R. has received travel grants and honoraria for lectures and advisory boards from Takeda, Sanofi-Genzyme, Amicus, Idorsia, and Chiesi. All other authors report no disclosures relevant to the contents of this article.
Supplementary Material
References
- 1. Mehta A, Hughes DA.. Fabry disease. In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K. et al. (eds). GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1292/ (10 July 2019, date last accessed). [Google Scholar]
- 2. Baig S, Edward NC, Kotecha D, Liu B, Nordin S, Kozor R. et al. Ventricular arrhythmia and sudden cardiac death in Fabry disease: a systematic review of risk factors in clinical practice. Europace 2017;20:f153–61. [DOI] [PubMed] [Google Scholar]
- 3. Nordin S, Kozor R, Medina-Menacho K, Abdel-Gadir A, Baig S, Sado DM. et al. Proposed stages of myocardial phenotype development in Fabry disease. JACC Cardiovasc Imaging 2018;12:1673–83. [DOI] [PubMed] [Google Scholar]
- 4. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, StöRk S. et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation 2009;119:524–9. [DOI] [PubMed] [Google Scholar]
- 5. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S. et al. Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry’s disease. N Engl J Med 2001;345:9–16. [DOI] [PubMed] [Google Scholar]
- 6. Moon JC, Sheppard M, Reed E, Lee P, Elliott PM, Pennell DJ.. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson 2006;8:479–82. [DOI] [PubMed] [Google Scholar]
- 7. Moon JCC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ. et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J 2003;24:2151–5. [DOI] [PubMed] [Google Scholar]
- 8. Krämer J, Niemann M, Störk S, Frantz S, Beer M, Ertl G. et al. Relation of burden of myocardial fibrosis to malignant ventricular arrhythmias and outcomes in Fabry disease. Am J Cardiol 2014;114:895–900. [DOI] [PubMed] [Google Scholar]
- 9. Sado DM, White SK, Piechnik SK, Banypersad SM, Treibel T, Captur G. et al. Identification and assessment of Anderson-Fabry disease by cardiovascular magnetic resonance noncontrast myocardial T1 mapping. Circ Cardiovasc Imaging 2013;6:392–8. [DOI] [PubMed] [Google Scholar]
- 10. Nordin S, Kozor R, Bulluck H, Castelletti S, Rosmini S, Abdel-Gadir A. et al. Cardiac Fabry disease with late gadolinium enhancement is a chronic inflammatory cardiomyopathy. J Am Coll Cardiol 2016;68:1707–8. [DOI] [PubMed] [Google Scholar]
- 11. Augusto JB, Nordin S, Vijapurapu R, Baig S, Bulluck H, Castelletti S. et al. Myocardial edema, myocyte injury, and disease severity in Fabry disease. Circ Cardiovasc Imaging 2020;13:e010171. [DOI] [PubMed] [Google Scholar]
- 12. Nordin S, Kozor R, Baig S, Abdel-Gadir A, Medina-Menacho K, Rosmini S. et al. Cardiac phenotype of prehypertrophic Fabry disease. Circ Cardiovasc Imaging 2018;11:e007168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Camporeale A, Pieroni M, Pieruzzi F, Lusardi P, Pica S, Spada M. et al. Predictors of clinical evolution in prehypertrophic Fabry disease. Circ Cardiovasc Imaging 2019;12:e008424. [DOI] [PubMed] [Google Scholar]
- 14. Vedder AC, Strijland A, Vd Bergh Weerman MA, Florquin S, Aerts J, Hollak C.. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis 2006;29:106–11. [DOI] [PubMed] [Google Scholar]
- 15. Vijapurapu R, Nordin S, Baig S, Liu B, Rosmini S, Augusto J. et al. Global longitudinal strain, myocardial storage and hypertrophy in Fabry disease. Heart 2019;105:470–6. [DOI] [PubMed] [Google Scholar]
- 16. Knott KD, Augusto JB, Nordin S, Kozor R, Camaioni C, Xue H. et al. Quantitative myocardial perfusion in Fabry disease. Circ Cardiovasc Imaging 2019;12:e008872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roudebush CP, Foerster JM, Bing OH.. The abbreviated PR interval of Fabry’s disease. N Engl J Med 1973;289:357–8. [DOI] [PubMed] [Google Scholar]
- 18. Namdar M, Kampmann C, Steffel J, Walder D, Holzmeister J, Lüscher TF. et al. PQ interval in patients with Fabry disease. Am J Cardiol 2010;105:753–6. [DOI] [PubMed] [Google Scholar]
- 19. Namdar M. Electrocardiographic changes and arrhythmia in Fabry disease. Front Cardiovasc Med 2016;3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmied C, Nowak A, Gruner C, Olinger E, Debaix H, Brauchlin A. et al. The value of ECG parameters as markers of treatment response in Fabry cardiomyopathy. Heart 2016;102:1309–14. [DOI] [PubMed] [Google Scholar]
- 21. Niemann M, Hartmann T, Namdar M, Breunig F, Beer M, Machann W. et al. Cross-sectional baseline analysis of electrocardiography in a large cohort of patients with untreated Fabry disease. J Inherit Metab Dis 2013;36:873–9. [DOI] [PubMed] [Google Scholar]
- 22. Namdar M, Steffel J, Jetzer S, Schmied C, Hürlimann D, Camici GG. et al. Value of electrocardiogram in the differentiation of hypertensive heart disease, hypertrophic cardiomyopathy, aortic stenosis, amyloidosis, and Fabry disease. Am J Cardiol 2012;109:587–93. [DOI] [PubMed] [Google Scholar]
- 23. Tse G, Gong M, Wong WT, Georgopoulos S, Letsas KP, Vassiliou VS. et al. The Tpeak–Tend interval as an electrocardiographic risk marker of arrhythmic and mortality outcomes: a systematic review and meta-analysis. Heart Rhythm 2017;14:1131–7. [DOI] [PubMed] [Google Scholar]
- 24. Maceira AM, Prasad SK, Khan M, Pennell DJ.. Normalized left ventricular systolic and diastolic function by steady state free precession cardiovascular magnetic resonance. J Cardiovasc Magn Reson 2006;8:417–26. [DOI] [PubMed] [Google Scholar]
- 25. Antzelevitch C. Cellular basis and mechanism underlying normal and abnormal myocardial repolarization and arrhythmogenesis. Ann Med 2004;36:5–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.