

Abstract
Purpose of review:
Type 2 diabetes mellitus (T2DM) is a risk factor for heart failure. The mechanistic target of rapamycin (mTOR) is a key mediator of the insulin signaling pathway. We will discuss the role of mTOR in myocardial dysfunction in T2DM.
Recent findings:
In T2DM, chronically activated mTOR induces multiple pathological events, including a negative feedback loop that suppresses IRS (insulin receptor substrate)-1. While short-term treatment with rapamycin, an mTOR inhibitor, is a promising strategy for cardiac diseases such as acute myocardial infarction and cardiac hypertrophy in T2DM, there are many concerns about chronic usage of rapamycin. Two mTOR complexes, mTORC1 and mTORC2, affect many molecules and processes via distinct signaling pathways that regulate cardiomyocyte function and survival.
Summary:
Understanding mechanisms underlying mTOR-mediated pathophysiological features in the heart is essential for developing effective therapies for cardiac diseases in the context of T2DM.
Keywords: mTOR, Cell Signaling, Diabetes Mellitus, Heart Failure, Cardiovascular Disease, Rapamycin
Introduction
At least 68% of people over 65 years of age with diabetes mellitus (DM) die of some form of heart disease in the United States [1]. It is well recognized that type 2 diabetes mellitus (T2DM) increases the risk of cardiovascular disease (CVD) 2- to 3-fold [2]. DM increases the risk of heart failure (HF) and adversely affects outcomes among patients with HF [1].
The mechanistic target of rapamycin (mTOR), a member of the PI3K (phosphatidylinositol 3-kinase)-related protein kinase family, is an important downstream molecule in the insulin and insulin-like growth factor 1 (IGF-1) signaling pathways and plays a crucial role in cell growth, metabolism, and cell proliferation [3••]. The mTOR protein is 289 kDa in size with multiple companion domains, and it forms two distinct complexes: mTOR complex 1 (mTORC1) and 2 (mTORC2). These two kinase complexes have mTOR and several binding proteins in common, but differ in other components and downstream signaling pathways [3••]. Previous reports, including ours, have demonstrated the significant role of mTOR in cardiac function, cell survival, and metabolism in normal and diabetic hearts [4–7]. In this review, we shed light on the mTOR signaling pathway and discuss how mTOR regulates insulin signaling and glucose metabolism, and links T2DM with DM-associated cardiac diseases that lead to HF, such as myocardial infarction (MI) and diabetic cardiomyopathy.
mTOR signaling pathway
Among the PI3K-related protein kinase family, mTOR is unique in terms of its FKBP12-rapamycin binding domain allowing it to bind directly rapamycin, an immunosuppressant [8]. mTOR, a ubiquitous serine/threonine protein kinase, exists in the PI3K/Akt signaling axis (Fig. 1). Although mTOR is encoded by a single gene in mammals, it binds to specific adaptor proteins to form two complexes: mTORC1 and mTORC2, both of which have functionally distinct mechanisms and effects. mTORC1 consists of multiple protein components: the catalytic mTOR subunit, RAPTOR (the regulatory-associated protein of mTOR) [9, 10], mLST8 (the mammalian lethal with sec-13 protein 8) [11], DEPTOR (the DEP domain containing mTOR-interacting protein) [12] and PRAS40 (the proline rich Akt substrate 40 kDa) [13] (Fig. 1). Despite their different functions, mTORC2 shares three components commonly with mTORC1: mTOR, mLST8, and DEPTOR. However, unlike mTORC1, mTORC2 contains other specific subunits: RICTOR (rapamycin-insensitive companion of mTOR) [14], and mSIN1 (mammalian stress-activated MAP kinase-interacting protein 1) [15] (Fig. 1). The Tel2 (telomere maintenance 2)/Tti1 (Tel2 interacting protein) complex is reported to be a critical factor in mTORC assembly for its stabilization [16, 17]. Tel2 binds to Tti1 and both Tel2 and Tti1 are necessary and sufficient to stabilize and activate both mTORC1 and mTORC2 signaling pathways [16, 18].
Fig 1. Signaling pathways of mTOR complex activation and regulation.
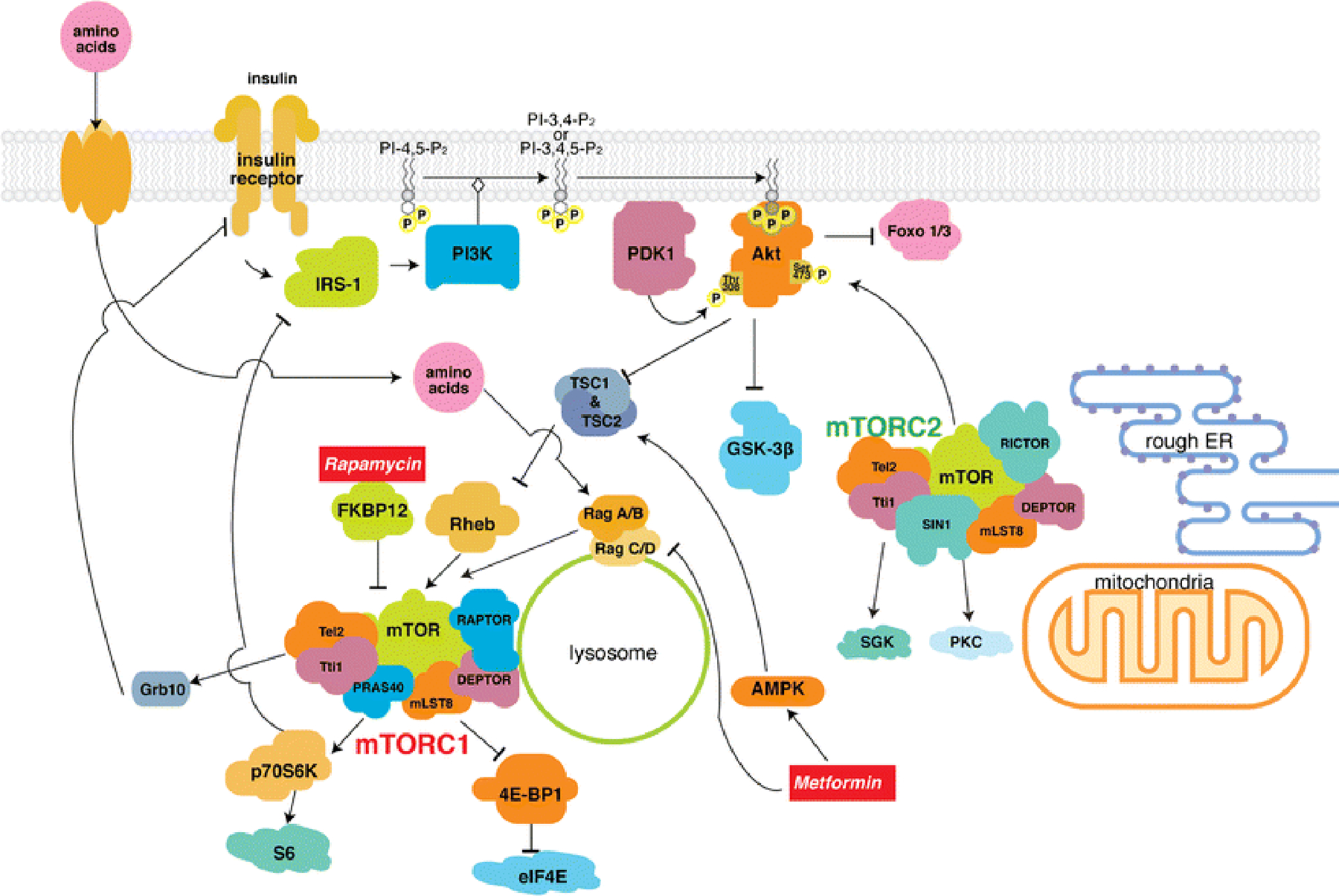
AMPK, AMP-dependent protein kinase; DEPTOR, DEP-domain-containing mTOR interacting protein; elF4E, eukaryotic translation initiation factor 4E; ER, endoplasmic reticulum; FKBP12, FK506-binding protein 12kDa; Foxo1/3, forkhead box protein O 1/3; Grb10, growth factor receptor-bound protein 10; GSK-3β, glycogen synthase kinase 3β; IRS-1, insulin receptor substrate 1; mLST8, mammalian lethal with Sec 13 protein 8; mTOR, mechanistic target of rapamycin; mTORC1/2, mTOR complex 1 and 2; PDK1, phosphoinositide-dependent kinase 1; PI3K, phosphatidylinositol 3-kinase; PKC, protein kinase C; PRAS40, proline-rich Akt/PKB substrate 40 kDa; RAPTOR, regulatory associated protein of mTOR; RICTOR, rapamycin-insensitive companion of mTOR; Rheb, Ras homologue enriched in brain; SGK, serum- and glucocorticoid-regulated kinase; SIN1, stress-activated protein kinase-interacting protein; Tel2, telomere maintenance 2; TSC1/2, tuberous sclerosis complex 1 and 2; Tti1, Tel2 interaction protein 1; 4E-BP1, 4E-binding protein 1.
Insulin and IGF-1 stimulate receptor tyrosine kinases and their cognate receptors, and subsequently activate the PI3K/Akt and Ras signaling pathways. The phosphorylated effector kinases – Akt and ERK1/2 (extracellular-signal-regulated kinase 1/2), directly phosphorylate and inactivate TSC1/2 (the tuberous sclerosis complex 1/ 2) heterodimer [19] [20]. TSC1/2 acts as a GTPase-activating protein (GAP) for Rheb (Ras homologue enriched in brain) GTPase, decreasing Rheb-GTP. Since Rheb-GTP stimulates mTORC1 kinase activity, insulin/IGF-1 signaling finally activates mTORC1 via the PI3K/Akt signaling axis and subsequent inhibition of TSC1/2 [21]. Akt can also stimulate mTORC1 kinase activity independently from TSC1/2 by directly phosphorylating and dissociating PRAS40 (mTORC1 inhibitor) from RAPTOR, which leads to the activation of mTORC1 [13]. Other states, such as low nutrients can lead to decreased ATP (adenosine triphosphate) levels, thus increasing the AMP (adenosine monophosphate)/ATP ratio, resulting in AMPK (AMP-dependent protein kinase) activation. AMPK phosphorylates TSC2 and increases its GAP activity toward Rheb [22, 23], as well as directly inactivating mTORC1 by phosphorylating RAPTOR, which causes 14-3-3 binding and the allosteric inhibition of mTORC1 [24]. GSK-3β (glycogen synthase kinese-3β) can also activate TSC2 and inhibit the mTORC1 signaling pathway during energy stress [25]. Amino acids (especially arginine and leucine) activate mTORC1 with Rag GTPases on the lysosome [26].
Among the downstream cellular processes of mTORC1, protein synthesis is the best characterized, and there are two well-known targets of mTORC1 [27]. Once activated, mTORC1 phosphorylates S6K1 (the ribosomal protein S6 kinase 1), which promotes various cellular processes, including: mRNA biogenesis, translation of ribosomal proteins, cell growth, and cell metabolism [28]. As discussed later, mTORC1 and S6K1 have an important negative feedback action to inhibit IRS-1 (the insulin receptor substrate-1) [29]. The other target of mTORC1 is 4E-BP1 [the eukaryotic translation initiation factor 4E (eIF4E)-binding protein], which accelerates its dissociation from eIF4E, thereby restoring conditions that favor protein synthesis [30]. Hence, phosphorylation of 4E-BP1 by mTORC1 is necessary to begin mRNA translation and protein synthesis. In addition, mTORC1 regulates cell growth and proliferation by inhibiting autophagy, a major process that maintains cellular metabolic homeostasis [31].
Compared to mTORC1, the regulation of mTORC2 remains largely unknown. The only known upstream activators are growth factors via the PI3K signaling pathway through ribosomal interaction [32]. Since mTORC2 has RICTOR instead of RAPTOR, it is less sensitive to acute treatment with rapamycin than mTORC1 because FKBP12-rapamycin cannot bind to it [33]. However, long-term treatment with rapamycin decreases mTORC2 activity by suppressing its subunits [34, 35]. mTORC2 controls cytoskeletal organization, cell size, cell survival, and cell metabolism. Well-known downstream substrates of mTORC2 include AGC kinase family members, such as Akt, SGK (serum- and glucocorticoid-regulated kinase), and PKC (protein kinase C) [36, 37]. Among these members, Akt is an especially important kinase since it has a role in the pathogenesis of cancer and diabetes. mTORC2 directly phosphorylates Akt at Ser473 in a hydrophobic motif [38], a site required for its maximal activation, before subsequent phosphorylation at Thr308 in the catalytic domain by PDK1 (phosphoinositide dependent protein kinase 1). Ser473- and Thr308-phosphorylated Akt is fully activated, and is able to phosphorylate many Akt targets, including Foxo1(the forkhead box protein O 1) and Foxo3 transcription factors [39]. Phosphorylation of Foxo1 and Foxo3 prevents them from translocating into the nucleus and activating apoptotic gene expression; thus, mTORC2 may contribute to cell survival.
mTOR and insulin resistance
Activating mTORC1 by TSC2 disruption in β-cells leads to hyperinsulinemia, expansion of β-cell mass, decreased blood glucose levels, and improved glucose tolerance [40, 41]. S6K1-deficient mice exhibit hypoinsulinemia, glucose intolerance, and diminished β-cell size [42]. Those findings suggest that the mTORC1/S6K1 axis positively regulates insulin secretion. However, it is paradoxical given that chronic hyper-activation of mTORC1/S6K1 signaling appears to exacerbate insulin resistance, thereby leading to development of T2DM [40]. Chronic mTORC1 activation contributes to development of insulin insensitivity by promoting lipogenesis in adipose, reducing glucose uptake in muscle and liver, and enhancing gluconeogenesis in the liver [43, 44]. Sustained activation of S6K1 through mTORC1 activity can lead to phosphorylation and dampening of the function of IRS-1, an adaptor protein that controls key downstream effectors of the insulin receptor [43]. Phosphorylation at Ser307- and Ser636/639- of IRS-1 initiates IRS-1 degradation, suppresses PI3Krecruitment, and leads to the blockade of PI3K/Akt signaling. This causes insulin desensitization via the mTORC1/S6K1-mediated negative feedback loop [29]. A substrate of mTORC, Grb10 (growth factor receptor-bound protein 10), is known to regulate the insulin signaling pathway [45, 46]. Grb10 is phosphorylated by mTORC1 and inhibits Thr-phosphorylation of insulin/IGF receptors, which blocks PI3K/Akt signaling and eventually results in insulin insensitivity [46]. Chronic hyperinsulinemia and hyperglycemia associated with T2DM can lead to an excess of mTORC1/S6K1 activity via insulin/PI3K/Akt signaling, and then chronic mTORC1/S6K1 activation may result in further worsening of hyperinsulinemia and hyperglycemia by these various responses towards insulin resistance. There is no doubt that too little mTORC1 activity results in hypoinsulinemia [42] whereas too much mTOR activation leads to insulin resistance [40], thus tight and well-regulated control of mTORC1 signaling is essential to maintain physiological energy homeostasis.
On the other hand, mTORC2 activates glucose uptake, positively regulates glycolysis, and suppresses hepatic gluconeogenesis [47, 35]. These all contribute to decreasing blood glucose levels. In addition, mTORC2 is an upstream positive regulator of Akt activity as discussed above [38], thus disruption of mTORC2 in β-cells results in diminishing Akt activity and activation of Foxo1 [38, 39]. That eventually leads to mild hyperglycemia and glucose intolerance due to a reduction in β-cell mass, proliferation, pancreatic insulin production, and glucose-stimulated insulin secretion [48].
Heart failure in patients with T2DM
Cardiovascular abnormalities in patients with T2DM include vascular endothelial dysfunction, which leads to atherosclerosis and eventually results in coronary artery disease [49]. This dysfunction also has detrimental effects on cardiomyocytes (CMs), which may link HF with diabetes. Local increases in angiotensin II (ANG II) in diabetes enhances oxidative damage by increasing apoptosis 85-fold and necrosis 4-fold in human CMs compared to non-diabetic CMs [50]. The Framingham Heart Study in the 1970s first revealed that T2DM independently confers roughly a 2-fold increase in risk of HF in men, and a 5-fold increase in women compared with age-matched control subjects [51]. In spite of its frequency, morbidity, and high mortality, HF is often excluded from the list of major diabetic complications and from the large-scale trials that focus mainly on cardio “vascular” outcomes in patients with DM [52, 53]. However, the etiology of HF in DM is characterized as several overlapping cardiotoxic and cellular maladaptive alterations, including hypertension and subsequent cardiac hypertrophy, myocardial ischemia, and diabetic cardiomyopathy [54–56]. According to this pathogenic model, atherosclerotic coronary artery disease is an additional component of HF in DM. In the following section, we will focus on CMs and explain the relationship between mTOR signaling and myocardial dysfunction caused by T2DM.
Role of the mTOR signaling pathway in myocardial dysfunction with T2DM
Hypertension and cardiac hypertrophy
The coexistence of cardiotoxic complications of T2DM seems to cooperatively contribute to biochemical, anatomical, and functional alterations in CMs and other tissues that impair cardiac function [55]. Among them, hypertension is reported as frequently comorbid in over two-thirds of patients with T2DM, and coincides with the development of hyperglycemia [57]. Hypertension may damage myocardial contractile proteins, induce myocardial fibrosis, and generate a hypertrophic state with systolic and diastolic dysfunction [58]. The mTOR signaling pathway is closely related to the development of both hypertension and cardiac hypertrophy [59••]. In aortic endothelial cells, activation of mTORC1/S6K1 by ANG II contributes to impairment of insulin-stimulated vasodilation through suppression of IRS-1 and nitric oxide synthase, eventually leading to vasoconstriction and hypertension [60]. In CMs, activation of mTORC1 signaling contributes to hypertrophic states in response to ANG II [61], IGF-1[62], and β-adrenergic stimulation [63]. Among upstream signals, the PI3K/Akt signaling axis contributes significantly to the activation of mTORC1, and enhancing the axis leads to eventual development of cardiac hypertrophy that can be reversed by pharmacological inhibition of mTORC1 with rapamycin [64]. Similarly, disruption of TSC1 in heart also induces early neonatal cardiac hypertrophy, resulting in heart failure that could be delayed by rapamycin [65].
Rapamycin ameliorates the exacerbation of cardiac hypertrophy in response to pressure overload [66]. Rapamycin also prevents cardiac hypertrophy and fibrosis in rats with spontaneous hypertension [67], and inhibits apoptosis in pressure-overloaded rat myocardium [68]. Moreover, overexpression of PRAS40, a component of mTORC1 and endogenous mTORC1 inhibitor, prevents the development of transverse aortic constriction (TAC)-induced cardiac hypertrophy, and delays established hypertrophy [69].
In contrast, inducible cardiac-specific deletion of mTOR and raptor leads to ventricular dilation and results in HF without an initial phase of compensatory hypertrophy [70, 5]. We reported that mice with cardiac-specific overexpression of wild-type mTOR (mTOR-Tg) were protected against pathological hypertrophy and HF following cardiac pressure overload induced by TAC [71]. Our data suggest that the cardioprotective effect of mTOR is mediated by inhibiting nuclear factor-κB (NF-κB)-regulated myocardial inflammation [71]. Interestingly, one key difference between mice with cardiac-specific overexpression of constitutively active Akt (myr-Akt-Tg) versus mTOR-Tg, is that myr-Akt-Tg mice exhibited massive cardiac hypertrophy [72], whereas mTOR-Tg mice displayed normal size [71]. At the molecular level, our mTOR-Tg mice showed activation of both mTORC1 and mTORC2 [71]. As discussed above, chronic activation of mTORC1 generates negative feedback on upstream molecules, such as IRS-1 [29]. The feedback system might contribute to maintaining normal heart size in chronic activation of mTOR, instead of letting hypertrophy occur unchecked. These results indicate that mTOR plays a crucial role in maintaining cardiac function and preventing cardiac dysfunction in developing cardiac hypertrophy induced by pressure overload.
Taken together, whereas cardiac mTOR is necessary and sufficient for cardiac function, inhibition of mTORC1 alone prevents the detrimental effects of hypertension and cardiac hypertrophy, which are major complications of T2DM.
Myocardial ischemia
For patients with T2DM, myocardial ischemia intimately correlates with hypertension and leads to severely dysfunctional myocardium and possible terminal HF [54]. Although preventive care for patients with T2DM has improved, and the rate of T2DM-related complication of acute myocardial infarction (AMI) has declined in recent decades [73], its frequency remains high because of the increasing population of T2DM patients [74]. The end result of coexisting hypertension and myocardial ischemia in T2DM is a fibrotic, non-compliant myocardium, that initially presents with diastolic dysfunction, and later systolic dysfunction [54].
During ischemic injury, inhibition of mTORC1 physiologically preserves energy homeostasis and promotes cell survival [59••]. Since mTORC1 inhibition reduces energy consumption and activates autophagy, genetic or pharmacological inhibition of mTORC1 augments the cardioprotective effect of mTORC1 on CMs and thus may be beneficial during ischemia and energy deprivation [75]. Inhibition of Rheb, a positive regulator of mTORC1, also protects CMs during ischemia and energy deprivation by activation of autophagy [75]. These data suggest that forced reactivation of Rheb/mTORC1 signaling promotes cell death in CMs both in vitro and in vivo, whereas inhibition of the Rheb/mTORC1 signaling pathway limits CM death during energy stress and reduces myocardial damage during ischemia, particularly in obese mice with HFD [75]. The same group reported that inhibition of GSK-3β exacerbates non-reperfusion myocardial ischemic injury by inhibiting autophagy following mTORC1 reactivation [76]. In addition, inhibition of AMPK in glucose-deprived CMs and ischemic mice hearts also diminished autophagy and increased CM death and ischemic injury [77].
Physiological activity of mTORC1 increases after ischemia, and contributes to adverse cardiac remodeling, to which mTORC1 inhibition does seem to have a therapeutic effect [78, 79]. Pharmacological mTORC1 inhibition with everolimus reduced cardiac infarct size and attenuated adverse left ventricular remodeling to improve post-MI cardiac function in rats [78]. PRAS40 overexpression inhibits mTORC1, reduces CM apoptosis and cardiac remodeling, and ameliorates cardiac function, while pharmacological inhibition of both mTORC1 and mTORC2 by Torin1 or a shift to dominant mTORC1 signaling by suppressing mTORC2 via rictor knockdown led to increased CM apoptosis and deterioration of cardiac function [79]. Taken together, prophylaxis of adverse cardiac remodeling after MI appears to be a complex challenge, requiring both mTORC1 inhibition and mTORC2 activation.
Compared to the scenarios of myocardial ischemia and cardiac remodeling as discussed above, the role of the mTOR signaling pathway in the context of ischemia/reperfusion (I/R) injury is still a controversial subject. While it is difficult to distinguish the initial acute myocardial infarct zone caused by ischemia from the subsequent infarct zone caused by reperfusion injury in the clinical settings, studies in animal models of AMI suggest that lethal reperfusion injury accounts for up to 50% of the final size of a myocardial infarct [80]. When administered before ischemia, rapamycin treatment reduces myocardial infarct size caused by I/R in murine ex vivo retro-grade perfused Langendorff hearts, and in in vivo left coronary artery ligation models by activating STAT3 (signal transducer and activator of transcription 3) [81]. A recent report showed that rapamycin therapy at the onset of reperfusion reduces infarct size in diabetic hearts through STAT3 signaling, and demonstrated this in HFD-induced obese mice and homozygous db/db T2DM mice [82]. On the other hand, another study demonstrated that rapamycin is not cardioprotective during I/R injury when administered before reperfusion [76]. One study suggested that autophagy may be protective during ischemia, whereas it may be detrimental during reperfusion [77]. By using ex vivo Langendorff-perfused heart and in vivo transient coronary artery ligation I/R models in mTOR-Tg mice, we demonstrated that mTOR overexpression preserved cardiac function and prevented CM necrosis [6]. In another study using HFD-induced obese mice, we showed that cardiac mTOR overexpression prevents the detrimental effects of diet-induced obesity on the heart after I/R injury by reducing cardiac dysfunction and myocardial scarring [7•]. In those studies, we showed that mTOR overexpression that induces both mTORC1 and mTORC2 activation inhibits necrosis rather than apoptosis in I/R injury. The formation of the mitochondrial permeability transition pore (mPTP) and subsequent permeabilization of the inner mitochondrial membrane is known to be a fundamental factor for necrotic cell death, especially in I/R injury (mPTP-dependent necrosis) [83]. Although the role of mTOR in regulated mPTP-dependent necrosis in CMs is not well characterized, a previous report demonstrated that the role of the mTORC2/Akt signaling axis in mitochondrial calcium influx [84]. Since high calcium concentration in the matrix is a potential trigger for mPTP opening [85], it is possible that the mTORC2/Akt signaling axis might negatively regulate mPTP-dependent necrosis of CMs in the setting of I/R injury.
Taken together, mTORC1 inhibition seems to be cardioprotective against myocardial ischemia and cardiac remodeling by activating autophagy and inhibiting protein synthesis. However, the role of mTORC1 during I/R injury is still ambiguous, whereas mTORC2 activation may have a beneficial effect. Therefore, further studies are required to better understand the role of mTOR – especially mTORC1, during I/R injury, and the role of mTORC2 in the close correlation between myocardial ischemia and T2DM.
Diabetic cardiomyopathy
The first clinical report of diabetic cardiomyopathy was published in 1972, which involved four patients with T2DM who had died of HF [86]. The anatomic pathology of their hearts demonstrated a cardiomyopathy characterized by abnormal myocardial structures, whereas there was no evidence of coronary artery disease, hypertension, or other explainable factors for HF [87]. Although it remains a contentious subject whether “diabetic cardiomyopathy” truly exists [88], ESC (European Society of Cardiology) guidelines in 2013 defined diabetic cardiomyopathy as a clinical condition diagnosed when ventricular dysfunction occurs in the absence of coronary atherosclerosis and hypertension [89]. ACCF/AHA (American College of Cardiology Foundation/American Heart Association) guidelines in 2013 also referred to the treatment of diabetic cardiomyopathy [90]. The diabetic cardiomyopathy observed in insulin-resistant or hyperinsulinemic states is characterized by impaired myocardial insulin signaling, mitochondrial dysfunction, endoplasmic reticulum stress, impaired calcium homeostasis, abnormal coronary microcirculation, activation of renin-angiotensin-aldosterone system/sympathetic nervous system, and maladaptive immune responses [87]. These pathophysiological changes lead to multiple toxic effects on CMs, and eventually result in HF [91]. However, compared to cardiac hypertrophy and myocardial ischemia, the role of mTOR in diabetic cardiomyopathy is not well characterized. A recent study showed that CM-specific PRAS40 overexpression inhibits mTORC1 and prevents the development of diabetic cardiomyopathy [92]. Although they did not use db/db mice throughout the experiments, and partially used HFD-fed obese mice as substitutes, PRAS40 overexpression induced by adeno-associated vector serotype 9 resulted in improved metabolic function, blunted hypertrophic growth, and preserved cardiac function [92]. Another recent study reported the association between autophagy and diabetic cardiomyopathy. They used db/db type 2 diabetic model mice and found suppression of AMPK activity and elevated expression of phosphorylated mTOR in T2DM hearts, leading to inhibited autophagy in the heart [93•]. They also showed that resveratrol, an autophagy enhancer, mitigates diastolic dysfunction, while chloroquine, an autophagy inhibitor, had opposite effects [93•]. Collectively, these data indicate that activating autophagy in CMs by inhibiting mTORC1 may prevent the exacerbation of developing diabetic cardiomyopathy, whereas the role of mTORC2 remains largely unknown. Despite the difficulty in creating a “genuine” model of diabetic cardiomyopathy without coronary artery disease or hypertension, we need additional studies to understand the correlation between mTOR signaling pathway and diabetic cardiomyopathy, which may explain the high incidence and poor prognosis of HF in T2DM patients.
Clinical implications
Everolimus, a derivative of rapamycin (rapalog), has been broadly used in drug-eluting coronary stents in patients with coronary diseases [94]. Rapamycin and rapalogs have both beneficial and detrimental effects on T2DM and coexisting myocardial dysfunction as discussed above. In terms of glucose metabolism, the duration of mTOR inhibition with rapamycin seems to be important for regulating metabolic homeostasis [95]. A human study showed that acute administration of rapamycin enhanced insulin-mediated glucose uptake by inhibiting IRS-1 phosphorylation and disrupting the mTORC1/S6K1-mediated negative feedback loop [96]. In contrast, chronic treatment with rapamycin worsened hyperglycemia and insulin resistance [97, 98]. Despite the high affinity of rapamycin for mTORC1 through FKBP12 during the acute phase, prolonged exposure can also inhibit the activity of mTORC2 [35]. Although it remains unknown whether this effect can occur in CMs, it is possible that long-term administration of rapamycin might exacerbate HF comorbid with T2DM by inhibiting mTORC2, especially in conditions of cardiac hypertrophy or I/R injury.
Metformin, which is a widely used FDA-approved first-line drug for treatment of T2DM, activates AMPK indirectly by inhibiting the mitochondrial complex I, thus inhibiting mTORC1 [99]. Unlike rapamycin, administration of metformin lowers blood glucose levels without detrimental side effects on glucose metabolism. In addition to its effects on AMPK, previous studies suggest that metformin also regulates mTORC1 via inhibition of Rag GTPases [100]. Moreover, since AMPK activation enhances autophagy by mTORC1-independent autophagy-regulatory proteins like Unc51-like kinase (ULK1) [31], administration of metformin might be beneficial for patients with diabetic cardiomyopathy.
Conclusions
In T2DM, chronically activated mTOR induces multiple pathological events, including a negative feedback loop that suppresses IRS-1. While short-term treatment with rapamycin is a promising strategy for cardiac diseases such as MI and cardiac hypertrophy in DM, there are many concerns about chronic usage of rapamycin. Two mTOR complexes, mTORC1 and mTORC2, affect many molecules and processes via distinct signaling pathways that regulate CM function and survival. Understanding mechanisms underlying mTOR-mediated pathophysiological features in the heart will be valuable in developing effective therapies against myocardial dysfunction in the context of T2DM.
Acknowledgments
This work was supported in part by a research grant from the Mitsukoshi Health and Welfare Foundation, Japan (TS), a research grant from Kochi Organization for Medical Reformation and Renewal, Japan (YB), and NIH training grant (T32HL115505 to BKS).
Footnotes
Conflict of Interest
Tomohiro Suhara, Yuichi Baba, Briana K. Shimada, Jason K. Higa, and Takashi Matsui declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
Since this is a review article it does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Fox CS, Coady S, Sorlie PD, D’Agostino RB Sr., Pencina MJ, Vasan RS et al. Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation. 2007;115(12):1544–50. doi: 10.1161/CIRCULATIONAHA.106.658948. [DOI] [PubMed] [Google Scholar]
- 3.••.Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15(3):155–62. doi: 10.1038/nrm3757. [DOI] [PubMed] [Google Scholar]; A comprehensive review of mTOR pathways, especially in metabolism and signaling crosstalk, providing new insight into the modulation of mTOR by other pathways.
- 4.Zhang P, Xu X, Hu X, van Deel ED, Zhu G, Chen Y. Inducible nitric oxide synthase deficiency protects the heart from systolic overload-induced ventricular hypertrophy and congestive heart failure. Circ Res. 2007;100(7):1089–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123(10):1073–82. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 6.Aoyagi T, Kusakari Y, Xiao CY, Inouye BT, Takahashi M, Scherrer-Crosbie M et al. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2012;303(1):H75–85. doi: 10.1152/ajpheart.00241.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.•.Aoyagi T, Higa JK, Aoyagi H, Yorichika N, Shimada BK, Matsui T. Cardiac mTOR rescues the detrimental effects of diet-induced obesity in the heart after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2015;308(12):H1530–9. doi: 10.1152/ajpheart.00008.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows the cardioportective effects of mTOR in diet-induced obesity with insulin resistance.
- 8.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. [DOI] [PubMed] [Google Scholar]
- 9.Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–75. [DOI] [PubMed] [Google Scholar]
- 10.Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–89. [DOI] [PubMed] [Google Scholar]
- 11.Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904. [DOI] [PubMed] [Google Scholar]
- 12.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137(5):873–86. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903–15. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 15.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127(1):125–37. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 16.Kaizuka T, Hara T, Oshiro N, Kikkawa U, Yonezawa K, Takehana K et al. Tti1 and Tel2 are critical factors in mammalian target of rapamycin complex assembly. J Biol Chem. 2010;285(26):20109–16. doi: 10.1074/jbc.M110.121699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takai H, Wang RC, Takai KK, Yang H, de Lange T. Tel2 regulates the stability of PI3K-related protein kinases. Cell. 2007;131(7):1248–59. doi: 10.1016/j.cell.2007.10.052. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Saiz V, Targosz BS, Lemeer S, Eichner R, Langer C, Bullinger L et al. SCFFbxo9 and CK2 direct the cellular response to growth factor withdrawal via Tel2/Tti1 degradation and promote survival in multiple myeloma. Nat Cell Biol. 2013;15(1):72–81. doi: 10.1038/ncb2651. [DOI] [PubMed] [Google Scholar]
- 19.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 20.Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002;4(9):658–65. doi: 10.1038/ncb840. [DOI] [PubMed] [Google Scholar]
- 21.Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–66. [DOI] [PubMed] [Google Scholar]
- 22.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–90. [DOI] [PubMed] [Google Scholar]
- 23.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–34. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126(5):955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 26.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14(3):133–9. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307–18. [DOI] [PubMed] [Google Scholar]
- 28.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123(4):569–80. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 29.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200–5. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 30.Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP et al. Regulation of eIF-4E BP1 phosphorylation by mTOR. J Biol Chem. 1997;272(42):26457–63. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144(5):757–68. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 33.Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–8. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 34.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 35.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335(6076):1638–43. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). The Biochemical journal. 2008;416(3):375–85. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- 37.Facchinetti V, Ouyang W, Wei H, Soto N, Lazorchak A, Gould C et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase C. EMBO J. 2008;27(14):1932–43. doi: 10.1038/emboj.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. [DOI] [PubMed] [Google Scholar]
- 39.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11(6):859–71. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 40.Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol. 2008;28(9):2971–9. doi: 10.1128/MCB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rachdi L, Balcazar N, Osorio-Duque F, Elghazi L, Weiss A, Gould A et al. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci USA. 2008;105(27):9250–5. doi: 10.1073/pnas.0803047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408(6815):994–7. doi: 10.1038/35050135. [DOI] [PubMed] [Google Scholar]
- 43.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell metabolism. 2006;3(6):393–402. [DOI] [PubMed] [Google Scholar]
- 44.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–6. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–22. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes. 2011;60(3):827–37. doi: 10.2337/db10-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tesfamariam B, Cohen RA. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263(2 Pt 2):H321–6. [DOI] [PubMed] [Google Scholar]
- 50.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A et al. Myocardial cell death in human diabetes. Circ Res. 2000;87(12):1123–32. [DOI] [PubMed] [Google Scholar]
- 51.Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. The American journal of cardiology. 1974;34(1):29–34. [DOI] [PubMed] [Google Scholar]
- 52.Nathan DM, Cleary PA, Backlund JY, Genuth SM, Lachin JM, Orchard TJ et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353(25):2643–53. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Group AC, Patel A, MacMahon S, Chalmers J, Neal B, Billot L et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358(24):2560–72. doi: 10.1056/NEJMoa0802987. [DOI] [PubMed] [Google Scholar]
- 54.Bell DS. Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes Care. 2003;26(8):2433–41. [DOI] [PubMed] [Google Scholar]
- 55.Bell DS. Diabetes: a cardiac condition manifesting as hyperglycemia. Endocr Pract. 2008;14(7):924–32. doi: 10.4158/EP.14.7.924. [DOI] [PubMed] [Google Scholar]
- 56.Gilbert RE, Krum H. Heart failure in diabetes: effects of anti-hyperglycaemic drug therapy. The Lancet. 2015;385(9982):2107–17. doi: 10.1016/s0140-6736(14)61402-1. [DOI] [PubMed] [Google Scholar]
- 57.Ferrannini E, Cushman WC. Diabetes and hypertension: the bad companions. The Lancet. 2012;380(9841):601–10. doi: 10.1016/s0140-6736(12)60987-8. [DOI] [PubMed] [Google Scholar]
- 58.Regan TJ, Wu CF, Yeh CK, Oldewurtel HA, Haider B. Myocardial composition and function in diabetes. The effects of chronic insulin use. Circ Res. 1981;49(6):1268–77. [DOI] [PubMed] [Google Scholar]
- 59.••.Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114(3):549–64. doi: 10.1161/CIRCRESAHA.114.302022. [DOI] [PMC free article] [PubMed] [Google Scholar]; In-depth review that demonstrates the role of mTOR signaling pathway in the regulation of cardiac pathophysiology such as cardiac homeostasis, hypertrophy, ischemia, and metabolic disorders.
- 60.Kim JA, Jang HJ, Martinez-Lemus LA, Sowers JR. Activation of mTOR/p70S6 kinase by ANG II inhibits insulin-stimulated endothelial nitric oxide synthase and vasodilation. Am J Physiol Endocrinol Metab. 2012;302(2):E201–8. doi: 10.1152/ajpendo.00497.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sadoshima J, Izumo S. Rapamycin selectively inhibits angiotensin II-induced increase in protein synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6 kinase in angiotensin II-induced cardiac hypertrophy. Circ Res. 1995;77(6):1040–52. [DOI] [PubMed] [Google Scholar]
- 62.Lavandero S, Foncea R, Perez V, Sapag-Hagar M. Effect of inhibitors of signal transduction on IGF-1-induced protein synthesis associated with hypertrophy in cultured neonatal rat ventricular myocytes. FEBS Lett. 1998;422(2):193–6. [DOI] [PubMed] [Google Scholar]
- 63.Simm A, Schluter K, Diez C, Piper HM, Hoppe J. Activation of p70(S6) kinase by beta-adrenoceptor agonists on adult cardiomyocytes. J Mol Cell Cardiol. 1998;30(10):2059–67. [DOI] [PubMed] [Google Scholar]
- 64.Proud CG. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc Res. 2004;63(3):403–13. doi: 10.1016/j.cardiores.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 65.Malhowski AJ, Hira H, Bashiruddin S, Warburton R, Goto J, Robert B et al. Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin. Hum Mol Genet. 2011;20(7):1290–305. doi: 10.1093/hmg/ddq570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107(12):1664–70. [DOI] [PubMed] [Google Scholar]
- 67.Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S et al. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54(6):1321–7. doi: 10.1161/HYPERTENSIONAHA.109.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harston RK, McKillop JC, Moschella PC, Van Laer A, Quinones LS, Baicu CF et al. Rapamycin treatment augments both protein ubiquitination and Akt activation in pressure-overloaded rat myocardium. Am J Physiol Heart Circ Physiol. 2011;300(5):H1696–706. doi: 10.1152/ajpheart.00545.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY et al. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci USA. 2013;110(31):12661–6. doi: 10.1073/pnas.1301455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang D, Contu R, Latronico MV, Zhang JL, Rizzi R, Catalucci D et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120(8):2805–16. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer-Crosbie M et al. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299(6):C1256–66. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH et al. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277(25):22896–901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 73.Gregg EW, Li Y, Wang J, Burrows NR, Ali MK, Rolka D et al. Changes in diabetes-related complications in the United States, 1990–2010. N Engl J Med. 2014;370(16):1514–23. doi: 10.1056/NEJMoa1310799. [DOI] [PubMed] [Google Scholar]
- 74.Nathan DM. Diabetes: Advances in Diagnosis and Treatment. JAMA. 2015;314(10):1052–62. doi: 10.1001/jama.2015.9536. [DOI] [PubMed] [Google Scholar]
- 75.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125(9):1134–46. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res. 2011;109(5):502–11. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T et al. Distinct Roles of Autophagy in the Heart During Ischemia and Reperfusion. Roles of AMP-Activated Protein Kinase and Beclin 1 in Mediating Autophagy. Circ Res. 2007;100(6):914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 78.Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol. 2009;54(25):2435–46. doi: 10.1016/j.jacc.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 79.Volkers M, Konstandin MH, Doroudgar S, Toko H, Quijada P, Din S et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128(19):2132–44. 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–35. doi: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 81.Das A, Salloum FN, Durrant D, Ockaili R, Kukreja RC. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J Mol Cell Cardiol. 2012;53(6):858–69. doi: 10.1016/j.yjmcc.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Das A, Salloum FN, Filippone SM, Durrant DE, Rokosh G, Bolli R et al. Inhibition of mammalian target of rapamycin protects against reperfusion injury in diabetic heart through STAT3 signaling. Basic Res Cardiol. 2015;110(3):31. doi: 10.1007/s00395-015-0486-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karch J, Molkentin JD. Regulated necrotic cell death: the passive aggressive side of Bax and Bak. Circ Res. 2015;116(11):1800–9. doi: 10.1161/CIRCRESAHA.116.305421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA. 2013;110(31):12526–34. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Finkel T, Menazza S, Holmstrom KM, Parks RJ, Liu J, Sun J et al. The ins and outs of mitochondrial calcium. Circ Res. 2015;116(11):1810–9. doi: 10.1161/CIRCRESAHA.116.305484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. The American journal of cardiology. 1972;30(6):595–602. [DOI] [PubMed] [Google Scholar]
- 87.Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12(3):144–53. doi: 10.1038/nrendo.2015.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Litwin SE. Diabetes and the heart: is there objective evidence of a human diabetic cardiomyopathy? Diabetes. 2013;62(10):3329–30. doi: 10.2337/db13-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Authors/Task Force M, Ryden L, Grant PJ, Anker SD, Berne C, Cosentino F et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur Heart J. 2013;34(39):3035–87. doi: 10.1093/eurheartj/eht108. [DOI] [PubMed] [Google Scholar]
- 90.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62(16):e147–239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 91.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell metabolism. 2012;15(6):805–12. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Volkers M, Doroudgar S, Nguyen N, Konstandin MH, Quijada P, Din S et al. PRAS40 prevents development of diabetic cardiomyopathy and improves hepatic insulin sensitivity in obesity. EMBO Mol Med. 2014;6(1):57–65. doi: 10.1002/emmm.201303183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.•.Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11(7):1146–60. doi: 10.1080/15548627.2015.1051295. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates and compares different autophagic adaptations seen in diabetic cardiomyopathy between type 1 and type 2 DM. It also reports AMPK suppression, mTORC1 activation, and autophagy inhibition in T2DM hearts.
- 94.Siontis GC, Stefanini GG, Mavridis D, Siontis KC, Alfonso F, Perez-Vizcayno MJ et al. Percutaneous coronary interventional strategies for treatment of in-stent restenosis: a network meta-analysis. Lancet. 2015;386(9994):655–64. doi: 10.1016/S0140-6736(15)60657-2. [DOI] [PubMed] [Google Scholar]
- 95.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell metabolism. 2014;19(3):373–9. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P et al. The Mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes. 2007;56(6):1600–7. doi: 10.2337/db06-1016. [DOI] [PubMed] [Google Scholar]
- 97.Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes. 2008;57(4):945–57. doi: 10.2337/db07-0922. [DOI] [PubMed] [Google Scholar]
- 98.Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59(6):1338–48. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J et al. Role of AMP-activated protein kinase in mechanism of metformin action. Journal of Clinical Investigation. 2001;108(8):1167–74. doi: 10.1172/jci13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell metabolism. 2010;11(5):390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]