

Abstract
Aim: The bioactive lipid, sphingosine-1-phosphate (S1P), has various roles in the physiology and pathophysiology of many diseases. There are five S1P receptors; however, the role of each S1P receptor in atherogenesis is still obscure. Here we investigated the contribution of S1P receptor 2 (S1P2) to atherogenesis by using a specific S1P2 antagonist, ONO-5430514, in apolipoprotein E-deficient ( Apoe −/− ) mice.
Methods: Apoe −/− mice fed with a western-type diet (WTD) received ONO-5430514 (30 mg/kg/day) or vehicle. To examine the effect on atherogenesis, Sudan IV staining, histological analysis, qPCR, and vascular reactivity assay was performed. Human umbilical vein endothelial cells (HUVEC) were used for in vitro experiments.
Results: WTD-fed Apoe −/− mice had significantly higher S1P2 expression in the aorta compared with wild-type mice. S1P2 antagonist treatment for 20 weeks reduced atherosclerotic lesion development ( p <0.05). S1P2 antagonist treatment for 8 weeks ameliorated endothelial dysfunction ( p <0.05) accompanied with significant reduction of lipid deposition, macrophage accumulation, and inflammatory molecule expression in the aorta compared with vehicle. S1P2 antagonist attenuated the phosphorylation of JNK in the abdominal aorta compared with vehicle ( p <0.05). In HUVEC, S1P promoted inflammatory molecule expression such as MCP-1 and VCAM-1 ( p <0.001), which was attenuated by S1P2 antagonist or a JNK inhibitor ( p <0.01). S1P2 antagonist also inhibited S1P-induced JNK phosphorylation in HUVEC ( p <0.05).
Conclusions: Our results suggested that an S1P2 antagonist attenuates endothelial dysfunction and prevents atherogenesis. S1P2, which promotes inflammatory activation of endothelial cells, might be a therapeutic target for atherosclerosis.
Keywords: Atherosclerosis, Inflammation, Endothelial function, S1P2 receptor, S1P
Abbreviations: Ach: acetylcholine, ANOVA: analysis of variance, Apoe −/− : apolipoprotein E-deficient, DMSO: dimethyl sulfoxide, HUVEC: human umbilical vein endothelial cell, ICAM-1: intercellular adhesion molecule-1, MCP-1: monocyte chemoattractant protein-1, MOMA2: monocyte/macrophage marker-2, qPCR: quantitative real-time PCR, S1P: sphingosine-1-phosphate, S1P2: S1P receptor 2, SNP: sodium nitroprusside, VCAM-1: vascular cell adhesion molecule-1, WTD: western-type diet
1. Introduction
Atherosclerosis, a chronic inflammatory disease, is characterized by thickening of the arterial wall and is the primary cause of cardiovascular disease and cerebrovascular accidents, two of the most common causes of morbidity and mortality worldwide 1 , 2) . A crucial factor in the development of atherosclerosis is vascular wall inflammation, which involves complex interactions of modified lipoproteins, various bioactive substances, inflammatory cells such as macrophages or foam cells, lymphocytes, endothelial cells, and smooth muscle cells 2 - 5) .
Sphingosine-1-phosphate (S1P), one of the bioactive lipids, has various physiological and pathophysiological roles 6 - 11) . Sphingosine-1-phosphate receptors, G-protein-coupled receptors, mediate these roles of S1P, contributing biological activities such as cell growth, differentiation, proliferation, migration, and barrier function in many cell types and tissues 12 - 18) . Numerous studies have shown the role of these S1P receptors in vascular function and development 19) . S1P receptors 1–3 are widely expressed in vascular cells, playing predominant roles in the vasculature 20) . Although some overlap is present, not only the expression pattern and signaling pathways but also the functions of these receptors are diverse 21 , 22) . Among these S1P receptors, S1P receptor 2 (S1P2) has various biological and pathobiological roles in the vasculature. Previous studies demonstrated that S1P2 regulates endothelial barrier integrity 23) and angiogenesis 24) . Several studies have also demonstrated a role of S1P2 in atherogenesis. S1P2 deficiency inhibited pro-inflammatory responses of macrophages and attenuated atherogenesis in apolipoprotein E-deficient ( Apoe −/− ) mice 6) . Moreover, the role of S1P2 in the regulation of vascular tone has been reported 25 - 28) . Because several studies reported both vasodilatory and vasoconstrictive effects, the role of S1P2 in vascular tone seems to be bidirectional and dependent on the vascular bed and disease context 25 - 28) . Vascular tone, which is partially mediated by endothelial function, is associated with the development of atherosclerosis. Therefore, to investigate the role of S1P2 in the development of endothelial dysfunction and atherosclerosis in a hypercholesterolemic mouse model, we administered a novel S1P2 antagonist to Apoe −/− mice in this study.
2. Materials and Methods
2.1. Animals and Drug Administration
Apoe −/− mice (C57BL/6J background), a hypercholesterolemic mouse model of atherosclerosis, and C57BL/6J (wild type) mice were originally purchased from The Jackson Laboratory and Japan SLC, Inc., respectively. S1P2 antagonist, ONO-5430514, was supplied by ONO Pharmaceutical Co., Ltd. Process of the development of ONO-5430514 was previously published 29 - 31) . In this study, we reported antagonistic action of ONO-5430514; however, we cannot open detail structure of it from the patent issue. From 8 weeks of age, male Apoe −/− mice were fed a western-type diet (WTD) and received S1P2 antagonist (30 mg/kg/day) by oral gavage for 20 weeks to examine its effects on atherogenesis. To investigate the effect of S1P2 antagonist on endothelial function at the early stage of atherosclerosis, the same dose of S1P2 antagonist was administered to 8-week-old male Apoe −/− mice for 8 weeks. Vehicle-treated animals with WTD served as the control. Mice were maintained under controlled lighting (12 h light/dark) and temperature (24℃) conditions. All animal experimental procedures conformed to the guidelines for animal experimentation of Tokushima University.
2.2. Antagonistic Action of ONO-5430514
Antagonistic action of ONO-5430514 was confirmed with the intracellular Ca 2+ assay 29) . Briefly, Chinese hamster ovary cells overexpressing human LPA1, S1P3, S1P2, or LPA2 were incubated in Fura2-AM solution (5 µM) [a Ham’s F12 medium containing 10% FBS, 20 mM HEPES buffer (pH 7.2–7.5), and 2.5 mM probenecid] at 37℃ for 60 min, and cells were then washed with Hank’s balanced saline containing 20 mM HEPES buffer (pH 7.2–7.5) and 2.5 mM probenecid. Cells were immersed in the same solution, and the intracellular calcium ion concentration was measured for 30 s without stimulation in a fluorescence-based drug screening system. ONO-5430514 (final concentration, 0.0025 µM to 0.08 µM) or a dimethyl sulfoxide (DMSO) solution was then applied to cells. Three minutes later, S1P (final concentration, 300 nM for S1P2 and S1P3) or oleoyl-L-α-lysophosphatidic acid sodium salt (18:1-LPA; final concentration, 100 nM for LPA1 and LPA2) was added to wells, and the increase in the intracellular calcium ion concentration before and 4 min after the addition of S1P was measured with an interval of 3 s (excitation wavelength, 340 nm and 380 nm; emission wavelength, 540 nm). The antagonistic activity of ONO-5430514 to each S1P or LPA receptor was calculated by using following formula, wherein A is a control value which was a peak value after addition of S1P (final concentration, 300nM) or 18:1-LPA (final concentration, 100nM) in the wells added DMSO and B is an increased amount after the addition of S1P or 18:1-LPA in the cells treated with ONO-5430514:
Suppression (%)=[(A-B)/A] x 100
IC50 values were calculated as the concentration of ONO-5430514, showing 50% suppression. A commercial available S1P2 antagonist, JTE-013 (Cayman Chemical Company), was used as the control for showing antagonistic action of ONO-5430514 to S1P2.
2.3. Blood Pressure and Laboratory Data
Blood pressure was measured as we described previously 32) . At the time of sacrifice, blood was collected from the heart, and plasma was separated and stored at −80℃ until required for further analysis. Plasma total cholesterol, high-density lipoprotein (HDL) cholesterol, and triglyceride levels were measured at LSI Medience Corporation (Japan).
2.4. Quantification of Atherosclerotic Lesions
The severity of atherosclerotic lesions in the aorta was assessed as previously described 32) . After perfusion with 0.9% sodium chloride solution at a constant pressure via the left ventricle, the heart and the whole aorta were immediately removed. After removing tissues surrounding the aorta, the thoracic aorta was opened longitudinally and fixed with 10% neutral buffered formalin. To quantify atherosclerotic lesions in the aortic arch, en-face Sudan IV staining was performed, and the percentage of Sudan IV-positive area in the aortic arch was measured. The remaining thoracic aorta and the abdominal aorta were removed and snap-frozen in liquid nitrogen for further analyses.
2.5. Histological and Immunohistochemical Analyses
Histological and immunohistochemical analyses were performed on frozen sections of the aortic root. The sections (at 5-µm intervals) were stained with Oil Red O to detect lipid deposition. Moreover, sections were incubated with monocyte chemoattractant protein-1 (MCP-1) antibody (BD Pharmingen), monocyte/macrophage marker 2 (MOMA2) antibody (Bio-Rad), or anti-vascular cell adhesion molecule-1 (VCAM-1) antibody (Abcam) followed by the avidin–biotin complex technique and stained with a Vector Red Substrate Kit (Vector Laboratories, Inc.). Each section was counterstained with hematoxylin. The ratio of positive area to plaque area was calculated in three valve lesions in the aortic root and used for comparison 32) .
2.6. Vascular Reactivity Assay
Analysis of vascular reactivity was performed as we described previously 33) . In brief, the descending thoracic aorta was cut into 2-mm rings with special care to preserve the endothelium and was mounted in organ baths filled with modified Krebs–Henseleit buffer (KHB; 118.4 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl 2 , 1.2 mM KH 2 PO 4 , 1.2 mM MgSO 4 , 25 mM NaHCO 3 , 11.1 mM glucose) aerated with 95% O 2 and 5% CO 2 at 37℃. The preparations were attached to a force transducer, and isometric tension was recorded on a polygraph. Vessel rings were primed with 31.4 mM KCl and were then pre-contracted with phenylephrine, producing submaximal (60% of maximum) contraction. After the plateau was attained, the rings were exposed to increasing concentrations of acetylcholine (Ach; 10 −9 –10 −4 M) and sodium nitroprusside (SNP; 10 −9 –10 −4 M) to obtain cumulative concentration–response curves.
2.7. Measurement of S1P
Measurement of S1P in plasma by liquid chromatography–tandem mass spectrometry (LC-MS/MS) was performed as in a previous study 34) with modifications. Plasma (10 µl) was mixed with 10 µl of S1P (d17:1, 1 µM, an internal standard), and 100 µl of ice-cold methanol was then added for extraction. The extract was centrifuged at 15,000 rpm for 10 min and transferred to an auto-injector vial for analysis. The system consisted of a Q-Trap 6500 (SCIEX) equipped with a Shimadzu LC-30AD HPLC system. A ZORBAX Eclipse Plus C18 column (100 mm×4.6 mm, 3.5 µm, Agilent Technologies) was used for sample separation. The mobile phase consists of (A) methanol/acetonitrile/water (1:1:3) and (B) isopropanol both containing 5 mM ammonium acetate, 500 nM EDTA, and 0.025% NH 3 water with gradient of 0% of B to 95% of B at a 0.4 ml/min flow rate. For monitoring and quantifying the level of S1P, the multiple reaction monitoring method was developed with signature ion pairs Q1 (parent ion)/Q3 (characteristic fragment ion), 366.1/250.1 for S1P (d17:1, internal standard) and 380.3/264.2 for S1P (d18:1).
2.8. Cell Culture Experiments
Human umbilical vein endothelial cells (HUVEC) were purchased from Life Technologies and cultured in EGM-2 (Lonza). HUVEC (passages 4–6) were incubated with 0.1 to 1 µM S1P2 antagonist or 0.01 to 0.1 µM JNK inhibitor, SP600125 (Sigma-Aldrich), for 2 h, and were then stimulated with 1 µM S1P in EBM-2 (Lonza) containing 2% FBS.
2.9. Quantitative RT-PCR
Total RNA was extracted from the aorta and HUVEC using a kit (illustra RNAspin RNA Isolation Kit, GE Healthcare). cDNA was synthesized (QuantiTect Reverse Transcription Kit, Qiagen), and quantitative real-time PCR (qPCR) was performed on an Mx3000P (Agilent Technologies) using Power SYBR Green PCR Master Mix (Applied Biosystems). Table 1 lists the sequences of primers. Data are expressed in arbitrary units normalized by β-actin or GAPDH.
Table 1. List of PCR primers.
| Sense | Antisense | |
|---|---|---|
| Mouse and Human | ||
| S1P1 | 5’-ATCATGGGCTGGAACTGCATCA-3’ | 5’-CGAGTCCTGACCAAGGAGTAGAT-3’ |
| S1P2 | 5’-CAGACGCTAGCCCTGCTCAAGA-3’ | 5’-TAGTGGGCTTTGTAGAGGA-3’ |
| S1P3 | 5’-ACAACCGCATGTACTTTTTCAT-3’ | 5’-TACTGCCCTCCCTGAGGAACCA-3’ |
| Mouse | ||
| MCP-1 | 5’-CCACTCACCTGCTGCTACTCAT-3’ | 5’-TGGTGATCCTCTTGTAGCTCTCC-3’ |
| VCAM-1 | 5’-CCCGTCATTGAGGATATTGG-3’ | 5’-GGTCATTGTCACAGCACCAC-3’ |
| F4/80 | 5’-TGCATCTAGCAATGGACAGC-3’ | 5’-GCCTTCTGGATCCATTTGAA-3’ |
| NOX2 | 5’-ACTCCTTGGGTCAGCACTGG-3’ | 5’-GTTCCTGTCCAGTTGTCTTCG-3’ |
| β-actin | 5’-CCTGAGCGCAAGTACTCTGTGT-3’ | 5’-GCTGATCCACATCTGCTGGAA-3’ |
| Human | ||
| MCP-1 | 5’-CCCCAGTCACCTGCTGTTAT-3’ | 5’-AGATCTCCTTGGCCACAATG-3’ |
| ICAM-1 | 5’-TGATGGGCAGTCAACAGCTA-3’ | 5’-GGGTAAGGTTCTTGCCCACT-3’ |
| VCAM-1 | 5’-GCTGCTCAGATTGGAGACTCA-3’ | 5’-CGCTCAGAGGGCTGTCTATC-3’ |
| GAPDH | 5’-TGGGTGTGAACCATGAGAAG-3’ | 5’-GCTAAGCAGTTGGTGGTGC-3’ |
2.10. Western Blot Analysis
RIPA buffer containing a protease inhibitor cocktail and phosphatase inhibitors was used for the preparation of protein lysates of HUVEC or aortic tissue. SDS-PAGE and transfer were performed as we described previously 33) . After blocking in 5% bovine serum albumin for 1 h at room temperature, membranes were incubated with primary antibody against either phospho-SAPK/JNK, SAPK/JNK (Cell Signaling Technology), or β-actin (Sigma) at 4℃ overnight. After washing in TBS containing 1% Tween 20, the membranes were incubated in horseradish peroxidase-conjugated secondary antibody (Chemicon) for 1 h. Antibody distribution was visualized with ECL Plus reagent (GE Healthcare) using a luminescent image analyzer (LAS-1000, Fujifilm). Expression of β-actin was used as an internal control to confirm equivalent total protein loading.
2.11. Statistical Analysis
All results are expressed as mean±SEM. Comparison of parameters between the two groups was performed by unpaired Student’s t -test. Differences between multiple groups were analyzed by one-way analysis of variance (ANOVA) followed by Scheffe’s post hoc analysis. Comparisons of dose–response curves were made by two-factor repeated measures ANOVA, followed by Tukey’s post hoc test for comparison between groups. A value of p <0.05 was considered statistically significant.
3. Results
3.1. Antagonistic Activity of ONO-5430514
The results of Ca 2+ assay demonstrated that ONO-5430514 had enough low IC50, 0.0057 µM. This is compatible with an authentic S1P2 antagonist, JTE-013. Moreover, IC50 of ONO-5430514 to other S1P or LPA receptors, such as LPA1, S1P3, and LPA2, was obviously higher than that to S1P2. Table 2 summarizes these data. These results indicated that ONO-5430514 has good S1P2 antagonistic activity.
Table 2. Antagonistic activity of ONO-5430514.
| ONO-5430514 | |||||
|---|---|---|---|---|---|
| (µM) | %Inhibition | AV | IC50 | ||
| S1P2/EDG-5 | 0.0025 | 39 | 37 | 38 | 0.0057 |
| 0.008 | 53 | 58 | 56 | ||
| 0.025 | 74 | 69 | 71 | ||
| 0.08 | 83 | 83 | 83 | ||
| LPA1/EDG-2 | 0.025 | −4 | 4 | 0 | > 0.8 |
| 0.08 | −10 | −8 | −9 | ||
| 0.25 | −10 | −5 | −8 | ||
| 0.8 | −2 | −4 | −3 | ||
| S1P3/EDG−3 | 0.025 | −13 | −6 | −10 | > 0.8 |
| 0.08 | 4 | 6 | 5 | ||
| 0.25 | 2 | 13 | 8 | ||
| 0.8 | −1 | 4 | 2 | ||
| LPA2/EDG−4 | 0.025 | 1 | −2 | 0 | > 0.8 |
| 0.08 | 8 | 4 | 6 | ||
| 0.25 | 4 | 1 | 2 | ||
| 0.8 | 0 | −3 | −1 | ||
| JTE-013 | |||||
| (µM) | %Inhibition | AV | IC50 | ||
| S1P2/EDG−5 | 0.0008 | 20 | 10 | 15 | 0.0053 |
| 0.0025 | 35 | 36 | 35 | ||
| 0.008 | 60 | 58 | 59 | ||
| 0.025 | 80 | 79 | 80 | ||
| 0.08 | 85 | 83 | 84 | ||
| 0.25 | 89 | 91 | 90 | ||
| 0.8 | 95 | 96 | 95 | ||
| 2.5 | 100 | 100 | 100 | ||
(µM)
3.2. S1P2 Antagonist Prevented Atherosclerosis in Apoe −/− Mice
S1P2 expression increased in Apoe −/− mice fed with WTD for 20 weeks compared with age- and sex-matched wild-type mice fed with normal chow ( p < 0.05). The expression of S1P1 and S1P3 was also higher in Apoe −/− mice; however, it did not reach statistical significance ( Fig.1A ) . S1P2 antagonist treatment for 20 weeks decreased the development of atherosclerotic lesions in the aortic arch compared with the vehicle group (43.9±0.42 vs 31.9±0.24%, p <0.05) ( Fig.1B ) . S1P2 antagonist treatment also decreased mRNA expression of inflammatory molecules (e.g., MCP-1 and VCAM-1), a macrophage marker, F4/80, and an NADPH subunit, NOX2, in the aorta ( Fig.1C ) . S1P2 antagonist did not affect metabolic parameters in Apoe −/− mice, as shown in Table 3 .
Fig. 1. S1P2 antagonist reduced atherosclerosis in Apoe-/- mice .
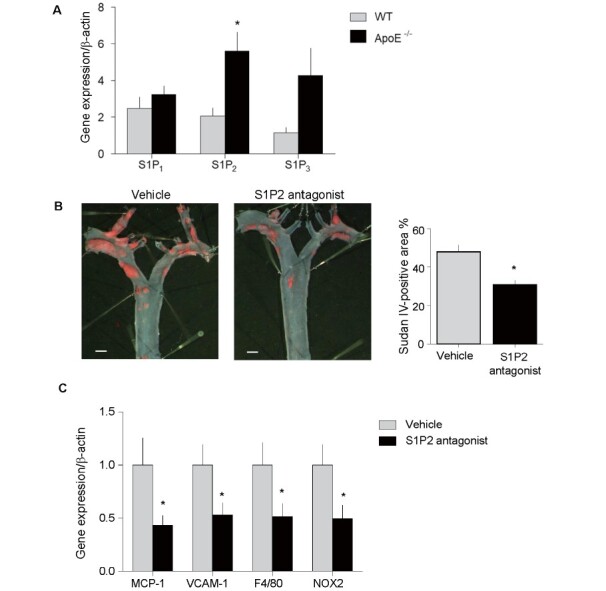
S1P2 expression increased in Apoe -/- mice fed with WTD compared with wild-type mice fed with normal chow. The expression of S1P1 and S1P3 was higher in Apoe -/- mice compared with wild-type mice; however, it did not reach statistical significance. (B) En-face Sudan IV staining showed that S1P2 antagonist administration for 20 weeks to Apoe -/- mice significantly decreased the development of atherosclerosis in the aortic arch ( n = 13–15, per group). (C) qPCR demonstrated that administration of S1P2 antagonist for 20 weeks decreased the expression of inflammatory molecules in atherosclerotic thoracic aorta compared with vehicle ( n = 13–15, per group). Bar, 1 mm. * ; p <0.05. All values are mean±SEM.
Table 3. Effects of S1P2 antagonist on metabolic parameters after 20 weeks of treatment.
| Control ( N = 13) | S1P2 antagonist ( N = 15) | p -value | |
|---|---|---|---|
| Body weight, g | 44.2±1.5 | 41.6±1.7 | 0.1 |
| Blood glucose, mg/dL | 142.7±8.3 | 128.25±8.3 | 0.22 |
| Total cholesterol, mg/dL | 1633.3±103.4 | 1462.73±101.4 | 0.25 |
| Triglyceride, mg/dL | 72.5±4.5 | 59.1±23.9 | 0.1 |
| HDL-cholesterol, mg/dL | 31.2±2.3 | 32.2±3.4 | 0.8 |
| Heart rate, bpm | 710.8±13.1 | 732.7±8.9 | 0.17 |
| Systolic BP, mmHg | 113.1±5.0 | 107.4±3.8 | 0.37 |
| Diastolic BP, mmHg | 77.33±3.6 | 77.47±2.0 | 0.98 |
HDL; high-density lipoprotein, BP; blood pressure. All values are mean±SEM.
3.3. S1P2 Antagonist Ameliorated Endothelial Dysfunction in Apoe −/− Mice
To investigate the mechanism by which S1P2 antagonist attenuates atherogenesis, we examined the effects of S1P2 antagonist on endothelial function and vascular inflammation, an initial steps in atherogenesis. Endothelium-dependent vasodilation in response to Ach was significantly impaired in Apoe −/− mice after 8-week WTD feeding compared with that in age- and sex-matched wild-type mice fed with normal chow. There was no difference in plasma S1P level between Apoe −/− mice and wild-type mice in this study (1.07± 0.14 vs. 1.11±0.06 µM, p =0.85). However, S1P2 antagonist administration ameliorated the impairment of endothelium-dependent vasodilation at this time point ( Fig.2A ) . On the other hand, endothelium-independent relaxation in response to SNP did not differ between the treated and vehicle groups ( Fig.2B ) . S1P2 antagonist inhibited the phosphorylation of JNK in the atherosclerotic aorta, suggesting that S1P2 antagonist inhibits JNK activation ( Fig.2C ) . Administration of S1P2 antagonist for 8 weeks did not cause a difference in metabolic parameters between the 2 groups ( Table 4 ) .
Fig. 2. S1P2 antagonist ameliorated vascular inflammation and endothelial dysfunction in Apoe-/- mice .
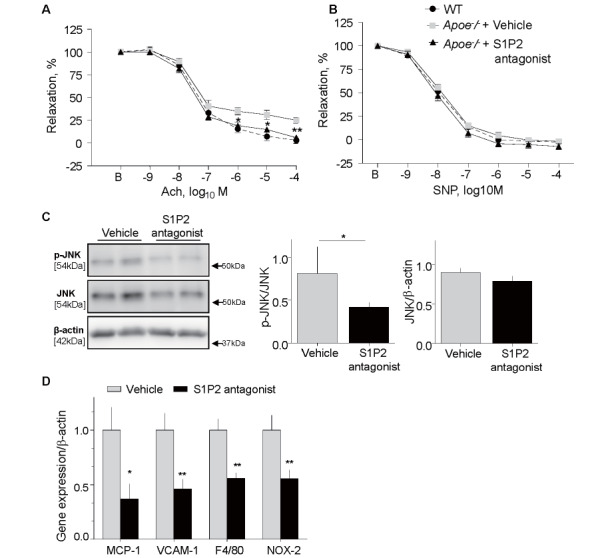
(A and B) Vascular reactivity of aortic rings to Ach (A) or SNP (B) was determined after 8 weeks of S1P2 antagonist or vehicle administration. Vehicle-treated Apoe -/- mice showed an impaired endothelial response compared with age- and sex-matched wild-type mice fed with normal chow. S1P2 antagonist significantly ameliorated endothelium-dependent vasodilation compared with vehicle-treated Apoe -/- mice.
Vasorelaxation in response to SNP did not differ among the three groups. ( n = 14, per group). (C) Western
blot analysis showed that S1P2 antagonist suppressed phosphorylation of JNK in the aorta of Apoe -/- mice compared with the vehicle group, while there was no difference in total JNK expression ( n = 16, per group).
(D) qPCR demonstrated that administration of S1P2 antagonist for 8 weeks decreased the expression of inflammatory molecules in atherosclerotic thoracic aorta compared with the vehicle ( n = 16, per group). * , p
<0.05, and ** , p <0.01 vs. vehicle. All values are mean±SEM.
Table 4. Effects of S1P2 antagonist on metabolic parameters after 8 weeks of treatment.
| Control ( N = 16) | S1P2 antagonist ( N = 16) | p -value | |
|---|---|---|---|
| Body weight, g | 30.3±0.4 | 30.1±0.4 | 0.68 |
| Blood glucose, mg/dL | 131.0±3.5 | 133.6±3.4 | 0.59 |
| Total cholesterol, mg/dL | 1068.1±106.2 | 1126.2±113.9 | 0.71 |
| Triglyceride, mg/dL | 82.6±14.46 | 106.5±12.6 | 0.22 |
| HDL-cholesterol, mg/dL | 14.8±1.7 | 19.1±2.1 | 0.12 |
| Heart rate, bpm | 693.3±8.7 | 687.6±18.3 | 0.77 |
| Systolic BP, mmHg | 109.8±2.8 | 110.2±3.1 | 0.93 |
| Diastolic BP, mmHg | 68.4±2.1 | 69.2±3.2 | 0.83 |
HDL; high-density lipoprotein, BP; blood pressure. All values are mean±SEM.
Administration of S1P2 antagonist for 8 weeks reduced mRNA expression of MCP-1 ( p <0.05), VCAM-1, F4/80, a macrophage marker, and NOX2, an NADPH subunit ( p <0.01, respectively) in the abdominal aorta compared with the vehicle group ( Fig.2D ) . Furthermore, histological analyses demonstrated that S1P2 antagonist attenuated the expression of MCP-1 and VCAM-1 in atherosclerotic plaques ( Fig.3A and 3B ) ( p <0.01, respectively). Associated with the reduction of a chemokine and an adhesion molecule, S1P2 antagonist reduced the accumulation of macrophages as determined by the expression of MOMA2 ( p <0.01) ( Fig.3C ) and lipid deposition in the lesions ( p <0.001) ( Fig.3D ) .
Fig. 3. Effect of S1P2 antagonist on characteristics of atherosclerotic plaques .
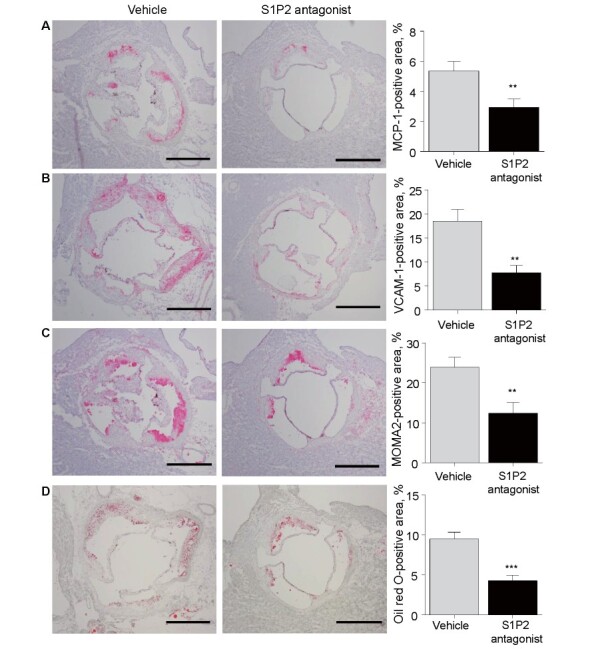
Histological analyses of atherosclerotic lesions in the aortic root of Apoe -/- mice treated with S1P2 antagonist or vehicle for 8 weeks. (A–C) Immunostaining against MCP-1 (A), VCAM-1 (B), and MOMA2
(C) demonstrated that S1P2 antagonist reduced the expression of MCP-1 and VCAM-1 and decreased the accumulation of macrophages ( n = 16, per group). (D) Oil Red O staining demonstrated that S1P2 antagonist reduced lipid deposition compared with the vehicle. Bar, 500 µm. ** , p <0.01, and *** , p <0.001. All values are mean±SEM.
3.4. S1P2 Antagonist Inhibited S1P-Induced Expression of Inflammatory Molecules in Endothelial Cells
The effects of S1P on the expression of inflammatory molecules in HUVEC were examined because the expression of S1P receptors on HUVEC was confirmed by qPCR ( Fig.4A ) . S1P promoted the expression of mRNA of inflammatory molecules such as MCP-1, VCAM-1, and ICAM-1 in HUVEC, while pretreatment with S1P2 antagonist inhibited these responses ( Fig.4B–D ) . The results of western blotting indicated that S1P stimulated the phosphorylation of JNK, suggesting that S1P activates JNK signaling in endothelial cells. S1P2 antagonist suppressed the JNK activation induced by S1P ( Fig.4E ) .
Fig. 4. S1P2 antagonist suppressed S1P-induced inflammation in endothelial cells .
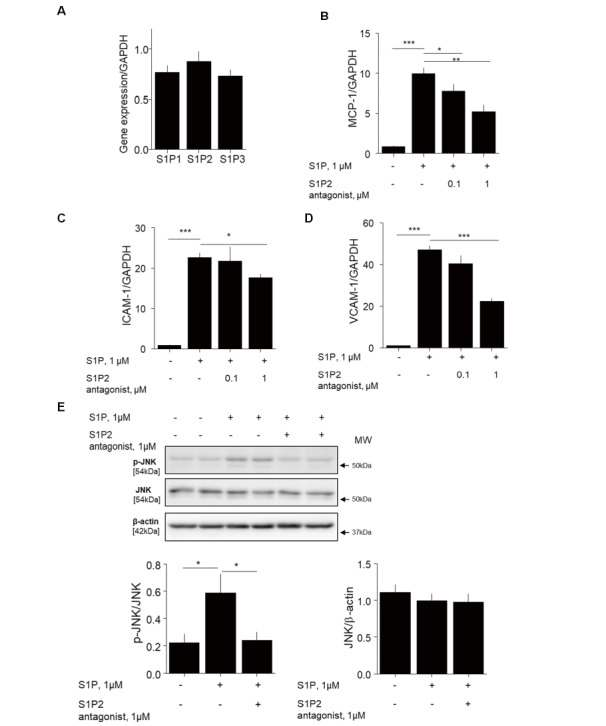
(A)Results of qPCR demonstrated that HUVEC express S1P receptors. (B–D) HUVEC preincubated with 0.1–1 µM S1P2 antagonist for 2 h were stimulated with 1 µM S1P for 4 h. qPCR demonstrated that S1P promoted the expression of inflammatory molecules such as MCP-1 (B), ICAM-1 (C), and VCAM-1 (D) in HUVEC, all of which were attenuated by S1P2 antagonist. (E) HUVEC preincubated with 1 µM S1P2 antagonist for 30 min were stimulated with 1 µM S1P for 4 h. S1P induced JNK phosphorylation in HUVEC, which was significantly inhibited by S1P2 antagonist ( n = 8, per group). There was no difference in total JNK expression in all conditions. * , p <0.05; ** , p <0.01; and *** , p <0.001. All values are mean±SEM.
3.5. Inhibition of JNK Ameliorated Endothelial Inflammation
To confirm the contribution of JNK signaling to S1P-induced inflammatory activation of endothelial cells, HUVEC were treated with S1P in the presence or absence of a JNK inhibitor (SP600125). S1P promoted the expression of VCAM-1, ICAM-1, and MCP-1, while SP600125 significantly reduced S1P-induced inflammatory molecule expression in HUVEC ( Fig.5A–C ) . This result suggested that S1P signaling involved JNK activation.
Fig. 5. Inhibition of JNK suppressed S1P-induced endothelial cell activation .

(A–C) HUVEC were incubated with 0.01 to 0.1 µM SP600125, a JNK inhibitor, for 2 h and were then stimulated with 1 µM S1P for 4 h. qPCR showed that JNK inhibitor decreased MCP-1 (A), ICAM-1 (B), and VCAM-1 (C) expression in HUVEC stimulated by S1P ( n = 8, per group). * , p <0.05; ** , p <0.01; and *** , p <0.001. All values are mean±SEM.
4. Discussion
Atherosclerosis is a chronic inflammatory disease that involves various cellular and molecular processes 1 , 2 , 4 , 35) . Vascular inflammation, which induces endothelial dysfunction and atherogenesis, is an attractive therapeutic target for atherosclerotic diseases 2 , 3) . In the present study, we demonstrated that treatment with S1P2 antagonist decreased the development of atherosclerotic lesions in Apoe −/− mice. S1P2 antagonist reduced inflammation in the aorta and improved endothelium-dependent vascular response, suggesting that S1P2 antagonist has protective effects on endothelial function in Apoe −/− mice. In fact, in vitro studies using HUVEC demonstrated that S1P promoted inflammatory activation of endothelial cells as determined by the expression of inflammatory molecules, which was blocked by S1P2 antagonist. These data together suggest that S1P2 may have a crucial role in vascular inflammation and atherogenesis and that S1P2 is a potential therapeutic target for atherosclerosis.
Previous studies have investigated the pathophysiological roles of S1P and S1P receptors in the development of atherosclerosis using animal experiments. In the vasculature, S1P1–3 are widely expressed and play dominant roles 20) . For example, S1P1 has atheroprotective effects. Stimulation of S1P1 induces a phenotypic change of macrophages to an anti-inflammatory phenotype 36) . Moreover, pharmacological activation of S1P1 attenuates the development of atherosclerosis in a hyperlipidemic mouse model 37) . On the other hand, S1P3 has proatherogenic effects. Activation of the S1P3 receptor promotes recruitment of monocytes/macrophages, accelerating inflammation and atherosclerosis 38) . Previous studies demonstrated that S1P2 also has proatherogenic effects. S1P2 receptor deficiency attenuated inflammatory activation of macrophages, leading to inhibition of the development of atherosclerotic lesions in Apoe −/− mice 6) . In addition, S1P2 regulates vascular tone, although the role of S1P2 in vascular tone is bidirectional and is dependent on the vascular bed and disease context 25 - 28) . Vascular tone, which is partially controlled by endothelial function, is associated with the development of atherosclerosis. Therefore, we investigated the role of S1P2 in the development of endothelial function and atherogenesis, using a novel S1P2 antagonist in Apoe −/− mice in this study.
In this study, S1P2 antagonist clearly attenuated the development of atherosclerotic lesions in Apoe −/− mice. This result is consistent with previous studies 6) . Other studies showed that inflammatory stimuli increase the expression of S1P2 in endothelial cells, which promotes vascular inflammation 39 , 40) . Moreover, a previous study demonstrated that senescent endothelial cells increase S1P2 expression 24) . We demonstrated an increase of S1P2 expression in the atherosclerotic aorta and a reduction of vascular inflammation and atherosclerosis by S1P2 antagonist. These results suggest that S1P2 is involved in atherogenesis, at least partially, in our mouse model and may be a potential therapeutic target for vascular inflammation and atherosclerosis.
We also demonstrated that S1P2 antagonist improved endothelium-dependent vasodilation in Apoe −/− mice. Using a different mouse model, another S1P2 antagonist (JTE-013) also attenuated endothelial dysfunction under high glucose conditions 41) . In this study, we demonstrated that S1P2 antagonist decreased the phosphorylation of JNK, which is an important regulator of vascular inflammation and endothelial function 42) , in treated animals. In in vitro experiments, S1P promoted the expression of inflammatory molecules in HUVEC, which was blocked by a JNK inhibitor, SP600125. S1P2 antagonist also inhibited S1P-induced JNK phosphorylation in HUVEC. Notably, the concentration of S1P used in in vitro experiment was compatible with plasma concentration of S1P in Apoe −/− mice. These results indicate that S1P2 signaling impaired endothelium-dependent vasodilation at least partially via JNK signaling. Previous studies showed that, in the vasculature, smooth muscle cells preferentially express S1P2, inducing vascular contraction 19 , 25) . However, in our present study, we did not observe an effect of S1P2 antagonist on endothelium-independent vasodilation induced by SNP. Previous studies indicated that the role of S1P2 in vascular tone is bidirectional and is dependent on the vascular bed and disease context 25 - 28) . Therefore, this might explain our present result. Further studies are needed to clarify the role and function of S1P2 signaling in the regulation of vascular tone.
In addition, we examined plasma level of S1P using a LC-MS/MS. In this study, the plasma level of S1P was not different between Apoe −/− mice and wild-type mice. Previous studies showed that hyperlipidemic mice had higher plasma S1P level and that plasma LDL level correlates with S1P level in humans 43 - 46) . There are several differences in experimental protocols between previous studies and our present study, which may explain the discrepancy. Moreover, the aorta of Apoe −/− mice and HUVEC express S1P receptors other than S1P2. There still are controversies regarding the role and level of S1P 47) ; thus, further studies are needed to establish therapeutic strategies targeting S1P signaling in atherosclerotic disease.
In summary, the results of our study demonstrated that S1P2 antagonist decreased the development of atherosclerotic lesions in Apoe −/− mice. Inhibition of S1P2 attenuated pro-inflammatory activation of endothelium. S1P-mediated S1P2 signaling in endothelial cells contributed to the expression of inflammatory molecules and the development of endothelial dysfunction, at least partially. Our results may provide novel therapeutic options for atherosclerosis.
Acknowledgments
The authors thank Shintaro Okamoto and Etsuko Uematsu (Tokushima University) for their technical assistance.
Competing Interests
The Department of Cardio-Diabetes Medicine, Tokushima University Graduate School is supported in part by unrestricted research grants from Boehringer Ingelheim. The authors declare that they have no conflict of interest.
Sources of Funding
This work was partially supported by JSPS Kakenhi Grants (Number 19K08584 to D.F. and Number 19H03654 to M.S.), Bristol-Myers Squibb Research Grants (D.F.), Takeda Science Foundation (M.S.), and the Vehicle Racing Commemorative Foundation (M.S.). The funders had no role in the study design, data collection and analysis, or preparation of the manuscript.
References
- 1).Rader DJ and Daugherty A . Translating molecular discoveries into new therapies for atherosclerosis. Nature, 2008; 451: 904- 913 [DOI] [PubMed] [Google Scholar]
- 2).Libby P . Inflammation in atherosclerosis. Nature, 2002; 420: 868- 874 [DOI] [PubMed] [Google Scholar]
- 3).Rao RM , Yang L , Garcia-Cardena G and Luscinskas FW . Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res, 2007; 101: 234- 247 [DOI] [PubMed] [Google Scholar]
- 4).Hansson GK . Inflammatory mechanisms in atherosclerosis. J Thromb Haemost, 2009; 7 Suppl 1: 328- 331 [DOI] [PubMed] [Google Scholar]
- 5).An S , Goetzl EJ and Lee H . Signaling mechanisms and molecular characteristics of G protein-coupled receptors for lysophosphatidic acid and sphingosine 1-phosphate. J Cell Biochem, 1998; 72 Suppl 30- 31: 147-157 [DOI] [PubMed] [Google Scholar]
- 6).Skoura A , Michaud J , Im DS , Thangada S , Xiong Y , Smith JD and Hla T . Sphingosine-1-phosphate receptor-2 function in myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler Thromb Vasc Biol, 2011; 31: 81- 85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Hla T . Physiological and pathological actions of sphingosine 1-phosphate. Semin Cell Dev Biol, 2004; 15: 513- 520 [DOI] [PubMed] [Google Scholar]
- 8).Pyne NJ and Pyne S . Sphingosine 1-phosphate and cancer. Nat Rev Cancer, 2010; 10: 489- 503 [DOI] [PubMed] [Google Scholar]
- 9).Ng ML , Wadham C and Sukocheva OA . The role of sphingolipid signalling in diabetesassociated pathologies (Review). Int J Mol Med, 2017; 39: 243- 252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Koch A , Pfeilschifter J and Huwiler A . Sphingosine 1-phosphate in renal diseases. Cell Physiol Biochem, 2013; 31: 745- 760 [DOI] [PubMed] [Google Scholar]
- 11).Mao-Draayer Y , Sarazin J , Fox D and Schiopu E . The sphingosine-1-phosphate receptor: A novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin Immunol, 2017; 175: 10- 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Moolenaar WH . Bioactive lysophospholipids and their G protein-coupled receptors. Exp Cell Res, 1999; 253: 230- 238 [DOI] [PubMed] [Google Scholar]
- 13).Igarashi Y and Yatomi Y . Sphingosine 1-phosphate is a blood constituent released from activated platelets, possibly playing a variety of physiological and pathophysiological roles. Acta Biochim Pol, 1998; 45: 299- 309 [PubMed] [Google Scholar]
- 14).Hla T , Lee MJ , Ancellin N , Liu CH , Thangada S , Thompson BD and Kluk M . Sphingosine-1-phosphate: extracellular mediator or intracellular second messenger? Biochem Pharmacol, 1999; 58: 201- 207 [DOI] [PubMed] [Google Scholar]
- 15).Garcia JG , Liu F , Verin AD , Birukova A , Dechert MA , Gerthoffer WT , Bamberg JR and English D . Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest, 2001; 108: 689- 701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Olivera A and Spiegel S . Sphingosine kinase: a mediator of vital cellular functions. Prostaglandins Other Lipid Mediat, 2001; 64: 123- 134 [DOI] [PubMed] [Google Scholar]
- 17).Pyne S and Pyne NJ . Sphingosine 1-phosphate signalling and termination at lipid phosphate receptors. Biochim Biophys Acta, 2002; 1582: 121- 131 [DOI] [PubMed] [Google Scholar]
- 18).Spiegel S and Milstien S . Exogenous and intracellularly generated sphingosine 1-phosphate can regulate cellular processes by divergent pathways. Biochem Soc Trans, 2003; 31: 1216- 1219 [DOI] [PubMed] [Google Scholar]
- 19).Alewijnse AE , Peters SL and Michel MC . Cardiovascular effects of sphingosine-1-phosphate and other sphingomyelin metabolites. Br J Pharmacol, 2004; 143: 666- 684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Yanagida K and Hla T . Vascular and Immunobiology of the Circulatory Sphingosine 1-Phosphate Gradient. Annu Rev Physiol, 2017; 79: 67- 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Blaho VA and Hla T . An update on the biology of sphingosine 1-phosphate receptors. J Lipid Res, 2014; 55: 1596- 1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Proia RL and Hla T . Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest, 2015; 125: 1379- 1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Cui H , Okamoto Y , Yoshioka K , Du W , Takuwa N , Zhang W , Asano M , Shibamoto T and Takuwa Y . Sphingosine-1-phosphate receptor 2 protects against anaphylactic shock through suppression of endothelial nitric oxide synthase in mice. J Allergy Clin Immunol, 2013; 132: 1205- 1214 e9 [DOI] [PubMed] [Google Scholar]
- 24).Estrada R , Zeng Q , Lu H , Sarojini H , Lee JF , Mathis SP , Sanchez T , Wang E , Kontos CD , Lin CY , Hla T , Haribabu B and Lee MJ . Up-regulating sphingosine 1-phosphate receptor-2 signaling impairs chemotactic, wound-healing, and morphogenetic responses in senescent endothelial cells. J Biol Chem, 2008; 283: 30363- 30375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Bischoff A , Czyborra P , Fetscher C , Meyer Zu Heringdorf D , Jakobs KH and Michel MC . Sphingosine-1-phosphate and sphingosylphosphorylcholine constrict renal and mesenteric microvessels in vitro. Br J Pharmacol, 2000; 130: 1871- 1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Lorenz JN , Arend LJ , Robitz R , Paul RJ and MacLennan AJ . Vascular dysfunction in S1P2 sphingosine 1-phosphate receptor knockout mice. Am J Physiol Regul Integr Comp Physiol, 2007; 292: R440- R446 [DOI] [PubMed] [Google Scholar]
- 27).Guan Z , Singletary ST , Cook AK , Hobbs JL , Pollock JS and Inscho EW . Sphingosine-1-phosphate evokes unique segment-specific vasoconstriction of the renal microvasculature. J Am Soc Nephrol, 2014; 25: 1774- 1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Salomone S , Potts EM , Tyndall S , Ip PC , Chun J , Brinkmann V and Waeber C . Analysis of sphingosine 1-phosphate receptors involved in constriction of isolated cerebral arteries with receptor null mice and pharmacological tools. Br J Pharmacol, 2008; 153: 140- 147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Kusumi K , Shinozaki K , Kanaji T , Kurata H , Naganawa A , Otsuki K , Matsushita T , Sekiguchi T , Kakuuchi A and Seko T . Discovery of novel S1P2 antagonists. Part 1: discovery of 1,3-bis(aryloxy)benzene derivatives. Bioorg Med Chem Lett, 2015; 25: 1479- 1482 [DOI] [PubMed] [Google Scholar]
- 30).Kusumi K , Shinozaki K , Yamaura Y , Hashimoto A , Kurata H , Naganawa A , Ueda H , Otsuki K , Matsushita T , Sekiguchi T , Kakuuchi A and Seko T . Discovery of novel S1P2 antagonists. Part 2: Improving the profile of a series of 1,3-bis(aryloxy)benzene derivatives. Bioorg Med Chem Lett, 2015; 25: 4387- 4392 [DOI] [PubMed] [Google Scholar]
- 31).Kusumi K , Shinozaki K , Yamaura Y , Hashimoto A , Kurata H , Naganawa A , Otsuki K , Matsushita T , Sekiguchi T , Kakuuchi A , Yamamoto H and Seko T . Discovery of novel S1P2 antagonists, part 3: Improving the oral bioavailability of a series of 1,3-bis(aryloxy)benzene derivatives. Bioorg Med Chem Lett, 2016; 26: 1209- 1213 [DOI] [PubMed] [Google Scholar]
- 32).Hara T , Fukuda D , Tanaka K , Higashikuni Y , Hirata Y , Nishimoto S , Yagi S , Yamada H , Soeki T , Wakatsuki T , Shimabukuro M and Sata M . Rivaroxaban, a novel oral anticoagulant, attenuates atherosclerotic plaque progression and destabilization in ApoE-deficient mice. Atherosclerosis, 2015; 242: 639- 646 [DOI] [PubMed] [Google Scholar]
- 33).Salim HM , Fukuda D , Higashikuni Y , Tanaka K , Hirata Y , Yagi S , Soeki T , Shimabukuro M and Sata M . Dipeptidyl peptidase-4 inhibitor, linagliptin, ameliorates endothelial dysfunction and atherogenesis in normoglycemic apolipoprotein-E deficient mice. Vascul Pharmacol, 2016; 79: 16- 23 [DOI] [PubMed] [Google Scholar]
- 34).SG BG , Ikeda K and Arita M . Facile determination of sphingolipids under alkali condition using metal-free column by LC-MS/MS. Anal Bioanal Chem, 2018; 410: 4793- 4803 [DOI] [PubMed] [Google Scholar]
- 35).Tabas I , Garcia-Cardena G and Owens GK . Recent insights into the cellular biology of atherosclerosis. J Cell Biol, 2015; 209: 13- 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Hughes JE , Srinivasan S , Lynch KR , Proia RL , Ferdek P and Hedrick CC . Sphingosine-1-phosphate induces an antiinflammatory phenotype in macrophages. Circ Res, 2008; 102: 950- 958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Poti F , Gualtieri F , Sacchi S , Weissen-Plenz G , Varga G , Brodde M , Weber C , Simoni M and Nofer JR . KRP-203, sphingosine 1-phosphate receptor type 1 agonist, ameliorates atherosclerosis in LDL-R-/- mice. Arterioscler Thromb Vasc Biol, 2013; 33: 1505- 1512 [DOI] [PubMed] [Google Scholar]
- 38).Keul P , Lucke S , von Wnuck Lipinski K , Bode C , Graler M , Heusch G and Levkau B . Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ Res, 2011; 108: 314- 323 [DOI] [PubMed] [Google Scholar]
- 39).Zhang W , An J , Jawadi H , Siow DL , Lee JF , Zhao J , Gartung A , Maddipati KR , Honn KV , Wattenberg BW and Lee MJ . Sphingosine-1-phosphate receptor-2 mediated NFkappaB activation contributes to tumor necrosis factor-alpha induced VCAM-1 and ICAM-1 expression in endothelial cells. Prostaglandins Other Lipid Mediat, 2013; 106: 62- 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Du J , Zeng C , Li Q , Chen B , Liu H , Huang X and Huang Q . LPS and TNF-alpha induce expression of sphingosine-1-phosphate receptor-2 in human microvascular endothelial cells. Pathol Res Pract, 2012; 208: 82- 88 [DOI] [PubMed] [Google Scholar]
- 41).Chen W , Xiang H , Chen R , Yang J , Yang X , Zhou J , Liu H , Zhao S , Xiao J , Chen P , Chen AF , Chen S and Lu H . S1PR2 antagonist ameliorate high glucose-induced fission and dysfunction of mitochondria in HRGECs via regulating ROCK1. BMC Nephrol, 2019; 20: 135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Manning AM and Davis RJ . Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov, 2003; 2: 554- 565 [DOI] [PubMed] [Google Scholar]
- 43).Chen W , Lu H , Yang J , Xiang H and Peng H . Sphingosine 1-phosphate in metabolic syndrome (Review). Int J Mol Med, 2016; 38: 1030- 1038 [DOI] [PubMed] [Google Scholar]
- 44).Kurano M , Tsukamoto K , Hara M , Ohkawa R , Ikeda H and Yatomi Y . LDL receptor and ApoE are involved in the clearance of ApoM-associated sphingosine 1-phosphate. J Biol Chem, 2015; 290: 2477- 2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Kowalski GM , Carey AL , Selathurai A , Kingwell BA and Bruce CR . Plasma sphingosine-1-phosphate is elevated in obesity. PLoS One, 2013; 8: e72449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Son DJ , Lee HW , Shin HW , Lee JJ , Yoo HS , Kim TJ , Yun YP and Hong JT . Enhanced release of sphingosine-1-phosphate from hypercholesterolemic platelets: role in development of hypercholesterolemic atherosclerosis. Prostaglandins Leukot Essent Fatty Acids, 2008; 78: 383- 390 [DOI] [PubMed] [Google Scholar]
- 47).Kurano M and Yatomi Y . Sphingosine 1-Phosphate and Atherosclerosis. J Atheroscler Thromb, 2018; 25: 16- 26 [DOI] [PMC free article] [PubMed] [Google Scholar]