

Abstract
Klotho-VS heterozygosity (KL-VShet) is associated with reduced risk of Alzheimer’s disease (AD). However, whether KL-VShet is associated with lower levels of pathologic tau, i.e., the key AD pathology driving neurodegeneration and cognitive decline, is unknown. Here, we assessed the interaction between KL-VShet and levels of beta-amyloid, a key driver of tau pathology, on the levels of PET-assessed neurofibrillary tau in 551 controls and patients across the AD continuum. KL-VShet showed lower cross-sectional and longitudinal increase in tau-PET per unit increase in amyloid-PET when compared to that of non-carriers. This association of KL-VShet on tau-PET was stronger in Klotho mRNA-expressing brain regions mapped onto a gene expression atlas. KL-VShet was related to better memory functions in amyloid-positive participants and this association was mediated by lower tau-PET. Amyloid-PET levels did not differ between KL-VShet carriers versus non-carriers. Together, our findings provide evidence to suggest a protective role of KL-VShet against amyloid-related tau pathology and tau-related memory impairments in elderly humans at risk of AD dementia.
Subject terms: Molecular imaging, Haplotypes, Dementia, Alzheimer's disease
The KL-VS haplotype of the Klotho gene has been associated with reduced risk of Alzheimer’s disease and dementia. Here the authors show an association between the KL-VS haplotype and amyloid-dependent tau accumulation using PET data.
Introduction
Klotho is a transmembrane protein that has been associated with enhanced longevity and better brain health in aging1,2. Klotho is expressed primarily in the kidney and brain, where it has been implicated in a number of vital cellular functions (for review see3). Loss-of-function mutations in transgenic mice are associated with reduced Klotho protein expression, accelerated aging phenotypes, and dramatically shortened life span1,4. In humans, two variants in the Klotho gene (KL, 13q13.1), rs9536314 (F352V) and rs9527025 (C370S), form a functional haplotype. Carrying one copy, but not two copies of the KL-VS haplotype, referred to as KL-VS heterozygosity (KL-VShet), has been previously linked to increased Klotho levels in the blood5,6. KL-VShet occurs in about 20–25% of the population5 and is associated with higher cognitive performance across the adult life span5,7–9, larger frontotemporal gray matter volume in cognitively normal individuals8, and lower mortality6,10. Together, these results suggest a crucial role of Klotho in the maintenance of cognitive abilities and brain integrity during aging.
Beyond the protective role of Klotho in normal aging, recent studies suggest an association between Klotho and reduced risk of Alzheimer’s disease (AD)11, the most frequent cause of dementia in the elderly12. A recent meta-analysis reported that KL-VShet was associated with reduced AD dementia risk and cognitive decline in elderly individuals carrying the ApoE ε4 allele13, i.e., the strongest genetic risk factor for AD dementia possibly through elevated levels of primary AD pathology including cortical beta-amyloid (Aβ) aggregation14,15. Importantly, KL-VShet was associated with reduced biomarker levels of Aβ deposition in ApoE ε4 carriers16 suggesting that KL-VShet may directly alter levels of primary AD pathology.
Yet, an open question is whether Klotho is associated with altered levels of fibrillary tangles containing pathologic tau, i.e., the key driver of disease progression in AD17. In the presence of Aβ deposition, i.e., the earliest primary pathology in AD18,19, neurofibrillary tangles spread from the medial temporal lobe to higher cortical areas20–22. The progressive development of neurofibrillary tangles in the presence of Aβ pathology is closely associated with gray matter atrophy23–25 and cognitive worsening20,26–28 and is more predictive of such alterations than Aβ29. Due to the high clinical relevance of tau pathology, it is pivotal to understand whether the KL-VShet variant attenuates the accumulation of neurofibrillary tangles at a given level of Aβ deposition, and thus a cognitive decline in AD. Studies in mouse models of Aβ and accelerated aging reported that enhancing KL expression was associated with reduced Aβ burden and phosphorylated tau11,30, although conflicting results were reported as well31. However, these mouse models fail to develop neurofibrillary tangles in the presence of Aβ and thus only incompletely recapitulate AD-specific tau pathology in humans.
Here, we examined whether KL-VShet attenuates the association between higher Aβ and higher fibrillar tau assessed via positron emission tomography (PET) in a group of 551 elderly asymptomatic and symptomatic individuals recruited within a large North American multicenter study on AD32. We found the KL-VShet variant to be associated with an attenuated increase in regional tau-PET at pathological levels of global amyloid-PET, suggesting that KL-VShet was potentially protective against Aβ-related increase in neurofibrillary tangles. This association was pronounced in ApoE ε4 carriers. The strength of the KL-VShet effect on region-specific tau-PET levels was correlated with the regional expression pattern of KL in the brain33,34 supporting the notion that the KL-VShet variant modulates the regional accumulation of tau pathology. Importantly, KL-VShet was associated with higher memory performance and this association was mediated by reduced tau-PET levels in KL-VShet carriers with the elevated amyloid-PET burden. For Aβ, we did not find the previously reported association between KL-VShet and lower Aβ pathology in the current sample, but confirmed this link in a larger sample including all individuals with amyloid-PET but not necessarily tau-PET assessment available indicating that the effect size of KL-VShet on Aβ was smaller than that on tau pathology.
Results
Detailed sample characteristics are presented in Table 1. Among the 551 participants (347 CN, 156 MCI, 48 ADD), there were 144 KL-VShet carriers and 407 non-carriers. Demographics (age, sex, and education) or ApoE ε4 status did not differ between KL-VShet carrier and non-carrier groups (all p > 0.05). Continuous values of global amyloid-PET uptake did not differ between KL-VShet carriers versus non-carriers (t(265.06) = 0.92, p = 0.373).
Table 1.
Sample characteristics.
| KL-VShet carriers | KL-VShet non-carriers | p value | |
|---|---|---|---|
| ADNI, all | |||
| N | 144 | 407 | |
| Age | 71.29 (6.61) | 71.39 (6.72) | 0.880 |
| Sex, F:M | 76:68 | 206:201 | 0.727 |
| Diagnosis, CN:MCI:ADD | 102:34:8 | 245:122:40 | 0.059 |
| Education, years | 16.20 (2.50) | 16.65 (2.51) | 0.065 |
| MMSE | 28.17 (2.43) | 28.11 (2.82) | 0.819 |
| ApoE ε4 status, neg:pos | 90:54 | 255:152 | 1.000 |
| Global amyloid-PET, CL | 28.85 (37.94) | 32.28 (40.30) | 0.373 |
| Amyloid-PET status, neg:pos | 89:55 | 232:175 | 0.365 |
| Longitudinal subsample | |||
| N | 52 | 148 | |
| Age | 70.93 (5.76) | 71.43 (6.56) | 0.631 |
| Sex, F:M | 28:24 | 69:79 | 0.462 |
| Diagnosis, CN:MCI:ADD | 36:12:4 | 77:55:16 | 0.097 |
| Education, years | 15.85 (2.58) | 16.56 (2.51) | 0.081 |
| MMSE | 28.00 (2.59) | 27.92 (2.48) | 0.841 |
| ApoE ε4 status, neg:pos | 25:27 | 81:67 | 0.506 |
| Global amyloid-PET, CL | 45.01 (41.44) | 42.73 (42.59) | 0.739 |
| Amyloid-PET status, neg:pos | 22:30 | 61:87 | 1.000 |
| Tau-PET follow-up, years | 1.54 (0.75) | 1.66 (0.80) | 0.330 |
CN cognitively normal, MCI mild cognitive impairment, ADD Alzheimer’s disease dementia, F female, M male, MMSE Mini-Mental State Examination, neg negative, pos positive, CL centiloid.
KL-VS heterozygosity is associated with lower amyloid-related tau accumulation
In the main analysis, we tested the hypothesis that KL-VShet modifies the association between Aβ and tau pathology (both assessed by continuous measures of PET uptake). In a region-of-interest (ROI)-based analysis, we focused on tau-PET in the inferior temporal cortex (i.e., ROI of early Aβ-related tau pathology20,26,35–37) and whole-brain tau-PET levels. The results of a linear regression analysis showed a significant KL-VShet × amyloid-PET interaction effect on tau-PET levels in both the inferior temporal ROI (standardized beta = −0.12, p = 0.009, N = 551, effect size measured by Cohen’s f = 0.112) and the global ROI (beta = −0.13, p = 0.008, N = 551, Cohen’s f = 0.114). For both tau-PET ROIs, the increase in tau-PET as a function of rising global amyloid-PET was attenuated in KL-VShet carriers versus non-carriers (Fig. 1a, b). The main effects of amyloid- on tau-PET for KL-VShet carriers and non-carriers are reported in Supplementary Table 1. All analyses were controlled for the main effects of the interaction terms, age, sex, diagnosis, education, and ApoE ε4 carrier status.
Fig. 1. Association between KL-VS heterozygosity, amyloid-, and tau-PET.

Scatterplots display the relationship between global amyloid-PET levels and a, b cross-sectionally assessed tau-PET levels or c, d longitudinally assessed tau-PET annual change rates measured in inferior temporal gyri (left panel) and globally in neocortical areas (right panel) as a function of KL-VShet variant. Blue and gray colors indicate individuals with heterozygous or non-heterozygous KL-VS alleles. Statistics of the KL-VShet × amyloid-PET interaction effect on tau-PET uptake were derived from multiple linear regression analyses, controlled for the main effects of KL-VShet and amyloid-PET levels as well as age, sex, diagnosis, education, and ApoE ε4 carrier status. Linear model fits are indicated together with 95% confidence intervals.
Next, we performed several secondary analyses. To ensure that our results were not driven by unequally sized KL-VShet groups, we repeated the main analyses in all 144 KL-VShet carriers and 144 out of 407 non-carriers who were selected based on propensity score matching for global amyloid-PET levels and diagnosis. Comparable KL-VShet × amyloid-PET interaction effects were found on tau-PET levels in both ROIs (inferior temporal ROI: beta = −0.20, p = 0.005, N = 288, Cohen’s f = 0.168; global ROI: beta = −0.21, p = 0.010, N = 288, Cohen’s f = 0.156; Supplementary Fig. 1a, b). In addition, we repeated the interaction analysis in 1000 bootstrapped samples (i.e., random sampling from the participant pool with replacement). As a reference, we also generated a null distribution by randomly reshuffling the KL-VShet labels on each iteration. The bootstrapped mean t-value of the interaction effect differed significantly from that of the null distribution (inferior temporal ROI: t(1901.9) = −47.43, p < 0.001; global ROI: t(1839.2) = −48.99, p < 0.001; Supplementary Fig. 2). Only the distribution of t-values based on the actual, unshuffled KL-VShet labels was significantly greater than zero (inferior temporal ROI: t(999) = −77.76, p < 0.001; global ROI: t(999) = −83.88, p < 0.001) and the 95% confidence intervals did not include zero (inferior temporal ROI: 95% CI = [−4.847, −0.563]; global ROI: 95% CI = [−4.763, −0.794]). Together, these results confirmed a robust association between KL-VShet and lower Aβ-associated tau accumulation.
In order to determine whether our findings were driven by clinical diagnosis, we repeated the analyses in 156 MCI patients (34 KL-VShet carriers and 122 non-carriers) and found comparable KL-VShet × amyloid-PET interaction effects on tau-PET uptake (inferior temporal ROI: beta = −0.26, p = 0.003, N = 156, Cohen’s f = 0.251; global ROI: beta = −0.25, p = 0.004, N = 156, Cohen’s f = 0.243; Supplementary Fig. 1c, d). Repeating the analysis in 347 CN participants (102 KL-VShet carriers and 245 non-carriers) yielded no KL-VShet × amyloid-PET interaction effects on tau-PET levels in either ROI (both p > 0.05; Supplementary Fig. 1e, f).
A few participants showed lower tau-PET levels in the ROIs than in the reference region resulting in a tau-PET standard uptake value ratio (SUVR) < 1 (2 participants for the inferior temporal ROI and 51 participants for the global ROI). Yet, the results of the KL-VShet × amyloid interaction analyses on tau-PET levels remained significant after excluding those participants (Supplementary Fig. 1g, f).
KL-VS heterozygosity is related to lower amyloid-dependent tau accumulation over time
In a subsample of 200 participants in whom longitudinal tau-PET data were available, we investigated whether KL-VShet attenuates the association between baseline amyloid-PET levels and the rate of change in tau-PET assessed over a time interval of 1.63 years on average (range: 1–4 years). We found a significant KL-VShet × amyloid-PET interaction effect on tau-PET annual change rates in the inferior temporal ROI (beta = −0.22, p = 0.039, N = 200, Cohen’s f = 0.148), but not in the global ROI (beta = −0.15, p = 0.176, N = 200, Cohen’s f = 0.098). KL-VShet carriers showed lower tau-PET increases in inferior temporal cortices over time as a function of rising global amyloid-PET levels (Fig. 1c, d) suggesting that the KL-VShet variant might be protective against Aβ-associated increase of tau pathology. The main effects of amyloid-PET on tau-PET change rates for KL-VShet carriers and non-carriers are reported in Supplementary Table 1.
Stronger protective effect of KL-VS heterozygosity in ApoE ε4 carriers
Previous studies have reported an ApoE ε4-genotype-dependent effect of KL-VShet on amyloid-PET16. Hence, we additionally explored whether ApoE ε4 carriers showed a stronger association between KL-VShet and lower tau accumulation than ApoE ε4 non-carriers, controlling for age, sex, education, diagnosis, and global amyloid-PET levels in the regression analyses. This analysis yielded a significant KL-VShet × ApoE ε4 interaction effect on tau-PET levels (inferior temporal ROI: beta = −0.11, p = 0.031, N = 551, Cohen’s f = 0.093; global ROI: beta = −0.10, p = 0.041, N = 551, Cohen’s f = 0.088; Supplementary Fig. 3).
Spatial match between KL mRNA expression and the effect of KL-VS heterozygosity on tau-PET
In order to estimate the spatial overlap between the strength of KL gene expression and the test statistic of the KL-VShet × amyloid-PET interaction on tau-PET, we obtained whole-brain mRNA expression levels of KL generated by post-mortem microarray assessments of six healthy brain donors and subsequently mapped to the Allen Brain Atlas33,34. We computed median scores of log2 mRNA expression of KL across the six donors within 34 left-hemispheric regions of the Freesurfer-based Desikan–Killiany brain atlas38. We focused on the left hemisphere since all donors had microarray assessment available for the left hemisphere and only two donors had an assessment for the right hemisphere. Furthermore, we estimated the KL-VShet × amyloid-PET interaction effect on tau-PET levels within the same 34 brain atlas regions using the aforementioned regression model. Surface mapping of both the KL-VShet × amyloid-PET interaction effect (which were all in the same direction) and KL mRNA expression is displayed in Fig. 2a–d. Spatial correlation analysis revealed a significant association (r = 0.46, p = 0.007; Fig. 2e). This result suggests that regions with higher KL mRNA expression levels were more likely to display lower Aβ-related tau-PET levels in KL-VShet carriers versus non-carriers. Visual inspection of the thresholded spatial maps indicated that those areas showing both a significant KL-VShet × amyloid-PET interaction effect (Fig. 2b; see Supplementary Table 2 for detailed statistical results) and high KL mRNA expression (log2 > 75th percentile) (Fig. 2d) were specifically located within the mesiotemporal and inferior and middle temporal brain regions and the posterior cingulum.
Fig. 2. Spatial patterns of KL-VShet-related attenuation of tau-PET and Klotho mRNA expression.

a Surface mapping of the interaction effect between KL-VShet and amyloid-PET levels on tau-PET accumulation within 34 left-hemispheric regions of the Desikan–Killiany atlas. Yellow colors indicate higher t-values reflective of a stronger interaction effect (all t-values inverted for illustration purpose; see Supplementary Table 2 for details statistical results). b Thresholded spatial map color-code only regions with a significant (p < 0.01, unadjusted for multiple comparisons) KL-VShet × amyloid-PET interaction effect. c Surface mapping of median KL mRNA expression (i.e., log2 derived from the Allen Brain Atlas) within the identical 34 atlas regions. Yellow colors indicate higher KL mRNA expression. d Thresholded spatial maps restricted to regions falling above the 75th percentile of KL mRNA expression. e Scatterplot depicting the association between ROI-based KL mRNA expression and KL-VShet × amyloid-PET interaction effect on regional tau-PET uptake. Statistical results are derived from the Pearson correlation (two-sided). Linear model fits are indicated together with 95% confidence intervals. Source data are provided as a Source Data file.
Tau mediates the association between KL-VS heterozygosity and less memory impairment
In the main analysis, we assessed whether KL-VShet is beneficial for memory functions via lowering tau pathology. Because the interaction effect of KL-VShet × amyloid-PET on tau-PET levels showed that KL-VShet is associated with lower tau accumulation at higher levels of amyloid-PET, we restricted our analysis to amyloid-positive participants. We used mediation analysis with 10,000 bootstrapping iterations in order to test whether KL-VShet is associated with better memory in individuals with elevated Aβ burden and whether this effect is mediated via reduced global tau-PET levels. Memory performance was measured by an established composite score based on participant’s results across multiple different memory tests (ADNI-MEM)39. The mediation analysis was controlled for age, sex, education, diagnosis, ApoE ε4 status, and global amyloid-PET levels. Supporting our hypothesis, we found KL-VShet to be associated with higher ADNI-MEM scores (beta = 0.13, p = 0.040, Cohen’s f = 0.104, N = 229, Fig. 3) and that this relationship was significantly mediated by lower tau-PET levels (bootstrapped average causal mediation effect: beta = 0.06, 95% CI = 0.01–0.12, p = 0.013, N = 229). This result indicates that, in individuals with an elevated Aβ burden, KL-VShet carriers showed less impaired episodic-memory abilities when compared to KL-VShetnon-carriers due to lower tau-PET levels in KL-VShet carriers. A path model of the mediation analysis is shown in Fig. 4.
Fig. 3. Association between KL-VS heterozygosity and memory in amyloid-positive individuals.
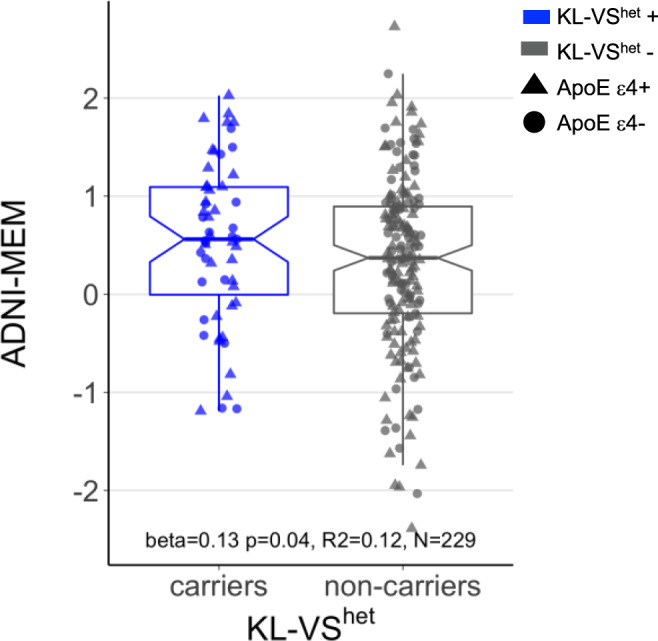
Boxplot shows memory performance as a function of KL-VShet variant in individuals with a positive amyloid-PET (SUVRFBP ≥ 1.11 or SUVRFBB ≥ 1.08). Blue and gray colors indicate individuals with heterozygous (N = 55) or non-heterozygous KL-VS alleles (N = 174). Memory was measured by an established composite score, ADNI-MEM, based on test performance across multiple different memory tests39. Statistical result of the main effect of KL-VShet on memory was derived from multiple linear regression analysis, controlled for age, sex, diagnosis, education, and ApoE ε4 carrier status. Boxplots show the 25th percentile, median, 75th percentile (box), 95% confidence intervals of the median (notch), and 1.5× IQR (whiskers).
Fig. 4. Lower tau-PET levels mediate the beneficial association of KL-VS heterozygosity and memory in individuals with the elevated amyloid-PET burden.
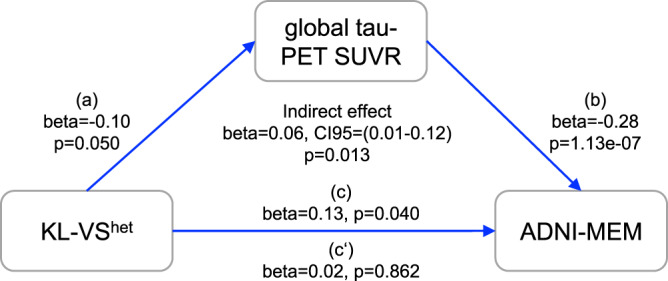
Path diagram of the mediation model (assessed only in amyloid-positive participants, N = 229), showing that the association between KL-VShet and better memory performance is mediated via lower global tau-PET uptake. Memory is measured by ADNI-MEM, i.e., an established memory composite score39. Path-weights are displayed as standardized beta values. All paths are controlled for age, sex, diagnosis, education, ApoE ε4 carrier status, and continuous global amyloid-PET levels. The significance of the indirect effect was determined using bootstrapping with 10,000 iterations as implemented in the mediation package in R.
In the secondary analysis, we addressed the question of whether KL-VShet exerts a beneficial influence on memory via lowered neurofibrillary tau in the whole sample without stratification based on amyloid-PET levels. To this end, we estimated the interaction effect between KL-VShet and amyloid-PET on ADNI-MEM scores, with and without controlling for global tau-PET levels as a covariate. We reasoned that if the association between KL-VShet on cognition is mediated by tau-PET, the interaction between KL-VShet and amyloid-PET on cognition should be diminished when controlling for tau-PET. Without controlling tau-PET levels, we found a significant KL-VShet × amyloid-PET interaction effect on memory functions (beta = 0.08, p = 0.037, N = 549, Cohen’s f = 0.090). Specifically, we observed that individuals with a high amyloid-PET burden showed better cognitive performance when being KL-VShet carriers compared to non-carriers (Supplementary Fig. 4). As hypothesized, the KL-VShet × amyloid-PET interaction on memory no longer reached significance level when global tau-PET levels were controlled for (beta = 0.06, p = 0.125, N = 549).
Besides our focus analysis of the beneficial effect of KL-VShet on memory, i.e., the cognitive domain affected early in AD, we also explored the effect on other cognitive domains including executive functions (composite score ADNI-EF), language (composite score ADNI-LAN) and visual-spatial perception (composite score ADNI-VS). In amyloid-positive participants, we found KL-VShet to be associated with higher ADNI-LAN scores (beta = 0.14, p = 0.027, Cohen’s f = 0.114, N = 229; Supplementary Fig. 5a), a trend-level association with higher ADNI-EF scores (beta = 0.12, p = 0.067, Cohen’s f = 0.095, N = 229) and no association with ADNI-VS scores (beta = 0.05, p = 0.468, N = 229). The beneficial association between KL-VShet and language abilities was mediated by lower global tau-PET levels in KL-VShet carriers versus non-carriers (bootstrapped average causal mediation effect: beta = 0.05, 95% CI = 0.01–0.12, p = 0.017, N = 229; Supplementary Fig. 5b).
Is KL-VS heterozygosity associated with lower Aβ accumulation?
A recent study found a protective influence of KL-VShet on longitudinal amyloid-PET in cognitively unimpaired ApoE ε4 carriers aged between 60 and 80 years, but not in ApoE ε4 non-carriers or older participants13. In contrast to this earlier report, we did not find an age-dependent KL-VShet effect on cross-sectional amyloid-PET levels acquired at the time of tau-PET assessment in the current sample (KL-VShet × age interaction: beta = 0.21, p = 0.597, N = 551; Supplementary Fig. 6a) or an ApoE ε4-dependent KL-VShet effect in the subsample of CN participants aged between 60 and 80 years (beta = 0.04, p = 0.586, N = 347, Cohen’s f = 0.030; Supplementary Fig. 6b). However, more subtle effects may have been overlooked in the current more restricted sample of individuals undergoing both amyloid- and tau-PET. Therefore, in a supplementary analysis, we included all participants with amyloid-PET (N = 1067) from ADNI, regardless of whether or not they underwent tau-PET assessment. We found a trend-level significant KL-VShet × age interaction effect one amyloid-PET that demonstrated that KL-VShet carriers in the lower age range (<80 years) displayed lower amyloid-PET levels than non-carriers (beta = 0.53, p = 0.046, Cohen’s f = 0.061, N = 1067; Supplementary Fig. 7a). Consistent with the earlier report, we found a significant KL-VShet × ApoE ε4 interaction effect on global amyloid-PET levels in CN participants aged between 60 and 80 years (beta = −0.121, p = 0.043, Cohen’s f = 0.095, N = 464; Supplementary Fig. 7b). The same analysis in MCI participants within the same age range showed no significant interaction effect (beta = 0.02, p = 0.780, N = 463, Cohen’s f = 0.013), suggesting that the association between KL-VShet and lower amyloid-PET uptake is restricted to a younger age and non-symptomatic cognitive status. See Supplementary Table 3 for detailed sample characteristics of the larger ADNI amyloid-PET sample compared to the current ADNI tau-PET sample. Thus, our results in the larger sample are consistent with those from Belloy et al.’s analysis on the effect of KL-VShet on amyloid-PET stratified by age and ApoE genotype while also showing that the effect size of KL-VShet on tau-PET is stronger than that on amyloid-PET.
Discussion
The heterozygous KL gene variant KL-VShet has been previously associated with higher longevity and cognition performance in adulthood and reduced AD dementia risk13. We demonstrate that elderly KL-VShet carriers with elevated Aβ burden, i.e., the earliest primary AD pathology, exhibited lower tau-PET levels and tau-PET annual change rates when compared to those in KL-VShetnon-carriers. In amyloid-positive participants, the KL-VShet variant was associated with better memory performance, and this relationship was mediated by lower tau-PET levels, suggesting that lower levels of pathologic tau in the KL-VShet carriers explained the association between KL-VShet and better memory performance. Although our findings do not implicate a causative mechanism of Klotho in AD, we provide evidence for a potential protective role of KL-VShet against Aβ-dependent tau pathology that is the key AD brain alteration linked to cognitive impairment.
To our knowledge, the current study is the first to date that evaluated the interaction between KL-VShet and Aβ on tau accumulation and cognitive decline in humans. There is a growing literature on protective genetic variants in AD40–42, but only a few studies have reported genetic variants to be associated with lower tau pathology in AD43. For the KL-VShet variant, previous studies reported an association with reduced Aβ accumulation in elderly ApoE ε4 risk-carriers13,16. We extend these previous findings by showing that the relationship between Aβ accumulation and fibrillar tau is modulated by KL-VShet, such that lower local and global tau-PET levels were observed per unit increase of global amyloid-PET burden in KL-VShet carriers when compared to those in non-carriers. This is important because Aβ deposition precedes the development of dementia symptoms by up to 20 years14, and as confirmed by a very recent longitudinal amyloid/tau-PET study, high baseline Aβ is associated with subsequent tau accumulation, while Aβ and tau in synergy lead to most pronounced subsequent cognitive decline21. The region showing one of the strongest interaction effects between KL-VShet and amyloid-PET on tau-PET was the inferior temporal gyrus (Fig. 2a), a brain area that typically shows an early Aβ-related increase in tau-PET26 before elevated tau-PET levels extend to other higher cortical brain areas20. The protective association between KL-VShet and tau-PET was present selectively in participants with abnormally elevated levels of amyloid-PET and more pronounced in ApoE ε4 carriers. Stratified analyses further revealed a significant KL-VShet effect in the MCI but not in the CN subgroup, which could potentially be due to a stage-dependent beneficial effect of Klotho. However, an alternative explanation is that the levels of both amyloid- and tau-PET are lower in CN compared to those in MCI, and thus any protective effect is likely to be of smaller size and would require a larger sample size to detect. Together, these results support the notion that KL-VShet is associated with an Aβ-related rather than age-related reduction of tau pathology.
In amyloid-positive individuals, we found KL-VShet to be associated with better memory performance, mediated by the effect of KL-VShet on tau-PET. Our results are broadly consistent with those from studies on healthy aging, reporting KL-VShet to be associated with better cognition5,8–10, and lower risk of conversion from cognitively normal to mild cognitive impairment or AD dementia in ApoE ε4 carriers13,16. Our findings suggest that the association between KL-VShet and lower neurofibrillary tau pathology is of central importance for the association found between KL-VShet and less cognitive impairment. A previously reported absence of an association between KL-VShet and cognitive decline in asymptomatic participants with elevated levels of Aβ44 did not assess the presence of abnormal neurofibrillary tau, which may have hampered to detect an effect of KL-VShet on cognitive decline in subjects at risk of AD35.
Previous studies reported KL-VShet to be associated with lower amyloid-PET in ApoE ε4 carriers (but not in ApoE ε4 non-carriers), which was strongest in the age range between 60 and 80 years13,16. In our primary analysis, we did not confirm age- or ApoE ε4-dependent effects of KL-VShet on tau-PET. By investigating a larger sample of all participants with available amyloid-PET regardless of the availability of tau-PET (N = 1067), we were able to substantiate those earlier findings13,16. Specifically, we showed reduced amyloid-PET burden in younger KL-VShet carriers (<80 years) and, in accordance with previous work, this association was mainly driven by cognitively unimpaired ApoE ε4 carriers rather than non-carriers or MCI patients. Comparing effect sizes, Cohen’s f = 0.061 for the association between KL-VShet and lower amyloid-PET versus f = 0.114 for the association with lower tau-PET, strengthens the important role of changes in tau pathology for understanding the role of Klotho in AD.
The mechanisms linking Klotho to tau pathology remain elusive. Klotho is a pleiotropic protein that has been implicated in multiple biological processes including insulin regulation4, growth factor functions, in particular of FGF2345, regulation of members of the redox system46, and calcium signaling47. One possibility of how the Klotho protein might be linked to reduced neurofibrillary tau is its involvement in autophagy48, a mechanism that is involved in the clearance of AD pathologies49. Lentiviral overexpression of Klotho protein in an APP-PS1 mouse model of Aβ deposition reduced Aβ plaque load in aged mice and rescued the impaired autophagy possibly by modulating the Akt/mTOR pathway11,50. Since APP-PS1 mice do not develop tau pathology, it remains, however, to be tested whether Klotho-induced autophagy reduces tau pathology. Those mechanistic explanations remain speculative at this point and the current work encourages future studies to investigate the mechanism that could underlie the protection Klotho exerts against the development of Aβ-related tau pathology.
Our findings of the spatial correspondence between the strength of the effect of KL-VShet on regional tau-PET and the spatial distribution of KL mRNA suggest a local effect of Klotho on the development of fibrillar tau, especially in temporal brain areas. Alternative splicing of the human KL mRNA results in both a membrane-bound and a secreted transcript of Klotho1,4, indicating that Klotho may act both in a cell-autonomous manner and as a humoral factor. Therefore, differences in gene expression in KL in the brain and/or different circulating levels of Klotho linked to KL-VShet5 may influence the development of pathological tau11, but this link remains to be investigated.
Our results have important implications for clinical trials in AD. Since tau pathology correlates more closely with clinical symptoms than Aβ, tau-targeted therapies seem a promising approach to arrest disease progression51. The common KL-VS genotype may inform those clinical trials that target tau pathology. Especially when anti-tau trials aim to include amyloid-positive or ApoE ε4 carrying participants, group differences in the KL-VShet variant may be taken into account when estimating the expected change in tau pathology over time, which would be useful in the computation of statistical power to detect a treatment effect. Furthermore, the current findings encourage future studies to test whether enhancing Klotho protein levels could reduce the development of tau pathology in amyloid-positive participants. The Klotho protein is druggable and could thus be made a target in the development of disease-modifying therapeutic approaches.
Several caveats should be considered when interpreting the current results. First, the human KL gene consists of three polymorphic variants. We decided to focus on the KL-VS haplotype given the existing evidence of its beneficial influence on Aβ and cognition in both mice and humans5,8–10,13,52. While the second variant C1818T (rs564481) is located on the fourth exon and likely has no functional consequences itself, the third variant G395A (rs1207568) is located in the promoter region and may be a potential regulatory site of KL. The two latter variants appear more frequent in Asian populations, where they have been linked to cardiovascular risk factors53. Related to the current research question, an investigation across three independent cohorts of oldest-old Danes found different polymorphic variants of KL, besides KL-VS, to be associated with better cognitive functions7. It has yet to be proven whether these other KL variants also support resilience in AD. Another caveat is that we did not measure Klotho protein levels in the serum or CSF. Circulating levels of Klotho decrease during aging54 and are associated with cognitive performance5 and gray matter volume55 in cognitively unimpaired individuals. In patients with AD, CSF levels of Klotho are reduced52, where the experimental reversal of reduced Klotho expression in transgenic mouse models exerted beneficial effects on Aβ and cognition8,11,30. While the KL-VShet variant has been associated with higher circulating levels of Klotho55, it remains to be investigated whether the association between KL-VShet and pathological tau are mediated by higher protein levels in the CSF and brain tissue.
In summary, our findings revealed a protective association of KL-VShet on tau accumulation that particularly manifested in amyloid-positive individuals, where lower tau pathology was related to better cognitive functions. These findings may be particularly informative for clinical anti-tau trials56 and may encourage future studies on enhancing Klotho protein levels as a therapeutic intervention to slow down the development of tau pathology and dementia in AD.
Methods
Sample characteristics
A total of 551 participants were selected from ADNI phase 3 (ClinicalTrials.gov ID: NCT02854033) based on the availability of KL-VS and ApoE ε4 genotyping, T1-weighted MRI, 18F-flortaucipir (FTP) tau-PET and 18F-florbetapir (FBP) or 18F-florbetaben (FBB) amyloid-PET. MR and PET imaging had to be acquired during the same study visit. In addition, a subsample of 200 participants with a follow-up tau-PET assessment was selected for the longitudinal analyses. The two single-nucleotide polymorphisms for KL-VS (rs9536314 for F352V, rs9527025 for C370S) and ApoE (rs429358, rs7412) were genotyped using DNA extracted by Cogenics from a 3 mL aliquot of EDTA blood. Participants were assigned to the heterozygous KL-VS group when they carried 1, but not 2, copies of the KL-VS haplotype. ApoE ε4 carriers were defined as individuals carrying at least one ε4 allele. Clinical classification was performed by the ADNI centers, dividing participants into cognitively normal (CN, Mini-Mental State Examination [MMSE] > 24, CDR = 0, non-depressed), mild cognitively impairment (MCI; MMSE > 24, CDR = 0.5, objective memory-loss on the education adjusted Wechsler Memory) or AD dementia (ADD; 19 < MMSE < 24, CDR = 0.5–1.0, NINCDS/ADRDA criteria for probable AD are fulfilled). All participants provided written informed consent and all work complied with ethical regulations for work with human participants.
MR and PET acquisition and preprocessing
All imaging data were downloaded from the ADNI loni image archive (https://ida.loni.usc.edu).
Structural T1-weighted images were acquired on 3T scanners using a 3D MPRAGE sequence with 1 mm isotropic voxel-size and a TR = 2300 ms (detailed scan protocols can be found on https://adni.loni.usc.edu/wp-content/uploads/2017/07/ADNI3-MRI-protocols.pdf). Structural images were processed using Freesurfer (version 5.3.0) and parcellated according to the Desikan–Killiany atlas57.
Tau-PET was assessed in 6 × 5 min blocks 75 min after intravenous bolus injection of 18F-FTP. Amyloid-PET scans were obtained during 4 × 5 min time frames measured 50–70 min post injection of 18F-FBP or 90–110 min post injection of 18F-FBB. For both tau- and amyloid-PET we downloaded partially preprocessed data (http://adni.loni.usc.edu/methods/pet-analysis-method/pet-analysis/).
All PET images were coregistered to the corresponding T1-weighted image to make use of Freesurfer-derived masks in participants’ high resolution, native space. SUVR scores were obtained by normalizing tau-PET images to the inferior cerebellar gray matter and amyloid-PET images to the whole cerebellum, following the previous recommendations58. In order to make FBP and FBB amyloid-PET measures comparable, we transformed SUVR scores into centiloid (CL) units using the established transformation formula (http://adni.loni.usc.edu/wp-content/themes/freshnews-dev-v2/documents/pet/ADNI Centiloids Final.pdf). For the analysis of longitudinal tau-PET, we additionally calculated annual tau-PET SUVR change rates as the difference between tau-PET SUVR scores measured at the follow-up versus baseline visit divided by the follow-up time in years.
Tau- and amyloid-PET regions of interest
For the analyses of tau-PET, we extracted mean SUVR scores from bilateral inferior temporal gyri marking Aβ-related increase of tau pathology to neocortical structures20,26,35–37. In addition, we assessed global tau-PET burden59 as the size-weighted mean SUVR score across all Freesurfer regions, excluding hippocampus, thalamus, and basal ganglia due to commonly reported tracer off-target binding60.
For the analysis of amyloid-PET images, we computed mean amyloid-PET levels from a global ROI spanning lateral and medial frontal, anterior and posterior cingulate, lateral parietal, and lateral temporal regions. Mean SUVR from these regions was also used for sample stratification into amyloid-positive participants based on established thresholds (SUVRFBP ≥ 1.11 or SUVRFBB ≥ 1.08; see “ADNI_UCBERKELEY_AV45_Methods_12.03.15.pdf” and “UCBerkeley_FBB_Methods_04.11.19.pdf” on the ADNI website).
mRNA expression levels of KL
Regional gene expression was obtained from publicly available microarray measurements of regional mRNA expression based on post-mortem data from the Allen Brain Atlas (http://human.brain-map.org). The Allen Brain atlas is based on more than 60,000 microarray probes collected from 3700 autopsy-based brain tissue samples from a total of six individuals aged 24–57 without a known history of neurological or psychiatric diseases33,34. Microarray-based log2 expression values of 20,737 genes within each of the 3700 samples were mapped back into MNI standard space by the Allen Brain Institute using stereotactic coordinates of the examined probes. The whole gene expression data have been recently mapped to the Freesurfer-based Desikan–Killiany atlas as median gene expression for probes falling within each of the 68 atlas ROIs38. Here, we specifically extracted median expression of KL mRNA within these Desikan–Killiany ROIs, to test a spatial correlation between KL expression and KL-VShet effects on local tau-PET uptake. Since microarray assessments and thus KL mRNA expression of all six Allen brain atlas subjects were available only for the left hemisphere (vs. two subjects for the right hemisphere), we restricted the analysis of KL mRNA expression data to the more robust estimates of the left hemisphere in line with previous studies61,62.
Neuropsychological assessment
The ADNI neuropsychological test battery contains multiple indicators for memory functions, on which basis a composite score (ADNI-MEM) has been established39. ADNI-MEM summarizes test performance on the Rey Auditory Verbal Learning Test, elements from the AD Assessment Scale-Cognitive Subscale, word recall from the MMSE, and the Wechsler Logical Memory Scale II. In the exploratory analysis, we also used established ADNI summary scores of executive functions (ADNI-EF), language (ADNI-LAN), and visual-spatial abilities (ADNI-VS) (see ADNI_Methods_UWNPSYCHSUM_March_2020.pdf on ADNI webpage). Note that 2 participants had no neuropsychological tests available resulting in a sample of 549 participants for this part of the analysis.
Statistical analysis
All statistical analyses were conducted with R statistical software (version 3.6.1). P values were considered significant when meeting a two-tailed alpha threshold of 0.05. Baseline tau-PET SUVR values were entered as log-transformed values into the statistical models to approximate normality. All interaction analyses were controlled for the main effect of the interaction terms. Group demographics were compared between KL-VShet carriers versus non-carriers using Welch T-tests for continuous measures and χ2 tests for categorical measures.
KL-VS heterozygosity × amyloid interaction on tau pathology
In our main analysis, we tested whether KL-VShet moderates the relationship between amyloid- and tau-PET. To this end, multiple linear regression analyses were used to estimate the interaction effects between KL-VShet and global amyloid-PET uptake on tau-PET levels in the inferior temporal ROI and the global ROI (N = 551). Age, sex, diagnosis, education, and ApoE ε4 carrier status were considered as covariates. In secondary analyses, we accounted for potential biases due to unequally sized KL-VShet groups by repeating the same interaction analysis in matched groups of equal size (N = 288). For this purpose, 144 out of 407 KL-VShetnon-carriers were selected based on propensity score matching for global amyloid-PET levels and clinical diagnosis using the matchit R package. To ensure that our results were not affected by the skewed distribution of PET data or outliers, we iteratively determined the t-statistic of the Kl-VShet × amyloid interaction effect on tau-PET levels using 1000 bootstrapping iterations (i.e., random sampling from the subject pool with replacement using the boot R package). As a reference, we generated a null distribution of the t-statistic using the same approach, but randomly reshuffling the KL-VShet labels on every iteration. We compared the mean t-value of the bootstrapped interaction effect to that of the null distribution using Welch T-tests. The significance of the bootstrapped interaction effect was determined by one-samplet-tests estimating whether the resulting distributions of t-values significantly differ from zero and by confirming that the 95% confidence intervals did not overlap with zero. In addition, we accounted for potential influences of clinical diagnoses by repeating the same interaction analyses in the subsample of only MCI (N = 156) or CN (N = 347) participants. Finally, we repeated the analysis in a subsample excluding participants with tau-PET SUVR values in the two ROIs smaller than the reference region (i.e., SUVR < 1).
Next, we tested whether KL-VShet moderates the relationship between amyloid- and tau-PET accumulation over time (N = 200). Separate linear regression models were used to estimate the KL-VShet × global amyloid-PET interaction effect on tau-PET annual change rates in the inferior temporal ROI and the global ROI, controlling for age, sex, and diagnosis.
KL-VS heterozygosity × ApoE interaction on tau pathology
Additional exploratory analyses were run to determine the influence of ApoE ε4 carrier status. To this end, we examined the interaction between KL-VShet and ApoE ε4 status on tau-PET levels in the whole sample (206 ApoE ε4 carriers and 345 non-carriers), controlling for age, sex, diagnosis, education, and global amyloid-PET uptake.
Spatial match between KL mRNA expression and the association of KL-VS heterozygosity on tau pathology
Next, we tested whether the favorable influence of KL-VShet on local tau-PET levels overlapped within those brain regions showing higher local KL mRNA expression levels. To this end, we determined KL mRNA expression using the Allen brain atlas data in all 34 left-hemispheric Desikan–Killiany atlas regions and determined the KL-VShet × amyloid-PET interaction effect on tau-PET uptake for corresponding anatomical regions. We then tested the ROI-to-ROI Pearson–moment correlation between regional KL mRNA expression and the interaction effect test statistic (not restricted to regions showing a significant interaction effect).
KL-VS heterozygosity–memory relationship and lower tau pathology as a mediator
We tested whether KL-VShet was associated with better memory functions, and whether this association was mediated by reduced tau-PET levels. For the main analysis, mediation analysis (causal mediation R package) was conducted in which KL-VShet variant was treated as a predictor, global tau-PET levels as a mediator, and ADNI-MEM scores as an outcome. Mediation analysis was performed in the subsample of amyloid-positive participants (N = 229) since we found KL-VShet to be associated with lower tau-PET levels specifically in individuals with elevated amyloid-PET uptake. Note that, since we conditioned the mediation effect on amyloid-PET levels, this is formally a moderated mediation analysis that we conducted only for one level of the moderator (amyloid status = positive) following our hypothesis. The significance of the mediation effect was determined using 10,000 bootstrapped iterations, where each path of the model was controlled for age, sex, diagnosis, education, ApoE ε4 carrier status, and global amyloid-PET levels.
We ran an alternative analysis strategy in the whole sample (including amyloid-positive and -negative participants, N = 549) that estimated the KL-VShet × amyloid-PET interaction effect on ADNI-MEM scores. Importantly, the interaction analysis was once run without and once with controlling global tau-PET levels. We specifically hypothesized that if the beneficial influence of KL-VShet is dependent on lowering tau accumulation, then the interaction effect should be diminished in the tau-controlled analysis. Other covariates considered in the multiple regression models were age, sex, diagnosis, education, and ApoE ε4 carrier status. In secondary analyses, we repeated the mediation analysis for ADNI summary scores of other cognitive domains.
Is KL-VS heterozygosity associated with lower Aβ accumulation?
Lastly, we performed an exploratory analysis with the aim to confirm previously observed age- and ApoE-dependent associations between KL-VShet and lower amyloid-PET burden13. For this purpose, we tested for a KL-VShet × age effect on global amyloid-PET levels in the current sample (N = 551) and in a larger ADNI sample (N = 1067) including all participants with amyloid-PET assessment and KL-VS status (regardless of whether or not they underwent tau-PET assessment). Sex, education, and diagnosis were considered as covariates. Comparable to the original report, we also investigated ApoE-dependent effects of KL-VShet on amyloid-PET levels in a subgroup including only CN participants aged between 60 and 80 years. Age, sex, and education were considered as covariates.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
Data used in the preparation of this manuscript were obtained from the ADNI database (adni. loni.usc.edu). As such, the investigators within the ADNI study contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this paper. The study was funded by DAAD post-doc fellowship (to J. N.), grants from the Alzheimer Forschung Initiative (AFI, Grant 15035 to M. E.), Legerlotz Stiftung (to M. E.), LMUexcellent (to M. E.), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant for major research instrumentation (DFG, INST 409/193-1 FUGG; to M. D.); Hertie Foundation for Clinical Neurosciences (to N. F.), LMU Förderung Forschung Lehre (Reg. 1032 to N. F.), European Union’s Horizon 2020 research and innovation programme (grant agreement No. 666881 [SVDs@target] and 667375 [CoSTREAM]; to M. D.), the DFG as part of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198) and the CRC 1123 (B3) to M. D.). M. B. received speaker honoraria from GE healthcare and LMI and is an advisor of LMI. ADNI data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging, and Bioengineering, and through contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org).
Source data
Author contributions
J.N.: study concept and design, data processing, statistical analysis, interpretation of the results, and writing the manuscript. N.F.: critical revision of the manuscript. A.R.: data processing and critical revision of the manuscript. M.D.: critical revision of the manuscript. M.B.: critical revision of the manuscript. R.M.: data processing and critical revision of the manuscript. M.E.: study concept and design, interpretation of the results, and writing the manuscript. ADNI provided all data used for this study.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
The data that support the findings of this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and are available from the ADNI database (adni.loni.usc.edu) upon registration and compliance with the data use agreement. A list including the anonymized participant identifiers of the currently used sample and the source file can be downloaded from the ADNI database (tau-PET data release in May 2020; UCBERKELEYAV1451_05_12_20.csv). The Allen Brain Atlas (http://human.brain-map.org) and Freesurfer-mapped transcriptomic data from the Allen Brain Atlas (http://figshare.com/articles/A_FreeSurfer_view_of_the_cortical_transcriptome_generated_from_the_Allen_Human_Brain_Atlas/1439749) are freely available online. Source data underlying Fig. 2 are provided with this paper.
Code availability
The R code pertaining to the figures in this manuscript is provided at https://github.com/njulianeitzel/NatCommun2021_KL-VS. Costume R code can be obtained from the first author upon request.
Competing interests
M.B. received speaker honoraria from GE healthcare and LMI and is an advisor of LMI. All other authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A list of authors and their affiliations appears at the end of the paper.
Contributor Information
Julia Neitzel, Email: j.neitzel@erasmusmc.nl.
Michael Ewers, Email: Michael.Ewers@med.uni-muenchen.de.
Alzheimer’s Disease Neuroimaging Initiative (ADNI):
Michael Weiner, Paul Aisen, Ronald Petersen, Clifford R. Jack, Jr., William Jagust, John Q. Trojanowki, Arthur W. Toga, Laurel Beckett, Robert C. Green, Andrew J. Saykin, John Morris, Leslie M. Shaw, Enchi Liu, Tom Montine, Ronald G. Thomas, Michael Donohue, Sarah Walter, Devon Gessert, Tamie Sather, Gus Jiminez, Danielle Harvey, Matthew Bernstein, Nick Fox, Paul Thompson, Norbert Schuff, Charles DeCArli, Bret Borowski, Jeff Gunter, Matt Senjem, Prashanthi Vemuri, David Jones, Kejal Kantarci, Chad Ward, Robert A. Koeppe, Norm Foster, Eric M. Reiman, Kewei Chen, Chet Mathis, Susan Landau, Nigel J. Cairns, Erin Householder, Lisa Taylor Reinwald, Virginia Lee, Magdalena Korecka, Michal Figurski, Karen Crawford, Scott Neu, Tatiana M. Foroud, Steven Potkin, Li Shen, Faber Kelley, Sungeun Kim, Kwangsik Nho, Zaven Kachaturian, Richard Frank, Peter J. Snyder, Susan Molchan, Jeffrey Kaye, Joseph Quinn, Betty Lind, Raina Carter, Sara Dolen, Lon S. Schneider, Sonia Pawluczyk, Mauricio Beccera, Liberty Teodoro, Bryan M. Spann, James Brewer, Helen Vanderswag, Adam Fleisher, Judith L. Heidebrink, Joanne L. Lord, Sara S. Mason, Colleen S. Albers, David Knopman, Kris Johnson, Rachelle S. Doody, Javier Villanueva Meyer, Munir Chowdhury, Susan Rountree, Mimi Dang, Yaakov Stern, Lawrence S. Honig, Karen L. Bell, Beau Ances, John C. Morris, Maria Carroll, Sue Leon, Mark A. Mintun, Stacy Schneider, Angela OliverNG, Randall Griffith, David Clark, David Geldmacher, John Brockington, Erik Roberson, Hillel Grossman, Effie Mitsis, Leyla deToledo-Morrell, Raj C. Shah, Ranjan Duara, Daniel Varon, Maria T. Greig, Peggy Roberts, Marilyn Albert, Chiadi Onyike, Daniel D’Agostino, II, Stephanie Kielb, James E. Galvin, Dana M. Pogorelec, Brittany Cerbone, Christina A. Michel, Henry Rusinek, Mony J. de Leon, Lidia Glodzik, Susan De Santi, P. Murali Doraiswamy, Jeffrey R. Petrella, Terence Z. Wong, Steven E. Arnold, Jason H. Karlawish, David Wolk, Charles D. Smith, Greg Jicha, Peter Hardy, Partha Sinha, Elizabeth Oates, Gary Conrad, Oscar L. Lopez, MaryAnn Oakley, Donna M. Simpson, Anton P. Porsteinsson, Bonnie S. Goldstein, Kim Martin, Kelly M. Makino, M. Saleem Ismail, Connie Brand, Ruth A. Mulnard, Gaby Thai, Catherine Mc Adams Ortiz, Kyle Womack, Dana Mathews, Mary Quiceno, Ramon Diaz Arrastia, Richard King, Myron Weiner, Kristen Martin Cook, Michael DeVous, Allan I. Levey, James J. Lah, Janet S. Cellar, Jeffrey M. Burns, Heather S. Anderson, Russell H. Swerdlow, Liana Apostolova, Kathleen Tingus, Ellen Woo, Daniel H. S. Silverman, Po H. Lu, George Bartzokis, Neill R. Graff Radford, Francine ParfittH, Tracy Kendall, Heather Johnson, Martin R. Farlow, Ann Marie Hake, Brandy R. Matthews, Scott Herring, Cynthia Hunt, Christopher H. van Dyck, Richard E. Carson, Martha G. MacAvoy, Howard Chertkow, Howard Bergman, Chris Hosein, Sandra Black, Bojana Stefanovic, Curtis Caldwell, Ging Yuek Robin Hsiung, Howard Feldman, Benita Mudge, Michele Assaly Past, Andrew Kertesz, John Rogers, Dick Trost, Charles Bernick, Donna Munic, Diana Kerwin, Marek Marsel Mesulam, Kristine Lipowski, Chuang Kuo Wu, Nancy Johnson, Carl Sadowsky, Walter Martinez, Teresa Villena, Raymond Scott Turner, Kathleen Johnson, Brigid Reynolds, Reisa A. Sperling, Keith A. Johnson, Gad Marshall, Meghan Frey, Jerome Yesavage, Joy L. Taylor, Barton Lane, Allyson Rosen, Jared Tinklenberg, Marwan N. Sabbagh, Christine M. Belden, Sandra A. Jacobson, Sherye A. Sirrel, Neil Kowall, Ronald Killiany, Andrew E. Budson, Alexander Norbash, Patricia Lynn Johnson, Thomas O. Obisesan, Saba Wolday, Joanne Allard, Alan Lerner, Paula Ogrocki, Leon Hudson, Evan Fletcher, Owen Carmichael, John Olichney, Charles DeCarli, Smita Kittur, Michael Borrie, T. Y. Lee, Rob Bartha, Sterling Johnson, Sanjay Asthana, Cynthia M. Carlsson, Steven G. Potkin, Adrian Preda, Dana Nguyen, Pierre Tariot, Stephanie Reeder, Vernice Bates, Horacio Capote, Michelle Rainka, Douglas W. Scharre, Maria Kataki, Anahita Adeli, Earl A. Zimmerman, Dzintra Celmins, Alice D. Brown, Godfrey D. Pearlson, Karen Blank, Karen Anderson, Robert B. Santulli, Tamar J. Kitzmiller, Eben S. Schwartz, Kaycee M. SinkS, Jeff D. Williamson, Pradeep Garg, Franklin Watkins, Brian R. Ott, Henry Querfurth, Geoffrey Tremont, Stephen Salloway, Paul Malloy, Stephen Correia, Howard J. Rosen, Bruce L. Miller, Jacobo Mintzer, Kenneth Spicer, David Bachman, Elizabether Finger, Stephen Pasternak, Irina Rachinsky, Dick Drost, Nunzio Pomara, Raymundo Hernando, Antero Sarrael, Susan K. Schultz, Laura L. Boles Ponto, Hyungsub Shim, Karen Elizabeth Smith, Norman Relkin, Gloria Chaing, Lisa Raudin, Amanda Smith, Kristin Fargher, and Balebail Ashok Raj
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-23755-z.
References
- 1.Kuro-o M, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 2.Zhu Z, et al. Klotho gene polymorphisms are associated with healthy aging and longevity: Evidence from a meta-analysis. Mech. Ageing Dev. 2019;178:33–40. doi: 10.1016/j.mad.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Kuro-o M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019;15:27–44. doi: 10.1038/s41581-018-0078-3. [DOI] [PubMed] [Google Scholar]
- 4.Kurosu H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dubal DB, et al. Life extension factor klotho enhances cognition. Cell Rep. 2014;7:1065–1076. doi: 10.1016/j.celrep.2014.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ. Res. 2005;96:412–418. doi: 10.1161/01.RES.0000157171.04054.30. [DOI] [PubMed] [Google Scholar]
- 7.Mengel-From J, et al. Genetic variants in KLOTHO associate with cognitive function in the oldest old group. J. Gerontol. A Biol. Sci. Med Sci. 2016;71:1151–1159. doi: 10.1093/gerona/glv163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yokoyama JS, et al. Variation in longevity gene KLOTHO is associated with greater cortical volumes. Ann. Clin. Transl. Neurol. 2015;2:215–230. doi: 10.1002/acn3.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deary IJ, et al. KLOTHO genotype and cognitive ability in childhood and old age in the same individuals. Neurosc Lett. 2005;378:22–27. doi: 10.1016/j.neulet.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 10.de Vries CF, et al. Klotho, APOEε4, cognitive ability, brain size, atrophy, and survival: a study in the Aberdeen Birth Cohort of 1936. Neurobiol. Aging. 2017;55:91–98. doi: 10.1016/j.neurobiolaging.2017.02.019. [DOI] [PubMed] [Google Scholar]
- 11.Zeng C-Y, et al. Lentiviral vector–mediated overexpression of Klotho in the brain improves Alzheimer’s disease–like pathology and cognitive deficits in mice. Neurobiol. Aging. 2019;78:18–28. doi: 10.1016/j.neurobiolaging.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Barker WW, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 2002;16:203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Belloy ME, et al. Association of Klotho-VS heterozygosity with risk of Alzheimer disease in individuals who carry APOE4. JAMA Neurol. 2020;7:849–862. doi: 10.1001/jamaneurol.2020.0414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jansen WJ, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA. 2015;313:1924–1938. doi: 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 16.Erickson CM, et al. KLOTHO heterozygosity attenuates APOE4-related amyloid burden in preclinical AD. Neurology. 2019;92:e1878–e1889. doi: 10.1212/WNL.0000000000007323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bejanin A, et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain. 2017;140:3286–3300. doi: 10.1093/brain/awx243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mattsson-Carlgren N, et al. Abeta deposition is associated with increases in soluble and phosphorylated tau that precede a positive Tau PET in Alzheimer’s disease. Sci. Adv. 2020;6:eaaz2387. doi: 10.1126/sciadv.aaz2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schöll M, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89:971–982. doi: 10.1016/j.neuron.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo, T. et al. Longitudinal cognitive and biomarker measurements support a unidirectional pathway in Alzheimer’s Disease pathophysiology. Biol. Psychiatry89, 786–794 (2021). [DOI] [PMC free article] [PubMed]
- 22.Jack CR, Jr, et al. Predicting future rates of tau accumulation on PET. Brain. 2020;143:3136–3150. doi: 10.1093/brain/awaa248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrison TM, et al. Longitudinal tau accumulation and atrophy in aging and alzheimer disease. Ann. Neurol. 2019;85:229–240. doi: 10.1002/ana.25406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaPoint MR, et al. The association between tau PET and retrospective cortical thinning in clinically normal elderly. Neuroimage. 2017;157:612–622. doi: 10.1016/j.neuroimage.2017.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanseeuw BJ, et al. Fluorodeoxyglucose metabolism associated with tau‐amyloid interaction predicts memory decline. Ann. Neurol. 2017;81:583–596. doi: 10.1002/ana.24910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson KA, et al. Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann. Neurol. 2016;79:110–119. doi: 10.1002/ana.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ossenkoppele R, et al. Associations between tau, Aβ, and cortical thickness with cognition in Alzheimer disease. Neurology. 2019;92:e601–e612. doi: 10.1212/WNL.0000000000006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aschenbrenner AJ, Gordon BA, Benzinger TL, Morris JC, Hassenstab JJ. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology. 2018;91:e859–e866. doi: 10.1212/WNL.0000000000006075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.La Joie R, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 2020;12:eaau5732. doi: 10.1126/scitranslmed.aau5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuang X, et al. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer’s disease mouse model. Neurobiol. Aging. 2014;35:169–178. doi: 10.1016/j.neurobiolaging.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 31.Dubal DB, et al. Life extension factor klotho prevents mortality and enhances cognition in hAPP transgenic mice. J. Neurosci. 2015;35:2358–2371. doi: 10.1523/JNEUROSCI.5791-12.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weiner MW, et al. The Alzheimer’s Disease Neuroimaging Initiative 3: continued innovation for clinical trial improvement. Alzheimers Dement. 2017;13:561–571. doi: 10.1016/j.jalz.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawrylycz MJ, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hawrylycz M, et al. Canonical genetic signatures of the adult human brain. Nat. Neurosci. 2015;18:1832. doi: 10.1038/nn.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pontecorvo MJ, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140:748–763. doi: 10.1093/brain/aww334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lockhart SN, et al. Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage. 2017;150:191–199. doi: 10.1016/j.neuroimage.2017.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sepulcre J, et al. In vivo tau, amyloid, and gray matter profiles in the aging brain. J. Neurosci. 2016;36:7364–7374. doi: 10.1523/JNEUROSCI.0639-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.French L, Paus T. A FreeSurfer view of the cortical transcriptome generated from the Allen Human Brain Atlas. Front Neurosci. 2015;9:323. doi: 10.3389/fnins.2015.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crane PK, et al. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Brain Imaging Behav. 2012;6:502–516. doi: 10.1007/s11682-012-9186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dumitrescu L, et al. Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain. 2020;143:2561–2575. doi: 10.1093/brain/awaa209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franzmeier, N. et al. The BDNF Val66Met SNP modulates the association between beta-amyloid and hippocampal disconnection in Alzheimer’s disease. Mol. Psychiatry26, 614–628 (2021). [DOI] [PMC free article] [PubMed]
- 42.Hohman TJ, Dumitrescu L, Cox NJ, Jefferson AL. Genetic resilience to amyloid related cognitive decline. Brain Imaging Behav. 2017;11:401–409. doi: 10.1007/s11682-016-9615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleineidam L, et al. PLCG2 protective variant p. P522R modulates Tau pathology and disease progression in patients with mild cognitive impairment. Acta Neuropathol. 2020;139:1025–1044. doi: 10.1007/s00401-020-02138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porter T, et al. Klotho allele status is not associated with Abeta and APOE epsilon4-related cognitive decline in preclinical Alzheimer’s disease. Neurobiol. Aging. 2019;76:162–165. doi: 10.1016/j.neurobiolaging.2018.12.014. [DOI] [PubMed] [Google Scholar]
- 45.Urakawa I, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 46.Zeldich E, et al. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J. Biol. Chem. 2014;289:24700–24715. doi: 10.1074/jbc.M114.567321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang Q, et al. The b-glucuronidase Klotho hydrolyzes and activates the TRPV5 channel. Science. 2005;310:490–493. doi: 10.1126/science.1114245. [DOI] [PubMed] [Google Scholar]
- 48.Fernandez AF, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uddin MS, et al. Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci. 2018;10:4. doi: 10.3389/fnagi.2018.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuang X, et al. Neuroprotective effect of ligustilide through induction of α-secretase processing of both APP and Klotho in a mouse model of Alzheimer’s disease. Front Aging Neurosci. 2017;9:353. doi: 10.3389/fnagi.2017.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Congdon EE, Sigurdsson EM. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018;14:399–415. doi: 10.1038/s41582-018-0013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semba RD, et al. Klotho in the cerebrospinal fluid of adults with and without Alzheimer’s disease. Neurosci. Lett. 2014;558:37–40. doi: 10.1016/j.neulet.2013.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rhee E, et al. Relationship between polymorphisms G395A in promoter and C1818T in exon 4 of the KLOTHO gene with glucose metabolism and cardiovascular risk factors in Korean women. J. Endocrinol. Invest. 2006;29:613–618. doi: 10.1007/BF03344160. [DOI] [PubMed] [Google Scholar]
- 54.Yamazaki Y, et al. Establishment of sandwich ELISA for soluble alpha-Klotho measurement: age-dependent change of soluble alpha-Klotho levels in healthy subjects. Biochem Biophys. Res Commun. 2010;398:513–518. doi: 10.1016/j.bbrc.2010.06.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yokoyama JS, et al. Systemic klotho is associated with KLOTHO variation and predicts intrinsic cortical connectivity in healthy human aging. Brain Imaging Behav. 2017;11:391–400. doi: 10.1007/s11682-016-9598-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cole MA, Seabrook GR. On the horizon—the value and promise of the global pipeline of Alzheimer’s disease therapeutics. Alzheimers Dement. 2020;6:e12009. doi: 10.1002/trc2.12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Desikan RS, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 58.Landau SM, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann. Neurol. 2012;72:578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maass A, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage. 2017;157:448–463. doi: 10.1016/j.neuroimage.2017.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marquié M, et al. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann. Neurol. 2015;78:787–800. doi: 10.1002/ana.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grothe MJ, et al. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain. 2018;141:2755–2771. doi: 10.1093/brain/awy189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franzmeier N, Rubinski A, Neitzel J, Ewers M. The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun. 2019;10:1–12. doi: 10.1038/s41467-019-09564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) and are available from the ADNI database (adni.loni.usc.edu) upon registration and compliance with the data use agreement. A list including the anonymized participant identifiers of the currently used sample and the source file can be downloaded from the ADNI database (tau-PET data release in May 2020; UCBERKELEYAV1451_05_12_20.csv). The Allen Brain Atlas (http://human.brain-map.org) and Freesurfer-mapped transcriptomic data from the Allen Brain Atlas (http://figshare.com/articles/A_FreeSurfer_view_of_the_cortical_transcriptome_generated_from_the_Allen_Human_Brain_Atlas/1439749) are freely available online. Source data underlying Fig. 2 are provided with this paper.
The R code pertaining to the figures in this manuscript is provided at https://github.com/njulianeitzel/NatCommun2021_KL-VS. Costume R code can be obtained from the first author upon request.