

Summary
Loss-of-function mutations in proline-rich transmembrane protein-2 (PRRT2) cause paroxysmal disorders associated with defective Ca2+ dependence of glutamatergic transmission. We find that either acute or constitutive PRRT2 deletion induces a significant decrease in the amplitude of evoked excitatory postsynaptic currents (eEPSCs) that is insensitive to extracellular Ca2+ and associated with a reduced contribution of P/Q-type Ca2+ channels to the EPSC amplitude. This synaptic phenotype parallels a decrease in somatic P/Q-type Ca2+ currents due to a decreased membrane targeting of the channel with unchanged total expression levels. Co-immunoprecipitation, pull-down assays, and proteomics reveal a specific and direct interaction of PRRT2 with P/Q-type Ca2+ channels. At presynaptic terminals lacking PRRT2, P/Q-type Ca2+ channels reduce their clustering at the active zone, with a corresponding decrease in the P/Q-dependent presynaptic Ca2+ signal. The data highlight the central role of PRRT2 in ensuring the physiological Ca2+ sensitivity of the release machinery at glutamatergic synapses.
Keywords: excitatory synaptic transmission, synchronous neurotransmitter release, SNARE complex, voltage-gated Ca2+ channels, P/Q-type Ca2+ channels, channel trafficking, probability of release, nerve terminals, active zone, PRRT2-linked paroxysmal disorders
Graphical abstract
Highlights
-
•
PRRT2 deficiency reduces the contribution of P/Q-type Ca2+ channels to the EPSC
-
•
PRRT2 deficiency decreases P/Q-type currents and membrane targeting of the channels
-
•
PRRT2 directly interacts with P/Q-type Ca2+ channels
-
•
PRRT2 deficiency reduces P/Q-channel clustering and Ca2+ signals in nerve terminals
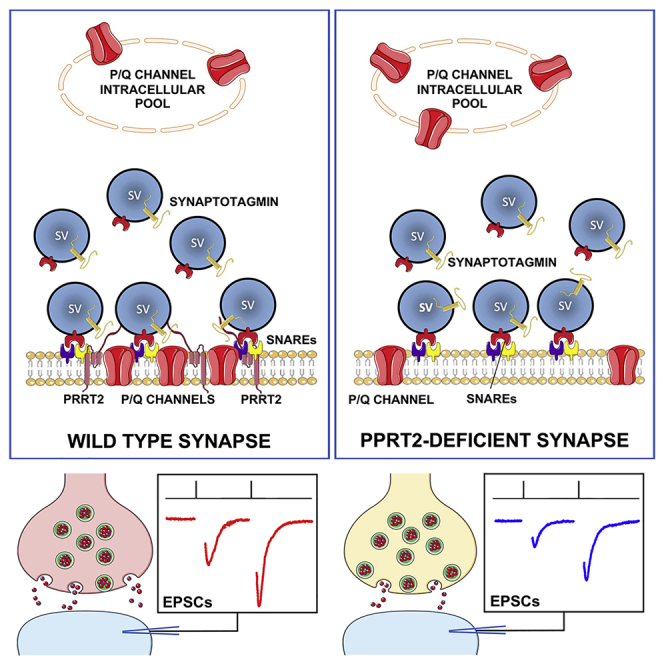
PRRT2 deficiency causes paroxysmal disorders associated with defective Ca2+ dependence of glutamatergic transmission. Ferrante et al. now find that, in the absence of PRRT2, the membrane targeting of P/Q-type Ca2+ channels is reduced, and the channels fail to concentrate at the nanodomain where the machinery for synchronous release is assembled.
Introduction
An array of paroxysmal disorders, namely benign infantile epilepsy, kinesigenic dyskinesia, infantile convulsions with choreoathetosis, and hemiplegic migraine are associated with mutations in the gene encoding for proline-rich transmembrane protein 2 (PRRT2; Heron and Dibbens, 2013; Valtorta et al., 2016; Ebrahimi-Fakhari et al., 2015, for review). Nonsense, missense, and frameshift mutations were identified, but the vast majority of patients (80%) carry the same frameshift single-nucleotide duplication c.649dupC that leads to a premature stop codon, resulting in a loss-of-function pathomechanism. Although the PRRT2-linked manifestations are pleiotropic, in the absence of clear genotype-phenotype correlations, the paroxysmal nature of PRRT2-linked diseases suggests that PRRT2 contributes to maintain the stability of neuronal networks.
PRRT2 is a neuron-specific, type 2 membrane protein with a C-terminal anchor that concentrates in synaptic domains, where it interacts with key components of the neurotransmitter release machinery (Lee et al., 2012; Liu et al., 2016; Rossi et al., 2016; Valente et al., 2016a). Protein interaction studies revealed that PRRT2 interacts with the SNARE proteins SNAP25 and VAMP/synaptobrevin and with the fast Ca2+ sensors synaptotagmins 1/2 (Syt1/2), suggesting a function in the Ca2+-dependent transition from synaptic vesicle (SV) priming to fusion (Valtorta et al., 2016, for review). Both PRRT2 knockdown (KD) by RNAi and constitutive inactivation of the PRRT2 gene in neurons induce an impairment of fast synchronous neurotransmitter release because of a sharp drop in release probability (Pr) and Ca2+ sensitivity of release (Fruscione et al., 2018; Valente et al., 2016a).
In principle, the PRRT2-dependent decrease in Pr can be due to either an uncoupling between the Ca2+ sensor and the SNARE complex or a decreased Ca2+ influx or both. The observation that PRRT2-depleted neurons are totally insensitive to an increase in the extracellular Ca2+ concentration ([Ca2+]o; Valente et al., 2016a) brought us to investigate the effects of PRRT2 deficiency on the properties of presynaptic voltage-gated Ca2+ channels (VGCCs) that convert action potential (AP) depolarization into a Ca2+ influx at the active zone, triggering synchronous SVs? fusion.
We found that PRRT2 interacts with P/Q-type VGCCs and that the loss-of-function of PRRT2 reduces the P/Q-type sensitive component of evoked excitatory postsynaptic currents (eEPSCs). This effect was associated with a decrease in the P/Q somatic currents and in the surface expression of P/Q channels. At nerve terminals, PRRT2 depletion reduced the P/Q-type channel clustering at the active zone and decreased the P/Q-dependent Ca2+ influx in response to APs. The results highlight the central role of PRRT2 in presynaptic Ca2+ dynamics, ensuring a physiological Ca2+ sensitivity of synchronous release at glutamatergic synapses.
Results
The eEPSC amplitude of PRRT2-deficient neurons is insensitive to extracellular Ca2+
We have previously shown that silencing PRRT2 impairs neurotransmitter release by markedly reducing Pr and that this reduction cannot be rescued by increasing the [Ca2+]o (Valente et al., 2016a). Thus, we tested the hypothesis that the loss of PRRT2 reduces the Ca2+ influx into the presynaptic terminal through VGCCs. To this end, we studied the dose-response relationship of eEPSC amplitude versus [Ca2+]o in autaptic excitatory neurons (11–15 days in vitro [DIV]) in which PRRT2 had been acutely silenced by RNA interference (PRRT2 KD; Valente et al., 2016a) or constitutively deleted (PRRT2 knockout [KO]; Michetti et al., 2017a). In these experiments, we used autaptic hippocampal neurons because they allow the recording of synaptic currents from isolated neurons, in response to the activation of a defined and homogeneous population of synapses (Valente et al., 2016b).
In both Scrambled RNA (ScrRNA)- and ShRNA-infected neurons, eEPSC amplitudes steeply increased with [Ca2+]o and saturated at > 4 mM (Figure 1A). Knocking down PRRT2 significantly decreased the amplitude of eEPSCs at [Ca2+]o > 1 mM without altering the shape of the dose-response curve (Figure 1B, upper panel). The Hill coefficient (Ca2+ cooperativity) and the dissociation constant (KD) remained unchanged, while the saturation level (Imax) was halved (Figure 1C, upper panel). A very similar effect on the Ca2+-dependence of excitatory transmission was observed after chronic deletion of PRRT2 (Figures 1B and 1C, lower panels).
Figure 1.

PRRT2 deficiency reduces the Ca2+-induced enhancement of eEPSC amplitude
(A) Representative eEPSC recordings evoked by a depolarizing pulse at +40 mV lasting 0.5 ms from a holding potential of −70 mV at increasing [Ca2+]o in autaptic hippocampal neurons (11–15 DIV), infected at 6 DIV with either Scr or Sh4 shRNAs. In all of the traces, stimulation artifacts were blanked for clarity.
(B) Means ± SEMs of eEPSC amplitude versus [Ca2+]o relationships in Scr/Sh4-infected neurons (upper panel) and in WT/PRRT2 KO neurons (lower panel). Curves were fitted using the Hill equation: I = Imax [Ca2+]n/([Ca2+]n + KD).
(C) Hill slope, saturation level (Imax), and half-maximal effective concentration (EC50) of eEPSCs in Scr/Sh4-infected neurons (upper panel) and WT/PRRT2 KO neurons (lower panel).
(D) Representative eEPSC recordings induced by the administration of 2 consecutive voltage stimuli (lasting 0.5 ms, at 40 mV from an Vh = −−70 mV) at an interpulse interval of 50 ms in Scr/Sh4-infected neurons (upper panel) and WT/PRRT2 KO neurons (lower panel).
(E) Means ± SEMs of the paired-pulse ratios (PPRs) as a function of [Ca2+]o in Scr/Sh4-infected neurons (upper panel) and WT/PRRT2 KO neurons (lower panel).
All data are means ± SEMs, with individual experimental points superimposed on the bar plots (n = 37 and 36 for Scr- and Sh4-infected neurons, respectively; n = 53 and 52 for WT and KO neurons, respectively). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, paired Student’s t test/Mann-Whitney U test.
We then examined paired-pulse ratio (PPR) in response to two consecutive stimuli with an interpulse interval of 50 ms and at different levels of [Ca2+]o in glutamatergic PRRT2 KD and PRRT2 KO neurons (Figure 1D). While a similar extent of facilitation was present at [Ca2+]o < 2 mM, both the acute and the constitutive deletion of PRRT2 significantly increased synaptic facilitation at higher [Ca2+]o (2–8 mM; Figure 1E).
PRRT2 deficiency decreases the contribution of presynaptic P/Q-type Ca2+ channels to glutamatergic transmission
The above results suggest that PRRT2 deficiency may reduce excitatory strength through a negative modulation of presynaptic VGCCs coupled with SV fusion. N- and P/Q-type VGCCs are the main actors mediating Ca2+-triggered glutamate release at the active zones of central synapses, while L-type channels do not significantly contribute to glutamatergic transmission under physiological conditions (Giansante et al., 2020; Nanou and Catterall, 2018; Reid et al., 1997; Takahashi and Momiyama, 1993).
To dissect the mechanism by which PRRT2 deficiency decreases Ca2+ sensitivity of glutamatergic transmission, we evaluated the contribution of individual VGCC subtypes to the excitatory strength by individually applying the subtype-specific antagonists nifedipine (Nife; 5 μM), Conotoxin-GVIA (Cono-GVIA; 3 μM) and Agatoxin-IVA (Aga-IVA; 0.2 μM) to block L-, N-, and P/Q-type VGCCs, respectively (Baldelli et al., 2005). A voltage step (from Vh = −70 to 40 mV, 0.5 ms) was applied at 0.1 Hz in glutamatergic autaptic neurons, and VGCC antagonists were added to the recording chamber after stabilization of the eEPSC amplitude (≈100 s) and maintained until the achievement of a new steady-state amplitude (Figures 2A, 2D, and 2G for Nife, Cono-GVIA, and Aga-IVA, respectively).
Figure 2.

PRRT2 deficiency specifically decreases the contribution of P/Q channels to evoked EPSCs
(A, D, and G) Representative eEPSCs recorded before (1, control) and during (2, toxins) application of Nife; (5 μM), Cono-GVIA (3 μM), or Aga-IVA (0.2 μM) in WT autaptic neurons. eEPSCs were elicited by clamping the cell at –70 mV and stimulating it with 0.5 ms voltage steps to +40 mV applied at 0.1 Hz. Toxins were applied after 100 s of baseline recording, only when the Ca2+ current rundown was lower than 10%. In all of the traces, the stimulation artifacts were blanked for clarity.
(B and C) Upper panels: eEPSC amplitude (means ± SEMs) recorded under control conditions and in the presence of Nife in Scr- (n = 6; red bars) and Sh4- (n = 7; blue bars) infected neurons (B) and in WT (n = 8; red open bars) and PRRT2 KO (n = 7; blue open bars) neurons (C). The line-connected symbols represent the individual responses before and after Nife application. Lower panels: individual data and means ± SEMs of the percent blockade of eEPSC amplitude (left) and of Nife-sensitive EPSC amplitude (right) in the corresponding experimental groups. p = 0.58 (Scr versus Sh4; % blockade), p = 0.68 (Scr versus Sh4; Nife sensitivity), p = 0.61 (WT versus KO; % blockade), p = 0.62 (WT versus KO; Nife sensitivity), paired Student’s t test.
(E and F) Recordings performed as in (B) and (C) under control conditions and in the presence of Cono-GVIA (3 μM) in the following experimental groups: Scr- (n = 11; red bars) and Sh4- (n = 13; blue bars) infected neurons (E) and in WT (n = 15; red open bars) and PRRT2 KO (n = 14; blue open bars) neurons (F). p = 0.32 (Scr versus Sh4; % blockade), p = 0.06 (Scr versus Sh4; Cono sensitivity), p = 0.36 (WT versus PRRT2 KO; % blockade), p = 0.12 (WT versus PRRT2 KO; Cono sensitivity), paired Student’s t test.
(H and I) Recordings performed as in (B) and (C) under control conditions and in the presence of Aga-IVA (5 μM) in the following experimental groups: Scr- (n = 15; red bars) and Sh4- (n = 10; blue bars) infected neurons (H) and in WT (n = 14; red open bars) and PRRT2 KO (n = 10; blue open bars) neurons (I). ∗∗p < 0.01 (Scr versus Sh4; % blockade), ∗∗∗p < 0.001 (Scr versus Sh4; Aga sensitivity), p = 0.2 (WT versus PRRT2 KO; % blockade), ∗∗p < 0.01 (WT versus PRRT2 KO; Aga sensitivity), paired Student’s t test.
As expected, L-type channels did not significantly contribute to glutamatergic transmission, and the lack of sensitivity of eEPSCs to Nife was not affected by either silencing or the constitutive deletion of PRRT2 (Figures 2B and 2C). By contrast, both Cono-GVIA (Figures 2E and 2F, upper panels) and Aga-IVA (Figures 2H and 2I, upper panels) efficiently decreased the eEPSC amplitude in all tested neurons by ∼35% and 40%, respectively.
However, while the loss of PRRT2 did not alter the Cono-GVIA sensitivity of eEPSCs (Figures 2E and 2F, lower panels), it significantly reduced the Aga-IVA sensitivity of eEPSCs in terms of both current amplitude and percentage blockade (Figures 2H and 2I, lower panels).
PRRT2 deficiency specifically affects P/Q-type Ca2+ currents in excitatory neurons
The above findings indicate that an impaired function of P/Q-type VGCCs could contribute to the reduced eEPSC amplitude observed in PRRT2-deficient neurons. On the basis of these data, we recorded somatic voltage-gated Ca2+ currents in low-density excitatory hippocampal neurons (14–17 DIV) that, having on average cell bodies that are smaller than autaptic neurons, improved the recording of voltage-gated currents. Neurons were voltage clamped at a holding potential of −70 mV and Ca2+ currents evoked by 200 ms voltage steps from −60 to +70 mV in 10 mV increments administered at 0.2 Hz (Figure 3A).
Figure 3.

PRRT2 deficiency specifically affects P/Q-type Ca2+ currents
Somatic voltage-gated Ca2+ currents were recorded in excitatory hippocampal neurons in which PRRT2 was acutely silenced (top panels) or constitutively deleted (bottom panels).
(A) Representative voltage-gated Ca2+ currents evoked by a voltage step at −10 mV lasting 200 ms from a holding potential of −70 mV and means ± SEMs current density (Idensity) recorded at −10 mV with superimposed individual data obtained in PRRT2-silenced (upper panel) and PRRT2 KO (lower panel) neurons.
(B) Ca2+ current density versus voltage relationships in PRRT2 KD (upper panel) and PRRT2 KO (lower panel) neurons. PRRT2 depletion did not affect membrane capacitance.
(C) Representative Ca2+ currents recorded in Scr- and Sh4-infected excitatory (GAD67-GFP−) hippocampal neurons (13 DIV). Holding potential −80 mV, steps between −70 and +60 mV for 20 ms in 10-mV increments, repolarization at −120 mV.
(D) Conductance-voltage (G/V) curves obtained in PRRT2 KD (upper panel) and PRRT2 KO (lower panel) neurons by measuring the amplitude of the tail currents on repolarization to −120 mV following step depolarizations from −70 to 60 mV (Vh = −80 mV). The tail Idensity, obtained by dividing the tail current by the cell capacitance, was further normalized to the peak tail Idensity. The G/V curves were fitted to the Boltzmann equation. The inset in the upper panel shows an enlargement of the Ca2+ current recordings shown in (C), highlighting the fast deactivation of the tail currents.
(E and H) Left: representative voltage-gated Ca2+ currents, evoked by a 200 ms voltage step at −10 mV (Vh = −70 mV) in Scr/Sh4-infected excitatory hippocampal neurons in the absence (Ctrl) or presence of either Cono-GIVA (E) or Aga-IVA (H) to identify N- and P/Q-type Ca2+ currents. Pure N- and P/Q-type peak Idensity, Idensity/V and G/V relationships were measured by subtracting the toxin-insensitive current recorded in the presence of either Cono-GIVA or Aga-IVA, from the current recorded under control conditions. Right: individual data and means ± SEMs and of N-type (E) and P/Q-type (H) Idensity in PRRT2-silenced (solid bars) and PRRT2 KO (open bars) neurons.
(F and I) Current density (Idensity) versus voltage (V) relationships for N-type (F) and P/Q-type (I) VGCC currents obtained in PRRT2-silenced (left) and PRRT2 KO (right) neurons.
(G and J) Normalized G/V curves of N-type (G) and P/Q-type (J) channels, obtained in PRRT2-silenced (left) and PRRT2 KO (right) neurons. The G/V curves were fitted to the Boltzmann equation.
∗p < 0.05, ∗∗p < 0.01, unpaired Student’s t test/Mann-Whitney U test. (A)–(D) n = 18 and 22 for Scr- and Sh4-infected neurons, respectively; n = 16 and 19 for WT and PRRT2 KO neurons, respectively. (E)–(G) n = 10, 10, 10, and 11 for Scr-infected, Sh4-infected, WT, and PRRT2 KO neurons, respectively. (H)–(J) n = 9, 10, 11, 10 for Scr-infected, Sh4-infected, WT, and PRRT2 KO neurons, respectively.
Interestingly, the macroscopic Ca2+ currents evoked at −10 mV were significantly decreased in neurons in which PRRT2 was either knocked down (Figure 3A, top) or constitutively deleted (Figure 3A, bottom). The analysis of the current density (Idensity; pA/pF) versus voltage (V) relationship revealed that in all recorded neurons, the Idensity/V curve peaked at −10 mV under both PRRT2-deficiency conditions (Figure 3B). Moreover, the voltage dependence of the VGCC conductance (G/V) was substantially unaffected in the two experimental groups (Figures 3C and 3D), indicating that the modulation of voltage-gated Ca2+ currents was not due to a change in the biophysical properties of the channels, but rather to a change in their membrane expression.
Based on the effects of PRRT2 deficiency on ePSC amplitude, we next investigated which of the VGCC subtypes expressed at the presynaptic level was affected by PRRT2. Pure N- and P/Q-type currents were obtained by subtracting the toxin-insensitive current recorded in the presence of either Cono-GIVA or Aga-IVA from the current recorded under control conditions (Figures 3E and 3H). Both the Idensity peak currents at −10 mV and the Idensity/V relationships of N-type somatic Ca2+ currents were not significantly altered in either PRRT2 KD or PRRT2 KO excitatory neurons, as compared to control conditions (Figures 3E and 3F). Conversely, both acute silencing and constitutive deletion of PRRT2 significantly decreased the Idensity of somatic P/Q-type Ca2+ conductance (Figures 3H and 3I). These results further confirm that the lack of PRRT2 mainly affects P/Q-type VGCCs, one of the two major Ca2+ channel subtypes that drive SV fusion at glutamatergic synapses. For both channel subtypes, the G/V curves were substantially unaffected (Figures 3G and 3J), suggesting a modulation of P/Q-type VGCC trafficking to the membrane, but not of their biophysical properties.
PRRT2 overexpression increases P/Q-type Ca2+ currents in excitatory neurons and HEK293 cells
To obtain further support for a PRRT2/P/Q-type VGCC interplay, we investigated the effects of PRRT2 overexpression. We recorded somatic voltage-gated Ca2+ currents in low-density excitatory hippocampal neurons (14–17 DIV) that had been previously transduced with lentiviral vectors expressing PRRT2 fused to the mCherry reporter. PRRT2 overexpression significantly increased the macroscopic Ca2+ currents evoked at −10 mV (Figure 4A) and the peak of the Idensity/V curve (Figure 4B), leaving the voltage dependence of the VGCC conductance (G/V; Figure 4C) unaffected.
Figure 4.

Expression of exogenous PRRT2 increases P/Q-type Ca2+ currents in excitatory neurons and HEK293 cells
(A) Somatic voltage-gated Ca2+ currents were recorded in excitatory hippocampal neurons transduced with PRRT2. Left: representative voltage-gated Ca2+ currents evoked by a voltage step at −10 mV lasting 200 ms from a holding potential of −70 mV. Right: individual data and means ± SEMs of current density (Idensity) recorded at −10 mV.
(B) Ca2+ current density versus voltage relationships in PRRT2-overexpressing neurons. PRRT2 overexpression did not affect membrane capacitance (58.31 and 55.45 pF for mCherry- and PRRT2-mCherry-infected neurons, respectively).
(C) Conductance-voltage (G/V) curves obtained in PRRT2-overexpressing neurons by measuring the amplitude of the tail currents on repolarization to −120 mV following step depolarizations from −70 to +70 mV (Vh = −80 mV). For further details, see Figure 3 legend.
(D) Representative fluorescence images of non-permeabilized HEK293 cells transiently co-transfected with Cav2.1-HA/α2δ-1/β4 and either mCherry alone or PRRT2-mCherry (red) and retrospectively stained with anti-HA antibodies (green). The white line, corresponding to the major axis of the cell, was used to measure the fluorescence intensity of Cav2.1-HA immunostaining. Scale bar, 10 μm.
(E) Intensity profiles of Cav2.1-HA fluorescence in non-permeabilized HEK293 cells expressing either mCherry (black lines) or PRRT2-mCherry (light blue lines).
(F) Normalized mean fluorescence intensity (n = 57 and 52 cells for mCherry and PRRT2-mCherry, respectively) of Cav2.1-HA immunoreactivity in HEK293 cells expressing either mCherry (black bars) or PRRT2-mCherry (light blue bars). PRRT2 significantly increases membrane expression of Cav2.1. Data are means ± SEMs.
(G) Left: representative traces of voltage-gated Ca2+ currents evoked by a 200 ms voltage step at 0 mV (Vh = −70 mV) 2 days after transfection of HEK293 cells with Cav2.1/α2δ-1/β4 and either mCherry alone (black) or PRRT2-mCherry (light blue). Right: individual data and means ± SEMs of current density (Idensity) values recorded in HEK293 cells expressing Cav2.1/α2δ-1/β4 in the absence (black) or presence (light blue) of PRRT2.
(H) Idensity versus voltage (V) relationships for HEK293 cells expressing Cav2.1/α2δ-1/β4 in the absence (black) or presence (light blue) of PRRT2.
(I) Representative Ca2+ currents recorded in HEK293 cells expressing Cav2.1/α2δ-1/β4 in the absence (black) or presence (light blue) of PRRT2. Holding potential −80 mV, steps between −70 and +60 mV for 20 ms in 10-mV increments, repolarization to −120 mV.
(J) Normalized conductance-voltage curves of HEK293 cells expressing Cav2.1/α2δ-1/β4 in the absence (black) or presence (light blue) of PRRT2. Curves were fitted to the Boltzmann equation.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 unpaired Student’s t test/Mann-Whitney U test (n = 19 for both mCherry- and PRRT2-mCherry-infected neurons; n = 51 and 53 for Cav2.1/α2δ-1/β4 expressing HEK293 cells in the absence or presence of PRRT2, respectively.
To investigate the effects of PRRT2 on P/Q-type channels in a simpler cellular environment, HEK293 cells were transiently transfected to express a hemagglutinin (HA)-tagged α1 subunit for Cav2.1 and the α2δ-1 and β4 auxiliary subunits, together with either PRRT2-mCherry or mCherry (Figure 4D). While PRRT2 did not alter the total expression levels of the channel (Figure S1), it enhanced its surface expression, as revealed by the staining for the exofacial HA-tag (Meyer et al., 2019) under non-permeabilizing conditions (Figures 4E and 4F). As in neurons, the expression of PRRT2 in HEK293 cells induced an increase in both peak currents and Idensity/V profiles (Figures 4G and 4H), without changing the G/V curves (Figures 4I and 4J). To probe the existence of a direct effect of PRRT2 on the Cav2.1 channel, we also investigated the macroscopic Ca2+ current in HEK293 cells expressing only the Cav2.1 α1 subunit. Although we observed smaller Ca2+ currents in the absence of auxiliary subunits, as previously reported (Altier et al., 2011; Gurnett et al., 1996), we confirmed the PRRT2 effects in both peak currents and Idensity/V profiles (Figure S2), indicating that the auxiliary subunits are not required for a PRRT2-dependent increase in Cav2.1 membrane expression. The above results indicate that the PRRT2 expression levels bidirectionally modulate P/Q-type Ca2+ currents in neurons.
PRRT2 interacts with P/Q-type Ca2+ channels and modulates their trafficking to the plasma membrane
To dissect the molecular basis of these results, the total expression level of Cav2.1 was analyzed in brain extracts obtained from wild-type (WT) and PRRT2 KO mice. No changes in the overall neuronal expression of P/Q-type Ca2+ channels were observed by quantitative western blotting analysis (Figure 5A). Given the significant decrease in the P/Q-type Ca2+ current density induced by PRRT2 deficiency in the absence of a detectable change in Cav2.1 expression, we used surface biotinylation to investigate the surface expression of P/Q-type VGCCs. PRRT2 KO neurons showed a significant decrease in the extracellular levels of P/Q-type VGCCs compared with WT neurons, with a concomitant significant increase in their intracellular fraction (Figure 5B). These results suggest that the lack of PRRT2 limits P/Q-type VGCC targeting to the plasma membrane, generating an intracellular accumulation of the P/Q-type channels.
Figure 5.

PRRT2 directly interacts with P/Q-type Ca2+ channels
(A) Left: representative immunoblots of the expression levels of Cav2.1 in total cortical lysates of WT and PRRT2 KO mice. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) immunoreactivity was included as a control of equal loading. Right: quantification of Cav2.1 expression in WT and PRRT2 KO mice normalized on GAPDH expression and expressed in percentage of the mean WT immunoreactivity (means ± SEMs with superimposed individual values; n = 3 independent preparations).
(B) Representative immunoblots of cell surface biotinylation performed in primary hippocampal WT and PRRT2 KO neurons (left panel). Total cell lysates (input), biotinylated (extracellular), and non-biotinylated (intracellular) fractions were analyzed by immunoblotting. Na/K-ATPase 1 and actin were included as markers of plasma membrane and cytosolic fractions, respectively. Individual values and means ± SEMs of total cell, extracellular, and intracellular Cav2.1 expression normalized on Na/K-ATPase 1 and actin expression (n = 5 independent preparations).
(C) Co-immunoprecipitation of PRRT2 with Cav2.1. Detergent extracts of mouse brain were immunoprecipitated (IP) with Cav2.1 antibodies or with an anti-GFP antibody, as indicated. After electrophoretic separation of the immunocomplexes and western blotting, membranes were probed with anti-Cav2.1 antibodies to test the immunoprecipitation efficiency, as well as with anti-PRRT2 antibodies to probe the interaction. Left: a representative immunoblot is shown. Right: quantification of the PRRT2 immunoreactive signal in the IP samples, normalized to the binding of the anti-GFP control (means ± SEMs with superimposed individual values, n = 6 independent experiments). Immunoglobulin G (IgG) HC, antibody heavy chain. Input, 10 μg total extract.
(D) Specificity of PRRT2 for Cav2.1 over Cav2.2. Extracts of HEK293 cells transiently expressing HA-tagged PRRT2 and either Cav2.1 or Cav2.2 were subjected to affinity precipitation with anti-HA agarose beads. Left panel: representative immunoblot showing input and pulled-down fractions from extracts of Cav2.2-expressing HEK293 cells. Center panel: representative immunoblot showing input and pulled-down fractions from extracts of Cav2.1-expressing HEK293 cells. VGCC (top) and PRRT2-HA (bottom) immunoblots are shown. Right panel: quantitative evaluation of the amounts of Cav2.1 and Cav2.2 immunoreactivities bound to PRRT2-HA, expressed in percentage of the respective input (means ± SEMs with superimposed individual values, n = 3 independent experiments).
(E) Proteomic screen using the C-terminal domain of the Cav2.1 as a bait. Left: volcano plot combining fold change (FC in log2) with statistical significance (p value in l-log10). PRRT2 is purified with FC > 4 versus control with a corresponding p = 0.012. Right: primary amino acid sequence of mouse PRRT2 with the exclusive spectra sequences identified by mass spectrometry analysis highlighted in yellow.
(F) Pull down of PRRT2 by the C-terminal domain of Cav2.1. A SNAP fusion protein of the C-terminal domain of Cav2.1 was used as a bait for pulling down PRRT2 from an extract of PRRT2-expressing HEK293 cells. Pull down with SNAP alone was used as an internal control. Left: a representative immunoblot is shown. Right: quantification of the PRRT2 immunoreactive signal in the pulled-down samples, expressed in percentage of the respective input (means ± SEMs with superimposed individual values, n = 3 independent experiments). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, unpaired Student’s t test/Mann-Whitney U test.
To confirm the presence of an interaction between PRRT2 and P/Q-type VGCCs in brain tissue, we performed co-immunoprecipitation experiments from neocortical extracts with antibodies specific for Cav2.1. PRRT2 was efficiently co-immunoprecipitated by the P/Q-type VGCCs, while an unrelated control antibody did not precipitate either protein (Figure 5C). Next, we assessed the specificity of the interaction between PRRT2 and Cav2.1 by performing PRRT2 pull-down experiments in HEK293 cells alternatively expressing Cav2.1 and Cav2.2 VGCCs. In line with the physiological data, PRRT2 pulled down 4-fold more Cav2.1 than Cav2.2, showing specificity for the former channel subtype (Figure 5D).
As additional proof of an interaction between PRRT2 and P/Q-type Ca2+ channels, we used a bait formed by the intracellular C-terminal domain of Cav2.1 fused to the SNAP protein (an engineered version of DNA repair protein O6-alkylguanine-DNA alkyltransferase) to pull down interacting proteins from brain synaptosomal fractions and screened the eluate for the presence of PRRT2. The volcano plot of the identified proteins revealed that PRRT2 was among the significant interactors (fold change [FC] > 4; p = 0.012), as also testified to by the wide sequence coverage from the PRRT2-derived peptides (Figure 5E). Finally, to verify that the interaction between PRRT2 and the C-terminal domain of Cav2.1 was not mediated by accessory proteins, we used the purified C terminus of Cav2.1 fused to SNAP to pull down PRRT2 expressed in HEK293 cells. The results of this experiment confirmed the proteomic data, indicating a direct interaction between the C terminus of Cav2.1 and PRRT2 (Figure 5F).
The absence of PRRT2 alters the presynaptic localization of P/Q-type Ca2+ channels
The data obtained so far suggest that PRRT2, by interacting with Cav2.1, plays a role in the correct membrane targeting of presynaptic P/Q-type VGCCs. In addition to the decreased membrane expression, PRRT2 deficiency could also alter their presynaptic localization, thus contributing to the impaired neurotransmitter release observed in PRRT2-lacking neurons. To investigate this possibility, we analyzed the presynaptic localization of P/Q-type VGCCs by immunofluorescence coupled to super-resolution confocal imaging.
Primary WT and PRRT2 KO hippocampal neurons (14 DIV) were double stained for Cav2.1 and Bassoon (Bsn), a presynaptic protein associated with the active zone (Figure 6A). The possible mislocalization of P/Q-type Ca2+ channels in PRRT2 KO terminals was analyzed in synaptic contacts unambiguously identified based on the overlap of Bsn/Cav2.1 staining using deconvolution-based super-resolution fluorescence microscopy (Leica SP8/HyD with the LAS-X Lightning deconvolution software, see Method details). In agreement with previous observations (Fruscione et al., 2018; Valente et al., 2016a, 2019), the constitutive deletion of PRRT2 moderately decreased the density of Bsn/Cav2.1 double-labeled synaptic boutons (Figure 6B). Interestingly, while in WT terminals, the Cav2.1 immunoreactivity area was smaller than the Bsn+ area, as expected from a channel strictly concentrated at active zones, in PRRT2 KO terminals, Cav2.1 spread out of the active zone, significantly increasing the total area of Cav2.1/Bsn overlap (Figures 6C–6E). Consistent with this result, PRRT2 deletion also induced an increase in the Manders’ coefficient, underlining the larger degree of overlap between Cav2.1+ and Bsn+ areas (Figure 6F). Moreover, due to the increased dispersion of the Cav2.1 subunit, both the absolute and the integrated fluorescence intensity of Cav2.1 within the Bsn+ areas were decreased in PRRT2 KO terminals (Figures 6G and 6H). Totally superimposable results were obtained using image scanning microscopy (ISM), an alternative super-resolution technique based on image acquisition with detector array (Castello et al., 2019; Figure S3).
Figure 6.

PRRT2 deletion disperses P/Q-type Ca2+ channels in presynaptic terminals
(A) Representative super-resolution confocal images of synaptic contacts from 14 DIV primary hippocampal neurons. Double-immunostaining for Bassoon (Bsn; red) and Cav2.1 (green) was used to analyze the localization of P/Q-type VGCCs at Bsn-labeled presynaptic boutons in WT (left) and PRRT2 KO (right) hippocampal neurons. Merge panels identify synaptic puncta in which Bsn and Cav2.1 colocalize (yellow). Scale bar, 2 μm.
(B) Quantification of the means ± SEMs linear density (puncta/μm) of double-stained synaptic boutons in WT (n = 29 independent coverslips) and PRRT2 KO (n = 30 independent coverslips) neurons, each from n = 3 independent neuronal preparations. Individual values are superimposed.
(C and D) Representative zoomed detail of single WT (left) and PRRT2 KO (right) synaptic boutons where Bsn and Cav2.1 stainings colocalize (C) and the corresponding distribution profile (D) of the immunofluorescence intensity for Cav2.1 (green) and Bsn (red).
(E–H) Quantitative analysis of the area of the Cav2.1 immunoreactivity (E), Manders’ coefficient (F), mean (G), and integrated (H) fluorescence intensities of Cav2.1 measured within Bsn+ areas. Means ± SEMs are shown with superimposed individual values. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, Student’s t test (n = 29 and 30 coverslips from n = 3 independent neuronal preparations for WT and PRRT2 KO neurons, respectively). From each coverslip, at least 10–15 fields were analyzed.
In addition to the overall decreased surface expression of P/Q-type channels, these observations indicate that the lack of PRRT2 also induces a mislocalization of presynaptic P/Q-type VGCCs that diffuse out of the active zone.
PRRT2 expression modulates the P/Q-type-dependent Ca2+ rise within nerve terminals
To determine whether the reduction in presynaptic expression of Cav2.1 associated with PRRT2 deficiency was accompanied by altered presynaptic Ca2+ transients in response to APs, we used the genetically encoded Ca2+ indicator SyGCaMP6s, in which GCaMP6s is fused to the cytoplasmic domain of the integral SV protein synaptophysin, allowing measurements of bulk volume-averaged Ca2+ transients in nerve terminals (Ding et al., 2014; Thalhammer et al., 2017).
Silencing PRRT2 in the absence of VGCC inhibitors did not significantly affect the overall presynaptic SyGCaMP6s responses over a wide range of stimulation intensities (1–50 APs; Figures 7A and 7B). However, when we applied Aga-IVA (0.3 μM) to block P/Q-type VGCCs, we noticed that the P/Q-type-dependent fluorescent transients were significantly smaller in PRRT2-deficient presynaptic boutons at high stimulation intensities (Figures 7C and 7D). Despite its highly non-linear behavior, SyGCaMP6s possesses ultra-sensitivity to Ca2+ dynamics, allowing the detection of presynaptic Ca2+ transients in response to single APs, even after partial blockade of Ca2+ entry. In response to a single AP, the presynaptic fluorescent response was highly variable from bouton to bouton; however, the percentage of Aga-IVA block was consistently lower in PRRT2 knocked down neurons (74.0% ± 6.0%) than in scramble control neurons (91.4%± 2.5%; Figures 7E and 7F). These data are in line with the immunolocalization experiments (Figure 6) and suggest that the loss of PRRT2 diminishes the contribution of P/Q-type VGCCs to presynaptic Ca2+ transients.
Figure 7.

PRRT2 knockdown decreases the contribution of P/Q-type Ca2+ channels to nerve terminal Ca2+ transients
(A and B) Presynaptic SyGCaMP6s fluorescence transients elicited by 1–50 APs at 40 Hz in primary hippocampal neurons transfected with either Scr (red traces/symbols) or Sh4RNA (shRNA) (blue traces/symbols). (A) AP-evoked fluorescence transients of individual boutons evoked by 50 APs. Traces are averages from individual fields of view (5–15 boutons). (B) Means ± SEMs peak amplitude of SyGCaMP6s transients from experiments as in (A) (n = 18 and 25 for Scr and Sh4 experimental groups, respectively).
(C and D) Effects of Aga-IVA on presynaptic SyGCaMP6s transients for experiments as in (A) and (B) (PRRT2 knockdown). (C) Representative fluorescence responses of individual boutons to electrical stimulation (50 APs) in hippocampal neurons transfected with either Scr (red traces) or Sh4 (blue traces) before and after application of Aga-IVA (0.3 μM). The Aga-sensitive fraction of Ca2+ transients is shown in red and blue color shading. (D) Means ± SEMs amplitude of Aga-sensitive SyGCaMP6s transients in response to 1–50 APs from experiments as in (C) (n = 8 and 10 for Scr and Sh4 experimental groups, respectively).
(E and F) SyGCaMP6s fluorescence transients in response to a single AP in primary hippocampal neurons transfected with either Scr (red traces/symbols) or Sh4 (blue traces/symbols) RNA before and after application of Aga-IVA (0.3 μM). (E) AP-evoked fluorescence transients of individual boutons evoked by 1 AP. Traces are averages from individual fields of view (5–15 boutons). (F) Means ± SEMs peak amplitude and individual experimental points of SyGCaMP6s transients from experiments as in (E) (n = 8 and 10 for Scr and Sh4 experimental groups, respectively).
(G and H) Presynaptic SyGCaMP6s fluorescence transients elicited by 1–50 APs at 40 Hz in primary hippocampal neurons transfected with either mCherry (black traces/symbols) or PRRT2-mCherry (Over; green traces/symbols). (G) Representative fluorescence responses of individual boutons evoked by 50 APs. Traces are averages from individual fields of view (5–15 boutons). (H) Means ± SEMs peak amplitude of SyGCaMP6s transients from experimnts as in (G) (n = 25 and 19 for Ctrl and Over experimental groups, respectively).
(I and J) Effects of Aga-IVA on presynaptic SyGCaMP6s transients for experiments as in (G) and (H) (PRRT2 overexpression). (I) Representative evoked fluorescence transients (50 APs) measured before and after application of Aga-IVA (0.3 μM) in primary hippocampal neurons transfected with mCherry alone (Ctrl; black traces) or PRRT2-mCherry (Over; green traces). (J) Means ± SEMs amplitude of Aga-sensitive SyGCaMP6s transients in response to 1–50 APs from experiments as in (I) (n = 12 and 10 for mCherry and PRRT2-mCherry groups, respectively).
Experimental data were obtained from from 5 independent cultures. ∗∗p < 0.01; ∗∗∗p < 0.001; 2-way repeated-measures followed by Sidak multiple comparison test. Scale bar, 1 μm.
We also investigated whether PRRT2 overexpression affected in an opposite fashion presynaptic Ca2+ transients elicited by AP trains. Strikingly, PRRT2 overexpression significantly increased the evoked presynaptic Ca2+ transients in response to 16 and 50 APs (Figures 7G and 7H), an effect that was mainly due to P/Q-type Ca2+ channels, as confirmed by the significant increase in the Aga-sensitive Ca2+ transients (Figures 7I and 7J).
Discussion
A large body of studies investigating the PRRT2 interactome revealed that PRRT2 is a component of important macromolecular complexes both in the axon and at the synaptic level, potentially regulating the trafficking of integral membrane proteins between the plasma membrane and the intracellular compartments (Lerche, 2018). At the presynapse, PRRT2 interacts with several components of the neurotransmitter release machinery, binding to SNARE proteins and modulating the assembly of the SNARE complex (Coleman et al., 2018; Lee et al., 2012; Mo et al., 2019; Stelzl et al., 2005; Tan et al., 2018; Valente et al., 2016a). In addition to the SNAREs, we found that PRRT2 specifically interacts with Syt1/2, the main fast Ca2+ sensors mediating the activation of synchronous release. These interactions provide a molecular basis for the dramatic decrease of EPSC amplitude and Pr under conditions of PRRT2 deficiency. This phenotype could not be rescued by increasing the [Ca2+]o, and the effect of intracellular Ca2+ chelators was fully occluded by the lack of PRRT2 (Valente et al., 2016a). Such results point to a key role of PRRT2 in increasing the sensitivity of the fusion machinery to the Ca2+ signal, an effect that can potentially be exerted at the level of the Ca2+ sensor and/or of the VGCCs mediating Ca2+ influx at the active zone.
In this work, we have investigated the effects of PRRT2 deficiency on the properties of the two major groups of VGCCs present in nerve terminals, namely N-type and P/Q-type channels, with the latter specifically involved in mediating the nanodomain increase in Ca2+ at the active zone responsible for synchronous release (Nanou and Catterall, 2018). We have found that (1) PRRT2 deficiency specifically decreases the contribution of P/Q-type channels to the EPSC amplitude; (2) PRRT2 deficiency/overexpression modulates in opposite directions P/Q-type Ca2+ currents; (3) PRRT2 specifically interacts with P/Q Ca2+ channels and its deficiency impairs their expression at the membrane surface; (4) PRRT2 deletion disperses P/Q-type Ca2+ channels in presynaptic terminals and decreases their contribution to nerve terminal Ca2+ transients evoked by APs. PRRT2 was not identified among the Cav2.1 interactors in a previous proteomic study using antibodies against the α1 and β subunits (Müller et al., 2010). The pull-down strategy adopted here, which furthers the enrichment of proteins specifically interacting with the C terminus of P/Q-type channels, and the refined proteomic techniques used in our study (see STAR Methods) allowed us to identify PRRT2.
The P/Q-type channel dysfunction contributes to the synaptic impairment of PRRT2-deficient neurons, resulting in paroxysmal phenotypes
The impairment of synchronous release due to a sharp decrease in Pr observed in PRRT2 silenced or constitutive deleted neurons (Valente et al., 2016a, 2019) translates into an enhanced facilitation of excitatory transmission that contributes to the hyperexcitability and instability of PRRT2-deficient networks (Fruscione et al., 2018; Michetti et al., 2017a, 2017b; Valente et al., 2016a, 2019).
Interestingly, other synaptic proteins regulating the assembly and Ca2+ sensitivity of the fusion machinery at the active zone are associated with a hyperexcitability phenotype. Mutations in the Rim-interacting protein PNKD causing paroxysmal non-kinesigenic dyskinesia also affect the regulation of various types of VGCCs (Shen et al., 2015). Mutations in another PRRT2 partner, the synaptosomal-associated protein 25kDa (SNAP-25), which is known to modulate the activity of various types of VGCCs (Condliffe et al., 2010; Pozzi et al., 2008), are associated with attention-deficit/hyperactivity disorder, absence epilepsy, and movement coordination disorders (Barr et al., 2000; Faraone et al., 2005; Feng et al., 2005; Kustanovich et al., 2003; Mill et al., 2004) that correlate with a condition of network hyperexcitability (Corradini et al., 2014).
Besides PRRT2, other genes encoding Ca2+ channels or interactors thereof (PNKD, Cav2.1, and TRPC3) have been associated with recurrent paroxysmal disorders in man and mouse. The dyskinetic/ataxic mice tottering, lethargic, rolling, and moonwalker (Becker et al., 2013; Khan and Jinnah, 2002; Matsushita et al., 2002) that share paroxysmal phenotypic traits with the PRRT2 KO mouse (Michetti et al., 2017a; Tan et al., 2018) bear mutations in Cav2.1, Cav2.1 β4 auxiliary subunit, and the non-selective cation channel TRPC3, respectively. Mutations in Cav2.1 in humans are also associated with episodic ataxia and hemiplegic migraine, two paroxysmal manifestations shared by PRRT2 patients. From these considerations, a picture emerges in which paroxysmal movement disorders can result from mutations in either ion channels or proteins regulating ion channel trafficking and functional assembly on the membrane.
PRRT2 may act as a multifunctional chaperone regulating the fate of voltage-dependent ion channels
For its ability to interact with a wide range of synaptic and axonal integral membrane proteins, PRRT2 has been defined as a “chameleon” protein (Lerche, 2018). It is known that intrinsically disordered proteins can function as hubs by assuming an ordered conformation when bound to their interactors and, notwithstanding the generally low binding affinity, their interactions can be highly specific (Tompa, 2002; Snead and Eliezer, 2019; Wright and Dyson, 2015). PRRT2 binds to Nav1.2 and 1.6, but not Nav1.1 (Fruscione et al., 2018), and, similarly, it interacts relatively specifically with Cav2.1 (this article). The specific binding activity of PRRT2 may be attributable to the concomitant presence of a proline-rich disordered cytoplasmic region, which can interact with several cytoplasmic loops of membrane proteins affecting their targeting, and of a peculiar transmembrane/intramembrane loop domain, which can affect the stability and membrane assembly of PRRT2 interactors (Rossi et al., 2016), such as SNARE proteins, Ca2+ sensors, Na/K-ATPase, Na+ channels (Stelzl et al., 2005; Lee et al., 2012; Fruscione et al., 2018; Mo et al., 2019; Coleman et al., 2018; Valente et al., 2016a; Tan et al., 2018; Sterlini et al., 2021) and VGCCs (this article). Although the results suggest that PRRT2 is devoted to regulating the trafficking of large integral membrane proteins, such as ion channels (Fruscione et al., 2018; this article), the detailed mechanism is still unknown. At the axonal level, PRRT2 negatively modulates the membrane exposure of Na+ channels (Nav1.2 and Nav1.6), resulting in a downregulation of intrinsic excitability (Fruscione et al., 2018; Valente et al., 2019). At the presynaptic level, it increases the membrane exposure of P/Q-type VGCCs at the active zone, increasing Pr in response to a single stimulus, but impairing the facilitation and enhancing the depression of excitatory transmission. Although the molecular mechanisms accounting for these opposite effects on ion channel trafficking deserve thorough investigation, their combined effects induce a stabilization of the activity of excitatory networks.
In conclusion, one can imagine a mechanistic model in which PRRT2 allows the correct targeting of P/Q-type channels to the membrane in excitatory terminals and contributes to the formation of a functional complex with the SNARE proteins and the Ca2+ sensor at the active zones. In the absence of PRRT2, the targeting of P/Q-type VGCCs to the membrane is reduced and, in parallel, the P/Q-type channels that have already reached the membrane fail to concentrate at the nanodomain in close association with the release machinery. This effect implicates PRRT2 in the regulation of the specific localization and function of fundamental membrane transducers in neurons and contributes to the synaptic impairment that characterizes PRRT2 loss-of-function mutations.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti - Actin Mouse Monoclonal | Sigma-Aldrich | Cat. #A4700; RRID:AB_476730 |
| Anti - Bassoon Guinea pig Polyclonal | Synaptic Systems | Cat. #141004; RRID:AB_2290619 |
| Anti - Cav 2.1 alpha-1A Rabbit Polyclonal | Synaptic Systems | Cat. #152203; RRID:AB_2619841 |
| Anti - Cav 2.2 alpha-1B Rabbit Polyclonal | Sigma-Aldrich | Cat. #C1478; RRID:AB_258742 |
| Anti - GFP Rabbit Polyclonal | Invitrogen | Cat. #A11122; RRID:AB_221569 |
| Anti - Na+/K+ ATPase 1 Mouse Monoclonal | Millipore | Cat. #05-369; RRID:AB_309699 |
| Anti - PRRT2 Rabbit Polyclonal | Sigma-Aldrich | Cat. #HPA014447; RRID:AB_1855786 |
| Anti - GAPDH Rabbit Polyclonal | Santa Cruz | Cat. #Sc-47724; RRID:AB_627678 |
| Anti - SNAP Rabbit Polyclonal | BioLabs | Cat. #P9310S; RRID:AB_10631145 |
| Anti-HA Rabbit Polyclonal | Invitrogen | Cat. #71-5500; RRID:AB_87935 |
| Anti-rabbit Alexa 488 | Invitrogen | Cat. #A11001; RRID:AB_2534069 |
| Anti-rabbit STAR 580 | Abberior | Cat. #ST580-1002 |
| Anti-guinea pig STAR RED | Abberior | Cat. #STRED-1006 |
| Anti-mouse Peroxidase conjugated | Bio-Rad | Cat. #1706516; RRID:AB_11125547 |
| Anti-rabbit Peroxidase conjugated | Bio-Rad | Cat. #1706515; RRID:AB_11125142 |
| Chemicals, peptides, and recombinant proteins | ||
| ω-Conotoxin GVIA | Peptide Institute | Cat. #4161-v |
| ω-Agatoxin IVA | Peptide Institute | Cat. #4256 s |
| Nifedipine | Sigma-Aldrich | Cat. #N7634 |
| Lipofectamine 2000 | Invitrogen | Cat. #11668019 |
| Tetrodotoxin (TTX) | Tocris | Cat. #1078 |
| D-AP5 | Tocris | Cat. #0106 |
| CNQX | Tocris | Cat. #0190 |
| CGP | Tocris | Cat. #1248 |
| Bicuculline (BIC) | Tocris | Cat. #2503 |
| Gentamicin solution | Sigma-Aldrich | Cat. #G1272 |
| Agarose | Sigma-Aldrich | Cat. #A9918 |
| Poly-L-lysine hydrobromide | Sigma-Aldrich | Cat. #P2636 |
| 0.25% Trypsin | GIBCO | Cat. #1505014 |
| Neurobasal-A Medium | GIBCO | Cat. #10888-022 |
| DMEM/F-12 1:1 (1X) | GIBCO | Cat. #11320033 |
| B-27 Supplement | GIBCO | Cat. #17504044 |
| TrypLE Express Enzyme (1X) | GIBCO | Cat. #12604013 |
| GlutaMAX -I (100X) | GIBCO | Cat. #35050-061 |
| Fetal Bovine Serum, qualified, heat inactivated | GIBCO | Cat. #10500064 |
| Penicillin-Streptomycin-Glutamine (100X) | GIBCO | Cat. #10378016 |
| KGluconate | Sigma-Aldrich | Cat. #P1847 |
| Glucose | Sigma-Aldrich | Cat. #G7021 |
| NaCl | Sigma-Aldrich | Cat. #793566 |
| CaCl2 | Sigma-Aldrich | Cat. #C8106 |
| BaCl2 | Sigma-Aldrich | Cat. #B0750 |
| CsCl | Sigma-Aldrich | Cat. #C4036 |
| BAPTA | Sigma-Aldrich | Cat. #A4926 |
| HEPES | AppliChem | Cat. #A3724,0500 |
| ATP | Sigma-Aldrich | Cat. #A6419 |
| GTP | Sigma-Aldrich | Cat. #G8877 |
| TEA-Cl, | Sigma-Aldrich | Cat. #T2265 |
| phosphocreatine | Sigma-Aldrich | Cat. #P7936 |
| MgCl2 | AppliChem | Cat. #A4425 |
| Sucrose | AppliChem | Cat. #A2211 |
| Tris Buffered Saline | Biorad | Cat #1706435 |
| EDTA | AppliChem | Cat. #A5097 |
| Phosphate buffered saline | Sigma-Aldrich | Cat. #P4417 |
| Triton X-100 | Sigma-Aldrich | Cat. #T9284 |
| Bovine serum Albumin (BSA) | Sigma-Aldrich | Cat. #A4503 |
| Paraformaldehyde | Sigma-Aldrich | Cat. #P6148 |
| Sodium deoxycholate | Sigma-Aldrich | Cat#D6750 |
| 1,4-dithiothreitol (DTT) | Sigma-Aldrich | Cat.#D9779 |
| Chloroacetamide | Sigma-Aldrich | Cat#C0267 |
| Tris(2-carboxyethyl)phosphine (TCEP) | Sigma-Aldrich | Cat#C4706 |
| Bovine serum Albumin (BSA) | Sigma-Aldrich | Cat. #A4503 |
| Critical commercial assays | ||
| Protease inhibitor cocktail | Cell Signaling | Cat # #5871 |
| Protein G Sepharose - GE Healthcare | Cytiva | Cat #17061801 |
| EDTA-free protease inhibitors (Complete) | Roche | Cat #11873580001 |
| Phosphatase Inhibitor cocktails 2 | Sigma | Cat #P5726 |
| Phosphatase Inhibitor cocktails 3 | Sigma | Cat #P0044 |
| BCA Assay | Thermo Scientific | Cat #23225 |
| Bradford Assay | Biorad | Cat #5000006 |
| Nitrocellulose membranes (Whatman) | Cytiva | Cat #GE10600001 |
| NeutrAvidin-conjugated agarose beads | Thermo Scientific | Cat #29200 |
| Anti-HA-agarose affinity resin | Sigma-Aldrich | Cat # A2095 |
| SNAP-capture magnetic beads | NEB | Cat. #S9145S |
| ECL | Thermo Scientific | Cat #32106 |
| EZ-Link Sulfo-NHS-LC-Biotin | Thermo Scientific | Cat # 21335 |
| Experimental models: cell lines | ||
| HEK293 | ATCC | CRL-3216 |
| Primary cell culture | Embryonic hippocampal neurons | n/a |
| Experimental models: organisms/strains | ||
| Mouse: PRRT2 KO C57BL7/N | EUCOMM/KOMP | n/a |
| Mouse: C57BL7/N | Charles River | n/a |
| Mouse: C57BL6/J | Charles River | n/a |
| Mouse: GAD67-GFP | Tamamaki et al., 2003 | n/a |
| Lentiviral vectors | ||
| pLKO.1-CMV-mCherry-Scr | Valente et al., 2016a | n/a |
| pLKO.1-CMV-mCherry-SH4 | Valente et al., 2016a | n/a |
| 743.pCCLsin.PPT.hPGK.CHERRY.Wpre | Valente et al., 2016a | n/a |
| 743.pCCLsin.PPT.hPGK.PRRT2-CHERRY.Wpre | Valente et al., 2016a | n/a |
| Recombinant DNA | ||
| Cav2.1 | Addgene | Cat. #26578 |
| Cav2.1-HA | Meyer et al., 2019 | n/a |
| Cav2.2 | Addgene | Cat. #26568 |
| 2δ-1 | Addgene | Cat. #26575 |
| β4 | Thalhammer et al., 2017 | n/a |
| Mouse PRRT2-HA | Rossi et al., 2016 | n/a |
| BAP-HA | Fruscione et al., 2018 | n/a |
| Human Cav2.1-C-termini bait | Muià, 2020 | n/a |
| pENTR4_SNAPf | Addgene | Cat #29652 |
| SyGCaMP6s | Thalhammer et al., 2017 | n/a |
| Software and algorithms | ||
| GraphPad Prism | GraphPad Softw. Inc | v.8.1.0 |
| Patchmaster | HEKA Inst | v.2x90.5 |
| Fitmaster | HEKA Inst | v.2x91 |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| LASX | Leica Microsystems | v.3.7.4 |
| MaxQuant | Cox and Mann, 2008 | https://www.maxquant.org/ |
| Perseus | Tyanova et al., 2016 | https://maxquant.net/perseus/ |
| ImageJ plugin Time Series Analyzer V3.0 | Schneider et al., 2012 | https://imagej.nih.gov/ij/plugins/time-series.html |
| Igor Pro | https://www.wavemetrics.com | v.6.03 |
| Origin2020 | OriginLab Corp., | 9.07.188 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fabio Benfenati, MD (fabio.benfenati@iit.it).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze datasets/code.
Experimental model and subject details
Mice
PRRT2 KO mice were generated by EUCOMM/KOMP using a targeting strategy based on the ‘knockout-first’ allele (Skarnes et al., 2011; Michetti et al., 2017b). Mutant animals in a C57BL/6N background were propagated as heterozygous colonies and were housed at constant temperature (22 ± 1°C) and humidity under a 12/12 h light/dark cycle and environmental enrichment conditions. in the IIT SPF facility. Genotyping was performed by PCR with primers Prrt2_F: AGGTAGACGGGCATTTGAGC, Prrt2_R: CGTGGGGAAGAGGAGACAAC; CAS_R1_Term: TCGTGGTATC GTTATGCGCC, that were used to detect the wild-type (WT) (Prrt2_F plus Prrt2_R product, 480 bp) and mutant (Prrt2_F plus Cas_R1_Term product, 200 bp) PRRT2 alleles and to genotype WT, heterozygous, and homozygous mice. The primer Prrt2_F, common to WT and mutant PCR, was designed in the intronic sequence between Prrt2 Exon 1 and Exon 2. The primers Prrt2_R and Cas_R1_Term were designed in the exon 2 of the PRRT2 gene and in the targeting cassette, respectively. For silencing experiments, either WT C57BL/6J mice or GAD67-GFP knock-in mice generated by inserting the cDNA encoding enhanced GFP into the GAD67 locus (Tamamaki et al., 2003) were used. Heterozygous GAD67-GFP males were mated with wild-type C57BL6/J females, and GFP-positive pups were identified at birth through a Dual Fluorescent Protein Flashlight (DFP-1, NIGHTSEA, Lexington, MA USA) and confirmed by genotyping. All experiments were carried out in accordance with the guidelines established by the European Communities Council (Directive 2010/63/EU of 4 March 2014) and were approved by the Italian Ministry of Health (authorizations n. 73/2014-PR, 1276/2015-PR and 22418/2020-N.REQ).
Primary neuronal cultures
Primary cultures of hippocampal neurons were prepared from WT C57BL6/J, C57BL6/N or PRRT2 KO mice of either sex at embryonic day 17 (E17), as previously described (Valente et al., 2016c) and from GAD67-GFP knock-in mice at postnatal day 0 (Prestigio et al., 2019; Tamamaki et al., 2003). Mice were sacrificed by CO2 inhalation, and 17/18-day embryos (E17-18) were removed immediately by caesarean section. In brief, hippocampi were dissociated by enzymatic digestion in 0.125% Trypsin for 20 min at 37°C and then triturated with a fire-polished Pasteur pipette. Dissociated neurons were plated at low-density (100 cells/mm2) on Petri dishes, previously pretreated with poly-L-lysine (0.1 mg/ml).
To study the effects of PRRT2 deficiency on the Ca2+-dependence and the contribution of presynaptic Ca2+ channels to glutamatergic transmission, autaptic neurons were used. This configuration offers the fundamental advantage of recording synaptic currents from isolated neurons, by activating a defined and homogeneous population of synapses and permits to precisely control the extracellular medium surrounding the cell under study, avoiding paracrine effects of neuromodulators released by neighboring neurons (Bekkers, 2003; Valente et al., 2016b). Hippocampal autaptic neurons were prepared as previously described (Valente et al., 2016b) with slight modifications. Dissociated neurons were plated at low density (40 cells/mm2) on microdots (diameter of about 40-300 μm) obtained by spraying a mixture of poly-L-lysine (0.1 mg/ml) on Petri dishes or glass coverslips, pretreated with 0.15% agarose the day before. Under this culture conditions, each Petri dish showed about 15-20 isolated single autaptic cells grown on poly-L-lysine microdots. Neurons were grown in a culture medium consisting of Neurobasal A (GIBCO) supplemented with 2% B-27 (Invitrogen), 1 mM Glutamax, and 5 μg/ml Gentamycin and maintained at 37°C in a humidified incubator with 5% CO2. No antimitotic drugs were added to prevent glia proliferation.
Cell lines
HEK293 cells obtained from ATCC (CRL-1573) were grown in DMEM supplemented with 5% fetal bovine serum, 1% L-glutamine, 100 units/ml penicillin, and 100 g/ml streptomycin (Life Technologies) and maintained at 37°C in 5% CO2.
Method details
Neuron transduction
For lentiviral infection experiments, low-density or autaptic hippocampal neurons were transduced with 5 MOI (multiplicity of infection) of lentiviral vectors added to the cell medium at 6 DIV. After 24 h, the medium was removed and replaced with equal volumes of fresh and conditioned medium (1:1). For silencing PRRT2 expression, the previously characterized Src or Sh4 sequences (Scr: GCCGACACTCGTCAGTTTAAA, Sh4: CATGTGGCCTGTCAACATTGT) were inserted into a pLKO.1-CMV-mCherry bicistronic lentiviral vector carrying the mCherry reporter. For PRRT2 overexpression experiments, PRRT2 fused to mCherry was inserted into the lentiviral vector 743.pCCLsin.PPT.hPGK.GFP.Wpre (gift from Dr. L. Naldini; Valente et al., 2016a).
For live imaging experiments, WT neurons were transfected at 7 DIV using 2 μL of Lipofectamine 2000 and 1 μg of plasmids; after 1 h the medium was removed and replaced with equal volumes of fresh and conditioned medium (1:1).
Cell transfection
HEK293 cells were grown in DMEM supplemented with 5% fetal bovine serum, 1% L-glutamine, 100 units/ml penicillin, and 100 g/ml streptomycin (Life Technologies) and maintained at 37°C in 5% CO2. All reagents were purchased from ThermoFisher Scientific. HEK293 cells were transfected with 2 μg of cDNA encoding either Cav2.1 (Addgene plasmid # 26578) or Cav2.1-HA (Meyer et al., 2019) together with α2δ-1 and β4 subunits (Addgene plasmid # 26575; Thalhammer et al., 2017) and either mCherry (743.pCCLsin.PPT.hPGK.CHERRY.Wpre) or PRRT2-mCherry (743.pCCLsin.PPT.hPGK.PRRT2-CHERRY.Wpre), in a ratio 1:1:1:1, with Lipofectamine 2000 (Invitrogen) following the recommendations of the manufacturer. For pull-down assays, HEK293 cells were transfected with Lipofectamine 2000 (Invitrogen) with 12 μg of cDNA encoding either Cav2.1 (Addgene plasmid # 26578) or Cav2.2 (Addgene plasmid #26568) and PRRT2-HA (pKH3 vector encoding for full length PRRT2 with HA tags at the C terminus; Rossi et al., 2016) or BAP-HA (pKH3 vector encoding bacterial alkaline phosphatase fused with HA tags; Fruscione et al., 2018). For testing the interactions of PRRT2 with the C terminus of Cav2.1, 12 μg of cDNA encoding either the C terminus of h.Cav2.1 fused to the SNAP protein (pENTR4_SNAPf, #29652, Addgene) or SNAP control and PRRT2-mCherry were co-transfected in a ratio 1:1 with Lipofectamine 2000 (Invitrogen), following the recommendations of the manufacturer.
Culture medium was changed 6 h after transfection and HEK293 cells were incubated at 37°C for 48 h before all experiments. Only for Cav2.1-HA cDNA, the transfection was conducted at 30°C for 6 h and cells were either incubated at 37°C for a further 48 h or transferred to 30°C for 72 h.
Electrophysiological recordings
Whole-cell patch-clamp recordings were made from autaptic or low-density hippocampal neurons, as previously described (Valente et al., 2012). The membrane potentials in whole-cell recordings were uncorrected for Donnan liquid junction potentials of 9 mV (Neher, 1992). All experiments were performed at room temperature (22-24°C). Patch pipettes prepared from thin-borosilicate glass (Hilgenberg) were pulled and fire-polished to a final resistance of 2-4 MΩ when filled with standard internal solution.
Evoked excitatory postsynaptic currents (eEPSCs) were recorded using an EPC-10 amplifier (HEKA Electronik) from autaptic neurons, maintained in a standard external solution containing (in mM): 140 NaCl, 2 CaCl2, 1 MgCl2, 4 KCl, 10 glucose, 10 HEPES (pH 7.3 with NaOH). For eEPSC recordings, the external solution was supplemented with: D-(-)-2-amino-5-phospho-nopentanoic acid (D-AP5; 50 μM; Tocris) and bicuculline (BIC, 30 μM, Tocris) to block N-methyl-D-aspartate (NMDA) and GABAA receptors, respectively. The standard internal solution was (in mM): 126 K Gluconate, 4 NaCl, 1 MgSO4, 0.02 CaCl2, 0.1 BAPTA, 15 glucose, 5 HEPES, 3 ATP, 0.1 GTP (pH 7.2 with KOH). The autaptic neurons under study were voltage-clamped at a holding potential (Vh) of −70 mV. Unclamped action potentials (APs) evoking EPSCs, were activated by a brief depolarization of the cell body to +40 mV for 0.5 ms at 0.1 Hz. eEPSCs were acquired at 10-20 kHz sampling rate and filtered at 1/5 of the acquisition rate with an 8-pole low-pass Bessel filter. Recordings with leak currents > 100 pA or series resistance > 15 MΩ were discarded. Data acquisition was performed using PatchMaster programs (HEKA Elektronik). eEPSCs were inspected visually, and only events that were not contaminated by spontaneous activity were considered. To calculate the peak current during an isolated stimulus or a train of stimuli, we first subtracted an averaged trace containing the stimulus artifact and the action potential current, lacking any discernable synaptic current (i.e., synaptic failures). Such traces were easily identified toward the end of a train of stimuli, when synaptic depression was maximal. These traces were averaged and scaled to the peak Na+ current contaminating the EPSCs. To analyze the paired-pulse ratio (PPR), 2 brief depolarizing pulses were applied to autaptic neurons with an inter-pulse interval of 50 ms. For each couple of EPSCs, PPR was calculated as the I2/I1 ratio, where I1 and I2 are the amplitudes of the PSCs evoked by the conditioning (1) and test (2) stimuli, respectively. To correctly estimate the amplitude of I2, the baseline of I2 was defined as the final value of the decay phase of I1 and the amplitude of I2 was calculated by subtracting the residual amplitude of I1 from the peak value of I2.
Voltage-clamp recordings of VGCCs after the acute knockdown/overexpression of PRRT2 were performed in primary cultures of hippocampal neurons prepared from postnatal GAD67-GFP knock-in mice, allowing the unambiguous identification of GFP-negative excitatory neurons (Tamamaki et al., 2003). For voltage-clamp recordings of VGCCs performed in WT and PRRT2 KO cultures, excitatory pyramidal neurons were morphologically identified by their teardrop-shaped somata and characteristic apical dendrite after 14-17 DIV (Pozzi et al., 2013). Voltage gated Ca2+ currents were acquired at 20 kHz and lowpass filtered at 4 kHz and series resistance was compensated 80% (2 μs response time). Recordings with either leak currents > 100 pA or series resistance > 20 MΩ were discarded. The external solution contained (in mM): 140 NaCl, 5 BaCl2, 1 MgCl2, 10 HEPES (pH 7.4 with NaOH 1M). TTX (1 μM), D-AP5 (50 μM), CNQX (10 μM), CGP (10 μM) and BIC (30 μM) were added to the external solution to block spontaneous APs, NMDA, non-NMDA, GABAA and GABAB receptors, respectively. The intracellular solution was composed of (in mM): 90 CsCl, 20 TEA-Cl, 10 EGTA, 10 glucose, 1 MgCl2, 4 ATP, 15 phosphocreatine (pH 7.4 with 1 M CsOH). The external solution containing the Ca2+ channel blockers: Nifedipine (Sigma, 5 μM), ω-Conotoxin GVIA (Peptide Institute, Code: 4161-v; 3 μM) and ω-Agatoxin IVA (Peptide Institute, Code: 4256 s; 0.2 μM), was added to the recording chamber after stabilization of the synaptic current of the neuron under study. In all voltage-clamp experiments, Vh was set at −70 mV. Voltage gated inward currents were evoked by stepping Vh from −70 to 60 mV for 200 ms with 10 mV increments with 2 s interpulse intervals. Linear capacity and leakage currents were eliminated by P/N leak subtraction procedure with a sub-pulse of –100 mV. The Ca2+ current density (Idensity = pA/pF) was obtained by dividing the peak inward current by the cell capacitance. The latter was estimated by the patch-clamp setting of Cslow (slow capacitance component) after on-line compensation of capacitive currents.
The Ca2+ conductance (G) of the total VGCCs was determined from the peak amplitude of the tail-currents on repolarization to −120 mV, following step depolarizations from −70 to 60 mV lasting 20 ms (10 mV step increment; Vh = −80 mV). To achieve a good voltage-clamp control and optimal dynamic recording conditions of VGCCs, we systematically chose to patch the neuronal body with reduced process branching from the soma. Moreover, VGCCs recordings were performed using electrodes with larger tips and electrode resistance ranging between 2 and 2.5 MΩ, allowing to obtain low access resistance and an optimal adjustment of the series resistance in whole-cell patch configuration. Finally, we used low Ba2+ concentrations (5 mM) to reduce IBa, and very high sampling frequency (100KHz) to better resolve the very fast tail currents.
Alternatively, the G/V relationships of pure N- and P/Q-type currents were calculated according to G = ICa / (V – ECa), where ICa is the peak Ca2+ current measured at potential V, and ECa is the measured Ca2+ equilibrium potential (65 mV). Data were normalized to maximum peak conductance (Gmax) and the conductance/voltage (G/V) relationships were fitted to a Boltzmann function in the form: G/Gmax = 1/{1+exp[(V-V0.5)/k]}, where V is the command pulse potential, V0.5 is the voltage required to activate the half-maximal conductance, and k is the slope of the curve at V0.5.
Biochemical assays
Co-immunoprecipitation
Mouse cortices were homogenized in ice-cold buffered sucrose solution plus 100 mM NaCl and protease inhibitor cocktail (Cell Signaling, Danvers, MA) and cleared by low-speed centrifugation (1,000 x g for 10 min at 4°C). The supernatant was centrifuged (12,100 x g for 15 min at 4°C), resuspended and incubated in lysis buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA and 1% Triton X-100 supplemented with protease inhibitor cocktail) at 4°C for 1 h and then centrifuged at 12,000 x g for 10 min. For immunoprecipitation, 5 μg of rabbit anti-P/Q (Synaptic Systems, n° 152203) channel or rabbit control antibody (anti-GFP, Invitrogen, n° A11122) were precoated with Protein G Sepharose (GE Healthcare) overnight and incubated with a mouse brain extracts. After extensive washes in lysis buffer and detergent-free lysis buffer, samples were resolved by SDS-PAGE and subjected to western blotting with anti-PRRT2 and anti-Cav2.1 antibodies.
Cav2.1 and Cav2.2 pull down
HEK293 cells were co-transfected with PRRT2-HA and either Cav2.1 or Cav2.2, as described above. Cells were harvested in lysis buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA and 1% Triton X-100 supplemented with protease inhibitor cocktail) and centrifuged at 10,000 x g for 10 min at 4°C. Kept an aliquot for input, the supernatant was incubated with 50 μL of monoclonal anti-HA-agarose affinity resin (Sigma-Aldrich) according to manufacturer’s instructions for 2 h at 4°C. After extensive washes in lysis buffer and detergent-free lysis buffer, samples were resolved by SDS-PAGE and subjected to western blotting with anti-HA, anti-Cav2.1 and anti Cav2.2 antibodies.
Affinity purification and proteomic analysis
As bait for affinity purification served the C-termini of h.Cav2.1 construct (aa 1808-2261 in O00555-3, UniProt) fused to the SNAP protein (pENTR4_SNAPf, #29652, Addgene) and cloned into pIRES2_EGFP. The construct was transfected into HEK293 cells. After 48 h cells were washed in ice-cold PBS, before harvesting them in RIPA buffer (100 mM NaCl, 50 mM Tris, 2 mM CaCl2, 1 mM MgCl2, 1% NP-40, 1% Triton X-100, EDTA-free protease inhibitors (Complete, Roche) and Phosphatase Inhibitor cocktails 2+3 (Sigma). After centrifugation at 15,000 x g for 15 min at 4°C, the supernatant was diluted 1:1 in immobilization buffer (50 mM Tris, 150 mM NaCl, 2 mM 1,4-dithiothreitol (DTT)) and covalently linked to SNAP-capture magnetic beads (S9145S, NEB) according to manufacturer’s instructions. SNAP without h.Cav2.1 terminus was used as negative control for the affinity purification. Adult murine cortices were homogenized on ice in RIPA buffer and homogenates cleared by centrifugation at 15,000 x g for 20 min at 4°C. The lysate was incubated with the bait-SNAP or SNAP only beads overnight at 4°C. After 3 washes in lysis buffer, proteins were eluted from the beads in elution buffer (1 M NaCl, 5 mM EDTA) and precipitated with acetone. After pull down, samples were lysed, reduced and alkylated in 30 ul 2% sodium deoxycholate (SDC), 40 mM chloroacetamide, 10 mM tris(2-carboxyethyl)phosphine (TCEP) and 100 mM Tris HCl pH 8 at 95°C for 10 min. Then samples were digested with 0.3 ug Trypsin over night at 37°C. Digested samples were processed by iST protocol (Kulak et al., 2014) and injected for analysis in a high-resolution LC/MS system composed of a Dionex Ultimate3000 UHPLC coupled to an Orbitrap Fusion Tribrid mass spectrometer (ThermoScientific). The raw data obtained were processed with the MaxQuant software default settings (version 1.6.6.0) and the statistical analysis was done with the Perseus software version 1.6.7.0. To double-check the results of the proteomic screen, pull-down assays were performed using the same C terminus h.Cav2.1-SNAP and SNAP control baits purified and immobilized on SNAP-capture magnetic beads (New England Biolabs) as described above. Baits were then incubated with extracts of PRRT2-transfected HEK293 cells in lysis buffer for 2 h at 4°C. After extensive washes in lysis buffer and detergent-free lysis buffer, samples were resolved by SDS-PAGE and subjected to western blotting with anti-SNAP and anti-PRRT2 antibodies.
Western blotting
For western blotting analysis, the sample protein concentration was determined using either the BCA or the Bradford assay and equivalent amounts of protein were subjected to SDS-PAGE on polyacrylamide gels and blotted onto nitrocellulose membranes (Whatman). Samples for VGCC detection were heated at 50°C for 5 min without boiling. Blotted membranes were blocked for 1 h in 5% milk in Tris-buffered saline (10 mM Tris, 150 mM NaCl, pH 8.0) plus 0.1% Triton X-100 and incubated overnight at 4°C with the appropriate primary antibody. Membranes were washed and incubated at room temperature for 1 h with peroxidase conjugated secondary antibodies. Bands were revealed with the ECL chemiluminescence detection system (ThermoFisher Scientific). Immunoreactive bands were quantified by densitometric analysis of the fluorograms obtained in the linear range of the emulsion response.
Surface biotinylation
Hippocampal neurons from WT or PRRT2 KO mice at 13 DIV were incubated with 1 mg/ml of EZ-Link Sulfo-NHS-LC-Biotin (Thermo Fisher, Waltham, MA USA) in cold phosphate-buffered saline (PBS) for 35 min at 4°C, constantly moving. Free biotin was quenched twice, with 100 mM Tris pH 8 and once with cold PBS to remove biotin excess. Cells were then lysed in lysis buffer. Total cell lysates were centrifuged at 10,000 x g at 4°C for 10 min. The supernatant fraction (0.5 mg protein) was incubated with 150 μl of NeutrAvidin-conjugated agarose beads at 4°C for 3 h, and the remaining supernatant was kept as input. After extensive washes of the beads, samples were eluted, resolved by SDS-PAGE and subjected to western blotting with anti-PRRT2 (Sigma-Aldrich, n° HPA014447), anti-P/Q (Synaptic Systems, n° 152203), anti-actin (Sigma-Aldrich, n°A4700) and anti- Na+/K+ ATPase 1 (Millipore, n° 05-369) antibodies.
Immunocytochemistry
HEK293 cells
Immunocytochemistry was carried out on HEK293 cells transfected with Cav2.1-HA, α2δ-1 and β4 together with mCherry or PRRT2-mCherry cDNAs. After transfection, cells were incubated at either 30°C or 37°C, as described, for 48-72 h before being fixed with 4% paraformaldehyde (PFA)/4% sucrose in PBS, pH 7.4 at RT for 10 min. When permeabilization was required, cells were incubated in PBS containing 0.2% Triton X-100 for 5 min at RT. Blocking was performed for 30 min at RT in PBS containing 20% FBS and 5% BSA. Primary antibodies were applied (diluted in PBS with 10% FBS and 2.5% BSA) for 1 h at RT followed by washing with PBS and final incubation with the indicated secondary antibodies in PBS containing 2.5% BSA and 10% FBS at RT for 1 h. To investigate the cell surface expression of Cav2.1, HEK293 cells were immunostained with anti-HA antibodies followed by Alexa 488 goat anti-rabbit secondary antibodies. For the analysis of the total Cav2.1 expression, permeabilized HEK293 cells were stained with an anti-Cav2.1 primary antibody followed by Alexa 488 goat anti-rabbit secondary antibodies.
Primary neurons
Primary hippocampal neurons (from WT and PRRT2 KO mice, 60 cells/mm2), were fixed at 15 DIV with 4% paraformaldehyde / 4% sucrose for 12 min at room temperature and then washed with phosphate buffered saline (PBS). Cells were permeabilized with methanol (−20°C; 10 min on ice) followed by 0.2% Triton X-100 for 10 min (Liao et al., 1999) and blocking with 5% FBS/0.1% BSA for 30 min in PBS. Incubation with primary antibodies in PBS/5% FBS/0.1% BSA was performed for 2 h, followed by washing with PBS and final incubation with secondary antibodies for 2 h. Neurons were immunostained with rabbit anti-Cav2.1 and guinea pig anti-Bassoon antibodies. Secondary antibodies were Abberior STAR 580 goat anti-rabbit IgGs and Abberior STAR RED goat anti-guinea pig IgGs.
Image analysis
HEK293 cells
Imaging was performed on LEICA SP8 confocal microscope at fixed microscope settings for all experimental conditions. Images were obtained using a 63x oil objective at a resolution of 2048 × 2048 pixels and an optical section of 0.5 μm. Images were analyzed using imageJ (imagej.net). Surface labeling of Cav2.1-HA was measured using the freehand line tool (25 pixels = 1 μm). Intracellular staining of Cav2.1 was measured using the freehand selection tool, excluding the nucleus and the plasma membrane. Background was subtracted by measuring the average intensity of areas without transfected cells.
Primary neurons
Images were acquired using a 63x objective and 3544x3544 pixels with a Leica SP8 confocal microscope equipped with Leica Hybrid Detectors (HyD). Images were analyzed by the adaptative deconvolution processing of the Leica LAS-X Lightning software that enables a resolution improvement of down to 120 nm in the lateral and 200 nm in the axial direction, according to the manufacturer’s specifications (https://www.leica-microsystems.com/science-lab/how-to-extract-image-information-by-adaptive-deconvolution/). As an alternative approach for super-resolution imaging, we used a two-color image-scanning microscope (ISM; Castello et al., 2019). The system uses a single-photon-avalanche diode (SPAD) array detector to implement the digital pixel-reassignment ISM (PRISM) principle (Sheppard, 1988). The PRISM system captures simultaneously (i.e., with a single scan) a collection of confocal images, which represent the sample from a different angle. The PRISM system recombines such images to increase the signal-to-noise ratio and to achieve a two-fold effective spatial resolution enhancement with respect to conventional microscopy. This strategy leads to resolution values as low as 120 nm, without the need of image deconvolution algorithms. The PRISM microscope is equipped with two pulsed diode lasers (LDH-D-C-640 and LDH-D-C-560, Picoquant) emitting at 640 nm and 560 nm and a 60x objective (CFI Plan Apo VC 60x, 1.4 NA, Oil, Nikon). Dual-color images were obtained thanks to a frame-by-frame acquisition scheme, using alternatively one laser source and a suitable fluorescence band pass emission filter (685/70 nm or 590/50 nm for the 640 nm and 560 nm excitation, respectively; AHF Analysentechnik). Analysis of fluorescence images was performed on bona fide synaptic boutons identified as those puncta where Cav2.1 and Bsn were colocalized along neuronal processes of ≈40-60 μm in length in manually selected fields. Measurements were carried out blind to the experimental condition. Synaptic density was measured by counting the total number of synaptic boutons divided by the dendritic length. The Cav2.1/Bsn overlap area was calculated as the number of pixels (converted in μm2) of Cav2.1 and Bsn dual-positive puncta. Colocalization between Bsn and Cav2.1 was estimated for the thresholded regions of interest (ROIs) with the Coloc2 plugin of ImageJ using the Manders’ coefficient: Σi Aicoloc/Σi Ai, where Σi Ai is the sum of intensities of all pixels above threshold for channel A (Cav2.1) and Σi Aicoloc is the sum of intensities of all pixels above threshold for channel B (Bsn). The Cav2.1 staining intensity was calculated as the average fluorescence intensity of the pixels contained into ROIs where Cav2.1 and Bsn were colocalized, divided by the background fluorescence of the field. The integrated fluorescence intensity was calculated as the product of the fluorescence intensity multiplied by the area of the ROIs where Cav2.1 and Bsn were colocalized. The number of samples (n) corresponds to the number of coverslips analyzed from three independent neuronal preparations. From each coverslip, at least 10–15 fields were analyzed.
Presynaptic Ca2+ imaging with SyGCaMP6s
Live imaging was performed as described in Thalhammer et al. (2017). Briefly, mouse primary cultures were imaged at room temperature in aCSF containing (in mM): 140 NaCl, 2.5 KCl, 2.2 CaCl2, 1.5 MgCl2, 10 D-glucose, 0.01 CNQX, 0.05 D-APV and 10 HEPES-NaOH (pH 7.38; osmolarity adjusted to 290 mOsm). Cultures were co-transfected with SyGCaMP6s and the required shRNA in a 1:1 DNA ratio 4-6 days prior to experiments and recorded at 14-16 DIV. Boutons were imaged using a cooled charge-coupled device (CCD) camera (ORCA-R2, Hamamatsu) mounted on an inverted microscope (DMI6000B, Leica) with a 40x, 1.25 NA oil immersion objective. A 200 W metal halide lamp (Lumen200Pro, Prior Scientific) and a filter set comprising a BP 470/40 nm excitation filter, a 500 nm dichroic mirror and a BP 525/50 emission filter (Leica) were used for illumination. Images were captured at 15 Hz with 50 ms integration times at a depth of 8 bits. APs were evoked by field stimulation (60 V, 1 ms pulses; Isolated Pulse Stimulator, A-M systems) before and after Agatoxin IVA application (0.3 μM, 2 min equilibration time) using a custom-made chamber with two parallel platinum wires 6 mm apart. Trains of APs were delivered at a frequency of 40 Hz every 18 s. Images were analyzed in ImageJ (https://rsb.info.nih.gov/ij) with the plugin Time Series Analyzer V3.0 (https://rsb.info.nih.gov/ij/plugins/time-series.html) and with customized routines in Igor Pro 6.03. Regions of interest (ROIs) with a diameter of 3.2 μm were positioned on all boutons responding to 1 AP. The intensity of a twin ROI positioned within 10 μm from the first was used to subtract the local background noise. Signals were quantified as ΔF/F0, where ΔF = F-F0, with F0 measured over 2 s period prior to stimulation.
Quantification and statistical analysis
Data are shown as means ± SEMs with superimposed individua experimental points. Normal distribution of data was assessed using the D’Agostino-Pearson’s normality test. The F-test was used to compare variance between two sample groups. To compare two normally distributed sample groups, the unpaired Student’s t test was used. To compare two sample groups that were not normally distributed, the Mann-Whitney’s U-test was used. The two-way repeated-measures test was followed by the Sidak’s multiple comparison test. A p value < 0.05 was considered significant. Statistical analysis was carried out using OriginPro-8 (OriginLab Corp., Northampton, MA, USA) and Prism (GraphPad Software, Inc.).
Acknowledgments
We thank Drs. Elisabetta Colombo, Caterina Michetti, and Riccardo Navone (Center for Synaptic Neuroscience, Istituto Italiano di Tecnologia, Genova, Italy) for help in breeding the mice and in confocal imaging; Silvia Casagrande (Department Experimental Medicine, University of Genova) in the preparation of primary cultures; and Luigi Naldini (Tiget, Milano, Italy) for lentiviral constructs and protocols. We also thank Genoa Instruments s.r.l. (Genova) for the support with the ISM measurements. The study was supported by research grants from the Compagnia di San Paolo Torino (2015.0546, to F.B., and 2017.20612, to P.B.); Era-Net Neuron 2017 Snaropathies (to F.B.); IRCCS Ospedale Policlinico San Martino (Ricerca Corrente and “5x1000” to A.C., P.V., P.B., and F.B.); and the Italian Ministry of University and Research (PRIN 2015-H4K2CR and 2017-A9MK4R, to F.B.). The support of Telethon-Italy (grant GGP19120, to F.B.) is also acknowledged.
Author contributions
D.F. carried out the electrophysiological and immunocytochemistry experiments and data analysis on neurons and HEK293 cells; C.P. contributed to the analysis of the immunocytochemistry data; B.S., A.M., F.O., and A.C. performed the biochemical experiments; A.P. and J.M. ran the proteomics experiments; G.T. and G.V. performed the ISM imaging experiments; A.T. and L.C. performed the Ca2+ imaging experiments; P.V. and P.B. analyzed and interpreted the electrophysiological data; F.B. and P.B. planned the experiments, wrote the paper, and funded the research; all of the authors contributed to the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 15, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109248.
Contributor Information
Fabio Benfenati, Email: fabio.benfenati@iit.it.
Pietro Baldelli, Email: pietro.baldelli@unige.it.
Supplemental information
References
- Altier C., Garcia-Caballero A., Simms B., You H., Chen L., Walcher J., Tedford H.W., Hermosilla T., Zamponi G.W. The Cavβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat. Neurosci. 2011;14:173–180. doi: 10.1038/nn.2712. [DOI] [PubMed] [Google Scholar]
- Baldelli P., Hernandez-Guijo J.-M., Carabelli V., Carbone E. Brain-derived neurotrophic factor enhances GABA release probability and nonuniform distribution of N- and P/Q-type channels on release sites of hippocampal inhibitory synapses. J. Neurosci. 2005;25:3358–3368. doi: 10.1523/JNEUROSCI.4227-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr C.L., Feng Y., Wigg K., Bloom S., Roberts W., Malone M., Schachar R., Tannock R., Kennedy J.L. Identification of DNA variants in the SNAP-25 gene and linkage study of these polymorphisms and attention-deficit hyperactivity disorder. Mol. Psychiatry. 2000;5:405–409. doi: 10.1038/sj.mp.4000733. [DOI] [PubMed] [Google Scholar]
- Becker F., Schubert J., Striano P., Anttonen A.-K., Liukkonen E., Gaily E., Gerloff C., Müller S., Heußinger N., Kellinghaus C. PRRT2-related disorders: further PKD and ICCA cases and review of the literature. J. Neurol. 2013;260:1234–1244. doi: 10.1007/s00415-012-6777-y. [DOI] [PubMed] [Google Scholar]
- Bekkers J.M. Synaptic transmission: functional autapses in the cortex. Curr. Biol. 2003;13:R433–R435. doi: 10.1016/s0960-9822(03)00363-4. [DOI] [PubMed] [Google Scholar]
- Castello M., Tortarolo G., Buttafava M., Deguchi T., Villa F., Koho S., Pesce L., Oneto M., Pelicci S., Lanzanó L. A robust and versatile platform for image scanning microscopy enabling super-resolution FLIM. Nat. Methods. 2019;16:175–178. doi: 10.1038/s41592-018-0291-9. [DOI] [PubMed] [Google Scholar]
- Coleman J., Jouannot O., Ramakrishnan S.K., Zanetti M.N., Wang J., Salpietro V., Houlden H., Rothman J.E., Krishnakumar S.S. PRRT2 Regulates Synaptic Fusion by Directly Modulating SNARE Complex Assembly. Cell Rep. 2018;22:820–831. doi: 10.1016/j.celrep.2017.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condliffe S.B., Corradini I., Pozzi D., Verderio C., Matteoli M. Endogenous SNAP-25 regulates native voltage-gated calcium channels in glutamatergic neurons. J. Biol. Chem. 2010;285:24968–24976. doi: 10.1074/jbc.M110.145813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini I., Donzelli A., Antonucci F., Welzl H., Loos M., Martucci R., De Astis S., Pattini L., Inverardi F., Wolfer D. Epileptiform activity and cognitive deficits in SNAP-25(+/-) mice are normalized by antiepileptic drugs. Cereb. Cortex. 2014;24:364–376. doi: 10.1093/cercor/bhs316. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Ding J., Luo A.F., Hu L., Wang D., Shao F. Structural basis of the ultrasensitive calcium indicator GCaMP6. Sci. China Life Sci. 2014;57:269–274. doi: 10.1007/s11427-013-4599-5. [DOI] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D., Saffari A., Westenberger A., Klein C. The evolving spectrum of PRRT2-associated paroxysmal diseases. Brain. 2015;138:3476–3495. doi: 10.1093/brain/awv317. [DOI] [PubMed] [Google Scholar]
- Faraone S.V., Perlis R.H., Doyle A.E., Smoller J.W., Goralnick J.J., Holmgren M.A., Sklar P. Molecular genetics of attention-deficit/hyperactivity disorder. Biol. Psychiatry. 2005;57:1313–1323. doi: 10.1016/j.biopsych.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Feng Y., Crosbie J., Wigg K., Pathare T., Ickowicz A., Schachar R., Tannock R., Roberts W., Malone M., Swanson J. The SNAP25 gene as a susceptibility gene contributing to attention-deficit hyperactivity disorder. Mol. Psychiatry. 2005;10:998–1005. doi: 10.1038/sj.mp.4001722. 973. [DOI] [PubMed] [Google Scholar]
- Fruscione F., Valente P., Sterlini B., Romei A., Baldassari S., Fadda M., Prestigio C., Giansante G., Sartorelli J., Rossi P. PRRT2 controls neuronal excitability by negatively modulating Na+ channel 1.2/1.6 activity. Brain. 2018;141:1000–1016. doi: 10.1093/brain/awy051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giansante G., Marte A., Romei A., Prestigio C., Onofri F., Benfenati F., Baldelli P., Valente P. Presynaptic L-Type Ca2+ Channels Increase Glutamate Release Probability and Excitatory Strength in the Hippocampus during Chronic Neuroinflammation. J. Neurosci. 2020;40:6825–6841. doi: 10.1523/JNEUROSCI.2981-19.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett C.A., De Waard M., Campbell K.P. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- Heron S.E., Dibbens L.M. Role of PRRT2 in common paroxysmal neurological disorders: a gene with remarkable pleiotropy. J. Med. Genet. 2013;50:133–139. doi: 10.1136/jmedgenet-2012-101406. [DOI] [PubMed] [Google Scholar]
- Khan Z., Jinnah H.A. Paroxysmal dyskinesias in the lethargic mouse mutant. J. Neurosci. 2002;22:8193–8200. doi: 10.1523/JNEUROSCI.22-18-08193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak N.A., Pichler G., Paron I., Nagaraj N., Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods. 2014;11:319–324. doi: 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Kustanovich V., Merriman B., McGough J., McCracken J.T., Smalley S.L., Nelson S.F. Biased paternal transmission of SNAP-25 risk alleles in attention-deficit hyperactivity disorder. Mol. Psychiatry. 2003;8:309–315. doi: 10.1038/sj.mp.4001247. [DOI] [PubMed] [Google Scholar]
- Lee H.-Y., Huang Y., Bruneau N., Roll P., Roberson E.D.O., Hermann M., Quinn E., Maas J., Edwards R., Ashizawa T. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. 2012;1:2–12. doi: 10.1016/j.celrep.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche H. Synaptic or ion channel modifier? PRRT2 is a chameleon-like regulator of neuronal excitability. Brain. 2018;141:938–941. doi: 10.1093/brain/awy073. [DOI] [PubMed] [Google Scholar]
- Liao D., Zhang X., O'Brien R., Ehlers M.D., Huganir R.L. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat. Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Liu Y.-T., Nian F.-S., Chou W.-J., Tai C.-Y., Kwan S.-Y., Chen C., Kuo P.-W., Lin P.-H., Chen C.-Y., Huang C.-W. PRRT2 mutations lead to neuronal dysfunction and neurodevelopmental defects. Oncotarget. 2016;7:39184–39196. doi: 10.18632/oncotarget.9258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita K., Wakamori M., Rhyu I.J., Arii T., Oda S., Mori Y., Imoto K. Bidirectional alterations in cerebellar synaptic transmission of tottering and rolling Ca2+ channel mutant mice. J. Neurosci. 2002;22:4388–4398. doi: 10.1523/JNEUROSCI.22-11-04388.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J.O., Dahimene S., Page K.M., Ferron L., Kadurin I., Ellaway J.I.J., Zhao P., Patel T., Rothwell S.W., Lin P. Disruption of the Key Ca2+ Binding Site in the Selectivity Filter of Neuronal Voltage-Gated Calcium Channels Inhibits Channel Trafficking. Cell Rep. 2019;29:22–33.e5. doi: 10.1016/j.celrep.2019.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michetti C., Castroflorio E., Marchionni I., Forte N., Sterlini B., Binda F., Fruscione F., Baldelli P., Valtorta F., Zara F. The PRRT2 knockout mouse recapitulates the neurological diseases associated with PRRT2 mutations. Neurobiol. Dis. 2017;99:66–83. doi: 10.1016/j.nbd.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michetti C., Corradi A., Benfenati F. PRRT2, a network stability gene. Oncotarget. 2017;8:55770–55771. doi: 10.18632/oncotarget.19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mill J., Richards S., Knight J., Curran S., Taylor E., Asherson P. Haplotype analysis of SNAP-25 suggests a role in the aetiology of ADHD. Mol. Psychiatry. 2004;9:801–810. doi: 10.1038/sj.mp.4001482. [DOI] [PubMed] [Google Scholar]
- Mo J., Wang B., Zhu X., Wu X., Liu Y. PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by influencing synaptic function in the primary motor cortex of rats. Neurobiol. Dis. 2019;121:274–285. doi: 10.1016/j.nbd.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Muià, J. (2020). Differential Protein-Protein Interactions of two P/Q-Type Ca2+ Channel Isoforms. Master's thesis in Medical-Pharmaceutical Biotechnology, University of Genoa.
- Müller C.S., Haupt A., Bildl W., Schindler J., Knaus H.-G., Meissner M., Rammner B., Striessnig J., Flockerzi V., Fakler B., Schulte U. Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc. Natl. Acad. Sci. USA. 2010;107:14950–14957. doi: 10.1073/pnas.1005940107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanou E., Catterall W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron. 2018;98:466–481. doi: 10.1016/j.neuron.2018.03.017. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods Enzymol. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Pozzi D., Condliffe S., Bozzi Y., Chikhladze M., Grumelli C., Proux-Gillardeaux V., Takahashi M., Franceschetti S., Verderio C., Matteoli M. Activity-dependent phosphorylation of Ser187 is required for SNAP-25-negative modulation of neuronal voltage-gated calcium channels. Proc. Natl. Acad. Sci. USA. 2008;105:323–328. doi: 10.1073/pnas.0706211105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi D., Lignani G., Ferrea E., Contestabile A., Paonessa F., D’Alessandro R., Lippiello P., Boido D., Fassio A., Meldolesi J. REST/NRSF-mediated intrinsic homeostasis protects neuronal networks from hyperexcitability. EMBO J. 2013;32:2994–3007. doi: 10.1038/emboj.2013.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestigio C., Ferrante D., Valente P., Casagrande S., Albanesi E., Yanagawa Y., Benfenati F., Baldelli P. Spike-Related Electrophysiological Identification of Cultured Hippocampal Excitatory and Inhibitory Neurons. Mol. Neurobiol. 2019;56:6276–6292. doi: 10.1007/s12035-019-1506-5. [DOI] [PubMed] [Google Scholar]
- Reid C.A., Clements J.D., Bekkers J.M. Nonuniform distribution of Ca2+ channel subtypes on presynaptic terminals of excitatory synapses in hippocampal cultures. J. Neurosci. 1997;17:2738–2745. doi: 10.1523/JNEUROSCI.17-08-02738.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi P., Sterlini B., Castroflorio E., Marte A., Onofri F., Valtorta F., Maragliano L., Corradi A., Benfenati F. A Novel Topology of Proline-rich Transmembrane Protein 2 (PRRT2): Hints for an intracellular function at the synapse. J. Biol. Chem. 2016;291:6111–6123. doi: 10.1074/jbc.M115.683888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Ge W.-P., Li Y., Hirano A., Lee H.-Y., Rohlmann A., Missler M., Tsien R.W., Jan L.Y., Fu Y.-H., Ptáček L.J. Protein mutated in paroxysmal dyskinesia interacts with the active zone protein RIM and suppresses synaptic vesicle exocytosis. Proc. Natl. Acad. Sci. USA. 2015;112:2935–2941. doi: 10.1073/pnas.1501364112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard C.J.R. Super-resolution in confocal imaging. Optik (Stuttg.) 1988;80:53–54. [Google Scholar]
- Skarnes W.C., Rosen B., West A.P., Koutsourakis M., Bushell W., Iyer V., Mujica A.O., Thomas M., Harrow J., Cox T. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead D., Eliezer D. Intrinsically disordered proteins in synaptic vesicle trafficking and release. J. Biol. Chem. 2019;294:3325–3342. doi: 10.1074/jbc.REV118.006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U., Worm U., Lalowski M., Haenig C., Brembeck F.H., Goehler H., Stroedicke M., Zenkner M., Schoenherr A., Koeppen S. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Sterlini B., Romei A., Parodi C., Aprile D., Oneto M Aperia A., Valente P., Valtorta F., Fassio A., Baldelli P. An interaction between PRRT2 and Na+/K+ ATPase contributes to the control of neuronal excitability. Cell Death Dis. 2021;12:292. doi: 10.1038/s41419-021-03569-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T., Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- Tamamaki N., Yanagawa Y., Tomioka R., Miyazaki J., Obata K., Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J. Comp. Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Tan G.-H., Liu Y.-Y., Wang L., Li K., Zhang Z.-Q., Li H.-F., Yang Z.-F., Li Y., Li D., Wu M.-Y. PRRT2 deficiency induces paroxysmal kinesigenic dyskinesia by regulating synaptic transmission in cerebellum. Cell Res. 2018;28:90–110. doi: 10.1038/cr.2017.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhammer A., Contestabile A., Ermolyuk Y.S., Ng T., Volynski K.E., Soong T.W., Goda Y., Cingolani L.A. Alternative Splicing of P/Q-Type Ca2+ Channels Shapes Presynaptic Plasticity. Cell Rep. 2017;20:333–343. doi: 10.1016/j.celrep.2017.06.055. [DOI] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Valente P., Casagrande S., Nieus T., Verstegen A.M.J., Valtorta F., Benfenati F., Baldelli P. Site-specific synapsin I phosphorylation participates in the expression of post-tetanic potentiation and its enhancement by BDNF. J. Neurosci. 2012;32:5868–5879. doi: 10.1523/JNEUROSCI.5275-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente P., Castroflorio E., Rossi P., Fadda M., Sterlini B., Cervigni R.I., Prestigio C., Giovedì S., Onofri F., Mura E. PRRT2 Is a Key Component of the Ca(2+)-Dependent Neurotransmitter Release Machinery. Cell Rep. 2016;15:117–131. doi: 10.1016/j.celrep.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente P., Orlando M., Raimondi A., Benfenati F., Baldelli P. Fine Tuning of Synaptic Plasticity and Filtering by GABA Released from Hippocampal Autaptic Granule Cells. Cereb. Cortex. 2016;26:1149–1167. doi: 10.1093/cercor/bhu301. [DOI] [PubMed] [Google Scholar]
- Valente P., Lignani G., Medrihan L., Bosco F., Contestabile A., Lippiello P., Ferrea E., Schachner M., Benfenati F., Giovedì S., Baldelli P. Cell adhesion molecule L1 contributes to neuronal excitability regulating the function of voltage-gated Na+ channels. J. Cell Sci. 2016;129:1878–1891. doi: 10.1242/jcs.182089. [DOI] [PubMed] [Google Scholar]
- Valente P., Romei A., Fadda M., Sterlini B., Lonardoni D., Forte N., Fruscione F., Castroflorio E., Michetti C., Giansante G. Constitutive Inactivation of the PRRT2 Gene Alters Short-Term Synaptic Plasticity and Promotes Network Hyperexcitability in Hippocampal Neurons. Cereb. Cortex. 2019;29:2010–2033. doi: 10.1093/cercor/bhy079. [DOI] [PubMed] [Google Scholar]
- Valtorta F., Benfenati F., Zara F., Meldolesi J. PRRT2: from Paroxysmal Disorders to Regulation of Synaptic Function. Trends Neurosci. 2016;39:668–679. doi: 10.1016/j.tins.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Wright P.E., Dyson H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.