

Abstract
Background
CD3-based bispecific T cell engagers (bsTCEs) are one of the most promising bispecific antibodies for effective cancer treatments. To elicit target-specific T cell-mediated cytotoxicity, these bsTCEs contain at least one binding unit directed against a tumor antigen and another binding unit targeting CD3 in T cell receptor complex. Development of CD3-based bsTCEs, however, has been severely hampered by dose-limiting toxicities due to cytokine release syndrome. To address this limitation, we developed a novel functionally trivalent T cell engager (t-TCE) antibody containing affinity-reduced CD3 binding unit positioned to ensure monovalent CD3 engagement, in combination with bivalent tumor antigen binding of carcinoembryonic antigen (CEA).
Methods
We modeled the variable region of anti-CD3 in the complementarity-determining regions of the heavy chain and obtained CD3 binders with reduced binding affinity. Two optimized versions CEA/CD3-v1 and CEA/CD3-v2 were identified and generated in tetravalent format, characterized and compared in vitro and in vivo for functional activity.
Results
Our lead candidate, CEA/CD3-v2, demonstrated subnanomolar binding and picomolar potency against a panel of CEA-expressing cancer cell lines. In addition, we detected reduced T cell cytokine release with potent cytotoxic activity. Our t-TCE CEA/CD3-v2 molecule demonstrated strong antitumor effect in a dose-dependent manner in human peripheral blood mononuclear cell (PBMC) xenograft model. Furthermore, combination of CEA/CD3-v2 with atezolizumab provided synergistic antitumor effect.
Conclusions
Because of its effective tumor cell killing in vitro and in vivo with reduced cytokine release, CEA/CD3 bsTCE may greatly benefit in CEA-positive cancer immunotherapy.
Keywords: CEA/CD3, T cell engager, bispecific antibody, cancer, toxicity
STATEMENT OF SIGNIFICANCE
Through optimization of CD3 binding affinity and tetravalent format with functional monovalent binding to CD3, t-TCE CEA/CD3–2 molecule not only retains high potency in vitro and in vivo, but also significantly reduces cytokine release.
INTRODUCTION
In recent years, immunotherapies utilizing T cell-dependent bispecific antibodies (bsAb) have revolutionized cancer treatments. These bsAbs recruit and redirect T cells to attack tumor cells and have shown tremendous potential for the treatment of both liquid and solid cancers [1]. One such bispecific T cell engager (bsTCE), blinatumomab, has been approved by the Food and Drug Administration for clinical use. A second bsTCE, catumaxomab, was approved in 2009 in the European Union, but withdrawn from the market in 2017 [2, 3]. A review of the clinicaltrials.gov database indicates that at the time of writing there are 48 active trials for CD3 bsAbs, with targets for both hematologic cancers, including CD19, CD20, BCMA, CD123, CD33, CD38 and solid tumors including EpCAM, Her2, Mucin16, NKG2D, CEA, GPC3 and DLL1. The development of bsTCEs, however, has run into several challenges that have slowed progress, particularly for solid tumors. These include insufficient potency, suboptimal tissue distribution and severe toxicity. Polyclonal and uncontrolled T cell activation driven by bsTCEs is the basis of dose-limiting toxicities and fatal cytokine release syndrome (CRS) for several compounds [4, 5]. CRS is caused by the overactivation of immune cells, leading to hypersecretion of cytokines by T cells and other immune cell types. Since cytokine release is intimately linked to T cell activation, high-affinity CD3 binding in the context of bsTCEs leads to increased cytokine release and a higher CRS risk. In addition, some evidence suggests that high-affinity CD3 binding impairs tumor antigen-dependent tissue distribution of bsAbs and leads to the accumulation of bsAbs in the T cell compartment instead [6]. Another challenge for developing TCEs is finding the suitable target antigens that are overexpressed on tumor cells with no or low expression in normal tissues. Even low level of target antigen expressed in nonmalignant primary tissues can trigger ‘on-target, off-tumor’ toxicity; fine tuning of the therapeutic window may be achieved by optimizing the binding affinity of the bsAb for both the target antigen and CD3 [7].
Carcinoembryonic antigen (CEA) is a cell surface glycoprotein with a molecular weight of ~180 kD and is a valuable clinical biomarker for gastrointestinal cancers [8, 9]. Overexpression of CEA has been observed in 90% of gastrointestinal malignancies, including colon, gastric, rectal and pancreatic tumors; 70% of lung cancers; ~ 30–50% of breast cancers and head–neck squamous cell carcinoma [10]. High expression of CEA on a variety of tumor types makes it an ideal target for therapeutic antibodies. In the Phase 1 clinical trial for Roche’s bsTCE against CEA, cibisatamab (CEA-TCB; RG7802), 45% of patients showed either a partial response or stable disease when treated with cibisatamab as monotherapy above 60 mg. Furthermore, treatment response increased to 82% when combined with Tecentriq (atezolizumab) [11, 12].
Here, we affinity-tuned an anti-CD3 antibody to optimize binding to CD3 on T cells. We modeled the heavy chain variable region of a CD3 antibody and identified hydrophobic or charged amino acids in the complementarity-determining regions (CDRs) to generate mutant variants. We incorporated an affinity-optimized anti-CD3 variant in a functionally trivalent TCE (t-TCE) bispecific format that bound CEA on tumor cells bivalently. In this functionally trivalent format, the two CD3 binding arms are situated to force functionally monovalent binding to T cells. We determined the functional potency of this novel, optimized t-TCE format both in vitro and in vivo.
MATERIALS AND METHODS
Generation of an anti-CEA/CD3 t-TCE and CEA-TCB
Anti-CEA antibody used here is a humanized and stability-engineered version of the T84.66 antibody [13]. Antibody that binds to human CD3 was derived from SP34, which is cross-reactive with cynomolgus monkey CD3 [14]. Targeted mutations to residues in the CDRs of the SP34 mAb were performed using SWISS-MODEL as previously described [15] and visualized with PyMOL (Schrödinger). DNA constructs were synthesized, cloned into pcDNA3.4 vector (Thermo Scientific) and transfected into Expi-CHO cells. CEA-TCB uses an asymmetric 2-to-1 molecular format and sequence information was obtained from Tnternational ImMunoGenoTics (IMGT) database (http://imgt.org/mAb-DB/mAbcard?AbId=795). The four DNA constructs encoding both heavy chain and light chain were synthesized, cloned into pcDNA3.4 vector and transfected into Expi-CHO cells together in different ratios. Supernatants were collected from shaker flasks and antibodies were purified by protein A affinity chromatography followed by size exclusion chromatography (SEC).
Cell lines and cell culture
LS-174T, KATO III, HPAC, A549, HT-29 and Jurkat Nuclear Factor of Activated T cell (NFAT) reporter cell lines were obtained from ATCC (Manassas, VA). MKN-45 cell line was purchased from JCRB Cell Bank. All cell lines were cultured in either RPMI-1640 or DMEM (both from Life Technologies, Carlsbad, CA) with 10% fetal bovine serum (FBS). Human PBMC were isolated from whole blood using a standard density gradient centrifugation technique. Cynomolgus monkey PBMCs were obtained from Biological Specialty Company (Medford, MA).
Cell based binding assays
To determine CEA or CD3 binding on the cell surface; CEA positive LS-174 and MKN-45 cell line or CD3-expressing Jurkat and human PBMCs were placed in a 96-well V-bottom plate (Corning, NY) and incubated with tested bsAbs on ice for 30 minutes. Cells were washed with fluorescence-activated cell sorting (FACS) buffer [phosphate-buffered saline (PBS), 2% FBS, 1 mM EDTA] and stained with goat anti-human-IgG-Phycoerythrin (PE) Fab (Jackson Immuno Research, West Grove, PA) on ice for 30 minutes. Cells were washed twice with FACS buffer and DAPI was added to the final suspension. Binding was assessed by flow cytometry and data were analyzed by FlowJo_v10.7.1.CL.
NFAT reporter assay
Jurkat NFAT-luciferase reporter cells (1 × 105/well) and CEA-expressing LS-174T tumor cells (1x105/well) were seeded in a 96-well plate and incubated in 100 μl complete media and a serial dilution of anti-CEA/CD3 bsAbs at 37°C for 6 hours. Bio-GloTm Reagent (Promega, Madison, WI) was added and luminescence was quantified using the BioMax Discover system. The data were fitted to a 4PL curve using GraphPad Prism 8 software.
In vitro cytotoxicity assay
Human PBMCs were isolated by Ficoll (STEMCELL Technologies, Vancouver, Canada) from blood of healthy donors. PBMCs and luciferase-expressing target cells were mixed at an effector to target (E:T) ratio of 20:1 in the presence of serially diluted anti-CEA/CD3 bsAbs or IC/anti-CD3 and incubated at 37°C for 48 hours. Bio-Glo™ was used to determine viability. Data were analyzed and cytotoxicity curves were generated with GraphPad Prism 8 software.
Cytokine measurement
Target cells and human PBMCs were plated in 96-well microplates at an E:T ratio of 20:1 and cocultured with various concentrations of anti-CEA/CD3. After 48 hours of incubation, cell-free supernatants were harvested, and the production of interleukin 2 (IL-2), interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) were measured by enzyme-linked immunosorbent assay (ELISA) assay (BD Biosciences) according to the manufacturer’s instructions.
Human tissue cross-reactivity study
Immunohistochemistry (IHC) was performed to evaluate tissue cross-reactivity of CEA/CD3 bsAb; conducted by Comparative Bioscience Inc. (Sunnyvale, CA). Briefly, formaldehyde-fixed paraffin-embedded (FFPE) tissue sections of several cancerous and normal tissues on slides were deparaffinized, hydrated and subjected to heated citrate buffer antigen retrieval for 20 minutes under low pressure. After PBS wash, sections were peroxide blocked (10 minutes), protein blocked (60 minutes) and incubated with primary antibody (60 minutes) followed by secondary antibody (60 minutes) at room temperature. Staining was visualized with diaminobenzidine peroxidase (in 1–2 minutes), and tissues were counterstained with hematoxylin. For the cell lines, the previously fixed cells were smeared on glass slides, air dried overnight and stored at 4°C. For analysis, the smears were fixed in fresh, cold 4% Paraformaldehyde (PFA) for 10 minutes followed by PBS wash. After washing, sections were treated for antigen retrieval followed by the same steps described for FFPE sections.
In vivo experiment
In vivo study was performed in a humanized PBMC/NCG model to evaluate the efficacy of CEA/CD3 bsAbs according to Institutional animal care and use committee (IACUC) guidelines. All the studies were conducted at Gempharmatech Co., Ltd (Nanjing, China). Briefly, LS-174T tumor cells were mixed with human PBMCs at different E:T ratios and injected subcutaneously on the rear flank of NOG Scid Gamma (NCG) mice. Anti-CEA/CD3 bsAbs or vehicle control were injected intravenously (i.v.) to the corresponding groups twice a week for 2 weeks starting from either day 7 or 9 postengraftation. Tumor size was measured biweekly. The study was terminated on day 24 or day 26.
Staining tumor-infiltrated cells by flow cytometry
Single-cell suspensions were prepared from tumor samples obtained from the antibody or vehicle control-treated mice by an incubation of 30 minutes with collagenase and DNase IV followed by tissue homogenization using a 70-μm cell strainer (Fisher scientific). Live cells were washed with FACS buffer. Cells were then stained with antibody cocktails against human CD45, CD3, CD4 and CD8. The samples were run by a Thermo Attune NxT flow cytometry and analyzed using FlowJo 10.7.1 software.
Statistical analysis
The Prism 8 software (GraphPad, San Diego, CA) was used to analyze the data. Results are reported as mean ± standard error of the mean (SEM). All the statistical comparisons were performed using the two-tailed Student’s t-test. Values of P < 0.05 were considered to be statistically significant.
RESULTS
Construction of anti-CEA/CD3 t-TCE
Among several well-characterized anti-CEA monoclonal antibodies (mAbs), T84.66 has previously been shown to bind CEA with high specificity and affinity and is widely explored for the diagnosis of colon cancer [16]. We utilized the humanized T84.66 sequence for generating the anti-CEA/CD3 bsAb [13]. Most T cell-dependent bsAbs developed to date rely on anti-CD3 arms derived from OKT3, UCHT1, SP34 and TR66 [17, 18]. Among these anti-CD3 antibodies, SP34 demonstrates subnanomolar binding affinity (0.17 nM) to CD3ε, and importantly, it shows cross-reactivity with cynomolgus monkey CD3, making in vivo toxicity studies in nonhuman primates feasible.
The bsAb-expressing plasmid constructs were transfected in Expi-CHO cells and the antibodies were purified by Protein A step followed by SEC. The analytical SEC showed one single peak correlated with the size of the molecule as compared with SEC protein standard (data not shown). We elected to use a 2:2 format because its structural symmetry benefits the manufacturability and stability of the antibody [19, 20]. In this format, two arms on the N-terminus of the bsAb are CEA binders and two anti-CD3 ScFvs are fused to the C-terminus of the anti-CEA light chains (Fig. 1A), which bind in a functionally monovalent manner to CD3 on T cells. To confirm whether CD3 arms in this format perform monovalent binding to CD3 on T cells, we generated two more constructs including bivalent anti-CD3 ScFv-Fc, and monovalent anti-CD3 ScFv-Fc heterodimer, in which one anti-CEA Fab was replaced with one anti-CD3-p ScFv, forming monovalent CEA and monovalent CD3 heterodimer antibody, as shown in Figure 1A. Apparent binding activity of these antibodies to CD3 was compared with our carcinoembryonic antigen/CD3-parental (CEA/CD3-p) bsAb by flow cytometry. Interestingly, the Bmax value of the bivalent anti-CD3 ScFv-Fc on CD3-expressing Jurkat cells was 6-fold higher than that of CEA/CD3-p bsAb (Fig. 1B). In contrast, we observed comparable binding of CEA/CD3-p and monovalent anti-CD3 ScFv-Fc heterodimeric antibody on Jurkat cells (Fig. 1B). These data supported the monovalent binding of CEA/CD3-p bsAb to CD3 on T cells. In addition, to disrupt Fc receptor binding interactions, we modified the Fc domain of anti-CEA/CD3 bsAb with the three-point mutations, i.e. L234A/L235A/P329A.
Figure 1.
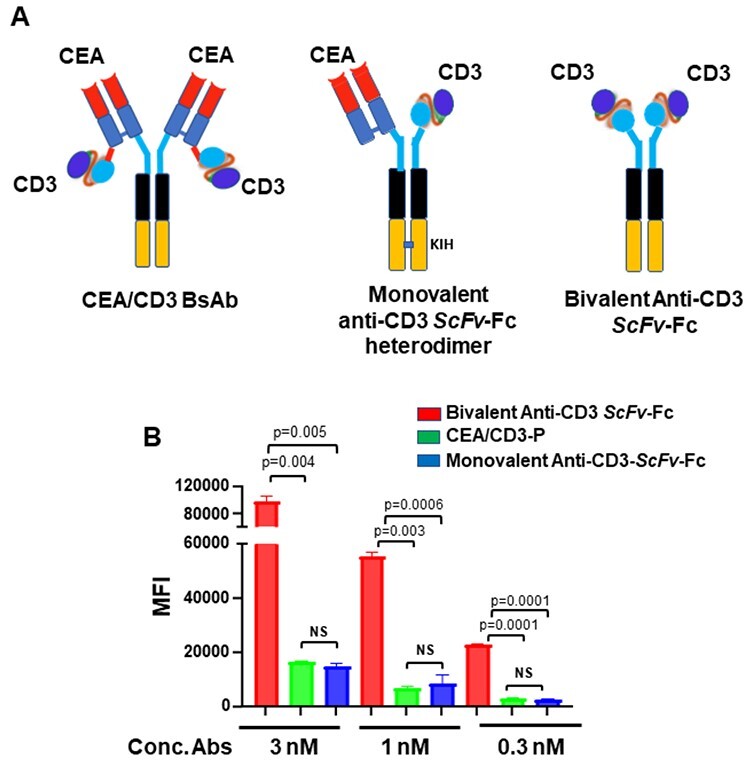
Generation of CEA/CD3 bsAb. (A) shows the t-TCE format of CEA/CD3 bsAb (left), schematic of monovalent anti-CD3 ScFv-Fc heterodimer format (middle) and bivalent anti-CD3 ScFv-Fc (right). (B). Cell binding of the monovalent and bivalent CD3 abs to CD3 on Jurkat cells. CD3-expressing Jurkat cells were stained with 3, 1 and 0.3 nM concentrations of the antibodies. Each point represents median fluorescence intensity (MFI) mean values of triplicate well and the ±SEM is represented by error bars.
Engineering of CD3 for reduced binding affinity
As higher CD3 binding leads to substantial cytokine release, we set out to fine tune target-dependent T cell antigen receptor (TCR) signaling, with the goal of maintaining killing activity but reduction of cytokine response. We modeled the variable region of anti-CD3 antibody and identified protruding, hydrophobic and charged amino acids in the CDRs of the heavy chain (Supplementary Fig. 1A and B). We generated six clones with mutations in the CDRs of heavy chain. The apparent binding activity of these anti-CD3 variants to human CD3 was measured by flow cytometry using Jurkat cells and healthy human donor-derived T cells. Anti-CD3-1a in the first set of clones showed comparable binding as CEA-CD3-p (CD3 SP34 parental) on Jurkat cells (Bmax: CEA/CD3-p: 74055 ± 1423 vs. CEA/CD3-1a: 72732 ± 6815), whereas anti-CD3-3b showed significant reduction in the binding to CD3 (Bmax: CEA/CD3-p: 74055 ± 1423 vs. CEA/CD3-3b: 39418 ± 6071) (Fig. 2A). These observations lead to the generation of combination variants based on mutations 1a, 3b and an additional 2b1 variant (Supplementary Fig. 1B). As expected, CD3 binding of anti-CD3-1a3b and anti-CD3-1a3b2b1, herein referred to as carcinoembryonic antigen/CD3-variant 1 (CEA/CD3-v1) and carcinoembryonic antigen/CD3-variant 2 (CEA/CD3-v2), respectively, had further declined as compared with CEA/CD3-3b by flow cytometry assay (Bmax: CEA/CD3-3b: 39418 ± 6071vs. CEA/CD3-v1: 32960 ± 3205 and CEA/CD3-v2: 29823 ± 5055). As shown in Figure 2, a comparable binding activity of CEA/CD3-v1 and CEA/CD3-v2 was observed on Jurkat cells (Fig. 2A) and human T cells (Fig. 2B) as well as cyno T cells (Supplementary Fig. 2). To assess whether TCR signaling triggered by the mutant anti-CEA/CD3 bsAbs correlated with the reduction in their apparent binding activity, we used Jurkat NFAT-luciferase reporter assay. Coculture of the luciferase reporter Jurkat cells with CEA-expressing LS-174T target cells in the presence of our t-TCE indicated lower NFAT-driven gene expression by CEA/CD3-v1 and CEA/CD3-v2 variants compared with CEA/CD3-p (Supplementary Fig. 3). Moreover, the NFAT activity mediated by CEA/CD3-v2 was even lower than CEA/CD3-v1 (Supplementary Fig. 3). Given these findings, we selected these two variants for further characterization.
Figure 2.
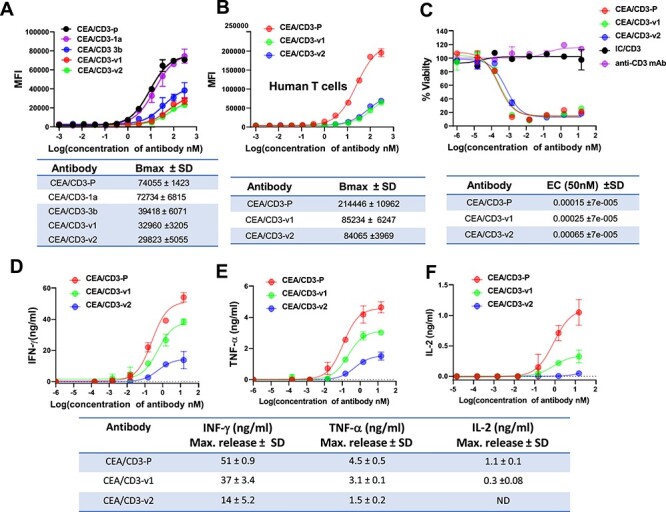
Functional evaluation of CEA/CD3 bsAbs with various CD3 affinity in vitro. (A) Jurkat cells were stained with serially diluted CEA/CD3 bsAbs. Cell surface-bound bsAbs were detected by PE-conjugated goat anti-human IgG1 Fab. Mean fluorescence intensities were determined by flow cytometry assay. (B) Human PBMCs were stained with the same protocol as (A). (C) In vitro cytotoxicity of T cell redirecting CEA/CD3 bsAbs against CEA-positive cell line MKN-45. Human PBMCs and MKN-45 cells (20:1) were cocultured in the presence of CEA/CD3 bsAbs for 48 hours. The specific cytotoxicity of CEA/CD3 bsAbs and IC/CD3 (an irrelevant tumor antigen binding arm with anti-CD3-v2) was estimated by luminescence unit. IFN-γ (D), TNF-α (E) and IL-2 (F) measurement in PBMC induced by CEA/CD3 bsAbs in the presence of MKN-45 tumor cells was compared. Supernatants of the coculture of PBMCs with MKN-45 tumor cells at E:T ratio of 20:1 were harvested at 48 hours and cytokine release was measured by ELISA. Data shown here are mean ± SD values from three independent triplicates values. The results are representative of at least three independent experiments.
Functional characterization of anti-CEA/CD3 variants
To evaluate functional properties of the CEA/CD3 t-TCE variants, we first assessed in vitro T cell-mediated killing using CEA-expressing tumor cell lines. Luciferase-expressing, CEAHi MKN-45 cells were cocultured with freshly isolated human PBMCs at an E:T ratio of 20:1 and specific lysis was determined after 48 hours of incubation by luminescence. Surprisingly, although the apparent binding activity of CEA/CD3-v1 and CEA/CD3-v2 to CD3 was reduced relative to CEA/CD3-p bsAb (Fig. 2A and B), comparable antibody-mediated cytotoxicity was observed (Fig. 2C). As expected, no cytotoxicity was observed using a t-TCE bsAb with an irrelevant tumor antigen binding arm/anti-CD3 (isotype control, refer as IC/CD3) and a canonical anti-CD3 mAb in the same assay (Fig. 2C). To assess potential differences in cytokine release, supernatants from coculture of hPBMC with MKN-45 target cells were collected after 48 hours, and cytokine release was measured by ELISA. The anti-CEA/CD3-p bsAb induced higher levels of IFN-γ, TNF-α and IL-2 release than CEA/CD3-v1 and CEA/CD3-v2 (Fig. 2D–F). Notably, maximum release of IFN-γ (14 ± 5.2 ng/ml) and TNF-α (1.5 ± 0.2 ng/ml) mediated by CEA/CD3- v2 were lower than that by CEA/CD3-v1 (37 ± 3.4 ng/ml for IFN-γ and 3.1 ± 0.1 ng/ml for TNF-α) (Fig. 2D and E). These results correlate with the Jurkat NFAT-luciferase reporter assay (Supplementary Fig. 3). These data together indicated that although binding affinity to CD3 was greatly reduced, CEA/CD3-v1 and CEA/CD3-v2 still demonstrated potent killing activity with reduced cytokine release in vitro. Due to even lower cytokine responses relative to CEA/CD3-v1, CEA/CD3-v2 was chosen as the lead candidate for further comparative analysis. Furthermore, to exclude the possibility that CEA/CD3-p and CEA/CD3-v2 may function as agonists, we investigated antibody-induced proliferation using CEA-expressing LS-174 and MKN-45 cells as well as CEA-negative tumor cells (MDA-MB-231 and HEK293). As shown in Supplementary Figure 4A–D, we did not observe any effects on tumor cell proliferation either with CEA/CD3-p or CEA/CD3-v2 on CEA-positive or CEA-negative cells. These findings confirmed no agonistic property of the bsAb in the absence of CD3 T cells.
Determining the threshold of effectiveness for CEA/CD3-v2 t-TCE
Although CEA is overexpressed in numerous cancer types, it is normally expressed in a variety of epithelial tissues, such as the urogenital, respiratory and gastrointestinal tracts [8, 9]. Therefore, it is critical to evaluate the minimum level of CEA expression that is required to elicit cytotoxicity mediated by CEA/CD3-v2. To do so, we selected multiple tumor cell lines and quantified CEA binding sites on the cell surface by analysis of immunofluorescence staining (Supplementary Table 1). Among the cell lines tested, our analysis revealed that CEA was highly expressed on the gastric tumor cell line MKN-45 and the lowest level of CEA expression was observed on A549 cells. Intermediate levels of CEA expression were detected on LS-174T, KATO III and HT-29 cell lines (Fig. 3C and Supplementary Table 1). To evaluate the effectiveness of our CEA/CD3-v2 against those target cells with various levels of CEA expression, we compared Cibisatamab (CEA-TCB) as a benchmark antibody as it has been shown encouraging antitumor activity in the treatment of metastatic colorectal cancers (CRCs) [11, 21]. First, we compared the binding affinity to the antigen molecules, CEA and CD3. As analyzed by flow cytometry, the apparent binding activity of CEA-TCB to CD3 expressed on human T cells was higher than CEA/CD3-v2 [Bmax: (CEA-TCB) 45116 ± 4103 vs. (CEA/CD3-v2) 17475 ± 4103, P < 0.0001] (Fig. 3A). We further compared CEA binding by CEA-TCB and CEA/CD3-v2 on CEA-positive tumor cells. The apparent binding activity of CEA-TCB to CEA on MKN-45 cell was lower than that of CEA/CD3-v2 as its maximus binding value showed 4-fold weaker relative to CEA/CD3-v2 [Bmax: (CEA/CD3-v2) 484430 ± 7973 vs (CEA-TCB) 123958 ± 7944, P < 0.0005] (Fig. 3B). Having demonstrated the differences in apparent binding activity for CEA and CD3 between CEA-TCB and CEA/CD3-v2, we next analyzed in vitro cytotoxic activity using high CEA expressing MKN-45 and LS-174T cells as targets (Fig. 3C). As shown in Figure 3D, CEA/CD3-v2 bsAb exhibited higher target-specific cytotoxicity against LS-174T and MKN-45 cells, and the half maximal effective concentration (EC50) killing of CEA/CD3-v2 was 53- and 55-fold lower than that of CEA-TCB, respectively (Fig. 3D). Interestingly, the further comparative studies showed that while treatment of KATO III cells with CEA/CD3-v2 in the presence of PBMCs was sufficient to drive antigen-specific cytolysis, no apparent target lysis was observed in treatment with CEA-TCB (Fig. 3D). Neither CEA-TCB nor CEA/CD3-v2 was able to trigger cytolytic activity against HT-29, which has considerably lower CEA expression (Fig. 3C and D and Supplementary Table 1).
Figure 3.
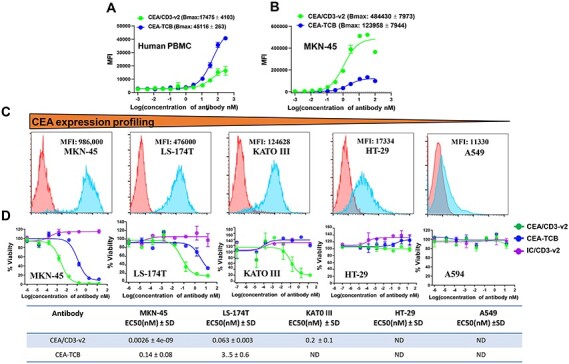
Comparative binding and in vitro cytotoxicity assessment of CEA/CD3-v2 bsAbs and CEA-TCB. Binding of CEA/CD3-v2 or CEA-TCB to CD3 on human PBMCs (A) and MKN-45 (B) tumor cells was analyzed by flow cytometry. Cells were stained with serially diluted CEA/CD3 bsAbs, followed by a PE-conjugated anti-human IgG Fab. On the y-axis, the MFI PE is plotted against the antibody concentration in log nM on the x-axis. Each point represents MFI mean values of triplicate values and the ±SEM is represented by error bars. (C). Tumor target cell lines: MKN-45, LS-174T, KATO III, HT29 and A594 was stained by CEA/CD3-v2 and CEA expression on the cell surface was determined by FACS assay. (D) The cytotoxicity of CEA/CD3-v2, CEA-TCB, IC/CD3-v2 (an irrelevant tumor antigen binding arm with anti-CD3-v2) was tested with human PBMCs in vitro with serial dilution of the antibodies. The tumor target cells: MKN-45 (MFI: 986 000), LS-174T (MFI: 476 000), KATO III (MFI: 124 628), HT-29 (MFI:17 334) or A549 (MFI:11 330) cocultured with human PBMCs at an effector-to target ratio of 20:1. Effector cytolytic activity was assessed after 48 hours. EC50 values with average ± SD are listed in the table under figure. All data depicted here are representative illustration of experiments carried out with at least three different donors.
Figure 4.
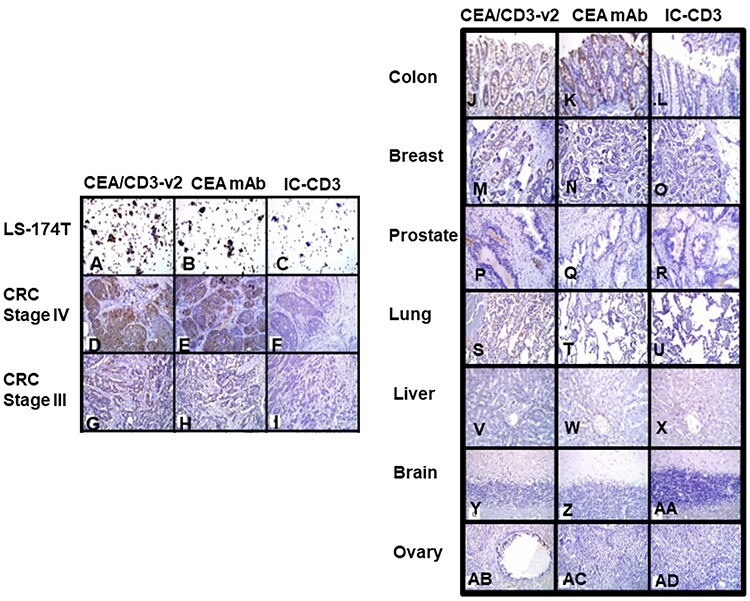
Assessment of tissue cross-reactivity of CEA/CD3-v2 bsAb in several normal and tumor tissues samples. Representative images of IHC staining of CEA/CD3-v2 bsAb, CEA mAb and IC/CD3 bsAb. At concentration of 3 μg/ml, LS-174-T cells (A–C), CRC stage IV (D–F) and CRC stage III (G–I) showed intense (grade 4+) staining with the CEA/CD3-v2 as well as anti-CEA MAb. The right panel showed staining of normal tissues with a magnification of ×200. Normal colon (J–I), breast (M–O), prostate (P–R), lung (S–U), liver (V–X), brain (Y–AA) and ovary (AB–AD) was also performed.
Evaluation of binding specificity and tissue cross reactivity of CEA/CD3-v2 bsAb
Targeted binding study was conducted to investigate if CEA/CD3-v2 bound to normal epithelium and more broadly to cancerous tissue that typically express CEA. To accomplish this, CEA/CD3-v2 and IC-CD3 antibodies were tested for their ability to bind cryosections of several normal and cancerous human tissues. The IC-CD3 control was used that bind to an irrelevant antigen but shared the anti-CD3 arm with CEA/CD3-v2 to determine CEA binding specificity in the IHC staining studies. At concentration of 3 μg/ml, strong (grade +4) staining was observed in CEA-expressing LS-174 cells (Fig. 4A), CRC stage IV tumor cells (Fig. 4D) as well as CRC stage III tumor cells (Fig. 4G) with CEA/CD3-v2 as well as the control anti-CEA mAb (Fig. 4B, E and H). The staining was distributed with multifocal and diffuse patterns in CRC stage IV and CRC stage III tissues, respectively. Both membranous and cytoplasmic staining were detected in the majority of the positively stained tumor cells with an overall moderate to high intensity (grade +2 to +4) (Fig. 4D, E, G and H). No specific staining was found in sections stained with the isotype control bsAb (Fig. 4C, F and I). The normal colon, lung and breast tissues showed a low-grade cytoplasmic staining with intensity of grade 1 (Fig. 4J, K, M, N, S and T). However, normal human prostate, liver, brain and ovary tissues did not show any off-target reactivity with the tested CEA/CD3-v2 and the control antibodies (Fig. 4P, Q, V, W, Y, Z, AB and AC). These studies indicated that staining intensity of CEA correlated with the stages of CRC and normal tissues showed lower or no staining by CEA/CD3-v2.
In vivo efficacy of CEA/CD3 bsAbs
Finally, we interrogated the in vivo efficacy of our CEA/CD3 t-TCE in xenograft model. NCG mice were injected subcutaneously on the right flank with a mixture of LS-174T cells and human PBMCs. Vehicle (PBS) or anti-CEA/CD3 t-TCE (1 or 3 mg/kg) were biweekly administered intraperitoneally (i.p.) day 7 after tumor engraft (Fig. 5A). No significant difference in body weight was observed between PBS and the antibody-treated mice during the 24-day observation period (data not shown). For antitumor efficacy studies, tumor volume was calculated weekly by caliper measurements. Consistent with the in vitro killing studies, the treatment of mice with anti-CEA/CD3-p at 3 or 1 mg/kg significantly inhibited growth of LS-174T cells, whereas the vehicle control showed no antitumor effect (Fig. 5B–D). Interestingly, in the CEA/CD3-v1 and CEA/CD3-v2 cohorts, we observed a significant reduction of tumor growth and near-complete tumor clearance at doses of both 3 and 1 mg/kg (Fig. 5B–E), with the exception of one mouse in the CEA/CD3-v2 cohort at a 1 mg/kg dose level.
Figure 5.
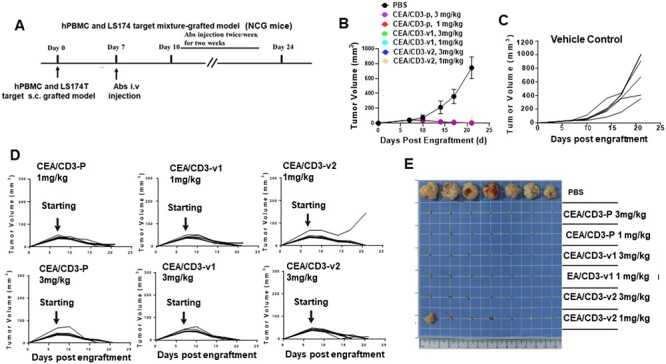
Potent T cell-mediated killing of tumor cells in CEA/CD3 bsAb-treated animal model. (A) Study design. A mixture of human PBMCs (1 × 107 cells) and LS-174T tumor cells (5 × 106 cells) were injected subcutaneously into NCG mice. The grafted mice were treated with different doses (1 or 3 mg/kg) of CEA/CD3 bsAbs and vehicle control i.v. twice/week starting from day 7 after PBMCs and LS-174T tumor cells were grafted. (B) Mice were monitored for tumor growth and results are expressed as mean tumor area. The tumor volume of the grafted NCG mice given vehicle control (C) or individual mouse in the same cohort treated with different doses of CEA/CD3-p, CEA/CD3-v1 and CEA/CD3-v2 (D) were expressed as mean tumor area. (E) Representative photographs of the remaining tumors from NCG mice implanted LS-174T cells at the study end point.
The strong efficacy of CEA/CD3-v2 observed prompted us to set up a dose-dependent in vivo study with lower E/T ratio to further evaluate antitumor activity. Human PBMCs were mixed with LS-174T tumor cells at a ratio of 1:1 before inoculation into NCG mice (Fig. 6A). As shown in Figure 6B, the antibody dose-dependent trend toward antitumor efficacy was observed, with the higher doses (3 and 1 mg/kg) led to 98.90% and 97.26% tumor growth inhibition (TGI), respectively, compared with the isotype control group and still significant tumor reduction for dose as low as 0.1 mg/kg (43.73% TGI).
Figure 6.
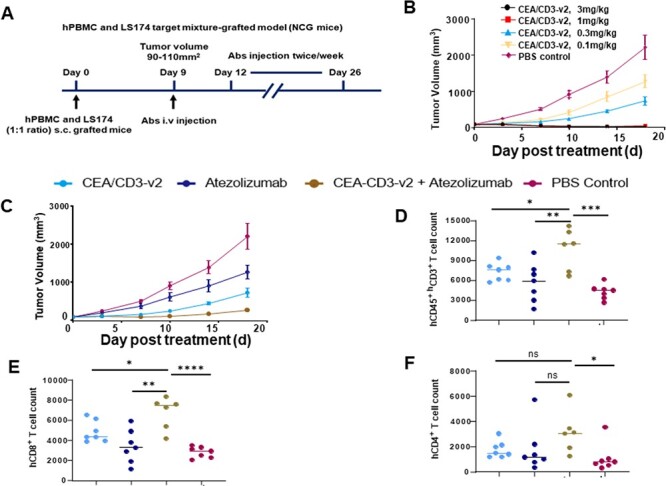
Treatment of CEA/CD3-v2 synergized with anti-PD-L1 antibody in hPBMC-NCG mice. (A) In vivo study design. (B) NCG mice were subcutaneously injected with mixture of hPBMCs (5 × 106) and LS-174T cells (5 × 106). Once tumor reaches between 90 and 110 mm3, the mice were randomly assigned (n = 6) and intravenous treatment of different doses of CEA/CD3-v2 (0.1, 0.3, 1 or 3 mg/kg) was initiated twice per week. Tumor growth was monitored every 3 days until the termination of the study. (C) Using the same protocol as (A), NCG mice were engrafted with mixture of hPBMCs and LS-174T cells and antibodies CEA/CD3-v2 (0.3 mg/kg) i.v. atezolizumab (20 mg/kg) i.p. alone or in combination was administered twice per week. The tumor growth curves showed mean ± SD. (D) Single live cell suspension of tumor samples from the antibody or vehicle control-treated mice were stained with antibody cocktails. The numbers of hCD45+CD3+(D), hCD8+ (E) and hCD4+ (F) TILs were calculated based on the frequencies of each cell types in 30 000 single live cells by flow cytometry assay. *P < 0.02, **P < 0.008, ***P < 0.0007, ****P < 0.000. t-test.
The synergistic effect of combination therapy of CEA-TCB and PD-L1 antibody was observed in the in vivo humanized model [22] as well as in the clinic trials [11], it would be interesting to evaluate enhanced efficacy using a suboptimal dose of CEA/CD3-v2 in combination with atezolizumab (anti-PD-L1) against LS174T tumor cells that are positive for both CEA and PD-L1 (data not shown) in hPBMC grafted-NCG model. The monotreatment of CEA/CD3-v2 (0.3 mg/kg) or atezolizumab (20 mg/kg) reduced TGI% by 66.83% and 43.05%, respectively, compared with vehicle control (Fig. 6C). However, combination of CEA/CD3-v2 and atezolizumab with the similar doses of the monotreatment synergistically enhanced the TGI (87.55%) (Fig. 6C). Moreover, tumor samples collected at study termination were further evaluated for tumor-infiltrating lymphocytes. Notably, the number of hCD45+CD3+ cells from the mice treated with CEA/CD3-v2 plus atezolizumab was significantly higher as compared with the monotreatment of either CEA/CD3-v2 or atezolizumab (Fig. 6D). Further FACS analysis of hCD45+CD3+ T cells demonstrated significantly increased number of hCD8+ T cells infiltrated into the tumor site when treated in combination than monotreatment of CEA/CD3-v2 or atezolizumab (Fig. 6E), which highlighted the strong correlation between hCD8+ TILs and antitumor efficacy. In addition, the trend of increased number of infiltrating hCD4+ T cells was also observed in the combination treatment (Fig. 6F). These results demonstrated that combination of CEA/CD3-v2 and atezolizumab can provide superior efficacy than monotherapy and may provide further benefits in the clinical setting.
DISCUSSION
CEA is an important biomarker in many tumors including CRC, and represents a promising tumor antigen for solid tumors. Roche’s CEA-TCB bsAb has shown initial proof of concept as monotherapy, as well as in combination with anti-PDL1 in mCRC [11, 22]. Here, we present a potent CD3 affinity-tuned t-TCE molecule with bivalent tumor targeting and functionally monovalent CD3 binding.
One of the major bottlenecks for the development of CD3-dependent bsTCEs is toxicity at higher doses due to CRS—with cytokine release often as the dose-limiting toxicity. This makes dose escalation difficult or impossible and greatly hinders application of CD3-dependent bsAbs in a clinical setting [5, 23]. By introducing mutations in the CDRs of the heavy chain of the anti-CD3, we were able to successfully reduce its apparent binding to CD3 on human T cells. It is interesting to note that the mutations in antibody clones 1a did not affect CD3 binding on T cells, whereas clone 3b reduced binding to CD3 as compared with CD3-p. Furthermore, apparent binding activity was further reduced when 1a and 3b mutations were combined (CEA/CD3-v1) or combined with an additional mutation, 2b1, which significantly reduced cytokine release in CEA-CD3-v2.
Natural TCR–pMHC interaction-induced T cell activation has been suggested to have different signaling thresholds for triggering cytotoxicity and cytokine production by T cells [5, 23, 24]. It was observed that T cell-mediated target cell killing required much less intensity of TCR engagement and exhibited a premature immunological synapse, whereas higher intensity of TCR engagement and mature synapse was required for T cell expansion and cytokine release [24]. In our study, reducing the CD3 binding to T cells did not affect significantly cytotoxic activity against CEA-expressing target cells, but cytokine release (IFN-γ, TNF-α and IL-2) was greatly reduced (3-fold or more). The decoupling of cytokine release from cytotoxicity in our findings supports the concept that dual threshold determines T cell-mediated killing and cytokine release. The potent cytotoxicity combined with low cytokines release appears to be a feature unique to our CEA/CD3-v2, which might result in a better safety profile in a clinical setting. Moreover, it is known that the reduction of TNF-α is of particular importance because of its influence on macrophage and monocyte production of IL-6 and IL-1β [5, 23]. Reduction in TNFα and other cytokines would represent an important step forward in mitigating CRS-related toxicities at therapeutic dose level. On the other hand, the high apparent affinity toward tumor antigen and low apparent affinity toward CD3 is not only important for reducing cytokine release, but also important in driving antibody biodistribution toward tumor tissue and being away from the T cell compartments, such as bone marrow and lymph nodes [25]. Our t-TCE format supports bivalent tumor antigen binding and functionally monovalent CD3 binding, as shown in Figure 1B.
We have also compared our CEA/CD3-v2 with CEA-TCB. The data indicated that our bsAb is more potent than CEA-TCB against high CEA-expressing tumor cell lines such as MKN-45 and LS-174T and effective against intermediate CEA-expressing cell line such as KATO III, whereas CEA-TCB appeared to be ineffective. This might be due to high binding affinity toward target cells and also the format of CEA/CD3-v2, which could mediate better tumor and T cell engagement [26] in CEA/CD3-v2 when comparing with CEA-TCB. For HT-29, where CEA binding sites on cell surface was calculated to be ~ 18 000 per cell, both antibodies lost cytolytic activity. This may provide a necessary safety margin in therapeutic setting.
Our CEA antibody is derived from T84.66, an antibody that is being clinically investigated as a diagnosis for the detection and tracking of CEA-positive tumor tissue. As shown in Figure 4. CEA/CD3-v2 showed high specificity toward CEA-expressing cancerous tissues, whereas either low or no reactivity toward normal tissues such as lung, brain, prostate, ovary and liver was observed. This study provided us valuable insights into the potential toxicity and helpful qualitative information for clinical trials.
The in vivo study in a setting of different E:T ratios demonstrated strong antitumor activity of CEA/CD3-v2 and tumor reduction was seen at low doses (0.1 mg/kg). In addition, combination of CEA/CD3-v2 with atezolizumab further enhanced TGI and improved T cell infiltration compared with mono-treatment, supporting a potential combination study in the clinical settings [11]. The current work provided preclinical validation for a potent CEA-CD3 candidate, CEA/CD3-v2, with strong antitumor potency and reduced cytokine release. This candidate will be pursued for clinical trials.
Supplementary Material
ACKNOWLEDGMENTS
The authors want to thank Xi-Yong (Sean) Fu and Paul Parren for valuable suggestions and feedback and Michael Harris for critical editing of the content. This work was funded by Luye Pharma Group.
Conflict of interest statement. None declared.
References
- 1. Labrijn, AF, Janmaat, ML, Reichert, JMet al. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 2019; 18: 585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 2. Newman, MJ, Benani, DJ. A review of blinatumomab, a novel immunotherapy. J Oncol Pharm Pract 2016; 22: 639–45. doi: 10.1177/1078155215618770. [DOI] [PubMed] [Google Scholar]
- 3. Seimetz, D, Lindhofer, H, Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM×anti-CD3) as a targeted cancer immunotherapy. Cancer Treat Rev 2010; 36: 458–67. doi: 10.1016/j.ctrv.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 4. Wilke, AC, Gökbuget, N. Clinical applications and safety evaluation of the new CD19 specific T-cell engager antibody construct blinatumomab. Expert Opin Drug Saf 2017; 16: 1191–202. doi: 10.1080/14740338.2017.1338270. [DOI] [PubMed] [Google Scholar]
- 5. Li, J, Piskol, R, Ybarra, Ret al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med 2019; 11: eaax8861. doi: 10.1126/scitranslmed.aax8861. [DOI] [PubMed] [Google Scholar]
- 6. Nie, S, Wang, Z, Moscoso-Castro, Met al. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib Ther 2020; 3: 18–62. doi: 10.1093/abt/tbaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vafa, O, Trinklein, ND. Perspective: designing T-cell engagers with better therapeutic windows. Front Oncol 2020; 10. doi: 10.3389/fonc.2020.00446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benchimol, S, Fuks, A, Jothy, Set al. Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion molecule. Cell 1989; 57: 327–34. doi: 10.1016/0092-8674(89)90970-7. [DOI] [PubMed] [Google Scholar]
- 9. Kotzev, AI, Draganov, PV. Carbohydrate antigen 19–9, carcinoembryonic antigen, and carbohydrate antigen 72–4 in gastric cancer: is the old band still playing? Gastrointest Tumors 2018; 5: 1–13. doi: 10.1159/000488240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thompson, JA, Grunert, F, Zimmermann, W. Carcinoembryonic antigen gene family: molecular biology and clinical perspectives. J Clin Lab Anal 1991; 5: 344–66. doi: 10.1002/jcla.1860050510. [DOI] [PubMed] [Google Scholar]
- 11. Tabernero, J, Melero, I, Ros, Wet al. Phase Ia and Ib studies of the novel carcinoembryonic antigen (CEA) T-cell bispecific (CEA CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients with metastatic colorectal cancer (mCRC). J Clin Oncol 2017; 35: 3002. doi: 10.1200/jco.2017.35.15_suppl.3002.28644773 [DOI] [Google Scholar]
- 12. Segal, NH, Saro, J, Melero, Iet al. Phase I studies of the novel carcinoembryonic antigen T-cell bispecific (CEA-CD3 TCB) antibody as a single agent and in combination with atezolizumab: preliminary efficacy and safety in patients (pts) with metastatic colorectal cancer (mCRC). Ann Oncol 2017; 28: v134. doi: 10.1093/annonc/mdx367.036. [DOI] [Google Scholar]
- 13. Yazaki, PJ, Sherman, MA, Shively, JEet al. Humanization of the anti-CEA T84.66 antibody based on crystal structure data. Protein Eng Des Sel 2004; 17: 481–9. doi: 10.1093/protein/gzh056. [DOI] [PubMed] [Google Scholar]
- 14. Blumberg, RS, Ley, S, Sancho, Jet al. Structure of the T-cell antigen receptor: evidence for two CD3 ε subunits in the T-cell receptor-CD3 complex. Proc Natl Acad Sci U S A 1990; 87: 7220–4. doi: 10.1073/pnas.87.18.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Waterhouse, A, Bertoni, M, Bienert, Set al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018; 46. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meyers, W. Limited improvement of tumour diagnosis by the simultaneous determination of carcinoembryonic antigen (CEA) and of a tumour-associated CEA-related antigen of Mr 128000 in serum. Clin Chem Lab Med 1989; 27. doi: 10.1515/cclm.1989.27.9.643. [DOI] [PubMed] [Google Scholar]
- 17. Spiess, C, Zhai, Q, Carter, PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol 2015; 67: 95–106. doi: 10.1016/j.molimm.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 18. Trabolsi, A, Arumov, A, Schatz, JH. T cell–activating bispecific antibodies in cancer therapy. J Immunol 2019; 203: 585–92. doi: 10.4049/jimmunol.1900496. [DOI] [PubMed] [Google Scholar]
- 19. Xu, H, Cheng, M, Guo, Het al. Retargeting T cells to GD2 pentasaccharide on human tumors using bispecific humanized antibody. Cancer Immunol Res 2015; 3: 266–77. doi: 10.1158/2326-6066.CIR-14-0230-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu, Z, Guo, HF, Xu, Het al. Development of a tetravalent anti-GPA33/anti-CD3 bispecific antibody for colorectal cancers. Mol Cancer Ther 2018; 17: 2164–75. doi: 10.1158/1535-7163.MCT-18-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bacac, M, Fauti, T, Sam, Jet al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res 2016; 22: 3286–97. [DOI] [PubMed] [Google Scholar]
- 22. Sam, J, Colombetti, S, Fauti, Tet al. Combination of T-cell bispecific antibodies with PD-L1 checkpoint inhibition elicits superior anti-tumor activity. Front Oncol 2020; 10. doi: 10.3389/fonc.2020.575737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santich, BH, Park, JA, Tran, Het al. Interdomain spacing and spatial configuration drive the potency of IgG-[L]-scFv T cell bispecific antibodies. Sci Transl Med 2020; 12: eaax1315. doi: 10.1126/scitranslmed.aax1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Faroudi, M, Utzny, C, Salio, Met al. Lytic versus stimulatory synapse in cytotoxic T lymphocyte/target cell interaction: manifestation of a dual activation threshold. Proc Natl Acad Sci U S A 2003; 100: 14145–50. doi: 10.1073/pnas.2334336100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mandikian, D, Takahashi, N, Lo, AAet al. Relative target affinities of T-cell–dependent bispecific antibodies determine biodistribution in a solid tumor mouse model. Mol Cancer Ther 2018; 17: 776–85. doi: 10.1158/1535-7163.MCT-17-0657. [DOI] [PubMed] [Google Scholar]
- 26. Reusch, U, Duell, J, Ellwanger, Ket al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19+ tumor cells. MAbs 2015; 7: 584–604. doi: 10.1080/19420862.2015.1029216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.