

Summary
Selective identification of newly synthesized proteins is challenging because all proteins, both existing and nascent, have the same amino acid pool and are therefore chemically indistinguishable. L-homopropargylglycine is an amino acid analog of methionine containing an alkyne moiety that can undergo a classic click chemical reaction with azide containing Alexa Fluor. Here, we present an integrated tool based on immunofluorescence staining to accurately trace and localize the newly synthesized protein in isolated primary mouse hepatocytes.
For complete details on the use and execution of this protocol, please refer to Shen et al. (2021).
Subject areas: Cell culture, Cell isolation, Cell-based Assays, Microscopy, Gene Expression, Molecular/Chemical Probes
Graphical abstract
Highlights
-
•
Isolate and prepare mouse hepatocytes for nascent protein measurement
-
•
Incubate the hepatocytes with HPG-containing medium along with the translation inhibitor
-
•
Perform the click reaction and detect nascent proteins by immunofluorescence
Selective identification of newly synthesized proteins is challenging because all proteins, both existing and nascent, have the same amino acid pool and are therefore chemically indistinguishable. L-homopropargylglycine is an amino acid analog of methionine containing an alkyne moiety that can undergo a classic click chemical reaction with azide containing Alexa Fluor. Here, we present an integrated tool based on immunofluorescence staining to accurately trace and localize the newly synthesized protein in isolated primary mouse hepatocytes.
Before you begin
The detecting newly synthesized protein protocol below describes the use of isolated primary mouse hepatocytes. However, we have also used this protocol in human neuroblastoma SK-N-BE(2) cells.
Preparation
Timing: 60 min for step 2
-
1.Mouse housing
-
a.All animal experiments were performed under the guidelines of the institutional Animal Care and Use Committee at Southeast University;
-
b.C57BL/6J mice were maintained in a barrier facility at 23°C–25°C, 40%–70% humidity, on a regular 12-h light and 12-h dark cycle;
-
c.Eight∼ten-week-old male mice were used in this protocol.
-
a.
-
2.Prepare the stock solutions
-
a.7% chloral hydrate: Dissolve 0.7 g chloral hydrate in distilled water to a final volume of 10 mL. Filter sterilize (0.22 μm filter), and store at 4°C at least for 1 year;
-
b.50 mM EGTA: Dissolve 0.951 g EGTA in 45 mL of distilled water, adjust pH to 7.4 at 37°C, and add distilled water to a final volume of 50 mL. Filter sterilize (0.22 μm filter), and store at 22°C–25°C at least for 1 year;
-
c.1 M CaCl2: Dissolve 5.55 g CaCl2 in 40 mL of distilled water, add distilled water to a final volume of 50 mL. Filter sterilize (0.22 μm filter), and store at 22°C–25°C at least for 1 year;
-
d.100 X Solution C stock solution:
Reagent Final concentration Amount KCl 0.48 M 35.79 g MgSO4·7H2O 0.12 M 29.58 g KH2PO4 0.12 M 16.33 g ddH2O n/a Up to 1 L Total n/a 1 L Note: Filter the Solution C stock solution with 0.22 μm filter and store it at 4°C at least for 6 months. -
e.10 mg/mL cycloheximide(CHX): Dissolve 0.1 g cycloheximide in 8 mL of distilled water, add distilled water to a final volume of 10 mL. Filter sterilize (0.22 μm filter), and store at −20°C at least for 1 year.
-
a.
-
3.Prepare buffers
-
a.Kreb Ringer (KR) Buffer with glucose:
Reagent Final concentration Amount NaCl 0.12 M 7 g NaHCO3 23.8 mM 2 g Glucose 20 mM 3.6 g HEPES (1 M) 5 mM 5 mL Solution C (100 X) 1 X 10 mL ddH2O n/a Up to 1 L Total n/a 1 L Note: Adjust the pH of the solution to 7.4; filter the solution with 0.22 μm filter then store it at 4°C at least for 6 months. -
b.Perfusion Buffer: Add 90 μL of EGTA (50 mM) in 45 mL of KR buffer before use.
-
c.Digestion Buffer:
Reagent Amount KR buffer 40 mL Collagenase 15∼18 mg CaCl2 (1 M) 54.88 μL
-
a.
Note: Prepare the digestion buffer before use and then filter it with 0.22 μm filter.
CRITICAL: The collagenase in the digestion buffer must dissolve completely.
-
4.Prepare hepatocyte culture medium
-
a.Growth medium:
Reagent Final concentration Amount DMEM 1 X 88 mL FBS 10% 10 mL 100 X penicillin-streptomycin 2 X 2 mL Total n/a 100 mL Note: Prepare the growth medium before use and store it at 4°C then use it up within 2 weeks. -
b.Modified L-methionine-free DMEM medium:
Reagent Final concentration Amount L-Cystine 200 μM 96.12 mg L-Glutamine (0.2 M) 2 mM 5 mL HEPES (1 M) 10 mM 5 mL 100 X penicillin-streptomycin 2 X 10 mL L-methionine-free DMEM 1 X UP to 500 mL Total n/a 500 mL
-
a.
Note: Penicillin-streptomycin is used to inhibit bacterial growth and avoid cell contamination.
Note: Prepare the modified L-methionine-free DMEM medium, then store it at 4°C and use it up within 2 weeks.
-
5.Prepare solutions for cell immunofluorescence staining
-
a.Fixation buffer (4% PFA): Dissolve 4 g paraformaldehyde (PFA) in 1× PBS at 65°C to a final volume of 100 mL. Cool it down and store at 4°C at least for 1 year;Note: Make the fixation buffer pre-warmed at 37°C about 10 min before use.
-
b.Permeabilization buffer (0.5% Triton® X-100 in 1 X PBS): Dissolve 0.5 g Triton® X-100 in 1 X PBS to a final volume of 100 mL. Filter sterilize (0.22 μm filter), and store at 22°C–25°C at least for 1 year;
-
c.Blocking buffer (3% BSA in 1 X PBS): Dissolve 3 g BSA in 1 X PBS to a final volume of 100 mL.Note: Prepare and filter the blocking buffer before use;
-
d.Wash buffer (0.3% Triton® X-100 in 1 X PBS): Dissolve 0.3 g Triton® X-100 in 1 X PBS to a final volume of 100 mL. Filter sterilize (0.22 μm filter), and store at 22°C–25°C at least about 1 year.
-
a.
-
6.Prepare Click-iT reaction stock buffers
-
a.Click-iT® HPG reaction buffer additive (Component E): Add 2 mL of sterilized deionized water to the vial and dissolve it thoroughly to make the 10 X stock solution of the Click-iT® HPG reaction buffer additive (Component E);CRITICAL: After use, store the remaining stock solution at −20°C, and this stock solution is stable for up to 1 year.
-
b.Click-iT® HPG reagent (Component C): Add 36 mL of sterilized deionized water to the Click-iT® HPG reagent (Component C) bottle and dissolve it thoroughly to make the 10 X Click-iT® HPG reaction buffer.
-
a.
Note: To make 1 X Click-iT® HPG reaction buffer, dilute the Component C bottle at 1:10 with sterilized deionized water.
-
7.Prepare pre-commissioning of pump
-
a.Prime the tubing with warm perfusion buffer from a 50 mL sterile syringe (pump speed 0.2 mL/s);Note: Avoid bubbles in the tubing.
-
b.When the irrigation finishes, prime the tubing with the pre-warmed digestion buffer from a 50 mL sterile syringe (pump speed 0.2 mL/s).
-
a.
Note: Remain the perfusion buffer in the intravenous needles tube before changing the 50 mL sterile syringe containing the digestion buffer.
Note: Avoid bubbles in the tubing.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Cycloheximide (CHX) | MCE | Cat#HY-12320 |
| Collagenase | Sigma | Cat#C-6885 |
| DMEM medium | Wisent Inc | Cat#319-005-CL |
| Fetal Bovine Serum (FBS) | Royacel | Cat#RY-F22-05 |
| 100 X penicillin-streptomycin | Wisent Inc | Cat#450-201-EL |
| PBS | Wisent Inc | Cat#311-010-CL |
| L-Glutamine | Sangon Biotech | Cat#E607004-0100 |
| L-Cystine | Sangon Biotech | Cat#A610088-0100 |
| DMEM L-methionine-free medium | Gibco | Cat#21013024 |
| BSA | Sangon Biotech | Cat#A602440-0050 |
| Triton X-100 | Sangon Biotech | Cat#A110694-0500 |
| NaCl | Sangon Biotech | Cat#A100241-0500 |
| NaHCO3 | Sangon Biotech | Cat#A610482-0500 |
| Glucose | Sangon Biotech | Cat#A501991-0500 |
| 1 M HEPES | Sangon Biotech | Cat#E607018-0100 |
| KCl | Sangon Biotech | Cat#A100395-0100 |
| MgSO4·7H2O | Sangon Biotech | Cat#A500864-0500 |
| KH2PO4 | Sangon Biotech | Cat#A100781-0100 |
| EGTA | Sangon Biotech | Cat#A600077-0025 |
| CaCl2 | Sangon Biotech | Cat#A100556-0250 |
| Antifade Mounting Medium with DAPI | Vector Laboratories | Cat#H-1500 |
| DAPI staining solution | Sangon Biotech | Cat#E607303 |
| Trypan blue solution | Sigma | Cat#T8154 |
| Paraformaldehyde (PFA) | Aladdin | Cat#C104188 |
| Chloral hydrate | Sangon Biotech | Cat# A600288 |
| Critical commercial assays | ||
| Click-iT® HPG Alexa Fluor® Protein Synthesis Assay Kits | Invitrogen | Cat#C10429 |
| Experimental models: cell lines | ||
| Primary hepatocytes | This paper | N/A |
| Experimental models: organisms/strains | ||
| C57BL/6J | Jackson Laboratory | Cat#JAX:000664, RRID:IMSR_JAX:000664 |
| Software and algorithms | ||
| Adobe Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| Zeiss LSM Image Browser | Zeiss | https://www.zeiss.com/microscopy/int/downloads/lsm-5-series.html |
| ImageJ software | ImageJ | https://imagej.nih.gov/ij/plugins/colocalization.html |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| Sterile syringe | Shengguang | Cat#20172150535 |
| Intravenous needles for single use (0.55 × 20 mm) | KDL | Cat#YZB/0692-2014 |
| Surgical scissor | JZ Surgical Instruments | Cat#JC2101 |
| Tweezer | JZ Surgical Instruments | Cat#J3C030 |
| Sterile filter membrane (0.22 μm) | Sangon Biotech | Cat#F513163-0001 |
| Poly-lysine coated coverslip | LYB | Cat#LY20-pll |
| Cell strainer | Sangon Biotech | Cat#F513441-0001 |
| 6-Well plates | Corning | Cat#3516 |
| High precision injection pump | CNYOHO | YH42BYGH60 401A |
| Centrifuge | Cence | L420 |
| Confocal microscope | Zeiss | LSM 700 |
| Stereomicroscope | COIC | 0800510 |
| CO2 incubator | ESCO | CCL-170B-8 |
| Clean bench | Airtech | SW-CJ-1FD |
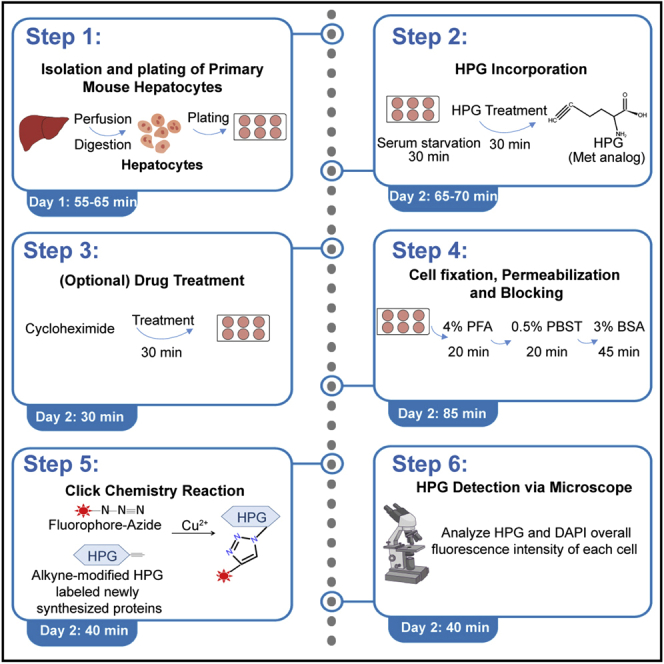
Step-by-step method details
Isolation and plating of primary mouse hepatocytes: Day 1
Note: Primary hepatocytes is an important physiological model of the liver in vitro, which has been widely used in the biomedical field (Bantubungi et al., 2014; Charni-Natan and Goldstein, 2020; Salem et al., 2018). The detailed preparation is as follows.
Mouse anesthesia and dissection: Day 1
-
1.
Anesthesia: Inject pre-warmed 7% chloral hydrate at 22°C–25°C to anesthetize 8–10-week old male C57BL/6J mice via intraperitoneal injection (150 μL/20 g body weight);
Note: Make sure the mouse is completely anesthetized. Observation indexes: When the mouse was lying on its back, the heartbeat and respiration are even, the muscles are relaxed, the limbs have no movements, the whiskers have no touching reaction, and the pedal reflex disappears. The mouse was considered to have achieved complete anesthesia.
-
2.
Positioning: Place the anesthetized mice on the edge of the dissecting tray with their heads protruding and their limbs fixed with needles;
-
3.
Dissection: Use surgical scissors to make a U-shaped incision to open the abdominal cavity of the mouse, exposing the intestine and liver, and then peel the intestine and liver to expose the inferior vena cava and portal vein (Figure 1).
Note: Make sure all the pieces of equipment are sterilized by 75% ethanol before use.
Figure 1.

Portal vein and vena cava exposure
Move the small intestine and other organs in the mouse's abdominal area to the right, and expose both portal vein and vena cava. Scale bar, 1 cm.
Inferior vena cava cannulation perfusion and liver digestion: Day 1
-
4.
Liver perfusion and digestion: Cut the portal vein, put a bent needle (0.55 × 20 mm) in inferior vena cava, inject pre-warmed 50 mL perfusion buffer and digestion buffer respectively at a constant speed of 0.2 mL/s via high precision injection pump (Figure 2). See Troubleshooting 1 and Troubleshooting 2. After the injection, use forceps to press the abdomen of the liver and observe: whether the press marks recover quickly, whether the liver becomes pale. When the liver appears in yellow and white, then the perfusion is successful;
Note: Mice should be 8–10-week old because veins in younger mice are too small, and veins in older mice have too much fat, making insertion more challenging.
-
5.
Liver cleaning and hepatocytes release: Collect mouse livers and remove the gallbladder, wash them with 10 mL pre-cooled PBS containing 1 × penicillin-streptomycin, and then transferred to 10 mL digestion buffer in Petri dishes at 37°C in a CO2 incubator about 5 min for digesting thoroughly. Then tear up the liver tissue with tweezers to obtain hepatocytes (Figure 3);
-
6.
Hepatocytes filtration and neutralization: Filter hepatocytes into a 50 mL centrifuge tube with 100 μm cell strainer in a cell culture hood, and the growth medium was added in the same volume for neutralization to stop the digestion of collagenase.
Note: Select the appropriate cell strainer according to the cell diameter.
Figure 2.

Pumping the perfusion or digestion buffer into the mouse liver
The needle's tip (bending the needle, almost at a 90-degree angle) is inserted horizontally into the vena cava.
Figure 3.

Hepatocytes release
(A) After digestion, pump the buffer into mouse liver, and transfer liver to a 10-cm dish.
(B) Thin-pointed forceps are used to tear the liver at several points along the liver's surface and gently release hepatocytes. Scale bar, 1 cm.
Hepatocytes collecting and plating: Day 1
-
7.
Hepatocytes collection: Centrifugate the filtered hepatocytes at 500 × g for 2 min, then discard the supernatants and then wash the cell pellets with 4 mL growth medium;
Note: wash cell pellets about three times with 4 mL of the growth medium.
-
8.
Check the viability of hepatocytes: Stain the hepatocytes by combining 50 μL of cell sample with 50 μL of a 0.2% trypan blue staining solution (for a final concentration of 0.1% trypan blue). Gently mix by pipetting up and down ten times, and count both dead (dark blue) and live cells using a hemocytometer on a bright-field microscope to calculate the survival rate. The resulting viability was about 85% (Figure 4);
Note: When a mouse liver is digested thoroughly, about 2.7 × 107 hepatocytes can be recovered.
-
9.
Hepatocytes Plating: Count hepatocytes and plate the cell on poly-lysine-coated cell culture plates/wells. The desired plating density is about 7 × 104 per well in a 12-well plate for the following immunofluorescence staining, which gives about 60%–70% confluence on the next day. Change the growth medium after 6 h, and culture hepatocytes for another 18 h before HPG incorporation (Figure 5).
Note: Hepatocytes should be plated evenly across the plate/well for the subsequent HPG incorporation and immunofluorescence staining. See Troubleshooting 4
Note: After 18 h of plating, incubate the plated hepatocytes with HPG. See Troubleshooting 5.
Figure 4.

Trypan blue viability assay for the hepatocytes
(A) Images were taken using a 5 × objective. Scale bar, 200 μm.
(B) Images were taken using a 10 × objective. Scale bar, 100 μm.
Figure 5.

Primary culture of hepatocytes
After 12 h of plating, the hepatocytes have adhered to the surface and acquire a spherical shape. After 18 h of plating, the plating hepatocytes acquire a typical hexagonal shape. Scale bar, 100 μm.
HPG incorporation: Day 2
-
10.
Serum starvation: Wash plated primary hepatocytes with warm PBS (37°C) twice, add 500 μL of DMEM medium without serum and incubate for 30 min for serum starvation ;
-
11.HPG incorporation.Note: HPG (L-homopropargylglycine) is a synthetic chemical molecule, which is a methionine (Met) analog and can substitute for Met in protein synthesis, and this protein-containing HPG can perform normal physiological functions. HPG contains an alkyne moiety, which can undergo a classic click chemical reaction with azide with Alexa Fluor. The newly synthesized proteins containing HPG can be labeled with fluorescence through a two-step linking reaction so that the newly synthesized proteins can be traced and located by immunofluorescence staining (Narita et al., 2011) (Figures 6A and 6B).
-
a.Prepare the working solution of Click-iT® HPG Component A: dilute the stock solution(50 mM) with a certain proportion in 10 mL of pre-warmed modified L-methionine-free DMEM medium to make a final working solution (50 μM);Note: Before use, briefly centrifuge Click-iT® HPG reagent Component A to maximize reagent recovery. The HPG-containing modified L-methionine-free DMEM medium should be pre-warmed.
-
b.Remove the DMEM medium and add 500 μL/well of medium with 50 μM Click-iT® HPG Component A working solution;
-
c.Incubate HPG for different times in a CO2 incubator at 37°C.
-
a.
Note: The HPG concentration and incubation time should be optimized for different cell types. See Troubleshooting 6.
Note: Contamination of another cell type was seen from bright-field images (Figure 6C).
Figure 6.

HPG labeling for newly synthesized proteins
(A) Chemical structure formula of Met and HPG.
(B) Diagram of click chemistry reaction.
(C) All hepatocytes can be stained with either HPG or DAPI dyes. The white arrows represent other cell types. Scale bar, 50 μm.
However, hepatocytes were easily distinguished. After 18 h of incubation, plated hepatocytes acquire a typical hexagonal shape, and most hepatocytes contain two nuclei, which are the typical features of hepatocytes.
(Optional) Drug treatment: Day 2
Note: Cycloheximide (CHX) is a protein synthesis inhibitor that inhibits peptide chain transfer by acting on the ribosome of the 80S, thereby inhibiting protein synthesis (Schneider-Poetsch et al., 2010).CHX can be used to identify whether HPG can label newly synthesized proteins.
-
12.
For CHX treatment, firstly, incubate hepatocytes with 500 μL/well DMEM medium containing 10 μg/mL CHX for 30 min during serum starvation and then remove CHX-containing DMEM medium. Secondry, add 500 μL/well modified L-methionine-free DMEM medium containing 50 μM Click-iT® HPG Component A working solution and 10 μg/mL CHX for 30 min (Figures 7A and 7B)
Note: Hepatocytes treated with 10 μg/mL CHX could still detect a weak fluorescent signal. The CHX concentration and incubation time should be optimized for different cell types.
Figure 7.

Identification of HPG labeled newly synthesized proteins
(A) Schematic diagram of CHX treatment procedure.
(B) Immunofluorescence images show the HPG incorporation in hepatocytes with or without CHX treatment. Scale bar, 50 μm.
Cell fixation, permeabilization and blocking: Day 2
-
13.
After HPG incubation, remove the HPG-containing modified L-methionine-free DMEM medium and wash cells once with 1 mL/well pre-warmed PBS, then remove PBS;
-
14.
Add 500 μL/well pre-warmed fixation buffer. Incubate for 20 min at 22°C–25°C, then remove the fixative;
Note: In order to maintain cell morphology, 4% PFA must be pre-warmed before cell fixation.
-
15.
After cell fixation, wash cells twice with 1 mL/well 1 × PBS and then add 500 μL/well of permeabilization buffer and incubate for 20 min at 22°C–25°C;
-
16.
Add 500 μL/well blocking buffer and incubate for 45 min at 22°C–25°C for blocking;
-
17.
Wash cells with 1 mL/well wash buffer for 2 times, 5 min/ time to remove non-incorporated HPG followed by Click-iT® HPG detection.
Click chemistry reaction: Day 2
Note: In our protocol, all materials needed in Click chemistry reaction were ordered from Thermo Fisher (Cat#C10429).
-
18.
Prepare 1 X Click-iT® HPG reaction buffer additive (Table 1): dilute the 10 X stock solution with deionized water ;
Note: The 1 X Click-iT® HPG reaction buffer additive should be prepared freshly and use the solution on the same day.
-
19.
Prepare Click-iT® reaction cocktail according to Table 1;
Note: The Click-iT® reaction cocktail must be used within 15 min.
-
20.
Add 500 μL/well Click-iT® reaction cocktail (Table 1) to each well and mix well;
-
21.
Incubate for 30 min at 22°C–25°C;
-
22.
Remove the Click-iT® reaction cocktail and wash with 500 μL/well of Click-iT® reaction rinse buffer (Component F) then remove the Click-iT® reaction rinse buffer;
Note: Make sure to wash at least 3 times and remove the non-incorporated HPG thoroughly. See Troubleshooting 7.
-
23.
Dilute DAPI stock solution at 1:5000 in PBS to obtain DAPI working solution, then add 500 μL/well of DAPI working solution for DNA staining. Protected from light and incubate for 10 min at 22°C–25°C;
-
24.
Remove the DAPI working solution, wash twice with 1 mL/well PBS, followed by mounting the coverslips for imaging.
Table 1.
Click-iT® reaction cocktail
| Reagent | Amount |
|---|---|
| 1×Click-iT® HPG reaction buffer | 860 μL |
| Copper (II) Sulfate (CuSO4) (Component D) | 40 μL |
| Alexa Fluor® azide (Component B) | 2.5 μL |
| 1×Click-iT® HPG buffer additive | 100 μL |
| Total | 1 mL |
HPG detection via microscope: Day 2
-
25.
Mount the coverslips on the glass slide with antifade mounting medium (20 μL/plate ), wait until the mounting medium fixed before taking images;
-
26.Obtain images with Zeiss LSM 700 confocal microscope;
-
a.Instrument settings: filter blocks used are SP 490 in channel 1 to detect the HPG signal, LP 560 in channel 2 to detect DAPI;
-
b.Adjust the laser energy, pinhole, gain, and exposure time to get the best signal/noise ratio, fix these settings throughout the acquisition;
-
c.Image acquisition: select the appropriate image resolution (at least 1024 X 1024), scan the sample frame by frame completely, then save and export the images in LSM format.
-
a.
Note: Ensure all sample surfaces were clear to avoid contaminating the lens, and the photomultiplier tube (PMT) detector can obtain sufficient signals. When acquiring images, keep laser exposure time as short as possible to avoid fluorescence quenching.
-
27.
Analyze the obtained images by Image J software: the overall fluorescence intensities of HPG and DAPI from each cell in the control or experimental groups were measured by Image J software. The average HPG: DAPI ratios from the control and experimental groups were calculated and presented as a bar graph using GraphPad software, indicating the levels of newly synthesized proteins.
Note: The location of newly synthesized proteins can be monitored using immunofluorescence as well. Proteins can be pulse-labeled with HPG and chased to monitor the degradation rate.
Expected outcomes
This protocol describes a method based on immunofluorescence staining to accurately trace and localize the newly synthesized proteins in isolated primary mouse hepatocytes. Our results show that HPG is incorporated into synthesizing proteins, and the inhibition of protein synthesis by CHX treatment decreases the Click-iT® HPG signal in plated hepatocytes (Figure 7B). This method may be applied to detect the changes in protein synthesis and degradation.
Limitations
This protocol was used to detect newly synthesized proteins via immunofluorescence, and it requires the cells maintained in good condition. When plated cells are too densely or too sparsely, downstream experiments may be compromised. It cannot separate the newly synthesized proteins for mass spectrometry to identify the protein sequences. Besides, the HPG incorporation protocol does not apply in vivo so far.
Troubleshooting
Problem 1
Bubbles appear in the tubing before pumping the digestion buffer into the mouse liver (inferior vena cava cannulation perfusion and liver digestion, step 4).
Potential solution
Gently pull the catheter back from the syringes without removing the needle completely, and leave little perfusion buffer in the catheter before the next syringes containing digestion buffer.
Problem 2
When infusing mouse liver, the blood clots (preparation, step 7 and inferior vena cava cannulation perfusion and liver digestion, step 4).
Potential solution
Increase the pump speed to avoid blood clotting.
Problem 3
After digestion, the mouse liver is not swollen, and no cells are released in the dish (preparation, step 3 and inferior vena cava cannulation perfusion and liver digestion, step 4).
Potential solution
Digestion buffer containing collagenase must be fully dissolved and filtered before anesthetizing mice, and the digestion buffer should be placed in a 37°C water bath at least 30 min before perfusion to obtain the best digestion efficiency of collagenase.
Problem 4
Plated cells are too densely or too sparsely, the HPG incorporation or immunofluorescence staining may be compromised. (hepatocytes collecting and plating, step 9).
Potential solution
The desired plating density is recommended about 7 × 104 per well in a 12-well plate for the following immunofluorescence staining, which gives about 60–70% confluence on the next day. When plated cells are too densely or too sparsely, HPG incorporation and immunofluorescence staining may be compromised.
Problem 5
The morphology of hepatocytes is not in typical hexagonal shape during the HPG incorporation stage (hepatocytes collecting and plating, step 9).
Potential solution
Increase the incubation time. After 12 h of incubation, the hepatocytes have adhered to the surface and acquire a spherical shape. After 18 h of incubation, the plated hepatocytes acquire a typical hexagonal shape. So we can proceed with the HPG incorporation after 18 h.
Problem 6
The HPG signal is too weak after incorporation (HPG incorporation, steps 10 and 11).
Potential solution 1
Fetal bovine serum and methionine affect the incorporation of HPG (Figure 8), so to achieve high incorporation efficiency of HPG, the cells should be starved without serum and maintained in a methionine-free medium before the incorporation of HPG treatment.
Figure 8.

HPG incorporation in hepatocytes under different treatment conditions
(A)Schematic diagram of the experiment process.
(B) IF images shows under different treatment conditions, the HPG incorporation in hepatocytes. Scale bar, 50 μm
Potential solution 2
According to the cell type, optimize the HPG concentration and the incubation time (Figure 9).
Figure 9.

HPG incorporation in hepatocytes with various HPG concentrations and incubation times
(A) Hepatocytes were incubated with Group I, 0 μM HPG for 10 min, 30 min and 60 min; Group II, 25 μM HPG for 10 min, 30 min and 60 min; Group III, 50 μM HPG for 10 min, 30 min and 60 min; Group IV, 100 μM HPG for 10 min, 30 min and 60 min. Scale bar, 50 μm.
(B) Quantification for A. Relative HPG incorporation is presented as the HPG: DAPI ratio ± SD, which were normalized to HPG(100 μM, 60 min) treatment (100%) (n ≥ 25).
Problem 7
Strong background in HPG immunostaining (click chemistry reaction, step 22).
Potential solution
Increase the wash times and make sure that the non-incorporated HPG is thoroughly washed out.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zi Chao Zhang; zhangzc@seu.edu.cn
Materials availability
We did not generate any new materials.
Data and code availability
We did not generate any dataset or code.
Acknowledgments
We thank all members of the Han Laboratory for their critical comments on the manuscript. This work was supported by the National Natural Science Foundation of China (81730034; 31671045 and 8190128), the Natural Science Foundation of Jiangsu Province (BK20170080), and Guangdong Key Project (2018B030335001).
Author contributions
Y.S. exerted the protocol, wrote the detailed procedure, and interpreted data; W.L. isolated the hepatocytes, obtained IF images with a microscope, and discussed data, J.Z. performed other images and discussed data, and Z.C.Z. and J.H. conceived this project and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Junhai Han, Email: junhaihan@seu.edu.cn.
Zi Chao Zhang, Email: zhangzc@seu.edu.cn.
References
- Bantubungi K., Hannou S.A., Caron-Houde S., Vallez E., Baron M., Lucas A., Bouchaert E., Paumelle R., Tailleux A., Staels B. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1alpha pathway. Diabetes. 2014;63:3199–3209. doi: 10.2337/db13-1921. [DOI] [PubMed] [Google Scholar]
- Charni-Natan M., Goldstein I. Protocol for primary mouse hepatocyte isolation. STAR Protoc. 2020;1:100086. doi: 10.1016/j.xpro.2020.100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M., Young A.R., Arakawa S., Samarajiwa S.A., Nakashima T., Yoshida S., Hong S., Berry L.S., Reichelt S., Ferreira M. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–970. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem E.S.B., Murakami K., Takahashi T., Bernhard E., Borra V., Bethi M., Nakamura T. Isolation of primary mouse hepatocytes for nascent protein synthesis analysis by non-radioactive l-azidohomoalanine labeling method. J. Vis. Exp. 2018:58323. doi: 10.3791/58323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T., Ju J., Eyler D.E., Dang Y., Bhat S., Merrick W.C., Green R., Shen B., Liu J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nature chemical biology. 2010;6:209–217. doi: 10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Zhang Z.C., Cheng S., Liu A., Zuo J., Xia S., Liu X., Liu W., Jia Z., Xie W. PQBP1 promotes translational elongation and regulates hippocampal mGluR-LTD by suppressing eEF2 phosphorylation. Mol. Cell. 2021;81:1425–1438.e10. doi: 10.1016/j.molcel.2021.01.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We did not generate any dataset or code.