

Abstract
Compared with the traditional forms of cell death—apoptosis, necrosis and autophagy, ferroptosis is a novel form of iron-dependent programmed cell death forms which is different from the above traditional forms of cell death. Brent R Stockwell, a Professor of Columbia University, firstly proposed that this from of cell death was named ferroptosis in 2012. The main characteristics of ferroptosis is increasing iron loading and driving a lot of lipid peroxide generated and ultimately lead to cell death. In this paper, the mechanism of ferroptosis, relationship between ferroptosis and common diseases and immune state of body are reviewed, and the inhibitors and inducers related to ferroptosis that have been found are summarized to provide medicine exploration targeted of ferroptosis and reference for the research in the future.
Keywords: Ferroptosis, Common clinical diseases, Tumor, Immune cells, Targeted therapy
Introduction
Cell death is definitely concern about mammalian growth, homeostasis regulation and development of various diseases. As is known to all, cell autophagy, apoptosis and necrosis are the traditional form of regulating cell death, in addition, ferroptosis is a fresh mode of iron-dependent death induced by small molecules such as Erastin and RSL3 proposed by Dixon in 2012, whose morphology, mechanism, induction and inhibition is different from traditional form of cell death [1]. The morphology of ferroptosis mainly shows that smaller mitochondria appear in the fine structure of the cell, and the mitochondrial membrane is shrunk, the mitochondrial cristae is reduced or disappeared, and the outer membrane is broken, but the morphological changes in the nucleus are not obvious. In the mechanism of occurrence, it does not depend on the Caspase9 family, but is closely related to lipid metabolism, amino acid metabolism, iron metabolism and gene regulation in vivo. Due to ferroptosis in recent years has been more and more gotten the attention of people, through deep researches in mechanism of ferroptosis, it was found ferroptosis and a variety of human diseases have inseparable relationship, including cardiac ischemic disease, kidney disease, liver damage, and degenerative disease, for example, ferroptosis of tumor cells can be induced to achieve the purpose of tumor treatment, and ferroptosis is also closely related to immunotherapy. Therefore, more and more drugs with ferroptosis as the therapeutic target can be studied. The compounds Erastin discovered by Dolma et al. [2] as early as 2003 are equivalent to the new compounds RSL3 and RSL5 that have similar functions to Erastin found in 2008 by Yang et al. [3], Yang et al. [3] also found that ferroptosis chelating agents, DFOM and vitamin E could inhibit this non-apoptotic cell death mode-ferroptosis. In this paper, mechanism of ferroptosis and its relationship with diseases and human immune state are described in detail, and the inhibitors and activators targeting of ferroptosis that have been found so far are summarized, hoping to provide a new perspective for the study of ferroptosis and explore other target medicine as a potential therapeutic target.
Mechanism of ferroptosis
Ferroptosis, metabolic cell death form mediated by amino acid metabolism, iron metabolism, and lipid metabolism, is complicated, and its complete mechanics are described in detail below and illustrated in Fig. 1.
Fig. 1.

Mechanism of ferroptosis
Amino acid metabolism
Glutathione (GSH)—Glutamate (GSH) is compounding from cysteine and glycine under the catalyze of GCLC enzyme and GSS enzyme, Glutathione peroxidase 4 (GPX4) oxidizes hydroperoxide and two molecules of reducing glutathione (GSH) into lipid alcohols and oxidizing glutathione (GSSG), acting as an antioxidant and free radical scavenger which can inhibit ferroptosis. Intracellular cysteine, a key substrate of synthesizing of GSH, can be obtained by de novo synthesis or by recovery through protein degradation, most of the cancer cells using cystine transporters system Xc − to get cystine from extracellular environment, and cysteine, which can be used in the synthesis of glutathione (and other biological molecules), is then produced through NADPH reduction reaction in its cytoplasm. Therefore, Glutamate (GSH) is a key target for controlling ferroptosis, and its synthesis processes (cysteine, GCLC enzyme and GSS enzyme), uptake processes (transporters system Xc -), and transport processes (GPX4) can regulate ferroptosis.
Sulfur REDOX protein (TXN)—In 1994, it was proposed that TXN reductase, the second most important substance in the mercaptan dependent antioxidant system [4], can catalyze the reduction of cystine to cysteine so as to maintain the activity of GPX4. SLC7A11 and TXN enzyme systems can synergistically supplement GSH. The TXN enzyme system of malignant tumor cell lines is usually very active, and inhibition of GSH and TXN pathways can effectively trigger tumor cell death. Therefore, TXN is also one of the key targets for regulating ferroptosis.
Glutamine metabolism—Glutamine is converted to glutamate in response to GSL1 and GSL2, if extracellular glutamate levels are high, the system Xc − is unable to transport intracellular glutamate out of the cell and unable to transport extracellular cysteine into the cell, which will inhibit glutathione synthesis and finally induce ferroptosis, so inhibiting glutamine metabolism can inhibit ferroptosis [5].
Glutathione peroxidase 4 (GPX4)—GPX4 is a selenoprotein [6]. As we all known, decoding selenocysteine requires special tRNA—selenocysteine tRNA (sec-tRNA), and synthesis of the isoprene group of sec-tRNA requires the intermediate isoprene pyrophosphate (IPP) from the MVA pathway, because IPP is one of the most significant products of the MVA approach [7]; hence, inhibitors of the MVA pathway can induce ferroptosis by inhibiting the synthesis of GPX4. Zhang et al. [8] found that GPX4 was largely expressed in tumor in comparison with normal tissues, and high expression of GPX4 was not conducive to the prognosis of patients.
System xc−—System xc− is made up of subunit SLC7A11 and subunit SLC3A2 [9], it transfers glutamate extracellular and cystine intracellular for GSH synthesis. Ferroptosis occurs when System XC − is inhibited, for example, Erastin induced ferroptosis through inhibiting the physical activity of System XC − [10]. A variety of regulatory factors can regulate ferroptosis by regulating SLC7A11, NFE2L2 [11] positively regulates SLC7A11, and tumor suppressor genes such as TP53 [12], BAP1 [13] and BECN1 [14] negatively regulate SLC7A11. Another alternative pathway for synthesize cysteine, the trans-sulfur pathway, synthesis cysteine by methionine, also can inhibits ferroptosis.
Other—Other intracellular anti-ferroptosis systems include coenzyme Q10 (CoQ10) produced by AIFM2, tetrahydrobiopterin (BH4) produced by GCH1 and the membrane repair system of EscRTIII, which play a critical part in anti-ferroptosis.
Briefly, SLC7A11-GSH-GPX4 pathway is identified as the most upstream process of regulating ferroptosis. The mechanism of amino acid metabolism regulating ferroptosis is mainly that cystine absorbed by System XC − is involved in the synthesis of glutathione (GSH), and GSH is reduced to GSSG under the action of GPX4 thereby restrain ferroptosis. The current targeted reagents of ferroptosis are mainly through target SLC7A11- GSH- GPX4 pathway, in addition, by adjusting the armor hydroxyl pentanoic acid pathway, sulfur transfer way, sulfur TXN REDOX proteins and glutamine metabolism to some extent, can also play the role of regulating ferroptosis.
Iron metabolism
As an indispensable trace element, Iron can adjust the occurrence of ferroptosis. Outside the cell, transferrin (TF) transport two Fe3 + to transferrin receptor 1 (TFR1) and form TF—(Fe3 +) 2—TFR1 compounds [15] into lysosomes in the cell, Basuli et al. [16] has shown that this process is significant increase in ovarian cancer, breast cancer and other cancers. After degradation of Fe3 + into Fe2 + under the action of iron reductase (STEAP3), Fe2 + is stored in Ferritin. As the most important protein in cells, Ferritin can maintain intracellular iron homeostasis and reduce oxidative stress caused by Fenton reaction, so as to achieve the purpose of protecting cells [3, 17, 18]. Xuexian Fang et al. [19] clarified the physiological effect of Ferritin inhibiting cardiac ferroptosis under iron overload. Fe2 +, which is stored in the ferritin, can also be mediated iron autophagy auxiliary by nuclear receptors activated factor 4 (NCOA4) and released into the cytoplasm [20], Gao et al. [21] knockout fibrosarcoma cells, pancreatic cells and other cells NCOA4 genes, the ferroptosis were significantly inhibited, and excess of Fe2 + will participate in fenton reaction to generate hydroxyl free radicals to induce ferroptosis, or induces ferroptosis by activating an enzyme that contains iron (e.g., lipoxygenase) [22, 23].
In addition, iron metabolism in the body is precisely regulated by hepcidin. Ferroportin1 (FPN1, SLC40A1), an iron channel, can transport iron from iron storage cells to extracellular and circulatory systems to maintain iron homeostasis in cells [24, 25]. Hepcidin promotes the endocytic degradation of FPN1 by binding to the membrane surface, thereby inhibiting iron uptake by intestinal epithelium and iron reuse by macrophages in aging RBC [26]. Hepcidin, as a hormone secreted by the liver, is regulated by multiple pathways: serum iron and intracellular iron of the liver upregulate Hepcidin through the BMP6/ Smad4 pathway [27]. Oxidative stress, hematopoiesis and energy metabolism downregulated Hepcidin through their respective pathways, respectively. In the process of pathogen infection and inflammatory response, many inflammatory factors upregulate the expression of Hepcidin through the JAK2/STAT3 pathway [28]. Excess hepcidin leads to iron retention in macrophages and decrease of free iron content in the circulatory system, which is one of the important mechanisms of the body against pathogen infection.
In a word, the mechanism of iron metabolism regulating ferroptosis is mainly that the TF-(Fe3 +)2-TFR1 complex is formed under the action of transferrin—transferrin receptor and enters into the cell lysosome, under the action of iron reductase (STEAP3), it is reduced to Fe2 + and stored in Ferritin. Excess Fe2 + oxidize lipids through Fenton reaction to produce ROS and lipid peroxides ultimately bring about ferroptosis. Therefore, the expression of iron overload, transferrin, transferrin receptor, ferritin and nuclear receptor coactivator4 ( NCOA4) can all be used as targets for regulating ferroptosis, ALOX or eGln proline hydroxylase (also known as PHD) are taking effect for the activity of lipid peroxidation and iron homeostasis, expression of hepcidin and FPN1 can also act as targets to regulate ferroptosis. Therefore, iron metabolism is one of the important pathways to inhibit or induce ferroptosis.
Lipid metabolism (oxidative stress)
What are the products of lipid peroxidation mainly included? There is no doubt that lipid hydroperoxides(LOOHS), malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are the most common products. Lipid peroxidation directly damages cell membrane and organelle membrane phospholipids, excessive reactive oxygen free radicals cause adverse damage to cell components such as nucleic acids; therefore, lipid peroxides accumulation and excessive active free radicals are the premise of ferroptosis [29].
The membrane and organelle membrane of living organisms are rich in PUFAs, which are substrates of lipoxygenase (LOXs), only in the way that they occur oxidation reaction and esterification reaction and turn into membrane phospholipids, will they become the signal of ferroptosis [30]. Under the action of ACSL4 and LPCAT3, free PUFAs react with phosphatidylethanolamine (PES) to synthesize PUFAS-PES [30, 31]. Because PUFAS-PES contains easily extracted diallyl hydrogen atoms, it is prone to lipid peroxidation.
Under the action of ACSL4, arachidonic acid (AA) and adrenal acid (ADA) bind with coenzyme A, respectively, generate to AA-CoA and ADA-CoA, which are their acyl coenzyme A esters, then bind to membrane phospholipids [30–33], it promotes the esterification of phosphatidylethanolamine to form AA-PE or ADA-PE after the action of LPCAT3 and finally resulting in ferroptosis [30, 31]. In addition, arachidonic acid (AA) generates prostaglandin (PEG2) and thromboxin under the action of cyctoxidase (COXS), and arachidonic acid (AA) generates leukotriene and hydroxy-eicosatetraenoicacid (Hetes) under the action of lipoxygenase (LOX), Arachidonic acid (AA) generates Hetes under the action of cytochrome P450 family enzymes. Both PEG2 and Hetes are different types of arachidonic acid. NADPH oxidase (NOXS), cytochrome P450 oxidoreductase (POR), and mitochondrial respiratory chain can produce ROS and also participate in lipid oxidation of iron death [34]. In addition, NADPH oxidase (NOXS), cytochrome P450 oxidoreductase (POR), and mitochondrial respiratory chain can produce ROS and also participate in lipid oxidation of ferroptosis [1, 35, 36].
In conclusion, the ACSL4-LPCAT3-ALOXS axis can promote lipid oxidation to form excessive lipid hydroperoxides and ROS, inducing cell membrane rupture and cell ferroptosis. In this metabolic process, GPX4, AA, ADA, PUFA, ACSL4, LPCAT3, ALOXS, and COXs all play a very important role and can be used as targets of ferroptosis to inhibit or induce ferroptosis. Therefore, lipid metabolism is also one of the important mechanisms for regulating ferroptosis.
Broadly speaking, deeply researching into the mechanism of ferroptosis has led to the discovery that the causing of resistance to previous treatments just because of the failure to link ferroptosis with treatment of many human diseases. Therefore, it is of great significance to link ferroptosis with disease by targeting the mechanism of ferroptosis. The following sections will elaborate on some of the diseases that have been found so far and the association between the body’s immune status and ferroptosis.
Relationship between ferroptosis and various diseases and immune cell
The association between ferroptosis and tumor
Studies pointed out that the use of the delta-2 agonist cimatinib and the tyrosine kinase inhibitor lapatinib in breast cancer cell lines induces ferroptosis by increasing iron levels [37]. Growing evidence revealed that the use of the rheumatoid arthritis drug phenofen in human hepatoma cell lines also induced ferroptosis and was saved by the ferroptosis-specific inhibitor FER-1 [38]. In addition, activated CD8+ T cells can release IFN-γ, which will inhibit the expression of SLC7A11 in tumor cells through the JAK1-STAT1 pathway, suggesting that targeted regulation of cysteine/cysteine levels in tumor cells may be a valid way of tumor immunotherapy [39]. For these reasons, one could envisage that the relationship between ferroptosis and tumors is inextricably linked.
Ferroptosis exerts a major influence in the development and treatment of many tumors. Previous works have shown ferroptosis sensitivity is significantly correlated with tumor gene mutations, such as RAS [40], TP53 mutation [41], HIF mutation, stress response pathways [42], epithelial-mesenchymal-transformation(EMT) [43] and other signaling pathways. On the one hand, ferroptosis inducers such as Erastin can cause ferroptosis and thus inhibit progression of tumor; on the other hand, ferroptosis can induce inflammation-related immunosuppressive tumor microenvironment to promote tumor growth. It is well known that the content of iron and glutathione in tumor microenvironment is higher than that in normal cells, so tumor cells should easily trigger ferroptosis. However, how do tumor cells regulate ferroptosis-sensitive microenvironment to protect themselves from being cleared by immune cells? Certainly the extent of ferroptosis’s influence on tumor biology remains to be further studied.
Relationship between ferroptosis and MDS (Mitochondrial DNA depletion syndrome)
MDS is caused by the reduction of DNA content by reason of the mutation of the nuclear gene that synthesizes mitochondrial DNA, leading to the dysfunction of multiple tissues and organs, the affected organs usually include liver, brain, muscle, etc. As the main iron storage organ in the human body, the liver is the first to be attacked when iron overload occurs. Mitochondrial DNA deletion widely exists in aging, degenerative diseases and other genetic diseases, so it has a wide range of potential pathological and therapeutic significance [44]. At least nine genetic mutations have been identified that cause MDS [45]. Deoxyguanosine kinase (DGUOK) role is largely synthetic purine nucleotide in the mitochondria, the mutation type most often lead to bone MDS [46], patients often died of severe liver failure within a year, in addition to liver transplantation, there is no other effective treatments [47], its important clinical phenotype is liver iron deposit, serological examination showed elevated serum ferritin and transferrin [48]. Studies have shown that hepatocytes of MDS patients are more sensitive to ferroptosis caused by iron deposition, first, mitochondrial DNA deletion in hepatocytes leads to mitochondrial dysfunction, decreased mitochondrial ATP synthesis, increased reactive oxygen species and glutathione depletion, and the iron in ferritin is released into the cytoplasm, causing lipid peroxidation to increase reactive oxygen species (ROS), and finally leading to ferroptosis of liver cells. Therefore, the regulation of iron metabolism pathway can provide a blueprint for the future development of therapeutic target for MDS liver failure, which is a novel pathological mechanism and potential therapeutic strategy.
Association of ferroptosis with cardiac ischemic disease (ischemia reperfusion injury)
Xuexian Fang et al. [49] found that Ferrostatin 1 (FER-1) significantly reduced cardiotoxicity induced by doxorubicin, HMOX1 is a key regulatory gene of ferroptosis in adriamycin cardiomyopathy; therefore, unlike previous approaches of preventing and treating cardiomyocytes, ferroptosis could be a promising therapeutic target. Marcus Conrad et al. [50] showed that the characteristics of cardiomyocyte is related to ferroptosis. A variety of examples demonstrated that it is clear that ferroptosis provides promising new ideas and new strategies of heart disease prevention and treatment, such as clinical myocardial infarction.
Ferroptosis and hepatocellular injury disease
Fu D.Wang et al. [51], whose study about that iron can induce ferroptosis in liver parenchymal cells and bone marrow macrophages, is the first study to be conducted in the world, and ferroptosis inhibitor (FEV-1) can effectively alleviates liver fibrosis and other indexes of liver injury by inhibiting ferroptosis. Yingying Yu et al. [52] for the first time clarified the physiological effect and molecular mechanism of Transferrin inhibiting liver fibrosis by regulating ferroptosis, and found that the metal ion transporter SLC39A14 in liver induced ferroptosis of liver chymal cells by absorbing non-transferrin Bound Iron (NTBI), thus leading to the occurrence of liver fibrosis. Several recent studies have shown that, ferroptosis not only well elucidates the pathological mechanism of liver diseases, but also provides a research basis of new drugs targeting diseases related to abnormal iron metabolism. As an important mechanism of liver fibrosis and an important target for the control and therapy of liver fibrosis, it provides ideas for the prevention of liver injury caused by iron overload in clinical practice. Ultimately, it is demonstrated that ferroptosis, as a new target for liver fibrosis, will bring good results.
Associations between ferroptosis and degenerative diseases
Alzheimer’s disease (AD). Alzheimer’s disease (AD) is the most common neurodegenerative disease among the elderly, in which the loss of synapses and neurons leads to memory loss. Current studies have shown that there is an exact relationship between excess iron accumulation in the brain and the occurrence and development of AD and learning and memory disorders [53]. There are many pathological changes associated with ferroptosis in AD patients and animal models, such as increased liposome oxidation, downregulation of GPX4, accumulation of iron ions, and increased extracellular glutamate concentration [22, 54]. Bao Wd et al. [55] for the first time systematically elucidates the characteristics of ferroptosis in hippocampal neurons in a mouse model of AD and identifies pathologic down regulation of Ferroportin (FPN) in both AD patients and mice, it has been confirmed that FPN deficiency can induce ferroptosis and lead to learning and memory impairment, restoration of FPN expression can improve ferroptosis characteristics of neurons and learning and memory impairment. The role of ferroptosis in neurodegenerative diseases has been widely concerned.
Parkinson's disease. Clinically, patients with Parkinson present with a large increase in iron and lipid peroxides, which are also the characteristics of ferroptosis. The unsaturated fatty acid chain of phospholipid in biofilm is continuously oxidized and repaired, if the repair is not timely, the accumulation of phospholipid peroxides may lead to ferroptosis of cells. Calcium-independent phospholipase A2β (IPLA2β, corresponding gene name pNPLA9), a Sn-2 acyl binding protein that specifically hydrolyzes phospholipids, take effect in the removal of oxidized phospholipids and the remodeling of phospholipids. One of the important pathological mechanisms, the accumulation of phospholipid oxidation in dopaminergic neurons, is caused by the loss of IPLA2β. Kagan VE et al. [33] found that 15-Lox can anchor to cell membrane under the action of companion molecule PEBP1, specifically oxidize phosphatidylethanolamine (PE) to produce oxidized phosphatidylethanolamine (ox-PE), among which PE oxidation product 15-HPET-PE is an important signal molecule for ferroptosis of cells. The anti-ferroptosis function of IPLA2β is mainly through scavenging ferroptosis signaling molecule 1-SA-2–15-HPET-P. The mutation of IPLA2β makes it conformational and cannot bind to the phospholipids of the cell membrane, thus reducing the enzymatic hydrolysis and oxidative capacity of the phospholipids.
Brain trauma and ferroptosis
In TBI patients, we can found iron content changes, suggesting that disruption of iron homeostasis may be an important pathophysiological process of TBI [56]. As an important biomarker, ferritin can be used to predict the time of injury after TBI and is associated with patient survival [57]. Tongyu Rui et al. [58] illustrated the time-history of ferroptosis-related protein expression and iron deposition after traumatic brain injury, found xCT, Cox2, Tfr1, Nox2 and Fpn protein expression is at its peak in 12 h to 24 h after trauma, then declining gradually close to or slightly lower than normal, and Alexandra bohne Fth, Ftl and 4 HNE protein expression increases in 3 days after trauma obviously, slightly down from 3 to 14 d, Pruslan staining confirmed a significant increase in iron deposition 7 days after trauma. After treatment with melatonin and ferroptosis inhibitor Liproxstatin 1 respectively, the expression levels of ferroptosis and iron metabolism-related proteins can be significantly reversed. Therefore, the anti-ferroptosis effect is supposed to be a newfound target for the brain injury treatment and provide a resultful means for the clinical treatment of brain injury.
The relationship between ferroptosis and Spontaneous intracerebral hemorrhage (ICH)
After intracerebral hemorrhage, the spilled hemoglobin degrades, releasing large amounts of iron ions which accumulate in the brain tissue around the hematoma. This excess iron accumulation can lead to increased oxidative stress, inducement of impaired cell function, and ultimately neuronal death. Bao WD et al. [59] systematically explained the regulatory effect of miR-124/Ferroportin signaling pathway on iron deposition and neuronal death in intracerebral hemorrhage, and revealed the influence of this signaling pathway on perifocal neuronal apoptosis and ferroptosis pathway. The regulation of ferroptosis can provide a suitable target of this disease, and it is definitely important in the study of the signaling pathway of iron ion regulation in the brain under the pathological state.
Influence and application of ferroptosis in immune cells and inflammatory diseases
Last several years, vast research evidences have shown that influence and application of ferroptosis in immune cells becomes more and more important, a summary of relationship of them is explained more in detail below.
The role of ferroptosis in T cell-mediated immunotherapy
CD8 + T cells can improve the antitumor effect in tumor cells by inducing ferroptosis. Interferon γ (INFγ) released by CD8 + T cells may activate the JAK1-STAT1 pathway to inhibit the expression of SLC7A11, sequentially reducing the absorb of cystine by tumor cells and leading to ferroptosis of tumor cells [39], so Interferon γ (INFγ) can be used as a target for regulating ferroptosis.
In addition, activated T cells have a high demand for iron, and the lack of iron inhibits the proliferation of T cells, iron overload leads to an imbalance in the proportion of CD4 and CD8 T cells, and iron overload also increases the ROS level in T cells and causes DNA damage [60]. In terms of molecular mechanism, iron can adjust the generation of cytokines such as GM-CSF after transcription, negatively regulate DNA methylation through Tet protein, and regulate mitochondrial function of T cells through the REDOX process of cytochrome C [61, 62].
Therefore, targeting T cell iron metabolism and interferon γ metabolism are potential strategies to raise efficiency of T cell immunotherapy. The role of ferroptosis in T cell-mediated immunotherapy is shown in Fig. 2.
Fig. 2.
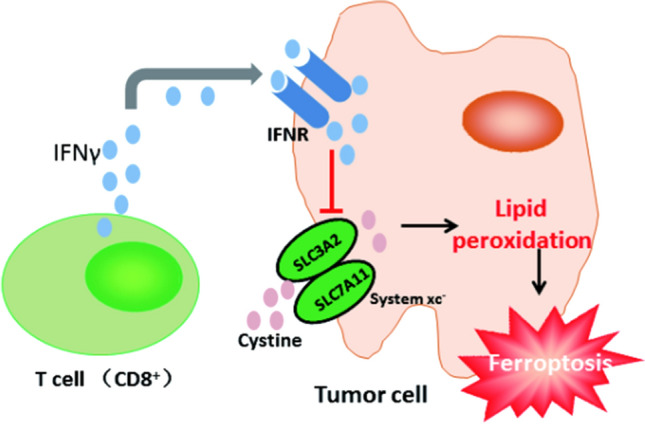
The role of ferroptosis in T cell-mediated immunotherapy
The role of ferroptosis in different B cell-mediated immunotherapies
Different B cell subsets showed different sensitivities to ferroptosis. The knockout of GPX4 gene in B1 and MZ B cells will trigger ferroptosis by inducing lipid peroxidation, finally affecting the immune response of B cells; however, deletion of GPX4 does not induce ferroptosis in follicular B cells (Fo B). The possible mechanism is that the content of fatty acid transporter CD36 in the plasma membrane of B1 and MZ B cells is significantly higher than that of Fo B cells, which leads to more acid and lipid droplets in B1 and MZ B cells, and thus more prone to lipid peroxidation [63].
Activated B cells also have a higher demand for iron. Iron supplementation can ameliorate the impaired proliferation and weakened antibody response of B cells that are deficient in iron. Studies have shown that iron ion regulates the expression of Cyclin E1 by regulating the activity of demethylase JMJC, thereby regulating the proliferation of B cells and antibody production [64].
All in all, inhibition of the fatty acid transporter CD36 on the plasma membrane of B1 and MZ B cells is an important target for regulating ferroptosis, and iron supplementation is also a feasible strategy for regulating ferroptosis. Figure 3 is a description of the role of ferroptosis in B1 and MZ B cells, and Fig. 4 is a description of the role of ferroptosis in Fo B cells.
Fig. 3.
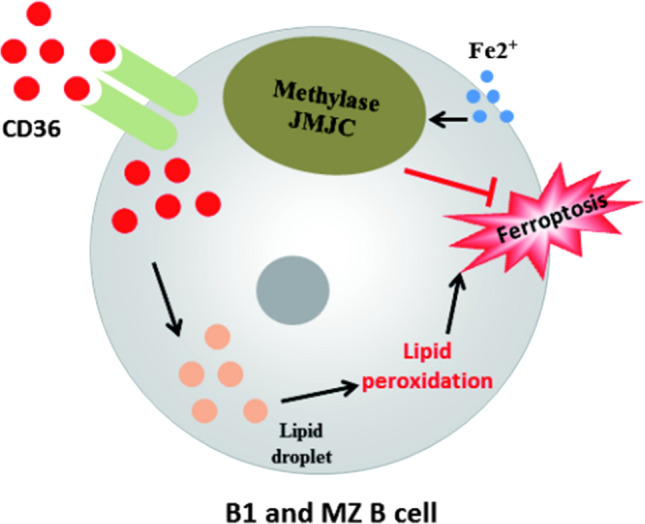
The role of ferroptosis in B1 and MZ B cells
Fig. 4.
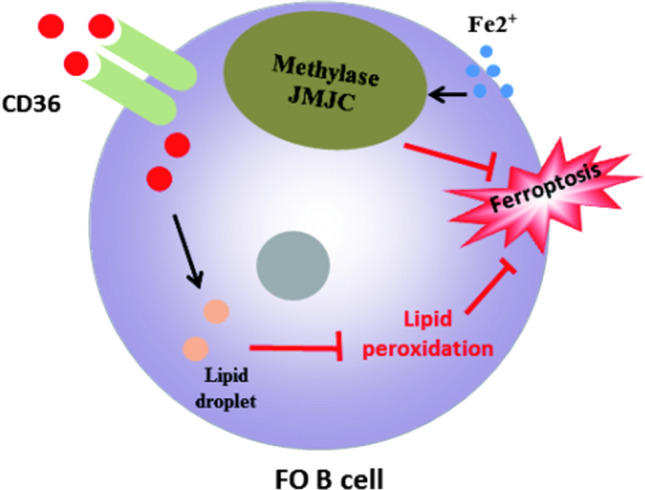
The role of ferroptosis in Fo B cells
The role of ferroptosis in different macrophage-mediated immunotherapies
D. Wang and his team demonstrated that iron citrate can induce ferroptosis in macrophages derived from bone marrow [51]. TGFβ1 secreted by macrophages regulates the expression of ferroptosis-related target genes ZEB1 [65, 66] and SLC7A11 [67] by activating SMAD signaling transcription, and SLC7A11 gene deletion promotes iron overload induced ferroptosis in macrophages. In other words, ferroptosis can be regulated by adjusting the expression of TGFβ1 in macrophages. He Rongrong’s team [68] found that macrophages can recognize the oxidized phospholipids (SAPE-OOH) on the surface of ferroptosis cells through the membrane receptor TLR2 (Toll-like receptor 2), which is a key signal for macrophages to recognize ferroptosis cells, thereby mediating the phagocytes to clear ferroptosis cells.
Iron content can regulate inflammatory cytokines in macrophages, but the direction and way of regulation are not unique. In Salmonella-infected macrophages, iron accumulation has been reported to decrease the generation of pro-inflammatory factors, which may be due to a fact that iron promotes the production of anti-inflammatory factors [69]. However, in vivo and in vitro experiments have shown that the gather of iron in macrophages facilitates the pro-inflammatory polarization and arises the expression of pro-inflammatory factors through the TLR4/TRIF pathway [70]. In addition, iron has also been reported to regulate the TCA cycle in macrophages, thereby regulating the production of cytokines [71].Based on the pro-inflammatory effect of iron, iron oxide nanoparticles have been reported to inhibit tumor growth by promoting the polarization of pro-inflammatory macrophages. That is to say, increasing the iron content in macrophages can achieve the effect of killing tumor cells.
In addition, similar to B cells, different subsets of macrophages have different sensitivities to ferroptosis. The content of inducer nitric oxide synthase (iNOS) in pro-inflammatory M1 macrophages is more than that in anti-inflammatory M2 macrophages, so there are higher nitric oxide free radicals in M1 cells, thus inhibiting lipid peroxidation. On the contrary, due to the low content of iNOS in M2 macrophages, less nitric oxide free radicals are produced, and the inhibition effect on lipid peroxidation is less [72].
In some cases, ferroptosis process and immune response may exist at the same time and interact with each other. And ferrroptosis can induce inflammation-related immunosuppressive tumor microenvironment to promote tumor growth. In conclusion, ferroptosis is extremely relevant to the immune state of the body, and exploring the relationship between ferroptosis and immunity is conducive to the development of more effective treatment strategies for many diseases, which is a very promising research direction. Figures 5 and 6 are respectively about the role of ferroptosis in M1 macrophages and M2 macrophages.
Fig. 5.
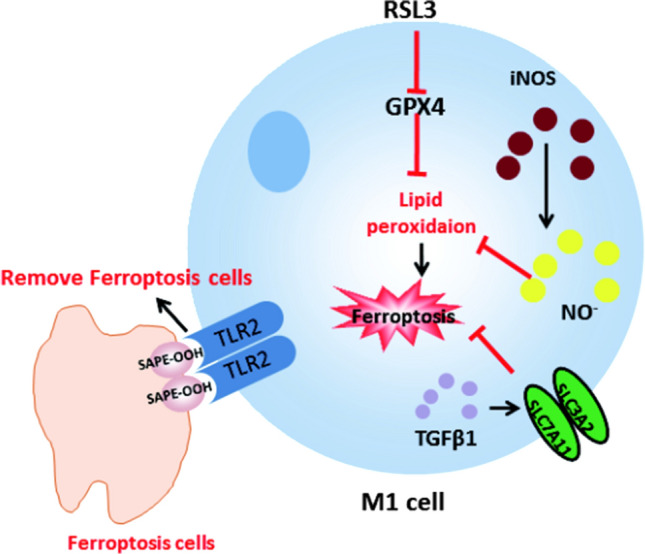
The role of ferroptosis in M1 macrophages
Fig. 6.
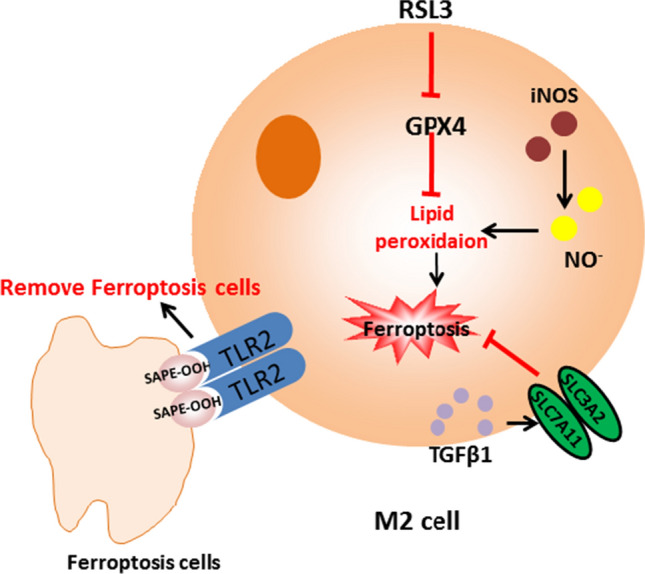
The role of ferroptosis in M2 macrophages
Overall, from the perspective of the biological molecular mechanism and occurrence mechanism of ferroptosis, it can well explain the onset and progression of many common diseases and the close relationship between the immune state of the body and ferroptosissy, systematically elucidating the molecular mechanism of ferroptosis, the functions and mechanisms of ferroptosis in various diseases and immune cells and immunotherapy, will be an extremely promising research in a fashion that provide a blueprint for the future development of new treatment strategies for many diseases. In addition, it also provides a way of thinking for the use of the inducers and inhibitors of ferroptosis that have been discovered at present; therefore, it is necessary to summarize the existing inducers and inhibitors of ferroptosis.
Ferroptosis inhibitors and inducers
Since ferroptosis has been studied, many substances have been found to induce ferroptosis, such as erastin, RSL3, sorafenib, salazopyridine, etc. At the same time, there are many kinds of ferroptosis inhibitors that can be used to inhibit ferroptosis, such as iron chelator, ferropstain-1, vitamin E, GSH, liproxstatin 1, etc. A summary of ferroptosis activators and inhibitors that have been found is respectively shown in Tables 1 and 2. Through a detailed review of the mechanism offerroptosis and its inhibitors and inducers, we hope to screen out targeted drugs that can effectively intervene ferroptosis to treat common human diseases.
Table 1.
Ferroptosis activators
| Classification | Compound/drug | Target/function |
|---|---|---|
| Activators | Cisplatin (CDDP) | Destroy DNA |
| Erastin | VDAC2/VDAC3 | |
| Gallic acid hydrate | COX-2 | |
| RSL3 ((1S,3R)-RSL3) | GPX4 | |
| TBHQ | Nrf2 | |
| Hemin | HO-1 | |
| Simvastatin (MK 733) | HMG-CoA reductase | |
| Pifithrin-α hydrobromide | P53 | |
| Lovastatin | HMG-CoA reductase | |
| PRIMA-1Met | P53; TrxR1 | |
| BAY 11–7085 (BAY 11–7083) | NF-κB; IκBα | |
| Atorvastatin hemicalcium salt (CI-981) | HMG-CoA reductase | |
| Sulfasalazine (NSC 667,219) | NF-κB | |
| Artesunate | STAT-3;EX1 (EXP1) | |
| L-Buthionine-(S,R)-sulfoximine | G-glutamylcysteine synthetase | |
| ML-210 | GPX4 | |
| iFSP1 | FSP1 (AIFM2) | |
| Piperlongumine | ERK1/2 signaling pathway | |
| Artemisinin (Qinghaosu) | Akt signaling pathway | |
| Brusatol (NSC 172,924) | Nrf2 | |
| Fluvastatin sodium | HMG-CoA reductase | |
| Withaferin A | NF-Kb; vimentin; EPCR | |
| Pravastatin sodium (CS-514 sodium) | HMG-CoA reductase | |
| DL-Buthionine-(S,R)-sulfoximine | Glutamylcysteine synthetase | |
| Matrine (Matridin-15-one) | Kappa opioid agonist | |
| Gallic acid | COX-2 | |
| L-Glutamic acid monosodium salt | Glutamate receptor | |
| (−)-Epicatechin | COX-1; iNOS | |
| Siramesine hydrochloride | The sigma-2 receptor agonist | |
| (S)-Glutamic acid | Glutamate receptors | |
| PD146176 (NSC168807) | 15-LO | |
| PRIMA-1 (NSC-281668) | TP53 | |
| L-Cystine | Cellular regulation | |
| Pseudolaric Acid B | T lymphocytes | |
| DL-Buthionine-(S,R)-sulfoximine hydrochloride | Glutamylcysteine synthetase | |
| Trigonelline | Nrf2 | |
| Chrysosplenetin | MDR1 and p-gp | |
| (E)-Ferulic acid | Remove ROS and inhibit lipid peroxidation | |
| Arteannuin B | Anti-SARS-CoV-2 | |
| Cerivastatin sodium | HMG-CoA reductase | |
| Siramesine(Lu 28–179) | Sigma-2 receptor agonist | |
| Cerivastatin | HMG-CoA reductase | |
| Dihydroartemisinic acid (Dihydroqinghao acid) | A natural product isolated from Artemisia annua |
Table 2.
Ferroptosis inhibitors
| Classification | Compound/drug | Target/function |
|---|---|---|
| Inhibitor | Ferrostatin-1 | Prevent membrane lipid damage |
| SP600125 | JNK | |
| Acetylcysteine (N-Acetylcysteine) | ROS; Cysteine | |
| Deferoxamine mesylate | Free iron | |
| Necrostatin-1 (Nec-1) | RIP1 kinase; IDO | |
| Rosiglitazone (BRL 49,653) | PPARγ agonist; TRPC5 and TRPM3 | |
| SB 202,190 | P38 and p38β2 | |
| Curcumin (Diferuloylmethane) | NF-κB and MAPKs | |
| (−)-Epigallocatechin Gallate | EGFRse;OXPHOS | |
| U-73122 |
Phospholipase C; 5-lipoxygenase (5-LO) |
|
| Bardoxolone methyl | Nrf2 and NF-κB | |
| Trolox | vitamin E | |
| L-Glutathione reduced | Scavenge oxygen free radicals | |
| Pioglitazone (U 72,107) | PPARγ | |
| Deferiprone | Iron chelator | |
| Troglitazone | PPARγ agonists | |
| Baicalein | Xanthine oxidase inhibitor | |
| Coenzyme Q10 | Electron transport chain | |
| Deferasirox (ICL 670) | Chelating excess iron ions | |
| α-Vitamin E (( +)-α-Tocopherol) | Fat-soluble antioxidant | |
| Zileuton | 5- lipoxygenase | |
|
L-Glutamine (L-Glutamic acid 5-amide) |
Metabolic processes | |
| Deferasirox | Iron ion chelating | |
| Vildagliptin (LAF237) | DPP-IV | |
| Nordihydroguaiaretic acid | 5LOX | |
| Idebenone | Coenzyme Q10 | |
| Pioglitazone hydrochloride | PPARγ agonists | |
|
Ciclopirox olamine (Ciclopirox ethanolamine) |
Antifungal agent | |
| Rosiglitazone maleate (BRL 49653C) | PPARγ;TRPM2;TRPM3 and TRPC5 | |
| Dp44mT | Iron Chelator | |
| Eugenol | Antioxidant | |
| DL-alpha-Tocopherol | Antioxidant | |
| Deferasirox Fe3 + chelate | Chelator of iron ions | |
| D-Glutamine | D—type stereoisomer of a cell permeable Glutamine | |
| Butylated hydroxytoluene | Antioxidant | |
| Docebenone (AA 861) | 5-LO | |
| Vildagliptin dihydrate (LAF237 dihydrate) | DPP-IV | |
| Pioglitazone D4 (U 72,107 D4) | PPARγ | |
| Curcumin D6 (Diferuloylmethane D6) | HATS; NF-κB and MAPKs |
Conclusion and perspectives
Ferroptosis is regulated by multiple pathways such as amino acid metabolism, lipid metabolism and iron metabolism, it is a new cell death mode that leads to lipid peroxidation of cell membrane by accumulating of lipid peroxides, the mechanism of ferroptosis, the various compounds that induce and inhibit ferroptosis, the correlation between ferroptosis and the onset and progression of various diseases and the immune state of the body have been thoroughly studied, but the existing reports on ferroptosis is still infantile and not profound enough, the study of cell death mode is still an important work to solve the problem of treating common human diseases. It is expected that there will be more and more related studies in the future, which will provide new ideas for the treatment plan with ferroptosis as the therapeutic target, and also will provide references for the development of more ferroptosis inhibitors and inducers. In addition, it will be a meaningful work to screen targeted drugs that regulate the metabolism of amino acids, lipids, reactive oxygen species and iron for the treatment of common human diseases such as tumor and heart disease. Through a detailed review of the mechanism of ferroptosis and its inhibitors and inducers, we hope to screen out targeted drugs that can effectively intervene ferroptosis to treat common human diseases, and provide reference for the future research and exploration of ferroptosis. It may also be a meaningful work that combining drug therapy with common treatments such as chemotherapy and radiation is a promising job.
Funding support
This review was supported by the National Natural Science Foundation of China (No. 82072340), the Major National Science and Technology Projects–Major New Drug Creation (2019ZX09301-132); Changjiang Scholars and Innovative Research Team in University (No. IRT_15R13); Guangxi Science and Technology Base and Talent Special Project (No. AD17129003).
Declarations
Competing interests
The authors have declared that no competing interest exists.
Human and animals rights
As this article is only a review of data already collected in the database, this article does not include any studies directly involving human participants.
Informed consent
For this type of study, formal consent is not required.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shengxian Li, Email: L914694127@126.com.
Yong Huang, Email: huangyong503@126.com.
References
- 1.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–96. [DOI] [PubMed]
- 3.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32(6):920–937. doi: 10.1016/j.cmet.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 5.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas JP, Geiger PG, Maiorino M, Ursini F, Girotti AW. Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim Biophys Acta. 1990;1045(3):252–260. doi: 10.1016/0005-2760(90)90128-K. [DOI] [PubMed] [Google Scholar]
- 7.Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–278. doi: 10.1038/cdd.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang X, Sui S, Wang L, et al. Inhibition of tumor propellant glutathione peroxidase 4 induces ferroptosis in cancer cells and enhances anticancer effect of cisplatin. J Cell Physiol. 2020;235(4):3425–3437. doi: 10.1002/jcp.29232. [DOI] [PubMed] [Google Scholar]
- 9.Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274(17):11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 10.Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165(1):20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D, Tavana O, Chu B, et al. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol Cell. 2017;68(1):224–232 e224. doi: 10.1016/j.molcel.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, Shi J, Liu X, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20(10):1181–1192. doi: 10.1038/s41556-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song X, Zhu S, Chen P, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr Biol. 2018;28(15):2388–2399 e2385. doi: 10.1016/j.cub.2018.05.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beguin Y, Aapro M, Ludwig H, Mizzen L, Osterborg A. Epidemiological and nonclinical studies investigating effects of iron in carcinogenesis–a critical review. Crit Rev Oncol Hematol. 2014;89(1):1–15. doi: 10.1016/j.critrevonc.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Basuli D, Tesfay L, Deng Z, et al. Iron addiction: a novel therapeutic target in ovarian cancer. Oncogene. 2017;36(29):4089–4099. doi: 10.1038/onc.2017.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pham CG, Bubici C, Zazzeroni F, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119(4):529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 18.Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM. Ferritin and the response to oxidative stress. Biochem J. 2001;357:241–247. doi: 10.1042/bj3570241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang X, Cai Z, Wang H, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127(4):486–501. doi: 10.1161/CIRCRESAHA.120.316509. [DOI] [PubMed] [Google Scholar]
- 20.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Zhang F, An P, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood. 2011;118(7):1912–1922. doi: 10.1182/blood-2011-01-330324. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. 2012;56(3):961–971. doi: 10.1002/hep.25746. [DOI] [PubMed] [Google Scholar]
- 26.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 27.Parrow NL, Fleming RE. Bone morphogenetic proteins as regulators of iron metabolism. Annu Rev Nutr. 2014;34:77–94. doi: 10.1146/annurev-nutr-071813-105646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 29.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]
- 33.Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu B, Kon N, Chen D, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21(5):579–591. doi: 10.1038/s41556-019-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie Y, Zhu S, Song X, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20(7):1692–1704. doi: 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 36.Yang WH, Ding CC, Sun T, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28(10):2501–2508 e2504. doi: 10.1016/j.celrep.2019.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Wang H, Yang X, et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct Target Ther. 2020;5(1):138. doi: 10.1038/s41392-020-00253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, Green M, Choi JE, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. doi: 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan MB, Corcoran RB. Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol. 2018;15(11):709–720. doi: 10.1038/s41571-018-0105-0. [DOI] [PubMed] [Google Scholar]
- 41.Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409–422 e421. doi: 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 42.Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer Cell. 2018;34(1):21–43. [DOI] [PMC free article] [PubMed]
- 43.Yang J, Antin P, Berx G, et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2020;21(6):341–352. doi: 10.1038/s41580-020-0237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo J, Duan L, He X, et al. A combined model of human iPSC‐derived liver organoids and hepatocytes reveals ferroptosis in DGUOK mutant mtDNA depletion syndrome. Adv Sci. 2021;8(10):2004680. [DOI] [PMC free article] [PubMed]
- 45.Suomalainen A, Isohanni P. Mitochondrial DNA depletion syndromes–many genes, common mechanisms. Neuromuscul Disord. 2010;20(7):429–437. doi: 10.1016/j.nmd.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Filosto M, Mancuso M, Tomelleri G, et al. Hepato-cerebral syndrome: genetic and pathological studies in an infant with a dGK mutation. Acta Neuropathol. 2004;108(2):168–171. doi: 10.1007/s00401-004-0872-9. [DOI] [PubMed] [Google Scholar]
- 47.El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10(2):186–198. doi: 10.1007/s13311-013-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pronicka E, Weglewska-Jurkiewicz A, Taybert J, et al. Post mortem identification of deoxyguanosine kinase (DGUOK) gene mutations combined with impaired glucose homeostasis and iron overload features in four infants with severe progressive liver failure. J Appl Genet. 2011;52(1):61–66. doi: 10.1007/s13353-010-0008-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conrad M, Proneth B. Broken hearts: Iron overload, ferroptosis and cardiomyopathy. Cell Res. 2019;29(4):263–264. doi: 10.1038/s41422-019-0150-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang H, An P, Xie E, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449–465. doi: 10.1002/hep.29117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu Y, Jiang L, Wang H, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi: 10.1182/blood.2019002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca LG. The role of iron in brain ageing and neurodegenerative disorders. The Lancet Neurology. 2014;13(10):1045–1060. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cardoso BR, Hare DJ, Bush AI, Roberts BR. Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry. 2017;22(3):328–335. doi: 10.1038/mp.2016.196. [DOI] [PubMed] [Google Scholar]
- 55.Bao WD, Pang P, Zhou XT, et al. Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer's disease. Cell Death Differ. 2021;28:1548–62. [DOI] [PMC free article] [PubMed]
- 56.Raz E, Jensen JH, Ge Y, et al. Brain iron quantification in mild traumatic brain injury: a magnetic field correlation study. AJNR Am J Neuroradiol. 2011;32(10):1851–1856. doi: 10.3174/ajnr.A2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ondruschka B, Schuch S, Pohlers D, Franke H, Dressler J. Acute phase response after fatal traumatic brain injury. Int J Legal Med. 2018;132(2):531–539. doi: 10.1007/s00414-017-1768-2. [DOI] [PubMed] [Google Scholar]
- 58.Rui T, Wang H, Li Q, et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J Pineal Res. 2021;70(2):e12704. doi: 10.1111/jpi.12704. [DOI] [PubMed] [Google Scholar]
- 59.Bao WD, Zhou XT, Zhou LT, et al. Targeting miR-124/Ferroportin signaling ameliorated neuronal cell death through inhibiting apoptosis and ferroptosis in aged intracerebral hemorrhage murine model. Aging Cell. 2020;19(11):e13235. doi: 10.1111/acel.13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shaw J, Chakraborty A, Nag A, Chattopadyay A, Dasgupta AK, Bhattacharyya M. Intracellular iron overload leading to DNA damage of lymphocytes and immune dysfunction in thalassemia major patients. Eur J Haematol. 2017;99(5):399–408. doi: 10.1111/ejh.12936. [DOI] [PubMed] [Google Scholar]
- 61.Wang Z, Yin W, Zhu L, et al. Iron Drives T Helper Cell Pathogenicity by Promoting RNA-Binding Protein PCBP1-Mediated Proinflammatory Cytokine Production. Immunity. 2018;49(1):80–92 e87. doi: 10.1016/j.immuni.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 62.Zhao B, Yang Y, Wang X, et al. Redox-active quinones induces genome-wide DNA methylation changes by an iron-mediated and Tet-dependent mechanism. Nucleic Acids Res. 2014;42(3):1593–1605. doi: 10.1093/nar/gkt1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 2019;29(9):2731–44. [DOI] [PubMed]
- 64.Jiang Y, Li C, Wu Q, et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat Commun. 2019;10(1):2935. doi: 10.1038/s41467-019-11002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu J, Minikes AM, Gao M, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature. 2019;572(7769):402–406. doi: 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Viswanathan VS, Ryan MJ, Dhruv HD, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547(7664):453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim DH, Kim WD, Kim SK, Moon DH, Lee SJ. TGF-beta1-mediated repression of SLC7A11 drives vulnerability to GPX4 inhibition in hepatocellular carcinoma cells. Cell Death Dis. 2020;11(5):406. doi: 10.1038/s41419-020-2618-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Luo X, Gong HB, Gao HY, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021;28(6):1971–89. [DOI] [PMC free article] [PubMed]
- 69.Nairz M, Schroll A, Haschka D, et al. Lipocalin-2 ensures host defense against Salmonella Typhimurium by controlling macrophage iron homeostasis and immune response. Eur J Immunol. 2015;45(11):3073–3086. doi: 10.1002/eji.201545569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang L, Harrington L, Trebicka E, et al. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J Clin Invest. 2009;119(11):3322–3328. doi: 10.1172/JCI39939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pereira M, Chen TD, Buang N, et al. Acute Iron Deprivation Reprograms Human Macrophage Metabolism and Reduces Inflammation In Vivo. Cell Rep. 2019;28(2):498–511 e495. doi: 10.1016/j.celrep.2019.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kapralov AA, Yang Q, Dar HH, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. 2020;16(3):278–290. doi: 10.1038/s41589-019-0462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]