

Abstract
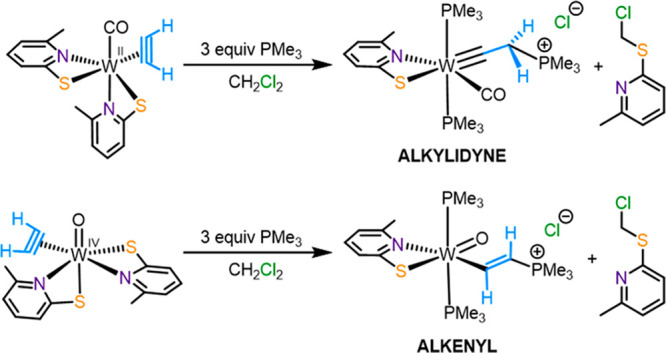
Inspired by the proposed inner-sphere mechanism of the tungstoenzyme acetylene hydratase, we have designed tungsten acetylene complexes and investigated their reactivity. Here, we report the first intermolecular nucleophilic attack on a tungsten-bound acetylene (C2H2) in bioinspired complexes employing 6-methylpyridine-2-thiolate ligands. By using PMe3 as a nucleophile, we isolated cationic carbyne and alkenyl complexes.
Short abstract
We report the first intermolecular nucleophilic attack on a tungsten-bound C2H2 in two bioinspired complexes differing only by the oxidation state of the metal center and one ligand. By using PMe3 as a nucleophile, we isolated cationic carbyne and alkenyl complexes.
The anaerobic bacterium Pelobacter acetylenicus can grow on acetylene (C2H2) as a single carbon and energy source. Its utilization is performed by the tungstoenzyme acetylene hydratase (AH), which catalyzes the hydration of acetylene to acetaldehyde.1−4 The coordination sphere of the tungsten(IV) center in the active site consists of four sulfur atoms from two molybdopterin cofactors, a thiolate from cysteine, and a water molecule.5 Although several experimental5,6 and computational studies7−10 have been carried out to shed light on the reaction mechanism, it remains unclear where C2H2 is located during hydration. Because of lower energetic barriers, density functional theory calculations favor a mechanism where C2H2 replaces the coordinated water and subsequently undergoes nucleophilic attack by a hydroxide (Scheme 1).7−10 Apart from investigations on [Et4N]2[WO(mnt)2] (mnt = maleonitriledithiolate),11−13 synthetic approaches to elucidate the mechanism of AH have only included systems with tungsten-coordinated C2H2.14,15 In a recent publication, the S,N-bidentate ligand pyridine-2-thiolate (PyS) was introduced to the tungsten(II) center to model the active site of AH as in Inter1 (Scheme 1).15 In contrast to previously reported structural model compounds,14 the coordination of a second C2H2 and subsequent insertion into the W–N bond occurred, showing that a second coordinated C2H2 is sufficiently activated to undergo a nucleophilic attack. A similar behavior was observed in molybdenum and tungsten complexes, where a nucleophilic attack on one of two coordinated hexafluorobut-2-yne moieties took place, yielding a η2-vinyl complex.16−20 Our aim is to facilitate an intermolecular nucleophilic attack by inhibiting the coordination of a second C2H2 and insertion as in the PyS system. Therefore, we anticipated the introduction of a methyl group next to the coordinating nitrogen atom in PyS (6-MePyS) so that the metal center is more shielded against the coordination of a second C2H2.
Scheme 1. Proposed Inner-Sphere Mechanism of the Hydration of C2H2 Performed by AH: Nucleophilic Attack of a Hydroxide on Coordinated C2H28.

For preparation of the redesigned tungsten complex [W(CO)(C2H2)(6-MePyS)2] (1), which contains the desired 6-MePyS ligands and only one C2H2, a previously developed procedure was modified.14,15,21 The reaction of [WBr2(CO)3(NCMe)2] with 2.1 equiv of Na(6-MePyS) in CH2Cl2 followed by stirring under a C2H2 atmosphere for 1 h allowed the isolation of 1 in 86% yield after silica gel filtration. The intermediately formed tricarbonyl complex [W(CO)3(6-MePyS)2] was characterized by single-crystal X-ray diffraction analysis (see the Supporting Information). Longer reaction times led to the insertion of a second C2H2 into the W–N bond forming [W(CO)(C2H2)(HCCH-6-MePyS)(6-MePyS)] (2), as previously observed in the unsubstituted analogue [W(CO)(C2H2)(HCCH-PyS)(PyS)].15 However, the additional methyl group significantly decreases intramolecular insertion because even after 24 h and repeated addition of C2H2 only partial conversion is observed. Furthermore, in the absence of additional C2H2, 2 reacts reversibly to 1 under the elimination of acetylene or polyacetylene depending on the solvent. Nevertheless, we were able to isolate 2 and unambiguously confirm its structure by single-crystal X-ray diffraction analysis (Figure 1) and by spectroscopic means. In CD2Cl2, the sterically hindered C2H2 protons of 1 resonate at 13.77 and 12.50 ppm and the carbon atoms at 205.73 and 204.14 ppm, suggesting that C2H2 acts as a four-electron donor.22 In 1H NMR spectra of 2 recorded in CD2Cl2, the η2-C2H2 protons appear as singlets at 12.90 and 12.03 ppm. The protons of the inserted C2H2 couple with each other thus appear as doublets (3J = 10.9 Hz) flanked with 183W satellites at 7.61 and 6.89 ppm. IR spectra of 1 and 2 show strong C≡O bands at 1891 and 1897 cm–1, respectively. Single-crystal X-ray diffraction analyses of 1 and 2 revealed almost identical W–C1, W–C2, and C1–C2 bond distances (Figure 1) compared to the literature values of tungsten(II) acetylene complexes.23,24 The inserted C2H2 in 2 is strongly activated and therefore has more ethylene character with a C–C bond length of 1.349(3) Å compared to 1.310(3) Å in η2-acetylene. Compared to the unsubstituted analogue [W(CO)(C2H2)(PyS)2] with W–N distances of 2.161 and 2.212 Å, 1 exhibits slightly longer bonds [2.197(3) and 2.259(4) Å].15 As was already observed in [MoO2(6-MePyS)2], the nitrogen atom in 6-MePyS is not able to bind to the metal center as tightly as it does in the unsubstituted version because of the methyl group in the ortho position.25
Figure 1.

Molecular structures of 1 (left), 2 (middle), and 3 (right) with probability ellipsoids drawn at the 50% probability level.
Considering that the tungsten center in AH is in the oxidation state +IV, we oxidized 1 with pyridine N-oxide to obtain the tungsten(IV) complex [WO(C2H2)(6-MePyS)2] (3) according to Scheme 2. After filtration to remove insoluble byproducts, 3 was crystallized in 84% yield. IR spectra show one strong band indicative of ν(W=O) at 924 cm–1.14,26−28 The 1H NMR spectrum of 3 in CD2Cl2 shows two equally sharp singlets flanked with 183W satellites for the C2H2 protons at 11.23 and 10.99 ppm. Thus, they are shifted upfield compared to 1. The same trend is observed in the 13C NMR spectrum, with resonances at 159.69 and 159.05 ppm being characteristic of a two- or three-electron-donor alkyne.29 Single crystals suitable for X-ray diffraction analysis were grown from a CH2Cl2/heptane solution. A molecular view of 3 is displayed in Figure 1. The C1–C2 bond [1.279(2) Å] is slightly shorter than that in 1 [1.306(7) Å], while the W–C bonds [W1–C1 of 2.022(5) Å for 1 vs 2.0693(15) Å for 3 and W1–C2 of 2.055(3) Å for 1 vs 2.1027(15) Å for 3] are essentially longer.
Scheme 2. Synthesis of Complexes 1–3.

With compounds 1 and 3 exhibiting related structures but different oxidation states of the metal center, a nucleophilic attack of PMe3 on the coordinated C2H2 was investigated. Of particular interest to us was the potential formation of the intermediate Inter2 (Figure 1). Treatment of a CH2Cl2 solution of 1 with 3 equiv of PMe3 led to an immediate color change from purple to orange-brown and full conversion of the starting material. An X-ray diffraction study on single crystals grown from a CH2Cl2/heptane solution revealed the product to be an ion pair consisting of the tungsten carbyne complex [W(CO)(CCH2PMe3)(PMe3)2(6-MePyS)]+ (4) and a chloride deriving from the solvent (Figure 2). Indeed, a nucleophilic attack of PMe3 at the coordinated C2H2 under the formation of a P–C bond had occurred. However, the attack also leads to the cleavage of one 6-MePyS, which reacts with CH2Cl2 to form 2-((chloromethyl)thio)-6-methylpyridine (6-MePySCH2Cl) and Cl–. The coordination surrounding of tungsten is completed by two PMe3 ligands preserving the 18e– character of the complex and explaining the need for 3 equiv of phosphine. After workup, pure 4 was obtained in 50% yield (Scheme 3).
Figure 2.
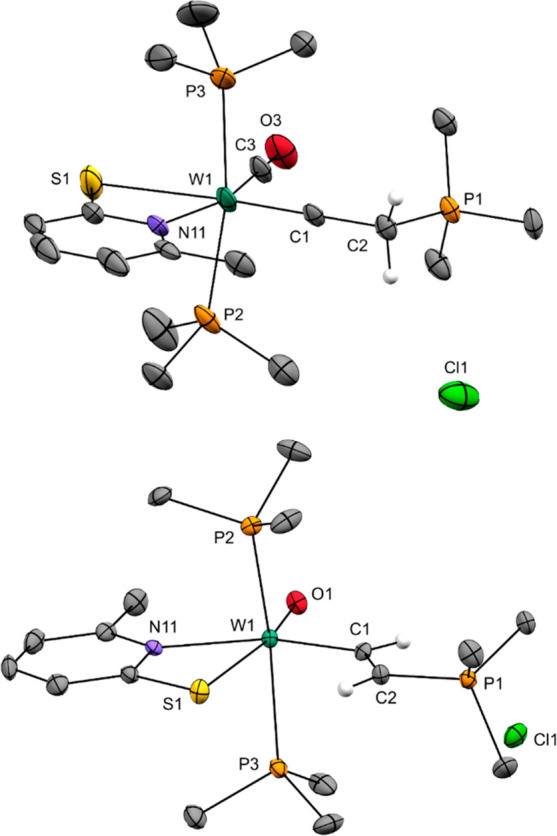
Molecular structures of 4 (top) and 5 (bottom) with probability ellipsoids drawn at the 30% (4) and 50% (5) probability levels, respectively.
Scheme 3. Reaction of 1 and 3 with 3 equiv of PMe3 in CH2Cl2 to Yield 4 and 5, Respectively.

The formation of 4 is also confirmed by 1H NMR spectroscopy, where the CH2 protons appear at 3.81 ppm as a doublet of triplets (2JHP = 19.4 Hz; 4JHP = 5.9 Hz) and the methyl groups of the two tungsten-coordinated PMe3 molecules at 1.44 ppm and those of the carbon-bound PMe3 at 2.12 ppm. To confirm that the carbyne carbon atom as well as the adjacent methylene group indeed derive from the coordinated C2H2, 1 was resynthesized using C2D2 to generate [W(CO)(C2D2)(6-MePyS)2] (1D). Upon reaction of 1D with PMe3, the signal at 3.81 ppm is absent, confirming a reaction of the coordinated C2H2 and no participation of CH2Cl2.30 While Cummins et al. reported the reaction of a molybdenum-bound (trimethylsilyl)acetylene with Li[BHEt3] to a η2-vinyl complex and the subsequent formation of carbyne only after heating to 80 °C for several hours, the η2-vinyl intermediate does not seem to be stable at all in our case.31 It is only observed in the 1H NMR spectrum when a reaction of 1 is carried out with less than 3 equiv of PMe3 directly in a J. Young NMR tube. The 31P{1H} NMR spectrum of 4 shows a triplet at 19.87 ppm for the carbon-bound PMe3 and a doublet at −17.94 ppm being flanked with 183W satellites (1JWP = 277.8 Hz) for the tungsten-bound PMe3. In the 13C NMR spectrum, the methylene carbon resonates at 46.80 ppm (d, 1JCP = 49.5 Hz). CO (q at 250.98 ppm) and W≡C (dt at 249.22 ppm) carbon atoms give signals of a similar shift and coupling pattern and could only be distinguished by heteronuclear multiple-bond correlation (HMBC) between the carbyne carbon and methylene protons. The W≡C resonance is similar to that of [W(CCH3)(PMe3)4Cl] (253.3 ppm)32 but shifted upfield compared to those of similar compounds like [W(CCH2Ph)(CO)2(dppe)Cl] (276.3 ppm),33 [Tp′(CO)2WCCH2W(CO)(C2Ph2)Tp] (312 ppm),34 and [Mo(CCH2B(C6F5)3)(dppe)2] (347.0 ppm).35 The byproduct 6-MePySCH2Cl was identified by 1H NMR and mass spectroscopy. When the reaction was carried out in CD2Cl2, the singlet for the methylene protons was absent. To a lesser extent, also bis((6-methylpyridin-2-yl)thio)methane [(6-MePyS)2CH2] was found, which was formed by the reaction of another 6-MePyS with the formerly generated 6-MePySCH2Cl. The W–C bond length of 1.793(4) Å in 4 confirms the triple-bond character, yet falls toward the shorter end of W≡C distances in recently published carbyne complexes (ca. 1.76–1.86 Å).36−42 The W1–C1–C2 angle of 179.4(4)° is almost perfectly linear, and the C1–C2 distance of 1.486(6) Å indicates a single bond.43 The P1–C2 bond [1.795(4) Å] is slightly longer than the other three P1–C bonds [1.761(5)–1.779(5) Å] but shorter than the remaining P–C bonds [1.807(6)–1.827(4) Å]. The W–S bond [2.6605(16) Å] is considerably longer than those in 1 [W1–S1 2.5834(12) Å; W1–S2 2.4073(12) Å], indicating a strong trans influence of the carbyne ligand.44
Treatment of a CH2Cl2 solution of 3 with 3 equiv of PMe3 led to an immediate color change from light yellow to dark green. After workup, the ethenyl complex [WO(CHCHPMe3)(PMe3)2(6-MePyS)]Cl (5; Scheme 3) was isolated in 90% yield as a black-green crystalline powder. Its structure was unambiguously identified by single-crystal X-ray diffraction analysis (Figure 2). Again, a nucleophilic attack at the coordinated C2H2 had occurred, forming a P–C bond. In contrast to 4, however, 5 remains with a coordinated ethenyl ligand exhibiting a W–C single bond. Similar reactions were already performed with chromium(0)-bound C2H2; however, treatment with PMe3 predominantly led to an exchange with the coordinated C2H2.45 The ethenyl protons of 5 are clearly identified by 1H NMR spectroscopy with resonances at 11.42 and 4.26 ppm and coupling to each other with 3J = 17.5 Hz. Upon reaction of [WO(C2D2)(6-MePyS)2] (3D) with PMe3 in CD2Cl2, the ethenyl proton resonances are absent. The 31P{1H} NMR spectrum of 5 shows two rather broad singlets at 4.73 and −22.14 ppm, with the latter being flanked with 183W satellites. The ethenyl carbon bound to the tungsten center resonates at 222.99 ppm and thus exhibits a significant downfield shift compared to the literature. The other gives a doublet at 96.83 ppm and is thus considerably shifted upfield compared to similar compounds.46,47 The W–C distance of 2.068(3) Å is shorter than that in rare examples of tungsten ethenyl complexes like [WO2(CHCH2)(Tp′)] (2.136 Å)46 and [W(Cp)(CHCHC(CH3)3)(η2-C(O)NR1R2)(NO)] (2.161 Å),47 while the C1–C2 distance of 1.363(4) Å is slightly longer compared to the aforementioned compounds (1.305 and 1.332 Å). The W1–C1–C2 [135.8(2)°] and C1–C2–P1 [123.4(2)°] angle confirm a slight deviation from sp2 hybridization on the carbon atoms. In contrast to 4, the P1–C2 bond [1.745(3) Å] is slightly shorter than the other three P1–C bonds [1.774(3)–1.796(3) Å].
In conclusion, we report the synthesis of tungsten acetylene complexes where intramolecular insertion of C2H2 into the W–N bond is prevented by steric adjustment at the ancillary ligand. This allowed the investigation of an intermolecular nucleophilic attack at the solely coordinated C2H2 using PMe3. Starting from the tungsten(II) complex 1, a tungsten ethylidyne complex is formed; hence, the four-electron-donor C2H2 converts to the four-electron-donor carbyne when assuming retention of the metal oxidation state. When the carbyne is considered to be a six-electron donor, oxidation to tungsten(IV) is formally taking place. Regarding the oxido ligand as a six-electron donor, the two-electron-donor C2H2 in the tungsten(IV) complex 3 converts to a two-electron-donor ethenyl moiety. In both cases, the reactions proceed under preservation of the 18e– character of the complexes. One 6-MePyS ligand is cleaved under reaction with CH2Cl2 to form 6-MePySCH2Cl and the counterion Cl– for the cationic tungsten complexes; thus, the presence of CH2Cl2 is crucial. We assume that cleavage of the sulfur ligand relieves the charge at tungsten accumulated as a result of the nucleophilic attack. The attack of PMe3 on the tungsten(IV)-bound C2H2 to form an ethenyl ligand resembles the step in which the Inter2 intermediate of the proposed mechanism of AH is formed.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.1c00643.
Experimental details, syntheses, crystallographic data, and NMR spectra of all compounds (PDF)
Accession Codes
CCDC 2050311–2050316 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
The manuscript was written through contributions of all authors.
Financial support by the Austrian Science Fund (Grant P31583) and NAWI Graz is gratefully acknowledged.
The authors declare no competing financial interest.
Notes
† Email: madeleine.ehweiner@uni-graz.at.
Notes
‡ Email: lydia.peschel@uni-graz.at.
Notes
§ Email: niklas.stix@edu.uni-graz.at.
Notes
⊥ Email: miljan.corovic@uni-graz.at.
Notes
∥ Email: ferdinand.belaj@uni-graz.at.
Supplementary Material
References
- ten Brink F. Living on acetylene. Met. Ions Life Sci. 2014, 14, 15–35. 10.1007/978-94-017-9269-1_2. [DOI] [PubMed] [Google Scholar]
- Meckenstock R. U.; Krieger R.; Ensign S.; Kroneck P. M. H.; Schink B. Acetylene hydratase of Pelobacter acetylenicus. Eur. J. Biochem. 1999, 264, 176–182. 10.1046/j.1432-1327.1999.00600.x. [DOI] [PubMed] [Google Scholar]
- Rosner B. M.; Schink B. Purification and Characterization of Acetylene Hydratase of Pelobacter acetylenicus, a Tungsten Iron-Sulfur Protein. J. Bacteriol. 1995, 177, 5767–5772. 10.1128/JB.177.20.5767-5772.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink B. Fermentation of acetylene by an obligate anaerobe, Pelobacter acetylenicus sp. nov. Arch. Microbiol. 1985, 142, 295–301. 10.1007/BF00693407. [DOI] [Google Scholar]
- Seiffert G. B.; Ullmann G. M.; Messerschmidt A.; Schink B.; Kroneck P. M. H.; Einsle O. Structure of the non-redox-active tungsten/[4Fe:4S] enzyme acetylene hydratase. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3073–3077. 10.1073/pnas.0610407104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Brink F.; Schink B.; Kroneck P. M. H. Exploring the active site of the tungsten, iron-sulfur enzyme acetylene hydratase. J. Bacteriol. 2011, 193, 1229–1236. 10.1128/JB.01057-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao R.-Z.; Himo F. Theoretical Study of the Chemoselectivity of Tungsten-Dependent Acetylene Hydratase. ACS Catal. 2011, 1, 937–944. 10.1021/cs200242m. [DOI] [Google Scholar]
- Liao R.-Z.; Yu J.-G.; Himo F. Mechanism of tungsten-dependent acetylene hydratase from quantum chemical calculations. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 22523–22527. 10.1073/pnas.1014060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent M. A.; Hillier I. H.; Periyasamy G.; Burton N. A. A DFT study of the possible role of vinylidene and carbene intermediates in the mechanism of the enzyme acetylene hydratase. Dalton Trans. 2010, 39, 3816–3822. 10.1039/b924800e. [DOI] [PubMed] [Google Scholar]
- Antony S.; Bayse C. A. Theoretical Studies of Models of the Active Site of the Tungstoenzyme Acetylene Hydratase. Organometallics 2009, 28, 4938–4944. 10.1021/om900230x. [DOI] [Google Scholar]
- Das S. K.; Biswas D.; Maiti R.; Sarkar S. Modeling the Tungsten Sites of Inactive and Active Forms of Hyperthermophilic Pyrococcus furiosus Aldehyde Ferredoxin Oxidoreductase. J. Am. Chem. Soc. 1996, 118, 1387–1397. 10.1021/ja9511580. [DOI] [Google Scholar]
- Yadav J.; Das S. K.; Sarkar S. A Functional Mimic of the New Class of Tungstoenzyme, Acetylene Hydratase. J. Am. Chem. Soc. 1997, 119, 4315–4316. 10.1021/ja970134l. [DOI] [Google Scholar]
- Schreyer M.; Hintermann L. Is the tungsten(IV) complex (NEt4)2WO(mnt)2 a functional analogue of acetylene hydratase?. Beilstein J. Org. Chem. 2017, 13, 2332–2339. 10.3762/bjoc.13.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschel L. M.; Belaj F.; Mösch-Zanetti N. C. Towards Structural-Functional Mimics of Acetylene Hydratase: Reversible Activation of Acetylene using a Biomimetic Tungsten Complex. Angew. Chem., Int. Ed. 2015, 54, 13018–13021. 10.1002/anie.201505764. [DOI] [PubMed] [Google Scholar]
- Vidovič C.; Peschel L. M.; Buchsteiner M.; Belaj F.; Mösch-Zanetti N. C. Structural Mimics of Acetylene Hydratase: Tungsten Complexes Capable of Intramolecular Nucleophilic Attack on Acetylene. Chem. - Eur. J. 2019, 25, 14267–14272. 10.1002/chem.201903264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu Bakar W. A. W.; Carlton L.; Davidson J. L.; Manojlović-Muir L.; Muir K. W. Metal-promoted insertion and cyclisation reactions of alkynes. J. Organomet. Chem. 1988, 352, C54–C58. 10.1016/0022-328X(88)83133-4. [DOI] [Google Scholar]
- Davidson J. L.; Wilson W. F.; Manojlović-Muir L.; Muir K. W. Isomerism and fluxional beviour in η2-vinyl complexes resulting in inversion of configuration at an asymmetric carbon atom. J. Organomet. Chem. 1983, 254, C6–C10. 10.1016/0022-328X(83)85127-4. [DOI] [Google Scholar]
- Davidson J. L.; Green M.; Stone F. G. A.; Welch A. J. Syntheses involving Co-ordinatively Unsaturated Cyclopentadienyl-molybdenum and -tungsten Complexes. J. Chem. Soc., Dalton Trans. 1977, 287–294. 10.1039/DT9770000287. [DOI] [Google Scholar]
- Davidson J. L.; Vasapollo G.; Manojlović-Muir L.; Muir K. W. Stabilisation of Vinylic Intermediates in the Addition, cyclisation and Oligomerisation Reactions of Alkynes by Co-ordination to Molybdenum and Tungsten. J. Chem. Soc., Chem. Commun. 1982, 0, 1025–1027. 10.1039/C39820001025. [DOI] [Google Scholar]
- Davidson J. L.; Murray I. E. P.; Preston P. N.; Russo M. V.; Manojlović-Muir L.; Muir K. W. Formation of novel η2-vinyl complexes by nucleophilic attack at co-ordinated hexafluorobut-2-yne; implications for the stereochemistry of addition and insertion reactions of co-ordinated acetylenes. J. Chem. Soc., Chem. Commun. 1981, 20, 1059–1061. 10.1039/C39810001059. [DOI] [Google Scholar]
- Peschel L. M.; Vidovič C.; Belaj F.; Neshchadin D.; Mösch-Zanetti N. C. Activation and Photoinduced Release of Alkynes on a Biomimetic Tungsten Center: The Photochemical Behavior of the W-S-Phoz System. Chem. - Eur. J. 2019, 25, 3893–3902. 10.1002/chem.201805665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton J. L. Four-Electron Alkyne Ligands in Molybdenum(II) and Tungsten(II) Complexes. Adv. Organomet. Chem. 1989, 29, 1–100. 10.1016/S0065-3055(08)60352-4. [DOI] [Google Scholar]
- Ricard L.; Weiss R.; Newton W. E.; Chen G. J.-J.; McDonald J. W. Binding and Activation of Enzymatic Substrates by Metal Complexes. 4. Structural Evidence for Acetylene as a Four-Electron Donor in W(CO)(C2H2)(S2CNEt2)2. J. Am. Chem. Soc. 1978, 100, 1318–1320. 10.1021/ja00472a062. [DOI] [Google Scholar]
- Helmdach K.; Ludwig S.; Villinger A.; Hollmann D.; Kösters J.; Seidel W. W. Synthesis and activation potential of an open shell diphosphine. Chem. Commun. 2017, 53, 5894–5897. 10.1039/C7CC02114C. [DOI] [PubMed] [Google Scholar]
- Ehweiner M. A.; Wiedemaier F.; Belaj F.; Mösch-Zanetti N. C. Oxygen Atom Transfer Reactivity of Molybdenum(VI) Complexes Employing Pyrimidine- and Pyridine-2-thiolate Ligands. Inorg. Chem. 2020, 59, 14577–14593. 10.1021/acs.inorgchem.0c02412. [DOI] [PubMed] [Google Scholar]
- Maatta E. A.; Wentworth R. A. D.; Newton W. E.; McDonald J. W.; Watt G. D. Reversible binding of acetylene by oxobis(diethyldithiocarbamate)molybdenum. J. Am. Chem. Soc. 1978, 100, 1320–1321. 10.1021/ja00472a063. [DOI] [Google Scholar]
- Maatta E. A.; Wentworth R. A. D. Simple alkyne adducts of MoO(S2CNR2)2. Inorg. Chem. 1979, 18, 524–526. 10.1021/ic50192a072. [DOI] [Google Scholar]
- Vidovič C.; Belaj F.; Mösch-Zanetti N. C. Soft Scorpionate Hydridotris(2-mercapto-1-methylimidazolyl) borate) Tungsten-Oxido and -Sulfido Complexes as Acetylene Hydratase Models. Chem. - Eur. J. 2020, 26, 12431–12444. 10.1002/chem.202001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward B. C.; Templeton J. L. Nuclear Magnetic resonance Studies of Alkynes as Four-Electron Donor Ligands in Monomeric Tungsten(II) Complexes. J. Am. Chem. Soc. 1980, 102, 1532–1538. 10.1021/ja00525a011. [DOI] [Google Scholar]
- Fürstner A.; Mathes C.; Lehmann C. W. Alkyne Metathesis: Development of a Novel Molybdenum-Based Catalyst System and Its Application to the Total Synthesis of Epothilone A and C. Chem. - Eur. J. 2001, 7, 5299–5317. . [DOI] [PubMed] [Google Scholar]
- Tsai Y.-C.; Diaconescu P. L.; Cummins C. C. Facile Synthesis of Trialkoxymolybdenum(VI) Alkylidyne Complexes for Alkyne Metathesis. Organometallics 2000, 19, 5260–5262. 10.1021/om000644f. [DOI] [Google Scholar]
- Atagi L. M.; Critchlow S. C.; Mayer J. M. Formation of tungsten-carbyne and tungsten-oxo-allyl complexes on reaction of WCl2(PR3)4 with unsaturated silanes. J. Am. Chem. Soc. 1992, 114, 1483–1484. 10.1021/ja00030a054. [DOI] [Google Scholar]
- Birdwhistell K. R.; Burgmayer S. J. N.; Templeton J. L. Tungsten vinylidenes and carbynes from terminal alkyne reagents. J. Am. Chem. Soc. 1983, 105, 7789–7790. 10.1021/ja00364a077. [DOI] [Google Scholar]
- Woodworth B. E.; White P. S.; Templeton J. L. Deprotonation and Oxidation of the W≡CCH2–W Bridge To Form a C2-Biscarbyne W≡C–C≡W Bridge. J. Am. Chem. Soc. 1998, 120, 9028–9033. 10.1021/ja980226q. [DOI] [Google Scholar]
- Stennett T. E.; Haddow M. F.; Wass D. F. Alkene to carbyne: tandem Lewis acid activation and dehydrogenation of a molybdenum ethylene complex. Angew. Chem., Int. Ed. 2013, 52, 11356–11359. 10.1002/anie.201305233. [DOI] [PubMed] [Google Scholar]
- Dewhurst R. D.; Hill A. F.; Smith M. K. Heterobimetallic C3 complexes through silylpropargylidyne desilylation. Angew. Chem., Int. Ed. 2004, 43, 476–478. 10.1002/anie.200352693. [DOI] [PubMed] [Google Scholar]
- Dewhurst R. D.; Hill A. F.; Willis A. C. The Interplay of Bis(tricarbido) and Dimetallaoctatetrayne Complexes of Platinum. Organometallics 2009, 28, 4735–4740. 10.1021/om900485b. [DOI] [Google Scholar]
- Frogley B. J.; Hill A. F. Flexible Platinum(0) Coordination to a Ditungsten Ethanediylidyne. Angew. Chem., Int. Ed. 2019, 58, 8044–8048. 10.1002/anie.201902116. [DOI] [PubMed] [Google Scholar]
- Caldwell L. M. Alkylidyne Complexes Ligated by Poly(pyrazolyl)borates. Adv. Organomet. Chem. 2008, 56, 1–94. 10.1016/S0065-3055(07)56001-6. [DOI] [Google Scholar]
- Tonzetich Z. J.; Lam Y. C.; Müller P.; Schrock R. R. Facile Synthesis of a Tungsten Alkylidyne Catalyst for Alkyne Metathesis. Organometallics 2007, 26, 475–477. 10.1021/om0610647. [DOI] [Google Scholar]
- Hill A. F.; Kong R. Y. High oxidation state bromocarbyne complexes. Chem. Commun. 2017, 53, 759–762. 10.1039/C6CC08946A. [DOI] [PubMed] [Google Scholar]
- Han Y.-S.; Hill A. F.; Kong R. Y. An unusual alkylidyne homologation. Chem. Commun. 2018, 54, 2292–2295. 10.1039/C8CC00119G. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Fu X.; Chen J.; Zhai H. Total synthesis of (−)-jiadifenin. Angew. Chem., Int. Ed. 2012, 51, 9825–9828. 10.1002/anie.201203176. [DOI] [PubMed] [Google Scholar]
- Colebatch A. L.; Hill A. F. Coordination chemistry of phosphinocarbynes: phosphorus vs. carbyne site selectivity. Dalton Trans. 2017, 46, 4355–4365. 10.1039/C6DT04770J. [DOI] [PubMed] [Google Scholar]
- Alt H. G.; Engelhardt H. E.; Filippou A. C. Acetylen als Baustein für Carben- und Vinylidenliganden am Chrom. J. Organomet. Chem. 1988, 355, 139–148. 10.1016/0022-328X(88)89017-X. [DOI] [Google Scholar]
- Crane T. W.; White P. S.; Templeton J. L. Deprotonation of Cationic Tungsten(IV) Aqua–Oxo–Alkyne Complexes To Form Dioxo–Vinyl Tungsten(VI) Complexes. Inorg. Chem. 2000, 39, 1081–1091. 10.1021/ic990994y. [DOI] [PubMed] [Google Scholar]
- Ipaktschi J.; Uhlig S.; Dülmer A. η2-Alkynyl and Vinylidene Transition Metal Complexes. Organometallics 2001, 20, 4840–4846. 10.1021/om010528s. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.