

Abstract
Cyclin D1 is a key mediator of cell cycle progression that is aberrantly regulated in multiple cancers, especially in breast cancers. A number of studies have indicated that a polymorphism in a splice donor site in the cyclin D1 gene is associated with alternative splicing and the production of the alternative cyclin D1b transcript. Furthermore, this polymorphism is selectively associated with disease outcomes. However, relatively little is known regarding the protein product of the alternatively spliced message, cyclin D1b. Using antibodies specific for cyclin D1b, it was found that this protein is readily detectable in a number of cancer cell lines and primary breast cancers. Whereas cyclin D1b interacts with cyclin-dependent kinase 4 (CDK4), it is relatively inefficient at mediating RB phosphorylation and cell cycle progression in model systems due to the lack of exon 5 of cyclin D1–encoded sequences. However, cyclin D1b protein levels are not significantly attenuated by DNA damage or antiestrogen treatment, indicating that the protein may have significant effect on the response to such therapeutic modalities. Whereas enforced expression of cyclin D1b was not sufficient to abrogate DNA damage checkpoint responses, it did efficiently overcome cell cycle arrest mediated by antiestrogen therapeutics. This action of cyclin D1b was not associated with effects on estrogen receptor activity, but was rather dependent on functional association with CDK4. Combined, these studies indicate that the cyclin D1b protein is aberrantly regulated and could contribute to therapeutic failure in the context of ER-positive breast cancer.
Introduction
Cyclin D1 is a key mediator of cell cycle progression that is aberrantly regulated in multiple cancers (1–4). This protein was initially identified based on complementation of yeast cyclin mutants and as a delayed early gene during mitogen stimulated proliferation (1, 4–6). It is now clear that cyclin D1 serves to integrate the activity of multiple mitogenic signaling cascades. This function of cyclin D1 is critical for appropriate cell cycle regulation as ectopic expression of cyclin D1 can promote the G1-S transition, whereas cyclin D1 deficiency compromises the ability of mitogenic signals to promote entry into the cell cycle (7, 8). Given this central role in modulating cellular proliferation, it is not surprising that deregulation of cyclin D1 is a common facet of human cancer. Cyclin D1 was found to be the site of Prad1 rearrangement in parathyroid cancer and is involved in a large fraction of translocations occurring in mantle cell lymphoma (1, 9, 10). Furthermore, amplification of the cyclin D1 gene is found in breast cancer at relatively high frequency and many primary breast tumors overexpress cyclin D1 protein in the absence of a clear underlying genetic mechanism (2, 4, 11). These studies indicate that cyclin D1 function may be particularly relevant in breast tumorigenesis. Consistent with this hypothesis, ectopic expression of cyclin D1 in mouse mammary models can drive tumor development, whereas deletion of the cyclin D1 gene potently protects against mammary tumorigenesis (12, 13).
Cyclin D1 is a critical regulator of cyclin-dependent kinase 4 (CDK4) function that contributes to cell cycle progression. The cyclin D1 protein directly associates with CDK4 and CDK6 and stimulates catalytic activity of the complex (14, 15). Interestingly, whereas most CDK/cyclin complexes phosphorylate multiple substrates harboring S-P or T-P motifs, cyclin D1–associated kinase activity is relatively specific toward the retinoblastoma tumor suppressor protein (RB), and the related proteins p107 and p130 (14, 16, 17). Cyclin D1-catalyzed phosphorylation leads to the disruption of RB activity, thus promoting cell cycle progression (16). RB represents a seminal target for cyclin D1 function, as RB-deficient cells efficiently bypass the requirement for D-type cyclins and CDK4 activity (18). Thus, it is believed that a principle means through which cyclin D1 acts to control cell cycle progression is via the phosphorylation of RB. However, a number of additional mechanisms of cyclin D1 function are known to contribute to cell cycle regulation and tumorigenesis. Particularly, cyclin D1 can serve as a transcriptional modulatory protein leading to the stimulation of specific transcription factors (e.g., estrogen receptor; refs. 19, 20), whereas mediating repression of others (e.g., androgen receptor; refs. 21, 22). Recent studies have suggested that such functions of cyclin D1 could be important in mammary gland development and hormone-dependent cancers (23, 24).
Whereas the conventional cyclin D1 protein that is encoded by five exons at the cyclin D1 locus has been extensively studied, it is now clear that variant cyclin D1 proteins are also encoded from this locus (1, 2, 4, 25). Specifically, an alternatively spliced form of cyclin D1 is produced, which has been termed cyclin D1b (26–28). This variant is encoded by the first four exons, but due to a lack of splicing excludes exon 5 encoded sequences and includes sequences derived from the fourth intron (26, 28). This splicing event is believed to be modulated by a polymorphism at the splice donor site and both the polymorphism and cyclin D1b expression have been associated with enhanced cancer risk and poor clinical outcome (4, 25, 26, 29).
Due to the importance of cyclin D1 in cell cycle control, the protein is subject to complex regulation of subcellular localization and stability. Primarily, protein localization and stability are modulated by phosphorylation of T286 (1, 30, 31), which is present in exon 5 encoded sequences of cyclin D1 and are thus absent in the cyclin D1b protein. As a result, the cyclin D1b protein remains exclusively nuclear and escapes this mode of regulation of cyclin D1 (32, 33). Prior studies had shown that mutation of T286 resulted in enhanced transformation in NIH-3T3 cells (31), and correspondingly, cyclin D1b harbors enhanced transforming potential relative to cyclin D1 (32, 33). These studies have fueled speculation that cyclin D1b expression could represent a potent event associated with tumorigenesis (4, 25).
Here, we probed the regulation of endogenous cyclin D1b protein and assessed the functional activity of cyclin D1b in the context of therapeutic modalities used in the treatment of breast cancers. These studies show that the regulation of cyclin D1b is distinct from that elicited on cyclin D1 and that cyclin D1b is a critical modulator of the response to estrogen antagonists used in the treatment of this disease.
Materials and Methods
Cell culture and drug treatment.
MCF-7, ZR751, T47D, SW13, C33A, SAOS-2, and U2OS cells were maintained in DMEM. BT549, A427, H596, H520, and H2172 cells were maintained in RPMI. HeLa cells were maintained in IMEM at 37°C in 5% CO2. Both DMEM and RPMI were supplemented with 10% fetal bovine serum (FBS), IMEM with 5% FBS, 100 unit/mL penicillin-streptomycin, and 2 mmol/L l-glutamine. For culture in steroid hormone-depleted media, MCF-7 cells were maintained in phenol red–free DMEM containing 10% charcoal dextran–treated (CDT) FBS with addition of 1 nmol/L 4-hydroxy tamoxifen (Sigma-Aldrich) or 1 μmol/L ICI 182,780 (Tocris Bioscience) for indicated time courses. MCF-7 cells were treated with a final concentration of 5 μmol/L MG132 (Sigma) for 6 h, cisplatin (CDDP; Redford) with a final concentration of either 16 or 32 μmol/L for 16 h, or PD33291 (Pfizer) at a final concentration of 250 nmol/L (provided by Pfizer).
Plasmids.
The RB, CDK4, H2B-GFP, GFP–cyclin D1, and GFP–cyclin D1b expression plasmids have been described previously (33). GFP–cyclin D1b–KE mutant was generated by the Quickchange Site-Directed Mutagenesis kit according to the manufacturer (Stratagene) using pEGFP–cyclin D1b as a template. GFP-cyclin D1-ΔI4 was generated by PCR amplification of cyclin D1 exon 1 to exon 4 sequences using pcDNA3.1-HA-cyclin D1 as template. The resulting fragment was inserted into pCR2.1 as directed by the manufacturer (Invitrogen). The fragment was subsequently removed from the pCR vector using BamH1/EcoR1 digestion and inserted into the BglII and EcoR1 sites of the pEGFP-C1 vector (Clontech). All mutants generated/used were confirmed by sequencing and primer sequences are available upon request. The ERE-Luciferase, cytomegalovirus β-galactosidase reporter plasmids and the human ERα plasmid have been described previously (34).
Transfection and 5-bromo-2’deoxyuridine incorporation.
Plasmids were transfected using the lipid-based transfection reagent FuGENE 6 (Roche Molecular Biochemical) in accordance with the manufacturer’s recommended protocol. In the 5-bromo-2’deoxyuridine (BrdUrd; Roche) incorporation experiments, 3 × 105 MCF-7 cells were plated in six-well culture dishes containing coverslips and incubated overnight. Cotransfections were done using 1.8 μg input DNA plus 0.2 μg H2B-GFP DNA. After 16 h, cells were washed with PBS and replaced in phenol red–free DMEM containing 10% CDT with either 1 nmol/L 4-hydroxy tamoxifen or 1 μmol/L ICI was added. Cells were then pulse labeled with 35 μg/mL BrdUrd for 6 h. Cells were fixed and BrdUrd-positive nuclei were visualized as previously described (35).
Adenoviral infections and bivariate flow cytometry and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide analyses.
MCF-7 cells were infected with adenovirus encoding cyclin D1 or cyclin D1b. GFP adenovirus, a kind gift from Gustavo Leone (Ohio State University), was used as a control. The infections were performed at a calculated multiplicity of infection of 50 to 100 for ~95% to 100% infection efficiency after 16 h (as determined by plaque assay in 293 cells). Cells were treated with CDDP (16 and 32 μmol/L) or vehicle for an additional 16 h or cells were washed with PBS extensively and switched to medium with 10% CDT serum in the presence of 1 nmol/L 4-hydroxy tamoxifen or 1 μmol/L ICI for 36 or 72 h. For flow cytometry analysis, Ad-GFP, Ad-D1, and Ad-D1b infected MCF-7 cells were pulse labeled with BrdUrd for 1 h. Bivariate flow cytometry analysis of DNA synthesis (BrdUrd incorporation) and DNA content (propidium iodide, PI) was performed. At least 20,000 gated events were collected for each sample. The percentage of BrdUrd-positive cells was quantified.
For measuring proliferation, 5,000 cells infected with cyclin D1 or cyclin D1b encoding adenoviruses were plated per well in 96-well flat-bottomed plates in quadruplicate. Cells were treated with tamoxifen or ICI at the time of plating to a total volume of 90 μL. At 24, 48, 72, 96, and 120 h postplating cell number/viability was determined by adding 3-(4,5-dimethylthiazol-2-yl)-2,5-dibenzyltetrozolium bromide (MTT) reagent (50 μg in 10 μL PBS) to each well. Cells were incubated for 4 h, then solubilized with 110 μL of 10% Triton X-100 (Amresco), 0.1 N HCl (Fisher) in anhydrous isopropanol (Fisher). Colorimetric analysis was performed at 570 nm.
Statistical analysis.
All statistical analysis was performed using Prism 4.0c software (GraphPad Software, Inc.) Results were analyzed for statistical significance using the Student’s unpaired t test and SE. For all analyses, P < 0.05 was considered significant.
PCR-RFLP analysis and reverse transcription–PCR.
Genomic DNA was extracted from cell culture lines. The isolated DNA was quantified, and 100 ng of DNA were amplified as described (36). Products were digested with ScrFI restriction enzyme (Biolab) and resolved by agarose gel electrophoresis. Transcript specific reverse transcription–PCR (RT-PCR) was as previously described (36).
Immunoblots and antibodies.
Immunoblotting was performed by following standard biochemical techniques. Antibodies against the following proteins were used: cyclin D1 (Ab-3; NeoMarkers), cyclin D1 (DSC-6; Cell Signaling), RB (BD Biosciences PharMingen), phosphorylated RB (Ser780; Cell Signaling), CDK4 (C-22; Santa Cruz), GFP (Santa Cruz), p21Cip1 (C-19; Santa Cruz), p27Kip1 (BD Biosciences PharMingen), β-tubulin (Santa Cruz), Lamin B (Santa Cruz), actin (Santa Cruz), and cyclin A (Santa Cruz). Cyclin D1b antibody against cyclin D1b–specific peptide sequence YRGRHLVPRKCRGWCQGPQG was generated by Bethyl Laboratories (37). The immunoactivity of cyclin D1b antibody was determined by Western blots using purified glutathione S-transferase (GST)–cyclin D1 and GST–cyclin D1b proteins. Quantification of resultant proteins was carried out using SDS-PAGE with bovine serum albumin (Sigma) as a standard curve. Band density was measured using SYPRO Red Protein Gel Stain kit (Cambrex Bio Science Rockland, Inc.). Known concentrations of GST-D1 and GST-D1b were subjected to SDS-PAGE and transferred to an Immobilon membrane (Millipore). GST-D1 and GST-D1b proteins were detected by cyclin D1 (DCS-6; Ab-3) and cyclin D1b specific antibodies. Proteins were visualized using horseradish peroxidase–conjugated secondary antibody (Pierce) along with enhanced chemiluminescence (Perkin-Elmer Life Sciences). The relative protein band intensities were determined using Image J software (version 1.24t).
Reporter assays.
MCF-7 cells were cotransfected with cytomegalovirus β-galactosidase reporter plasmid, ERE-Luciferase reporter plasmid, ERα, and cyclin D1 and cyclin D1b expression plasmids or empty vector at a 0.5:1:1:1.5 ratio. At 24 h posttransfection, cells were washed with PBS, media were replaced, and cells were treated with the indicated concentration of estradiol (E2) or ethanol vehicle for 24 h. After stimulation, cells were harvested, and luciferase activity was determined and normalized against β-galactosidase activity for transfection efficiency. Data were collected from three independent experiments.
Immunohistochemistry and automated quantitative analyses.
Immunohistochemistry analyses were performed at two different array platforms with slightly different staining and quantitation protocols:.
First, sections of a tissue array generated by cutting edge matrix assembly (38) that contained 80 invasive breast carcinoma tissues and 20 normal breast tissue were used. Briefly, after deparaffinization and rehydration of array sections, antigen retrieval was performed by microwave treatment in citrate buffer (pH 6; DAKO). Sections were blocked with 10% goat serum and followed by incubation of primary cyclin D1b at a dilution of 1:50 for 1 h. Sections were then washed thrice with TBS and subsequently incubated with a mouse cytokeratin antibody (DAKO) for 1 h. The cyclin D1b antibodies were detected using an antirabbit horseradish peroxidase–conjugated secondary antibody (DAKO EnVision-Plus), followed by incubation with Tyramide-Cy5 (Perkin-Elmer). Cytokeratin was visualized by further incubating the sections with a mouse secondary antibody conjugated to Alexa 488 (Molecular Probes). Finally, all sections were stained with 4′,6-diamidino-2-phenylindole (DAPI; Vector) for nuclear visualization. Automated quantitative analysis was performed using the AQUA/PM2000 Imaging Platform (HistoRx) as described (39). Tissue array slides were scanned and images of each breast cancer tissue were captured at different channels detecting FITC/Alexa 488, Cy5, or DAPI. AQUA software was then used to identify epithelial masks based on FITC-positive cytokeratin-expressing cells. AQUA scores for cyclin D1b representing average signal intensities within epithelial cells.
Second, immunohistochemistry for cyclin D1b was performed on formalin-fixed paraffin-embedded tissue microarrays (TMA) containing 1-mm diameter cores derived from 175 patients with invasive ductal carcinoma of no special type treated at St. Vincent’s Hospital. Sctions (4 μm thick) were mounted on Superfrost Plus adhesion slides (Lomb Scientific). Slides were baked at 78°C before being deparaffinized in xylene and rehydrated through serial alcohol solutions (100%, 95%, and 70%). Endogenous peroxidase activity was blocked using 3% H2O2 for 5 min, followed by protein block serum–free (DAKO Corporation) for 30 min. Antigen retrieval was performed in a pressure cooker at pH 9.0 (Dako retrieval solution S2367) for 10 s. Sections were incubated with cyclin D1b primary antibody at room temperature at a dilution of 1:150 for 30 min, followed by detection using Envision+ Rabbit (DAKO) for 30 min at room temperature and visualization with 3,3′-diaminobenzidine+ (DAKO) for 10 min. Sections were then counterstained with Mayer’s hematoxylin (DAKO), dehydrated, and mounted for analysis. The negative control used concentration-matched nonspecific rabbit IgG in place of the primary antibody. Stained slides were assessed by an experienced breast pathologist (E.K.A.M.) and scored for the percentage and intensity of cells which showed nuclear staining: 0 negative, 1+ weak, 2+ moderate, 3+ strong. An average intensity score was calculated as the mean intensity from two to six cores per patient.
Results
Cyclin D1b expression in cancer cells and primary breast cancer.
The cyclin D1 and cyclin D1b proteins differ only in their distinct COOH terminal domains (26). In the case of cyclin D1, the exon 5 encoded sequence is the epitope for many of the commonly used commercial antibodies. Conversely, cyclin D1b harbors a unique COOH terminal domain that is encoded by intron 4 containing sequences (Fig. 1A). A 20–amino acid peptide from intron 4 encoded sequences was used to immunize rabbits, and resultant sera was affinity purified. To test the specificity and relative sensitivity of this reagent (YW2 antibody), GST–cyclin D1 and GST–cyclin D1b proteins were purified from bacteria to serve as a source of known antigen. The DCS-6 antibody recognizes an epitope common to cyclin D1 and cyclin D1b and recognized both proteins with equivalent sensitivity (Fig. 1B, top). The Ab-3 antibody recognizes an epitope in the COOH terminus of cyclin D1 and correspondingly only recognized the GST–cyclin D1 protein under these conditions. In contrast, the YW2 antibody specifically recognized cyclin D1b (Fig. 1B). Having confirmed the specificity of the antibodies, the relative sensitivity of Ab-3 and YW2 antibodies was determined by blotting the same membrane with both antibodies simultaneously (Fig. 1B, bottom). These analyses indicated that the cyclin D1b–specific antibody had heightened reactivity for cyclin D1b, as compared against the AB3 antibody that recognizes cyclin D1.
Figure 1.

Cyclin D1b expression in different cancer cell lines and association with CDK4 in MCF-7 cells. A, diagram depicting genomic structures and splicing of cyclin D1 gene; exons indicated by boxes and introns by lines. The sequence VSEGDVPGSLAGAYRGRHLVPRKCRGWCQGPQG was derived from intron 4. Cyclin D1b antibody was derived from the sequence indicated in the underlined portion. B, YW2-D1b specific antibody efficiently recognizes cyclin D1b protein. Different amounts of GST-D1 and GST-D1b were resolved by SDS-PAGE. GST-D1 and GST-D1b were detected by immunoblotting with cyclin D1 (DCS-6) antibody (top) and YW2-D1b–specific antibody (middle) alone. YW2-D1b and D1 (Ab3) antibodies were used to blot the membranes separately. The separated membranes were exposed together (bottom). C, total protein isolated from the indicated cancer cell lines was resolved by SDS-PAGE. Cyclin D1b (first panel) or cyclin D1 (second panel) expression was detected by immunoblotting with cyclin D1b or D1 (Ab-3) antibody, respectively. Equal loading was confirmed by β-tubulin immunoblot (third panel). Genotype of exon 4–intron 4 boundary polymorphism (bottom). DNA was isolated from indicated cell lines and analyzed as described in Materials and Methods. D, endogenous cyclin D1b and D1 expression in MCF-7, BT549, and Zr-75-1 cells was determined by immunoblot. SAOS-2 cells were used as negative control (top). Equal loading was confirmed by lamin B immunoblot (bottom).
Using the YW2 and Ab3 antibody, the levels of cyclin D1b and D1 proteins were evaluated in multiple cell lines (Fig. 1C, top). There was clear heterogeneity in the levels of cyclin D1 and cyclin D1b between cell lines. Interestingly, relative levels of expression were not a direct reflection of the A-870 polymorphism that is believed to influence the production of cyclin D1b (Fig. 1C, bottom). Strikingly, the breast cancer lines MCF-7, BT549, and Zr-75-1 expressed detectable levels of cyclin D1b, suggesting that cyclin D1b could be particularly relevant in reference to breast cancer biology. To compare the relative amount of cyclin D1 and cyclin D1b in a given tumor line, the antibody preparation as used for Fig. 1B was used. Surprisingly, although cyclin D1b is clearly detected in MCF-7 cells, based on the higher affinity of the cyclin D1b antibody, it represents <10% of the cyclin D1 protein present in the cell (Fig. 1D). Similarly, in Zr-75-1 cells, cyclin D1b represents a minor portion of the total cyclin D1 protein. This finding may explain the relative difficulty in detecting endogenous cyclin D1b with reagents that also detect cyclin D1 (27, 28). Interestingly, BT549 cells express undetectable levels of cyclin D1, as has been previously reported. However, expression of cyclin D1b is clearly detectable. Thus, a heterogenous expression of cyclin D1b is found in breast cancer cell lines and can, in specific circumstances, represent a significant fraction of the cyclin D1 protein present in the cell.
To determine the expression of cyclin D1b in primary tumors, a cohort of 19 normal biopsy specimens and 65 breast cancers specimens were analyzed by immunohistochemistry quantitatively using AQUA technology. As shown in Fig. 2A, relatively low levels of cyclin D1b were observed in normal mammary tissue. However, cyclin D1b was broadly expressed in breast cancer specimens and its expression was significantly increased in primary tumors (P < 0.001). Consistent results were observed in an independent cohort of 175 invasive ductal carcinomas, which exhibited heterogeneous levels of cyclin D1b expression (Fig. 2C). Thus, cyclin D1b expression is a feature of breast cancer and its relative level of expression could be related to pathologic features.
Figure 2.

Cyclin D1b is expressed in primary breast cancer. A, a TMA section consisting of normal tissue and breast cancer tissue was costained for pan-cytokeratins (green) and cyclin D1b (red) expression and visualized by immunofluorescence. Representative images of normal mammary tissue and two tumor sections with high or low D1b expression are shown. Summary of automated quantitative imaging of cyclin D1b levels within cytokeratin-positive benign or malignant epithelial cells. B, an independent tissue array containing two to six cores of invasive ductal carcinoma from 175 patients was stained for cyclin D1b expression by immunohistochemistry and scored by a pathologist. Representative images depicting the absence of staining (a) and intensity scores of 1+ (b), 2+ (c), and 3+ (d) are shown. The percentage of breast cancers exhibiting a given intensity score was quantified.
The inefficient catalytic activity of cyclin D1b is due to a lack of exon 5 encoded sequences.
Because cyclin D1 proteins are believed to function in concert with CDK4, the interaction of endogenous cyclin D1b with CDK4 was analyzed by coimmuno-precipitation (Fig. 3A). Whereas the negative control antibody (DBF4) failed to precipitate CDK4, the cyclin D1b antibody resulted in significant precipitation of CDK4. Thus, these analyses show that the endogenous cyclin D1b protein does associate with CDK4 and thus could contribute to cell cycle control in tumor cells by functioning through the canonical cyclin D1–RB pathway. To probe the function of cyclin D1b in catalyzing RB phosphorylation and cell cycle progression with CDK4, the SAOS-2 cell culture model was used. SAOS-2 cells are a well established model for analyzing cyclin function (16, 33, 40) and lack the expression of RB and correspondingly express very low/undetectable levels of cyclin D1 and cyclin D1b (Fig. 1C). As such, it is an ideal model for evaluating the functional relationships regarding the control of RB phosphorylation by exogenous cyclin D1 proteins. SAOS-2 cells were cotransfected with RB, CDK4, and the indicated GFP–cyclin D1 expression plasmid (Fig. 3C). The use of GFP fusions allowed us to clearly determine that the level of expression in all transfected populations was both of equal level (Fig. 3C) and confirm that percentage of transfected cells was equivalent (not shown). The ectopic expression of CDK4 and cyclin D1 induced phosphorylation of RB with high efficiency, such that virtually all of the RB protein was hyperphosphorylated (Fig. 3C, lane 1), and cell cycle progression was stimulated (Fig. 3D). Consistent with previously published work (33), cyclin D1b proved to be a poor mediator of RB phosphorylation (Fig. 3C, lane 2). The differing activity between cyclin D1 and cyclin D1b could be due to several possible mechanisms. A stimulatory motif in the exon 5 encoded sequences could be responsible for the enhanced activity of cyclin D1. Conversely, an inhibitory motif encoded in intron 4 could be mediating the reduced activity of cyclin D1b. To differentiate between these possibilities, we developed the cyclin D1-ΔI4 allele, which encodes only exons 1 to 4 (Fig. 3B). This mutant behaved in a manner virtually identical to cyclin D1b (Fig. 3C, lane 4). Thus, the exon 5 encoded sequences are important for stimulating cyclin D1 activity against RB. Cyclins harbor multiple mechanisms through which they can mediate phosphorylation. To determine if the effects observed with cyclin D1b were occurring through CDK4 interactions, cyclin D1b was engineered to contain the K112E (KE) mutation (Fig. 3B). This mutation limits the ability of cyclin D1 to interact with CDK4 (41, 42). The cyclin D1b KE mutant was severely compromised for the phosphorylation of RB (Fig. 3C, lane 3). The ability to phosphorylate RB directly correlated with cell cycle progression, as measured by BrdUrd incorporation, in this model (Fig. 3D). Whereas both CDK4/cyclin D1b promoted BrdUrd incorporation, this activity is compromised relative to CDK4/cyclin D1 (P < 0.05). In contrast, cyclin D1b KE was incapable of promoting cell cycle progression (Fig. 3D). Thus, the activity of cyclin D1b in this model is apparently dependent on CDK4 activity. To confirm that CDK4 activity is critical for the effects of cyclin D1b in this model, PD-332991 (a highly specific inhibitor of CDK4) was used. As shown in Fig. 3E, PD-332991 strongly inhibited CDK4/cyclin D1b induced RB phosphorylation and cell cycle progression. Together, these data indicate that cyclin D1b acts through CDK4 to modulate cell cycle progression.
Figure 3.
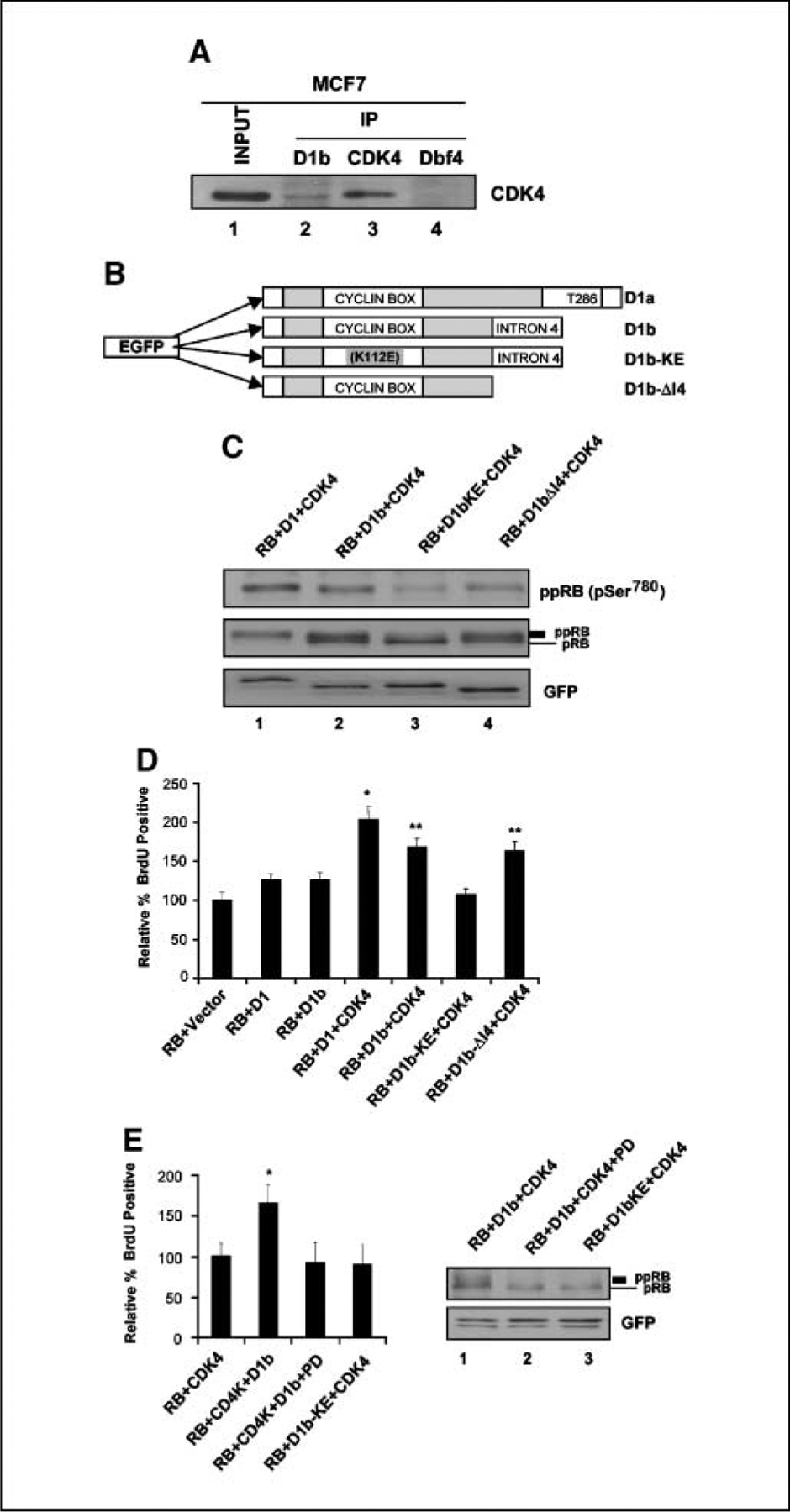
The functions of cyclin D1b strongly associate with CDK4 and its intron 4 regions. A, MCF-7 cells were harvested, and cell lysate was prepared and subjected to immunoprecipitation (IP) with specific cyclin D1b, CDK4, and Dbf4 antibodies. Input lysate and the resultant precipitated proteins were resolved by SDS-PAGE. CDK4 proteins were detected by immunoblotting. B, diagram depicting the mutant structures of cyclin D1b gene. The cyclin D1b–KE is a mutation of K112E. The mutant D1b-ΔI4 is encoded by the first four exons of cyclin D1 gene and the COOH terminal intron 4 has been deleted. Both D1b mutants are NH2 terminal GFP tagged proteins. C, SAOS-2 cells were cotransfected with the indicated expression plasmids. Total cellular protein was resolved by SDS-PAGE. RB, hyperphosphorylated RB, GFP-D1, D1b, and D1b mutant proteins were detected by immunoblotting. D, SAOS-2 cells were cotransfected with RB, CDK4, and the GFP-D1b, GFP-D1, and GFP-D1b mutants for 40 h and labeled with BrdUrd for 6 h. Cells were fixed and immunostained for relative BrdUrd incorporation to monitor S phase progression. Data shown are from two independent experiments with at least 200 cells counted for each experiment. *, P < 0.01 compared with RB + Vector; **, P < 0.05 compared with RB + D1 + CDK4. E, SAOS-2 cells were cotransfected and treated with vehicle (DMSO) or PD-0332991 as indicated. At 40 h posttransfections, cells were labeled with BrdUrd for 6 h. Cells were fixed and relative BrdUrd incorporation was determined from two independent experiments; *, P < 0.05 compared with RB + CDK4 (left). Lysates were prepared and RB detected by immunoblotting (right).
Cyclin D1b is subjected to aberrant regulation.
The aforementioned studies indicate that the exon 5 encoded sequences of cyclin D1 are critical for catalytic activity. However, these same sequences are also important in the regulation of cyclin D1 protein levels and localization (1, 31). Therefore, modulation of endogenous cyclin D1b protein levels was analyzed in response to stresses that influence the stability and regulation of cyclin D1 protein. The cyclin D1 protein has a relatively short half-life and its abundance can be dramatically enhanced via the inhibition of proteasomal degradation (43, 44). Consistent with these published studies, treatment of MCF-7 cells with the proteasome inhibitor MG132 led to accumulation of both endogenous cyclin D1 and a GFP–cyclin D1 introduced into the cells via transfection (Fig. 4A, top). In contrast, MG132 had little effect on cyclin D1b abundance, either with the endogenous protein or ectopically expressed GFP–cyclin D1b (Fig. 4A, bottom).
Figure 4.
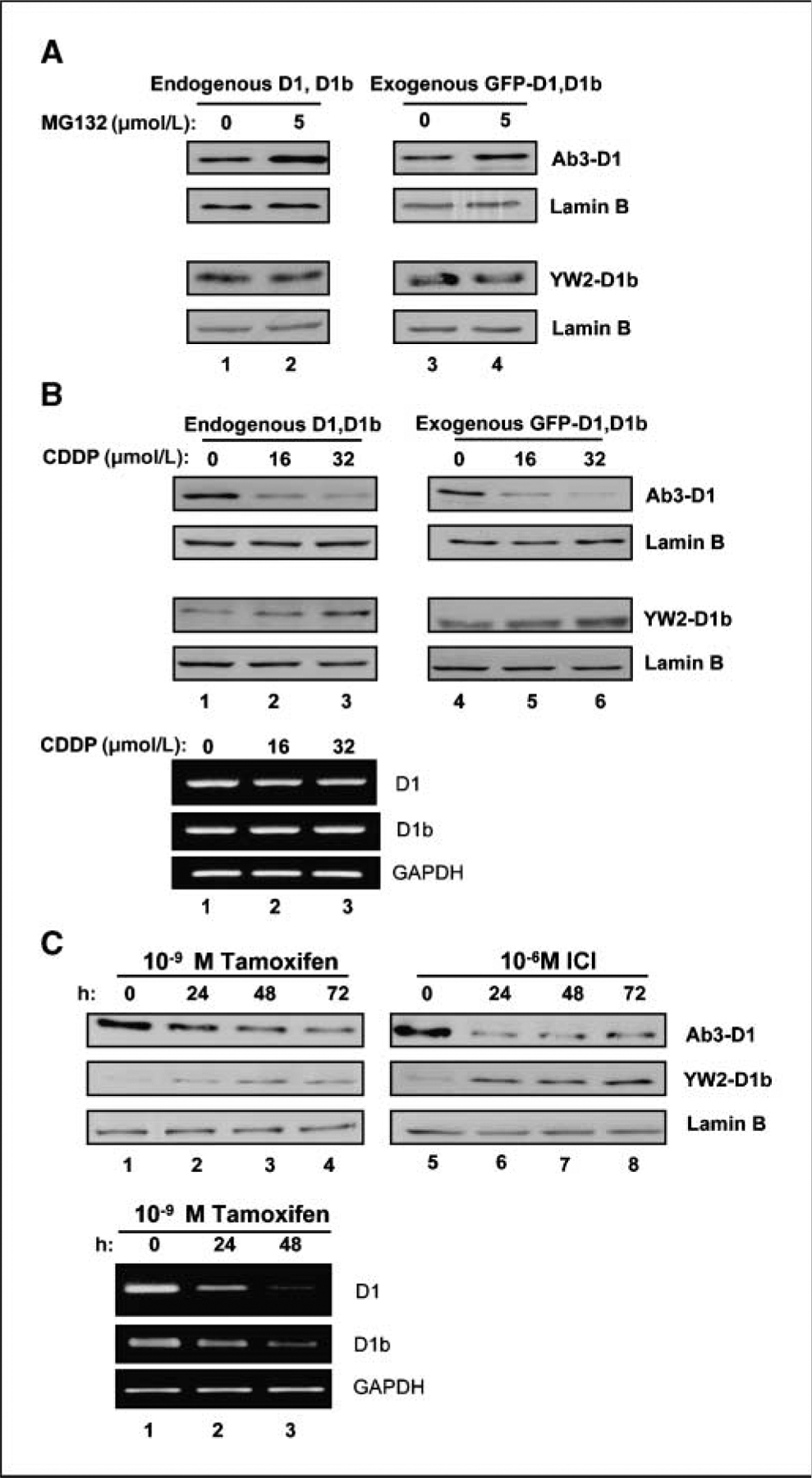
Distinct behaviors of cylin D1b protein compared with cyclin D1. A, MCF-7 cells were transfected with GFP–cyclin D1b or D1 expression plasmids. Cells were treated with 5 μm MG-132 for 6 h. Total cell lysate was resolved by SDS-PAGE. Endogenous D1 (left, top) and D1b (left, low) proteins were detected by immunoblotting. Exogenous GFP–cyclin D1 (right, top) and GFP–cyclin D1b (right, low) were blotted with GFP antibody. Lamin B was used as equal loading control. B, total protein or RNA was isolated from MCF-7 cells treated with CDDP for 16 h and resolved by SDS-PAGE. Western blot analysis for endogenous D1 (left, top), D1b (left, bottom), and exogenous GFP-D1 (right, top), GFP-D1b (right, bottom) was performed. Lamin B was used as a control for equal loading. RNA levels were evaluated by transcript specific RT-PCR, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a control for equal loading (bottom). C, MCF-7 cells were treated with either 10−9 mol/L Tam or 10−6 mol/L ICI for up to 72 h. Total cell lysates were harvested at indicated intervals. The effects of tamoxifen (left) and ICI (right) upon expression of cyclin D1 and cyclin D1b protein were detected by Western blot. Lamin B was used as equal loading control. RNA levels were evaluated by transcript specific RT-PCR, GAPDH is a control for equal loading (bottom).
In contrast with proteasomal inhibitors, DNA damage has been shown to mediate cyclin D1 degradation via NH2 terminal regulatory motifs, which are present in both cyclin D1 and cyclin D1b (43). As shown in Fig. 4B, cyclin D1 levels were efficiently down-regulated as a consequence of cisplatin-induced DNA damage (top). These results were apparent with both endogenous and exogenously expressed GFP–cyclin D1 protein. Surprisingly, neither endogenous cyclin D1b nor GFP–cyclin D1b levels were altered after cisplatin exposure (Fig. 4B, bottom). The influence of CDDP on protein levels was not associated with alteration in RNA levels, as there was no effect of CDDP on the RNA levels of either cyclin D1 transcript. Thus, cyclin D1b evades turnover downstream from DNA damage signaling pathways.
Lastly, we determined the influence of hormonal regulation on cyclin D1 variant expression. MCF-7 cells are an ER-positive breast cancer cell line and dependent on estrogen and ER activity for proliferation (45, 46). Estrogen withdrawal and estrogen antagonists,, such as tamoxifen and ICI, result in the attenuation of cyclin D1 expression (2, 45–47), which is believed to be important for the cell cycle inhibition achieved by such therapeutic modalities. Correspondingly, levels of cyclin D1 were diminished with both tamoxifen and ICI treatment (Fig. 4C). Surprisingly, during this same time course of estrogen antagonism, the levels of cyclin D1b were not significantly reduced and in fact became elevated. Consistent with published studies, cyclin D1 RNA levels were diminished in response to estrogen antagonists (47–51). Similarly, cyclin D1b RNA levels were attenuated with estrogen antagonists. Thus, the levels of cyclin D1b protein are not directly associated with its RNA levels and indicate that regulation of translation/protein stability are critical modulators of cyclin D1b protein levels. Combined, these studies indicate that the regulation of cyclin D1b protein levels occurs in a manner largely distinct from cyclin D1.
Cyclin D1b does not influence cisplatin DNA damage response but does specifically deregulate response to ER-antagonists.
Because cyclin D1b is refractory to degradation elicited by cisplatin damage, the ability of cyclin D1b to override the cisplatin induced checkpoints in MCF-7 cells was evaluated. MCF-7 cells were infected with adenoviruses that express GFP (control), cyclin D1, or cyclin D1b proteins. At 24 hours, postinfection cells were exposed to cisplatin at the indicated doses and harvested 16 hours posttreatment. As expected in the cells infected with GFP, cisplatin treatment lead to the specific loss of cyclin D1 protein while retaining cyclin D1b (Fig. 5A). Under this condition, RB is dephosphorylated, suggesting that the relatively low levels of cyclin D1b present in such cells are not sufficient to promote cell cycle progression. Ectopic expression of cyclin D1 was used to mimic the overproduction of cyclin D1 proteins observed in tumors. As expected, the overall levels of cyclin D1 protein increased, but the accumulated protein was efficiently degraded, consistent with our findings using GFP–cyclin D1. In the case of cyclin D1b, the exogenously expressed cyclin D1b was refractory to the effects of cisplatin. However, the protein was incapable of maintaining RB phosphorylation. Furthermore, neither cyclin D1 nor cyclin D1b was sufficient to overcome the CDDP-mediated checkpoint (Fig. 5B and C). Analyses of p21Cip1 levels revealed a significant up-regulation of p21Cip1 (Fig. 5A), suggesting that the action of this CDK inhibitor may mediate the cessation of cell cycle progression even in the presence of significant cyclin D1b protein.
Figure 5.

Cyclin D1b overexpression in MCF-7 cells does not affect the DNA damage checkpoint. A, MCF-7 cells were infected with adenovirus encoding cyclin D1b, cyclin D1, and GFP (control) for 24 h. Cells were then treated with CDDP for 16 h. Total cell lysates were harvested and analyzed by immunoblotting with antibodies to RB, cyclin A, cyclin D1, cyclin D1b, and p21Cip1. Lamin B was used as a loading control. B, MCF-7 cells were treated as described in A of same figure. After treatment with CDDP, cells were pulse labeled with BrdUrd for 1 h, followed by BrdUrd staining and propidium iodide staining. Bivariate flow cytometry analysis of DNA synthesis (BrdUrd incorporation) and DNA content (PI) was performed. Shown are representative histograms of 20,000 gated events from two independent experiments. Quantitation of the percentage of BrdUrd-positive cells are from two independent bivariate flow cytometry experiments.
To further define a role for cyclin D1b in response to antimitogenic therapeutics, the response to estrogen antagonists was determined. For these analyses, MCF-7 cells were infected with adenoviruses encoding GFP, cyclin D1, and cyclin D1b and subjected to estrogen deprivation in combination with tamoxifen or ICI exposure. Consistent with the literature and previous findings (45–51), both estrogen antagonists led to down-regulation of cyclin D1 levels concomitant with RB dephosphorylation in control (GFP-infected) cells (Fig. 6A). This effect of estrogen antagonists was similarly associated with the cessation of cell cycle progression as monitored via bivariate flow cytometry (Fig. 6B and C). The ectopic expression of cyclin D1 was maintained in the presence of tamoxifen and ICI (Fig. 6A). However, this event was not sufficient to maintain RB phosphorylation, and correspondingly, downstream target gene expression was repressed (e.g., cyclin A). Consistent with these outcomes, cell cycle inhibition was elicited by estrogen antagonists in the presence of cyclin D1 expression (Fig. 6B and C). In contrast, exogenous expression of cyclin D1b maintained RB phosphorylation in the presence of both tamoxifen and ICI. Consistent with these findings, cyclin D1b expression maintained RB target gene expression (e.g., cyclin A levels) in the presence of tamoxifen and ICI. These molecular findings were further corroborated as cyclin D1b expressing cells efficiently bypassed the cell cycle inhibition mediated by tamoxifen or ICI (Fig. 6B and C). Whereas p21Cip1 is implicated in mediating DNA damage response pathways, p27Kip1 is crucial for response to estrogen antagonists (52). Analyses of p27Kip1 expression showed significant up-regulation in both GFP and cyclin D1–transduced cells with tamoxifen or ICI exposure (Fig. 6A). In contrast, cyclin D1b ectopic expression reduced the levels of p27Kip1 in MCF-7 cells and blunted the up-regulation observed upon treatment of estrogen antagonists (Fig. 6A). To determine whether the cyclin D1b transduced cells retained the capacity to proliferate in the presence of estrogen antagonists, MTT analysis was performed. MCF-7 cells were infected with cyclin D1 and cyclin D1b encoding adenoviruses. Cells were then cultured either in the presence of full steroid (FBS) or in the absence of steroids and supplemented with tamoxifen or ICI. As shown in Fig. 6D, infected cells expressing cyclin D1 or cyclin D1b proliferated with similar kinetics in the presence of estrogen. However, in the presence of estrogen antagonists cyclin D1b provided a significant proliferative advantage over cyclin D1 (Fig. 6D). Thus, exogenous expression of cyclin D1b is capable of potently promoting both cell cycle progression and proliferation in the presence of estrogen antagonism.
Figure 6.
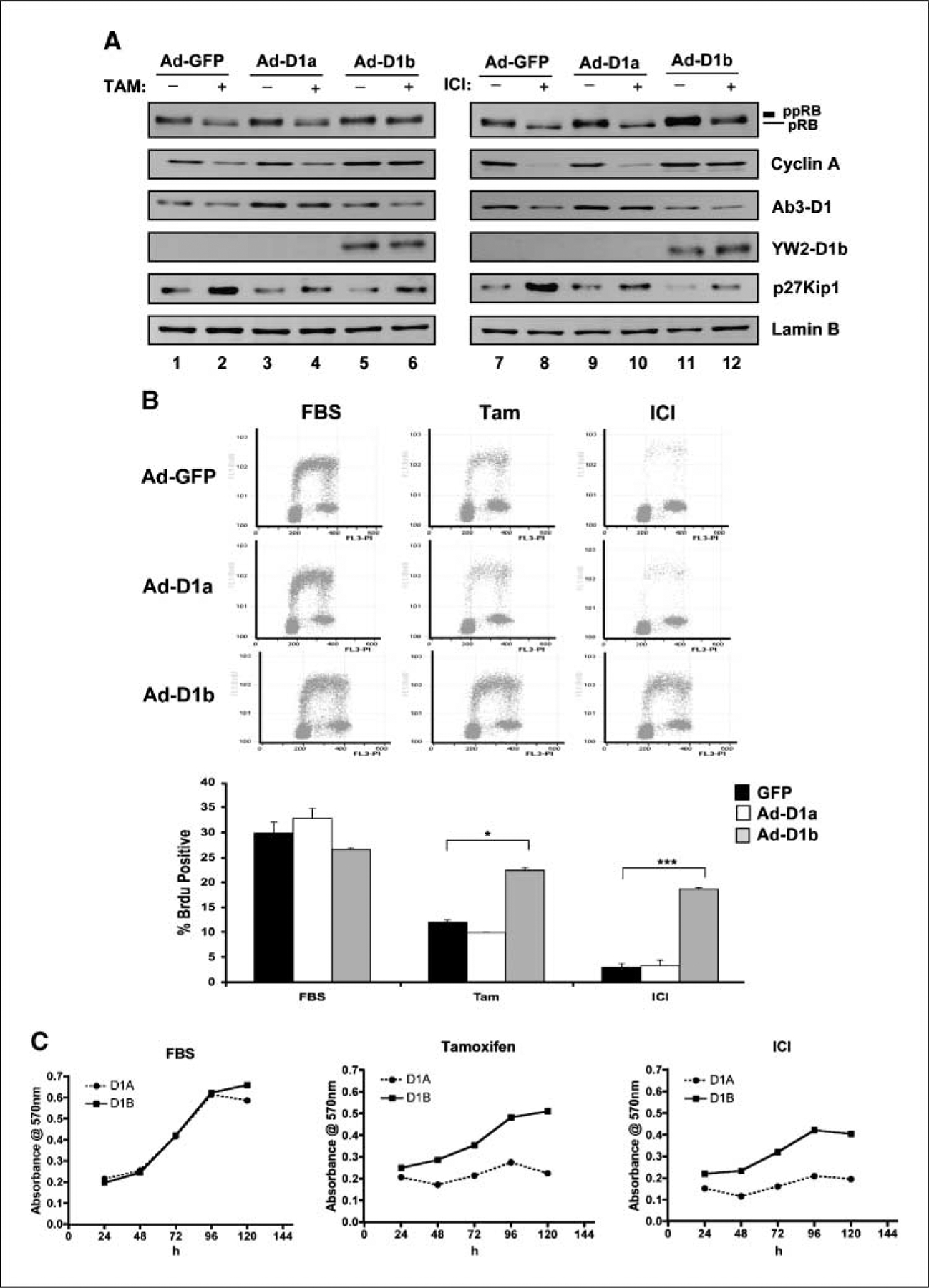
Cyclin D1b overexpression in MCF-7 cells imparts resistance to antiestrogen therapy. A, MCF-7 cells were infected with adenovirus encoding cyclin D1b and cyclin D1, GFP (control) for 24 h. Next, cells were cultured in complete medium, CDT/Tam (left), or CDT/ICI (right) for another 36 h. Cells were harvested and analyzed by immunoblotting with antibodies to RB, cyclin A, cyclin D1, cyclin D1b, and p27Kip1. Lamin B was used as a loading control. B, MCF-7 cells were treated as described in A of the same figure. Bivariate flow cytometry was performed. Representative histograms of 20,000 gated events from two independent experiments. Quantitation of the percentage of BrdUrd-positive cells are from two independent bivariate flow cytometry experiments; *, P < 0.05; ***, P < 0.001 compared with GFP. C, MCF-7 cells infected with adenoviruses encoding either cyclin D1 or cyclin D1b were plated and cultured in FBS, CDT/Tam, or CDT/ICI for the indicated time and then processed for MTT analyses. Four replicates were performed, and the SD in the data are shown.
Bypass of tamoxifen response is dependent on CDK4 interaction.
The cyclin D1 protein has been postulated to contribute to deregulation of estrogen dependence via two mechanisms. First, cyclin D1 can function as an estrogen receptor coactivator, and it has been suspected that this function could contribute to tamoxifen resistance (19, 20, 53). Therefore, the influence of cyclin D1 and cyclin D1b on ER activity was evaluated. Using reporter assays, it was confirmed that cyclin D1 cooperates to stimulate the transcriptional activity of the ER (19). However, cyclin D1b does not share this activity and is thus not competent in ER coactivation (Fig. 7A). This result is not surprising, as cyclin D1b lacks the LXXL motif that has been shown to be critical for cyclin D1 function in ER regulation. Thus, these findings suggest that cyclin D1 function in this system could involve CDK4 activity (3, 54). Therefore, cyclin D1b mutants were used to assess the effect of CDK4 interaction on overcoming estrogen antagonists. MCF-7 cells were transfected with expression plasmids encoding the indicated proteins and subsequently challenged by culture in tamoxifen or ICI containing media. Under this condition, vector transfected cells incurred an inhibition of BrdUrd incorporation. Consistent with the preceding data, the cell cycle inhibitory effects of tamoxifen or ICI could be largely reversed via the expression of cyclin D1b. The cyclin D1ΔI4 protein behaved in a manner consistent with cyclin D1b. The KE mutant, which fails to stimulate CDK4 activity, was deficient in bypassing tamoxifen-mediated or ICI-mediated cell cycle inhibition (Fig. 7B). Therefore, these data suggest that cyclin D1b uses CDK-dependent mechanisms to stimulate cell cycle progression in the presence of estrogen antagonists.
Figure 7.

Effects of cyclin D1b on the transcriptional activity of ER and requirement for CDK4 activity in response to antiestrogen therapies. A, MCF-7 cells were cotransfected and treated as described in Materials and Methods with cytomegalovirus β-galactosidase reporter and ERE-Luciferase reporter plasmids. Cells were harvested and assayed for luciferase activity, which was normalized to β-galactosidase activity. Data are from three independent experiments. B, MCF-7 cells were cotransfected with H2B-GFP and indicated plasmids for 16 h. Then, cells were washed with PBS and replaced in phenol red–free DMEM containing 10% CDT with either 10−9 mol/L 4-hydroxy tamoxifen or 10−6 mol/L ICI for 36 h. Cells were pulsed with BrdUrd for 6 h, and BrdUrd incorporation was monitored via indirect immunofluoresence. Two independent experiments with at least 200 cells counted per experiment. Error bars, SD. *, P < 0.05 compared with vector.
Discussion
The cyclin D1 locus has been critically implicated in tumorigenesis (1, 2, 4, 25). Whereas the activity of the canonical encoded cyclin D1 protein is well established, surprisingly little is known regarding the cyclin D1b protein, which is produced as a result of alternative splicing. The polymorphism that is believed to contribute to the expression of cyclin D1b transcript has an allele frequency of 40%, and thus could represent a significant participant in cancers driven by cyclin D1 gene amplification or translocations (4, 25). Interestingly, using RT-PCR approaches, it has become clear that cyclin D1b transcript levels are quite high in tumors, supporting the speculation that cyclin D1b may function in tumorigenesis (29, 55). Here, we quantitatively interrogated the affinity and specificity of an antibody specific to cyclin D1b. Utilizing this reagent, it is apparent that cyclin D1b protein is expressed in a significant fraction of tumor cell lines. Furthermore, cyclin D1b protein levels are clearly elevated in primary breast cancers. Interestingly, in a number of asynchronously proliferating cell lines, the levels of cyclin D1b represent <10% of the total cyclin D1 protein in the cell line. This result is surprising given the relatively similar levels of cyclin D1 and cyclin D1b transcripts were detected in cell lines and tumor specimens (26, 56). Thus, it would seem that the alternatively spliced cyclin D1b transcript, while produced, is not efficiently translated. This finding also explains why the detection of cyclin D1b protein is difficult in the absence of specialized reagents for detection (27, 32). However, there are clear exceptions, wherein cyclin D1b seems to be the predominant cyclin D1 species in specific cell lines (e.g., BT549). Strikingly, the levels of cyclin D1b could play significant roles in tumor behavior under specific conditions, wherein translation of the cyclin D1b transcript could become more favorable or the levels of the protein become predominant.
The regulation of the cyclin D1 protein levels is subject to complex transcriptional and posttranscriptional mechanisms. Mitogenic signaling pathways are believed to effect cyclin D1 protein levels primarily via the regulation of cyclin D1 RNA levels and modulation of protein stability via the T286 phosphorylation site (1, 4, 25, 30, 57). This regulatory motif is compromised in tumors harboring specific mutations and in synthetic alleles which surgically mutate the T286 site (31, 58, 59). Therefore, it is perhaps not surprising that regulation of cyclin D1b protein stability is distinct from that observed with cyclin D1. However, it has been reported that sites in the NH2 terminus of cyclin D1 modulate protein stability after DNA damage (43). As such, it was surprising that cyclin D1b escaped degradation mediated by cisplatin damage, whereas cyclin D1 was readily degraded under identical conditions. Presumably, this enhanced stability is not a reflection of differences in nucleo/cytoplasmic shuttling as the T286A allele of cyclin D1, which is exclusively nuclear, is sensitive to DNA damage–mediated degradation (43). Thus, the mechanism through which cyclin D1b evades such signaling is at present unknown. It is well appreciated that cyclin D1 levels are regulated by multiple mechanisms via estrogen antagonists in ER-positive breast cancer cells (45–51). Surprisingly, cyclin D1b also evades this form of regulation and in fact cyclin D1b levels are stimulated in the presence of estrogen antagonists. As such, the ratio of cyclin D1b to cyclin D1 can be dramatically altered in various conditions of challenge (4.5-fold increased in DNA damage response and 11-fold increased with estrogen antagonist). However, the level of endogenous cyclin D1b is apparently not sufficient to preclude cell cycle arrest mediated by estrogen antagonists or DNA damaging agents in MCF-7 cells as these cells arrest although the expression of cyclin D1b is retained.
The cyclin D1 locus is subject to amplification in primary breast cancer and overexpression of cyclin D1 through additional mechanisms is observed in >50% of breast cancer (2, 49). Based on our data in primary tumors, overexpression of cyclin D1b could contribute to the pathology of breast cancer and may be particularly relevant under those conditions wherein D1b would be the predominant cyclin D1 moiety. Similar results were previously observed with primary prostate cancer specimens, wherein the cyclin D1b transcript was specifically induced in tumor tissue versus matched nonneoplastic controls. Under conditions of cisplatin treatment, MCF-7 cells incur a strong antiproliferative arrest accompanied by the degradation of cyclin D1 and dephosphorylation of RB. The status of RB is critically important for this response as MCF-7 cells rendered RB-deficient fail to elicit cell cycle arrest in the presence of cisplatin damage (60). Strikingly, overexpression of cyclin D1b is not sufficient to overcome the effects of cisplatin at the molecular level or bypass the DNA damage checkpoint. As such, the alterations in cyclin D1b stability are not apparently influencing the response to this form of challenge. However, the overexpression of cyclin D1b did efficiently overcome the effects of estrogen antagonists. This finding suggests that high levels of cyclin D1b expression could have a deleterious effect on response to hormone therapies. Additional studies will be required to define this action of cyclin D1b in primary tumors.
The mechanism through which cyclin D1b exerts its influence in cancer has remained largely obscure. Prior studies indicate that cyclin D1b is a more potent oncogene in classic fibroblast transformation models and has a propensity to impart anchorage-independent growth to cells (32, 33). This response is associated with the constitutive nuclear localization of the protein. However, the mechanism through which this occurs is not clear, particularly because cyclin D1b has impaired catalytic activity relative to cyclin D1 (33). We show that this impairment is due to the lack of the COOH terminal region. Thus, paradoxically, the exon 5 encoded sequences of cyclin D1 impart enhanced catalytic activity against critical substrates of CDK4, yet also enhance susceptibility to degradation mediated via a variety of antimitogenic signaling cascades. Whereas it is appealing to speculate that the effect of cyclin D1b on susceptibility to estrogen antagonists could be through a direct effect on ER function, we find that cyclin D1b is not an effector of ER, and conversely the interaction of cyclin D1b with CDK4 is crucial for this activity. These results are in agreement with structure function analyses carried out in T47D cells (61) and suggest that aberrant modulation of the CDK4 assembled complexes underlies the action of cyclin D1b proteins on therapeutic response. Combined, these studies underscore the aberrant regulation and function of cyclin D1b and indicate that this protein could be particularly important in the context of hormonal therapy for ER-positive breast cancer.
Acknowledgments
Grant support:
Supported by NIH grants R01-CA104213 (to E.S. Knudsen), R01-CA101841 (to H. Rui), and CA099996 (to K.E. Knudsen); National Health and Medical Research Council of Australia, The Cancer Institute NSW, Australian Cancer Research Foundation, RT Hall Trust, and The Petre Foundation (to R.L. Sutherland).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Diehl JA. Cycling to cancer with cyclin D1. Cancer Biol Ther 2002;1:226–31. [DOI] [PubMed] [Google Scholar]
- 2.Arnold A, Papanikolaou A. Cyclin D1 in breast cancer pathogenesis. J Clin Oncol 2005;23:4215–24. [DOI] [PubMed] [Google Scholar]
- 3.Sherr CJ. D-type cyclins. Trends Biochem Sci 1995;20:187–90. [DOI] [PubMed] [Google Scholar]
- 4.Knudsen KE, Diehl JA, Haiman CA, Knudsen ES. Cyclin D1: polymorphism, aberrant splicing and cancer risk. Oncogene 2006;25:1620–8. [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ. Mammalian G1 cyclins and cell cycle progression. Proc Assoc Am Physicians 1995;107:181–6. [PubMed] [Google Scholar]
- 6.Xiong Y, Connolly T, Futcher B, Beach D. Human D-type cyclin. Cell 1991;65:691–9. [DOI] [PubMed] [Google Scholar]
- 7.Lukas J, Pagano M, Staskova Z, Draetta G, Bartek J. Cyclin D1 protein oscillates and is essential for cell cycle progression in human tumour cell lines. Oncogene 1994;9: 707–18. [PubMed] [Google Scholar]
- 8.Lukas J, Bartkova J, Bartek J. Convergence of mitogenic signalling cascades from diverse classes of receptors at the cyclin D-cyclin-dependent kinase-pRb-controlled G1 checkpoint. Mol Cell Biol 1996;16: 6917–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnold A, Motokura T, Bloom T, et al. PRAD1 (cyclin D1): a parathyroid neoplasia gene on 11q13. Henry Ford Hosp Med J 1992;40:177–80. [PubMed] [Google Scholar]
- 10.Motokura T, Bloom T, Kim HG, et al. A novel cyclin encoded by a bcl1-linked candidate oncogene. Nature 1991;350:512–5. [DOI] [PubMed] [Google Scholar]
- 11.Bartkova J, Lukas J, Muller H, Lutzhoft D, Strauss M, Bartek J. Cyclin D1 protein expression and function in human breast cancer. Int J Cancer 1994;57: 353–61. [DOI] [PubMed] [Google Scholar]
- 12.Yu Q, Geng Y, Sicinski P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001;411: 1017–21. [DOI] [PubMed] [Google Scholar]
- 13.Wang TC, Cardiff RD, Zukerberg L, Lees E, Arnold A, Schmidt EV. Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature 1994;369:669–71. [DOI] [PubMed] [Google Scholar]
- 14.Matsushime H, Ewen ME, Strom DK, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 1992;71:323–34. [DOI] [PubMed] [Google Scholar]
- 15.Matsushime H, Quelle DE, Shurtleff SA, Shibuya M, Sherr CJ, Kato JY. D-type cyclin-dependent kinase activity in mammalian cells. Mol Cell Biol 1994;14: 2066–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ewen ME, Sluss HK, Sherr CJ, Matsushime H, Kato J, Livingston DM. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 1993;73:487–97. [DOI] [PubMed] [Google Scholar]
- 17.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev 1993;7:331–42. [DOI] [PubMed] [Google Scholar]
- 18.Lukas J, Bartkova J, Rohde M, Strauss M, Bartek J. Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol Cell Biol 1995;15:2600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neuman E, Ladha MH, Lin N, et al. Cyclin D1 stimulation of estrogen receptor transcriptional activity independent of cdk4. Mol Cell Biol 1997;17:5338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997; 88:405–15. [DOI] [PubMed] [Google Scholar]
- 21.Knudsen KE, Cavenee WK, Arden KC. D-type cyclins complex with the androgen receptor and inhibit its transcriptional transactivation ability. Cancer Res 1999; 59:2297–301. [PubMed] [Google Scholar]
- 22.Petre CE, Wetherill YB, Danielsen M, Knudsen KE. Cyclin D1: mechanism and consequence of androgen receptor co-repressor activity. J Biol Chem 2002;277: 2207–15. [DOI] [PubMed] [Google Scholar]
- 23.Lamb J, Ramaswamy S, Ford HL, et al. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell 2003;114:323–34. [DOI] [PubMed] [Google Scholar]
- 24.Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 2006;9: 13–22. [DOI] [PubMed] [Google Scholar]
- 25.Knudsen KE. The cyclin D1b splice variant: an old oncogene learns new tricks. Cell Div 2006;1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene 1995;11:1005–11. [PubMed] [Google Scholar]
- 27.Hosokawa Y, Joh T, Maeda Y, Arnold A, Seto M. Cyclin D1/PRAD1/BCL-1 alternative transcript [B] protein product in B-lymphoid malignancies with t(11;14)(q13;q32) translocation. Int J Cancer 1999;81: 616–9. [DOI] [PubMed] [Google Scholar]
- 28.Hosokawa Y, Gadd M, Smith AP, Koerner FC, Schmidt EV, Arnold A. Cyclin D1 (PRAD1) alternative transcript b: full-length cDNA cloning and expression in breast cancers. Cancer Lett 1997;113:123–30. [DOI] [PubMed] [Google Scholar]
- 29.Bala S, Peltomaki P. CYCLIN D1 as a genetic modifier in hereditary nonpolyposis colorectal cancer. Cancer Res 2001;61:6042–5. [PubMed] [Google Scholar]
- 30.Gladden AB, Diehl JA. Location, location, location: the role of cyclin D1 nuclear localization in cancer. J Cell Biochem 2005;96:906–13. [DOI] [PubMed] [Google Scholar]
- 31.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev 2000;14:3102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res 2003;63:7056–61. [PubMed] [Google Scholar]
- 33.Solomon DA, Wang Y, Fox SR, et al. Cyclin D1 splice variants. Differential effects on localization, RB phosphorylation, and cellular transformation. J Biol Chem 2003;278:30339–47. [DOI] [PubMed] [Google Scholar]
- 34.Hess-Wilson JK, Boldison J, Weaver KE, Knudsen KE. Xenoestrogen action in breast cancer: impact on ER-dependent transcription and mitogenesis. Breast Cancer Res Treat 2006;96:279–92. [DOI] [PubMed] [Google Scholar]
- 35.Strobeck MW, Fribourg AF, Puga A, Knudsen ES. Restoration of retinoblastoma mediated signaling to Cdk2 results in cell cycle arrest. Oncogene 2000;19: 1857–67. [DOI] [PubMed] [Google Scholar]
- 36.Sawa H, Ohshima TA, Ukita H, et al. Alternatively spliced forms of cyclin D1 modulate entry into the cell cycle in an inverse manner. Oncogene 1998;16:1701–12. [DOI] [PubMed] [Google Scholar]
- 37.Burd CJ, Petre CE, Morey LM, et al. Cyclin D1b variant influences prostate cancer growth through aberrant androgen receptor regulation. Proc Natl Acad Sci U S A 2006;103:2190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.LeBaron MJ, Crismon HR, Utama FE, et al. Ultrahigh density microarrays of solid samples. Nat Methods 2005; 2:511–3. [DOI] [PubMed] [Google Scholar]
- 39.Dolled-Filhart M, Ryden L, Cregger M, et al. Classification of breast cancer using genetic algorithms and tissue microarrays. Clin Cancer Res 2006; 12:6459–68. [DOI] [PubMed] [Google Scholar]
- 40.Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992;70:993–1006. [DOI] [PubMed] [Google Scholar]
- 41.Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci U S A 1994;91:709–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA. Physical interaction of the retinoblastoma protein with human D cyclins. Cell 1993;73:499–511. [DOI] [PubMed] [Google Scholar]
- 43.Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 2000;102: 55–66. [DOI] [PubMed] [Google Scholar]
- 44.Bagui TK, Mohapatra S, Haura E, Pledger WJ. P27Kip1 and p21Cip1 are not required for the formation of active D cyclin-cdk4 complexes. Mol Cell Biol 2003;23: 7285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster JS, Henley DC, Bukovsky A, Seth P, Wimalasena J. Multifaceted regulation of cell cycle progression by estrogen: regulation of Cdk inhibitors and Cdc25A independent of cyclin D1–4 function. Mol Cell Biol 2001;21:794–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prall OW, Sarcevic B, Musgrove EA, Watts CK, Sutherland RL. Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J Biol Chem 1997;272:10882–94. [DOI] [PubMed] [Google Scholar]
- 47.Watts CK, Brady A, Sarcevic B, deFazio A, Musgrove EA, Sutherland RL. Antiestrogen inhibition of cell cycle progression in breast cancer cells in associated with inhibition of cyclin-dependent kinase activity and decreased retinoblastoma protein phosphorylation. Mol Endocrinol 1995;9:1804–13. [DOI] [PubMed] [Google Scholar]
- 48.Musgrove EA, Hamilton JA, Lee CS, Sweeney KJ, Watts CK, Sutherland RL. Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol Cell Biol 1993;13: 3577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buckley MF, Sweeney KJ, Hamilton JA, et al. Expression and amplification of cyclin genes in human breast cancer. Oncogene 1993;8:2127–33. [PubMed] [Google Scholar]
- 50.Carroll JS, Prall OW, Musgrove EA, Sutherland RL. A pure estrogen antagonist inhibits cyclin E-Cdk2 activity in MCF-7 breast cancer cells and induces accumulation of p130–2F4 complexes characteristic of quiescence. J Biol Chem 2000;275:38221–9. [DOI] [PubMed] [Google Scholar]
- 51.Hui R, Finney GL, Carroll JS, Lee CS, Musgrove EA, Sutherland RL. Constitutive overexpression of cyclin D1 but not cyclin E confers acute resistance to antiestrogens in T-47D breast cancer cells. Cancer Res 2002;62: 6916–23. [PubMed] [Google Scholar]
- 52.Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A 2000;97:9042–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zwijsen RM, Buckle RS, Hijmans EM, Loomans CJ, Bernards R. Ligand-independent recruitment of steroid receptor coactivators to estrogen receptor by cyclin D1. Genes Dev 1998;12:3488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sherr CJ. Cell cycle control and cancer. Harvey Lect 2000;96:73–92. [PubMed] [Google Scholar]
- 55.Howe D, Lynas C. The cyclin D1 alternative transcripts [a] and [b] are expressed in normal and malignant lymphocytes and their relative levels are influenced by the polymorphism at codon 241. Haematologica 2001;86:563–9. [PubMed] [Google Scholar]
- 56.Hosokawa Y, Arnold A. Mechanism of cyclin D1 (CCND1, PRAD1) overexpression in human cancer cells: analysis of allele-specific expression. Genes Chromosomes Cancer 1998;22:66–71. [DOI] [PubMed] [Google Scholar]
- 57.Alao JP. The regulation of cyclin D1 degradation: roles in cancer development and the potential for therapeutic invention. Mol Cancer 2007;6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Benzeno S, Lu F, Guo M, et al. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene 2006;25:6291–303. [DOI] [PubMed] [Google Scholar]
- 59.Moreno-Bueno G, Rodriguez-Perales S, Sanchez-Estevez C, et al. Cyclin D1 gene (CCND1) mutations in endometrial cancer. Oncogene 2003;22:6115–8. [DOI] [PubMed] [Google Scholar]
- 60.Bosco EE, Wang Y, Xu H, et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest 2007;117:218–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bindels EM, Lallemand F, Balkenende A, Verwoerd D, Michalides R. Involvement of G1-S cyclins in estrogen-independent proliferation of estrogen receptor-positive breast cancer cells. Oncogene 2002;21:8158–65. [DOI] [PubMed] [Google Scholar]