

Abstract
Introduction
Women or girls with haemophilia (WGH) represent a group of female symptomatic carriers who experience bleeding events more frequently than non‐carriers. Bleeding events include spontaneous/traumatic bleeds and prolonged bleeding related to surgery, menstruation and pregnancy. Challenges for the treatment of WGH include lack of screening, diagnosis and treatment guidelines.
Aim
Evaluate clinical characteristics, haemostasis management and clinical outcomes regarding menstruation, childbirth, dental procedures, surgeries and other bleeding events in WGH.
Methods
A retrospective, non‐interventional review of medical records from WGH among three haemophilia treatment centres (HTCs) was conducted in the United States (2012–2018). Patients with ≥2 visits to the HTC and who had undergone intervention for haemostasis management with the outcome documented were included. Descriptive statistics were used.
Results
Of 47 women and girls included in the chart review (37 with factor VIII deficiency, 10 with factor IX deficiency), median age at diagnosis was 22.6 years. Approximately 79% (n = 37) were diagnosed with mild haemophilia. Events of interest were primarily managed by factor concentrates or antifibrinolytics. Most treatment approaches were successful across clinical scenarios, except for heavy menstrual bleeding being insufficiently controlled in 8 (57%) of the 14 patients who experienced it.
Conclusions
Bleeding events in WGH, such as excessive and prolonged bleeding during menstruation, demonstrate a unique burden and require specific medical intervention. These results highlight the importance of assessing the need for haemostasis management in WGH and may contribute to future prospective study designs.
Keywords: bleeding, carriers, factor IX, factor VIII, female, haemophilia, treatment
1. INTRODUCTION
Female carriers of haemophilia, an X‐linked recessive disorder, have one normal and one abnormal allele of the factor VIII (FVIII; haemophilia A) or factor IX (FIX; haemophilia B) gene. 1 , 2 Carriers may be obligate or possible carriers of haemophilia, depending on whether they have inherited the haemophilia gene from their father or mother, respectively. 3 Furthermore, this population may be diagnosed with mild FVIII or FIX deficiency and yet have normal factor levels, depending on when they are tested.
Carriers may be clinically asymptomatic, although those with low (<0.40 IU/ml) and mildly reduced (0.41–0.60 IU/ml) clotting factor levels typically bleed more than women and girls who are not haemophilia carriers. 1 , 4 One‐third of carriers are reported to have factor levels <60% of normal, resulting in an increased bleeding tendency. 3 Additionally, haemophilia A carriers with normal clotting factor levels may have an increased bleeding tendency. 4 , 5 , 6 , 7 , 8
The term women and girls with haemophilia (WGH) describes a population of symptomatic carriers. 9 Bleeding manifestations may include easy bruising, epistaxis and prolonged or serious bleeding after surgery, dental procedures or trauma. 3 , 5 , 6 , 7 Additionally, WGH may experience problems with their reproductive health, including heavier and more prolonged bleeding during menstruation, childbirth and postpartum. 3 , 10 , 11 WGH often have reduced health‐related quality of life, adverse psychosocial effects and increased time lost from work compared with women and girls who are not haemophilia carriers. 12 , 13 , 14
Current clinical challenges for WGH include a lack of screening, diagnosis, clear nomenclature and treatment guidelines. In general, bleeding disorders in women and girls have, to date, been under‐recognised 11 , 13 ; thus, the prevalence of factor deficiency among women and girls is unknown. There are a number of initiatives aimed at addressing these issues, including the Centers for Disease Control and Prevention Community Counts programme, 15 My Life, Our Future 16 and the Cornerstone Initiative. 17
The objective of this study was to describe the characteristics of WGH and evaluate haemostasis management and clinical outcomes during menstruation, childbirth, dental procedures, surgeries and other spontaneous bleeding episodes.
2. METHODS
This was a non‐interventional, retrospective, multicentre review of medical records of WGH from three haemophilia treatment centres (HTC) in the United States between 1 April 2012 and 15 November 2018.
WGH with or without genetic testing were included, and patients had to have had at least two visits to the HTC and undergone medical or surgical intervention for haemostasis management, with the outcome documented in the medical chart. Women and girls who were obligate or potential carriers of haemophilia were determined by a history of excessive bleeding and supported by, if possible, the presence of at least one of the following: family history of biological father and/or sons diagnosed with FVIII or FIX deficiency, positive genetic test for FVIII or FIX genes or persistent FVIII or FIX deficiency. WGH could be diagnosed with mild FVIII or FIX deficiency and have normal factor levels at the time of their testing. Patients with any other coagulation disorder were excluded, with the exception of low von Willebrand factor (VWF), VWF antigen or VWF ristocetin cofactor 0.4–0.5 IU/ml to allow the inclusion of patients with blood type O and those who had low VWF without von Willebrand disease. Institutional review board approval was obtained where relevant, and de‐identified patient data are presented.
The primary endpoint was the clinical presentations and assessments of menstruation, peripartum/postpartum (pregnancy), surgery and major dental procedures, as well as spontaneous, traumatic, joint and minor dental bleeds. The secondary endpoints included efficacy of medical management during haemostatic challenges in this cohort. Descriptive statistics were used to summarise results, with no inferential statistics applied.
3. RESULTS
3.1. Patient population
Forty‐seven patients were identified as WGH among three combined paediatric and adult HTCs in the United States (Table 1), and among these, 37 were diagnosed with FVIII deficiency and 10 were diagnosed with FIX deficiency. The median (range) age at diagnosis was 22.6 (0–72) years (median age was 25.3 and 5.7 years for WGH with FVIII deficiency and FIX deficiency, respectively). The median (range) age at first visit to the HTC was 28.1 (0.4–72) years (median age was 28.3 and 6.7 years for WGH with FVIII deficiency and FIX deficiency, respectively), indicating that there was a gap between diagnosis and initial visit to the HTC. Of the 26 WGH with known referring sources, haematologists outside the HTC, family doctors and paediatricians constituted 81% (21/26) of the known referrals. All WGH with FIX deficiency (100%) and 70% with FVIII deficiency were obligate carriers.
TABLE 1.
Demographics and disease characteristics of WGH cohort as documented at the first visit to the HTC a
| Overall (N = 47) | FVIII deficiency (n = 37) | FIX deficiency (n = 10) | |
|---|---|---|---|
| Median (range) age at the first HTC visit, years | 28.1 (0.4–72.0) | 28.3 (0.4–72.0) | 6.7 (0.6–57.5) |
| Genotype, n (%) | |||
| Obligate carrier b | 36 (76.6) | 26 (70.3) | 10 (100.0) |
| Potential carrier b | 11 (23.4) | 11 (29.7) | 0 (0.0) |
| Median (range) age at diagnosis, years | 22.6 (0.0–72.0) | 25.3 (0.1–72.0) | 5.7 (0.0–57.7) |
| Diagnosis confirmation by, n (%) | |||
| Family history c | 41 (87.2) | 31 (83.8) | 10 (100.0) |
| Genetic testing c | 19 (40.4) | 16 (43.2) | 3 (30.0) |
| Laboratory results c | 38 (80.9) | 29 (78.4) | 9 (90.0) |
| Not confirmed | 1 (2.1) | 1 (2.7) | 0 (0.0) |
| Menarche status | |||
| Reached | 38 (80.9) | 31 (83.8) | 7 (70.0) |
| Not reached yet | 7 (14.9) | 5 (13.5) | 2 (20.0) |
| Unknown | 2 (4.3) | 1 (2.7) | 1 (10.0) |
| Age at menarche, years | |||
| n | 11 | 8 | 3 |
| Median (range) | 12 (11–14) | 12 (11–14) | 12 (12–12) |
| Family history of haemophilia, n (%) | |||
| Yes | 42 (89.4) | 32 (86.5) | 10 (100.0) |
| No | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Unknown | 5 (10.6) | 5 (13.5) | 0 (0.0) |
| Family history of platelet dysfunction, n (%) | |||
| Yes | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| No | 25 (53.2) | 16 (43.2) | 9 (90.0) |
| Unknown | 22 (46.8) | 21 (56.8) | 1 (10.0) |
| Family history of other bleeding disorder, n (%) | |||
| Yes | 11 (23.4) | 10 (27.0) | 1 (10.0) |
| No | 18 (38.3) | 10 (27.0) | 8 (80.0) |
| Unknown | 18 (38.3) | 17 (45.9) | 1 (10.0) |
| Factor level at diagnosis, IU/dl | |||
| n | 43 | 33 | 10 |
| Median (range) | 32.0 (0.5–101.1) | 32.0 (0.5–101.1) | 30.5 (13.0–45.0) |
| Severity of disease based on factor level at diagnosis, n (%) | |||
| Normal range (≥50%) | 3 (6.4) | 3 (8.1) | 0 (0.0) |
| Mild haemophilia (>5% to <50%) | 37 (78.7) | 27 (73.0) | 10 (100.0) |
| Moderate haemophilia (≥1% to ≤5%) | 2 (4.3) | 2 (5.4) | 0 (0.0) |
| Severe haemophilia (<1%) | 1 (2.1) | 1 (2.7) | 0 (0.0) |
| Unknown | 4 (8.5) | 4 (10.8) | 0 (0.0) |
| Comorbidities, n (%) c | |||
| No comorbidity | 29 (61.7) | 20 (54.1) | 9 (90.0) |
| Hepatitis C | 1 (2.1) | 1 (2.7) | 0 (0.0) |
| Obese/overweight | 2 (4.3) | 1 (2.7) | 1 (10.0) |
| Hypertension | 1 (2.1) | 0 (0.0) | 1 (10.0) |
| Hypercholesterolaemia | 2 (4.3) | 2 (5.4) | 0 (0.0) |
| Diabetes | 1 (2.1) | 1 (2.7) | 0 (0.0) |
| Renal disease | 1 (2.1) | 1 (2.7) | 0 (0.0) |
| Other comorbidity | 15 (31.9) | 15 (40.5) | 0 (0.0) |
| Haemoglobin, g/dl d | |||
| n | 36 | 30 | 6 |
| Median (range) | 13.0 (5.4–15.0) | 13.0 (5.4–15.0) | 13.2 (12.3–13.8) |
| von Willebrand factor antigen, IU/ml d | |||
| n | 22 | 20 | 2 |
| Median (range) | 0.8 (0.5–1.3) | 0.8 (0.5–1.3) | 1 (0.9–1) |
| von Willebrand factor ristocetin cofactor, IU/ml d | |||
| n | 22 | 20 | 2 |
| Median (range) | 0.7 (0.4–1.5) | 0.7 (0.4–1.5) | 1.0 (0.9–1.1) |
| Number of documented bleeds in the first year coming to the HTC e | |||
| n | 32 | 27 | 5 |
| Median (range) | 0.5 (0–24) | 1 (0–24) | 0 (0–3) |
| Number of patients without bleeds f | 16 | 13 | 3 |
Abbreviations: FIX, factor IX; FVIII, factor VIII; HTC, haemophilia treatment centre; WGH, women and girls with haemophilia.
Data taken from patient records available at the first visit to the HTC.
Obligate and possible carriers were determined by a history of excessive bleeding and supported by, if possible, the presence of at least one of the following: family history of biological father and/or sons diagnosed with FVIII or FIX deficiency, positive genetic test for FVIII or FIX genes or persistent FVIII or FIX deficiency.
Patients may have been counted in more than one category.
Laboratory values were collected from data at the first clinic visit and, if not available, data from the next visit to the clinic prior to any treatments for study bleeding.
The first year at the HTC may or may not overlap with the study period.
The number is the subset of n for this variable indicating patients with no bleeds.
Most WGH had diagnosis confirmed by either family history and/or laboratory results. At diagnosis, 6% (n = 3) of WGH had normal factor levels. Seventy‐nine per cent (n = 37) of WGH were diagnosed with mild haemophilia based on factor levels, whereas 4% (n = 2) and 2% (n = 1) were diagnosed with moderate or severe haemophilia, respectively. WGH diagnosed with moderate or severe haemophilia had FVIII deficiency. The factor level at diagnosis for the remaining 4 WGH was not known. Overall, 42 (89%) WGH had a family history of haemophilia, although documentation of histories of other bleeding disorders was largely unavailable in patient charts. VWF antigen assessment performed closest to the first HTC visit prior to treatment revealed median (range) VWF antigen levels of 0.8 (0.5–1.3) IU/ml among patients with FVIII deficiency (n = 20) and 1.0 (0.9–1.0) IU/ml in patients with FIX deficiency (n = 2).
The most common reason for visiting the HTC for the first time was family history/genetic counselling, followed by spontaneous or traumatic bleeds (Figure 1). Of the 17 WGH whose reason for their first HTC visit was spontaneous or traumatic bleeds, 10 (59%) had easy bruising, 6 (35%) had epistaxis, 5 (29%) had joint bleeds, 4 (24%) had excessive dental bleeding and 4 (24%) had other bleeds. Eight (17%) WGH went to the HTC for the first time for treatment of haemophilia A or B for a family member. At the first visit to the clinic, 4 (9%) WGH reported concern around fertility and childbirth and 2 (4%) reported avoiding sports. Ten (21%) WGH reported pain, with a median (range) pain score of 4.5 (2–5) on a visual analogue scale (n = 4). Further data regarding the impact of haemophilia on the lives of WGH included in this chart review were not commonly available. Within their first year coming to the HTC, the median (range) number of documented spontaneous or traumatic bleeds was 0.5 (0–24) and nearly one‐third of patients had no documented bleeds (16/47).
FIGURE 1.
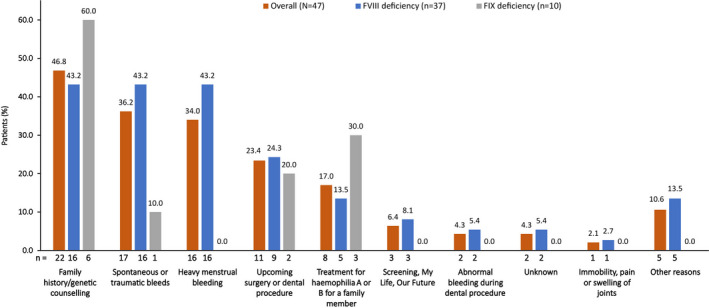
WGH reasons for the first visit to the clinica. FIX, factor IX; FVIII, factor VIII; WGH, women and girls with haemophilia.aPatients may have been counted in more than one category.
3.2. Heavy menstrual bleeding
Within the overall population, 38 (81%) WGH were known to have reached menarche. Fourteen WGH were treated for their heavy menstrual bleeding (HMB) as reported in the charts, which included 11 WGH with FVIII deficiency and 3 with FIX deficiency. The overall median (range) number of days of bleeding per month was 7.0 (6.0–28.0), and the median (range) number of days of heavy bleeding per month was 3.0 (2.0–12.0). WGH with FVIII deficiency had a median (range) of 7.0 (6.0–28.0) days of bleeding and 3.0 (2.0–12.0) days of heavy bleeding per month. WGH with FIX deficiency had a median (range) of 7.0 (6.0–8.0) days of bleeding and 3.0 (2.0–7.0) days of heavy bleeding per month.
Overall, antifibrinolytics were the most commonly prescribed medication for WGH with HMB (Table 2), which was received by 7 (50%) WGH to treat HMB. Desmopressin was used in 4 (29%) WGH, FVIII concentrate in 2 (14%) WGH and oral contraceptives in 1 (7%) WGH. Six WGH out of 14 received more than one treatment for HMB. Two WGH received FVIII replacement as part of an ongoing prophylaxis treatment, 1 of whom received it related to a bleeding event that was uncontrolled by the end of the study. Interventions used for HMB often led to a reduction in bleeding, although 8 WGH still experienced bleeding worse than expected during normal menstruation (Table 2).
TABLE 2.
Haemostasis management in WGH during first treatment of heavy menstrual bleeding a
| Overall (n = 14) b | FVIII deficiency (n = 11) | FIX deficiency (n = 3) | |
|---|---|---|---|
| Medication, n | |||
| Yes | 14 | 11 | 3 |
| No | 0 | 0 | 0 |
| Type of intervention, n (%) c | |||
| Antifibrinolytic | 7 (50.0) | 6 (54.5) | 1 (33.3) |
| Desmopressin | 4 (28.6) | 4 (36.4) | 0 (0.0) |
| FVIII concentrate | 2 (14.3) | 2 (18.2) | 0 (0.0) |
| Oral contraceptive | 1 (7.1) | 0 (0.0) | 1 (33.3) |
| Hormonal IUD | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Surgery | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Endometrial balloon tamponade | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Other | 1 (7.1) | 1 (9.1) | 0 (0.0) |
| Clinical outcomes, n (%) | |||
| Bleeding stopped | 1 (7.1) | 1 (9.1) | 0 (0.0) |
| Bleeding with sufficient control | 5 (35.7) | 3 (27.3) | 2 (66.7) |
| Bleeding with insufficient control | 8 (57.1) | 7 (63.6) | 1 (33.3) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; IUD, intrauterine device; WGH, women and girls with haemophilia.
Treatment information was collected from the first treatment for heavy menstrual bleeding during the study period. Patients may have received multiple treatments.
Patients with heavy menstrual bleeding during the study period and outcomes available during the study.
No medication was specified for 1 patient who was a carrier of FIX deficiency.
3.3. Pregnancy and childbirth
Twenty‐five WGH had a lifetime history of pregnancy, for a total of 49 births (Table 3). In a subset of WGH for whom data on the most recent delivery were available (n = 7), all were live births, with most (71%) newborns being delivered by Caesarean section. Of the 7 newborns, 2 males were diagnosed with haemophilia and 1 each with haemophilia A or haemophilia B. The newborn with haemophilia B had bruising/haematoma at birth and received medication to treat the bleeding.
TABLE 3.
Summary of lifetime pregnancy and birth event history in WGH
| Lifetime births, n (%) | Overall (N = 47) | FVIII deficiency (n = 37) | FIX deficiency (n = 10) |
|---|---|---|---|
| Number of patients who gave birth at least once | 25 | 21 | 4 |
| Number of births | 49 | 41 | 8 |
| Blood transfusion during delivery and postpartum | |||
| Number of patients, % | 7 (28.0) | 4 (19.0) | 3 (75.0) |
| Number of events, a % | 9 (18.3) | 5 (12.1) | 4 (50.0) |
| Clotting factor during delivery and postpartum | |||
| Number of patients, % | 6 (24.0) | 5 (23.8) | 1 (25.0) |
| Number of events, a % | 11 (22.4) | 9 (21.9) | 2 (25.0) |
| Excessive bleeding during delivery and postpartum | |||
| Number of patients, % | 11 (44.0) | 8 (38.1) | 3 (75.0) |
| Number of events, a % | 19 (38.8) | 15 (36.5) | 4 (50.0) |
| Miscarriage or stillbirth | |||
| Number of patients, % | 5 (20.0) | 4 (19.0) | 1 (25.0) |
| Number of events, % | 13 (26.5) | 9 (21.9) | 4 (50.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; WGH, women and girls with haemophilia.
Number of births with documentation of blood transfusion/clotting factor/excessive bleeding.
Factor concentrates were administered to the mother in preparation for or during delivery in 4 (57%) WGH (3 with FVIII deficiency and 1 with FIX deficiency) (Table 4). Doses prescribed were 50 IU/kg for patients with FVIII deficiency and 40 IU/kg for the patient with FIX deficiency (Table 5). Within 6 weeks postpartum, factor concentrates and antifibrinolytics were the most frequently administered medications (Table 4). Blood transfusions were needed for 2 patients during the postpartum period. When factor concentrates were given before (or in preparation for) or during delivery, bleeding was often reduced to levels that are expected for a normal delivery. Those treated within 6 weeks postpartum experienced bleeding cessation (1 patient), reduced bleeding as expected for a normal postpartum period (2 patients) or bleeding that was worse than expected (2 patients).
TABLE 4.
Haemostasis management in WGH for the most recent births during the study period
| In preparation for/during delivery | Within 6 weeks postpartum | |||||
|---|---|---|---|---|---|---|
| Overall (n = 7) a | FVIII deficiency (n = 6) | FIX deficiency (n = 1) | Overall (n = 7) a | FVIII deficiency (n = 6) | FIX deficiency (n = 1) | |
| Medication, n | ||||||
| Yes | 4 | 3 | 1 | 5 | 4 | 1 |
| No | 2 | 2 | 0 | 2 | 2 | 0 |
| Unknown | 1 | 1 | 0 | 0 | 0 | 0 |
| Type of medication, n (%) | ||||||
| Antifibrinolytic | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (28.6) | 1 (16.7) | 1 (100.0) |
| FVIII concentrate | 3 (42.9) | 3 (50.0) | NA | 2 (28.6) | 2 (33.3) | NA |
| FIX concentrate | 1 (14.3) | NA | 1 (100.0) | 1 (14.3) | NA | 1 (100.0) |
| Other | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (14.3) | 1 (16.7) | 0 (0.0) |
| Blood transfusion, n (%) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (28.6) | 1 (16.7) | 1 (100.0) |
| Iron supplementation, n (%) | NA | NA | NA | 3 (42.9) | 2 (33.3) | 1 (100.0) |
| Clinical outcomes, n (%) b | ||||||
| Bleeding stopped | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (20.0) | 1 (25.0) | 0 (0.0) |
| Bleeding with sufficient control c | 3 (75.0) | 3 (100.0) | 0 (0.0) | 2 (40.0) | 1 (25.0) | 1 (100.0) |
| Bleeding with insufficient control d | 1 (25.0) | 0 (0.0) | 1 (100.0) | 2 (40.0) | 2 (50.0) | 0 (0.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; NA, not applicable; WGH, women and girls with haemophilia.
Patients for whom data on the most recent delivery were available. Patients may have received multiple treatments.
Patients in the subset who received medical haemostasis management in preparation for/or during the most recent delivery and/or within 6 weeks postpartum.
Bleeding as normally expected for a normal pregnancy was considered bleeding with sufficient control.
Bleeding control worse than normally expected for normal pregnancy was considered bleeding with insufficient control.
TABLE 5.
Dose of clotting factors received to manage haemostasis in WGH
| FVIII deficiency | FIX deficiency | |||
|---|---|---|---|---|
| EHL a | SHL b | EHL a | SHL c | |
| Most recent delivery | ||||
| In preparation for/during | ||||
| n | 0 | 3 | 0 | 1 |
| Median (range) loading dose, IU/kg | 50.0 (50.0–50.0) | 40.0 (NA) | ||
| n | 0 | 3 | 0 | 0 |
| Median (range) total dose, IU | 3000.0 (2900.0–4000.0) | |||
| Within 6 weeks postpartum | ||||
| n | 0 | 2 | 1 | 0 |
| Median (range) dose, IU/kg | 37.5 (25.0–50.0) | 50.0 (NA) | ||
| Most recent surgery/major dental procedure | ||||
| In preparation for/during | ||||
| n | 2 | 9 | 0 | 2 |
| Median (range) loading dose, IU/kg | 50.0 (50.0–50.0) | 47.8 (20.0–51.4) | 72.0 (50.0–94.0) | |
| n | 1 | 9 | 0 | 2 |
| Median (range) total dose, IU | 830.0 (NA) | 2500.0 (1500.0–4000.0) | 4200.0 (1000.0–7400.0) | |
| Within 2 weeks after | ||||
| n | 1 | 2 | 0 | 0 |
| Median (range) dose, IU/kg/wk | 50.0 (NA) | 41.0 (32.0–50.0) | ||
| Spontaneous, traumatic or joint bleeds, and/or bleeds from minor dental procedure | ||||
| For first bleed | ||||
| n | 9 | 3 | ||
| Median (range) loading dose, IU/kg | 26.0 (11.0–50.0) | 50.0 (40.0–60.0) | ||
| n | 7 | 3 | ||
| Median (range) total dose, IU | 2000.0 (1750.0–14,400.0) | 6000.0 (60.0–6000.0) | ||
| For most recent bleed | ||||
| N | 6 | 1 | ||
| Median (range) loading dose, IU/kg | 25.4 (13.0–75.0) | 50.0 (NA) | ||
| N | 4 | 1 | ||
| Median (range) total dose, IU | 3700.0 (2000.0–5898.0) | 3270.0 (NA) | ||
Abbreviations: EHL, extended half‐life; FIX, factor IX; FVIII, factor VIII; NA, not applicable; rFIX, recombinant factor IX; rFIXFc, recombinant factor IX Fc fusion protein; rFVIII, recombinant factor VIII; rFVIIIFc, recombinant factor VIII Fc fusion protein; SHL, standard half‐life; WGH, women and girls with haemophilia.
EHLs included either rFVIIIFc (ELOCTATE®, Sanofi Genzyme) or rFIXFc (Alprolix®, Sanofi Genzyme) for those with FVIII or FIX deficiency, respectively.
SHLs included rFVIII (Kogenate®, Bayer HealthCare LLC; Advate®, Baxalta US Inc.; Helixate® FS, Bayer HealthCare LLC; Nuwiq®, Octapharma AB; Xyntha®, Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc) or antihemophilic factor/von Willebrand factor complex (Humate‐P®, CSL Behring GmbH).
SHL was rFIX (BeneFIX®, Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc).
3.4. Surgery
Overall, 20 WGH (15 with FVIII deficiency and 5 with FIX deficiency) had a history of surgery and/or major dental procedures (Table 6). Surgeries included tooth extractions, hysterectomy, laparoscopic cholecystectomy, ovarian cyst surgery and central venous access device placement, among others. Among these WGH, 5 (25%) were hospitalised following the procedure for a median (range) of 2.0 (2.0–94.0) days and 1 (5%) with FVIII deficiency experienced excessive blood loss prior to or during surgery. None of the WGH with FIX deficiency and 73% (n = 11) of those with FVIII deficiency did not experience excessive blood loss. Neither group of WGH experienced excessive blood loss within 2 weeks after surgery.
TABLE 6.
Surgery and major dental procedure characteristics in WGH a
| Overall (n = 20) | FVIII deficiency (n = 15) | FIX deficiency (n = 5) | |
|---|---|---|---|
| Type of surgery, n (%) | |||
| Major surgery b | 10 (50.0) | 10 (66.7) | 0 (0.0) |
| Minor surgery b | 5 (25.0) | 3 (20.0) | 2 (40.0) |
| Major dental procedure c | 5 (25.0) | 2 (13.3) | 3 (60.0) |
| Emergency or elective surgery, n (%) | |||
| Elective | 18 (90.0) | 13 (86.7) | 5 (100.0) |
| Emergency | 2 (10.0) | 2 (13.3) | 0 (0.0) |
| Hospitalisation, n (%) | |||
| Yes | 5 (25.0) | 4 (26.7) | 1 (20.0) |
| Median (range) duration of stay, days | 2.0 (2.0–94.0) | 2.5 (2.0–94.0) | 2.0 (NA) |
| No | 8 (40.0) | 4 (26.7) | 4 (80.0) |
| Unknown | 7 (35.0) | 7 (46.7) | 0 (0.0) |
| Excessive blood loss prior to or during surgery, n (%) | |||
| Yes | 1 (5.0) | 1 (6.7) | 0 (0.0) |
| No | 16 (80.0) | 11 (73.3) | 5 (100.0) |
| Unknown | 3 (15.0) | 3 (20.0) | 0 (0.0) |
| Excessive blood loss within 2 weeks after surgery, n (%) | |||
| No | 18 (90.0) | 13 (86.7) | 5 (100.0) |
| Unknown | 2 (10.0) | 2 (13.3) | 0 (0.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; NA, not applicable; WGH, women and girls with haemophilia.
Number of patients with surgery data assessed in this study. Surgery data were collected from the most recent event during the study period.
Major surgery was defined as any surgical procedure that involved general anaesthesia and/or respiratory assistance in which a major body cavity was penetrated and exposed, or for which there was substantial impairment of physical function. Minor surgery was defined as any surgical procedure that did not involve general anaesthesia and/or respiratory assistance.
Dental procedures were defined as procedures that involved extraction, abscess removal, apicectomy, etc.
The most commonly received medications prior to or during surgery were factor concentrates (Table 7). FVIII concentrates and antifibrinolytics were the most commonly administered medications within 2 weeks after surgery; no blood transfusions were reported during that time. One patient with FVIII deficiency was reported to have ongoing prophylaxis treatment. Interventions generally led to bleeding control as expected for surgery and led to stopped or reduced to levels as expected within 2 weeks after surgery (Table 7).
TABLE 7.
Haemostasis management in WGH during and after surgery or major dental procedures during the study period
| Prior to or during procedure | Within 2 weeks after procedure | |||||
|---|---|---|---|---|---|---|
| Overall (n = 20) a | FVIII deficiency (n = 15) | FIX deficiency (n = 5) | Overall (n = 20) a | FVIII deficiency (n = 15) | FIX deficiency (n = 5) | |
| Medication, n b | ||||||
| Yes | 20 | 15 | 5 | 11 | 8 | 3 |
| No | 0 | 0 | 0 | 9 | 7 | 2 |
| Type of medication, n (%) | ||||||
| FVIII concentrate | 12 (60.0) | 12 (80.0) | NA | 5 (25.0) | 5 (33.3) | NA |
| FIX concentrate | 4 (20.0) | NA | 4 (80.0) | 0 (0.0) | NA | 0 (0.0) |
| Desmopressin | 3 (15.0) | 3 (20.0) | 0 (0.0) | 1 (5.0) | 1 (6.7) | 0 (0.0) |
| Antifibrinolytic | 2 (10.0) | 1 (6.7) | 1 (20.0) | 5 (25.0) | 2 (13.3) | 3 (60.0) |
| Blood transfusion, n (%) b | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Clinical outcomes, n (%) b | ||||||
| Bleeding stopped | 1 (5.0) | 0 (0.0) | 1 (20.0) | 5 (45.5) | 4 (50.0) | 1 (33.3) |
| Bleeding with sufficient control c | 17 (85.0) | 13 (86.7) | 4 (80.0) | 5 (45.5) | 3 (37.5) | 2 (66.7) |
| Bleeding with insufficient control d | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Bleeding continued uncontrolled | 1 (5.0) | 1 (6.7) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Unknown | 1 (5.0) | 1 (6.7) | 0 (0.0) | 1 (9.1) | 1 (12.5) | 0 (0.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; NA, not applicable; WGH, women and girls with haemophilia.
Patients with data for surgery assessed in this study. Patients may have received multiple treatments.
Patients who received medical homeostasis management in preparation for/or during surgery/major dental procedure and/or within 2 weeks after surgery.
Bleeding as normally expected for surgery/dental procedure was considered bleeding with sufficient control.
Bleeding control worse than normally expected for surgery/dental procedure was considered bleeding with insufficient control.
3.5. Spontaneous, traumatic, joint or minor dental procedure bleeds
Overall, 23 (49%) WGH (18 with FVIII deficiency and 5 with FIX deficiency) had spontaneous, traumatic or joint bleeds during the study period (Table 8). Traumatic bleeds were the most common (44%) followed by joint bleeds (39%). Epistaxis was the most common spontaneous bleed, and muscle bleeds were the most common traumatic bleeds.
TABLE 8.
Spontaneous, traumatic, joint and minor dental procedure bleed characteristics in WGH
| Overall (n = 23) | FVIII deficiency (n = 18) | FIX deficiency (n = 5) | |
|---|---|---|---|
| History, n (%) | |||
| Spontaneous (non‐joint) | 4 (17.4) | 4 (22.2) | 0 (0.0) |
| Muscle | 1 (4.3) | 1 (5.6) | NA |
| Oral | 1 (4.3) | 1 (5.6) | NA |
| Epistaxis | 2 (8.7) | 2 (11.1) | NA |
| Traumatic | 10 (43.5) | 7 (38.9) | 3 (60.0) |
| Muscle | 3 (13.0) | 2 (11.1) | 1 (20.0) |
| Skin/Mucosa | 1 (4.3) | 1 (5.6) | 0 (0.0) |
| Internal | 1 (4.3) | 0 (0.0) | 1 (20.0) |
| Soft tissue | 2 (8.7) | 1 (5.6) | 1 (20.0) |
| Other | 3 (13.0) | 3 (16.7) | 0 (0.0) |
| Bleeds from minor dental procedures | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Joint bleeds | 9 (39.1) | 7 (38.9) | 2 (40.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; NA, not applicable; WGH, women and girls with haemophilia.
Among those who experienced a joint bleed (n = 9) (Table 9), the median factor level at diagnosis for those with FVIII deficiency (n = 4) was 23% (20%–27%); for 2 WGH with FIX deficiency, factor levels at diagnosis were 13% or 44.6%. Data were not available for the remaining 3 WGH with joint bleeds. For those WGH with repeat bleeds (n = 11), 5 (46%) had repeat joint bleeds and 6 (55%) had repeat non‐joint bleeds.
TABLE 9.
| n (%) | First bleed | Repeat bleed d | ||||
|---|---|---|---|---|---|---|
| Overall (n = 9) | FVIII deficiency (n = 7) | FIX deficiency (n = 2) | Overall (n = 11) | FVIII deficiency (n = 9) | FIX deficiency (n = 2) | |
| Right knee | 1 (11.1) | 1 (14.3) | 0 (0.0) | 2 (18.2) | 2 (22.2) | 0 (0.0) |
| Left knee | 2 (22.2) | 1 (14.3) | 1 (50.0) | 1 (9.1) | 1 (11.1) | 0 (0.0) |
| Right ankle | 1 (11.1) | 1 (14.3) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Left ankle | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Right elbow | 3 (33.3) | 2 (28.6) | 1 (50.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Left elbow | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Other joint | 2 (22.2) | 2 (28.6) | 0 (0.0) | 2 (18.2) | 1 (11.1) | 1 (50.0) |
| Non‐joint repeat bleed | – | – | – | 6 (54.5) | 5 (55.6) | 1 (50.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; WGH, women and girls with haemophilia.
Represents data for joint bleeds and non‐joint repeat bleeds during the study period (1 April 2012–15 November 2018).
First joint bleed in the study period that required haemostasis management and for which the outcome is available.
Column may not add up to 100% owing to rounding.
Repeat bleeds for the most recent joint or non‐joint bleeds in the study period that required haemostasis management and for which the outcome is available.
The most commonly received medications for bleeds were factor concentrates (Table 10). At the time of data cut‐off, 4 WGH with FVIII deficiency were receiving ongoing treatment for bleeding events (1 was receiving a prophylactic regimen, 2 were receiving on‐demand treatment and another was receiving ongoing treatment for an unresolved bleed). Interventions generally led to a reduction in bleeding, although continued bleeding was present in 1 patient at the end of the study period (Table 10).
TABLE 10.
Haemostasis management in WGH for the first treatment of spontaneous, traumatic or joint bleed events and/or bleeding from minor dental procedures a
| Overall (n = 23) b | FVIII deficiency (n = 18) | FIX deficiency (n = 5) | |
|---|---|---|---|
| Medication, n | |||
| Yes | 23 | 18 | 5 |
| No | 0 | 0 | 0 |
| Type of medication, n (%) | |||
| FVIII concentrate | 10 (43.5) | 10 (55.6) | NA |
| FIX concentrate | 5 (21.7) | NA | 5 (100.0) |
| Desmopressin | 5 (21.7) | 5 (27.8) | 0 (0.0) |
| Antifibrinolytic | 1 (4.3) | 1 (5.6) | 0 (0.0) |
| Other | 2 (8.7) | 2 (11.1) | 0 (0.0) |
| Clinical outcomes, n (%) | |||
| Bleeding stopped | 12 (52.2) | 8 (44.4) | 4 (80.0) |
| Bleeding reduced | 9 (39.1) | 8 (44.4) | 1 (20.0) |
| Bleeding continued | 2 (8.7) | 2 (11.1) | 0 (0.0) |
Abbreviations: FIX, factor IX; FVIII, factor VIII; NA, not applicable; WGH, women and girls with haemophilia.
Treatment information was collected from the first treatment for spontaneous or traumatic bleed events during the study period. Patients may have received multiple treatments.
Patients with spontaneous, traumatic, joint bleeds or bleeding from minor dental procedures in the study period that required haemostasis management and for which outcome is available.
4. DISCUSSION
This retrospective chart review found that the majority of WGH who presented to HTCs in this cohort did so in the context of a family member's haemophilia or for genetic counselling. In addition, HMB and spontaneous or traumatic bleeds were other common reasons for the first visit to the clinic. The patient group was heterogeneous with diverse demographics, including clinical characteristics and symptom presentation. Patients included in this study had factor levels ranging from normal to severe deficiency of clotting factor, although the majority of WGH (79%) included in this chart review had mild haemophilia based on clotting factor levels. Regarding those with normal factor levels (n = 3), it is possible that these patients had false laboratory results, considering they experienced bleeding symptoms. As such, repeated testing in these situations would be recommended to verify factor deficiency.
The median (range) clotting factor level in the symptomatic carrier population presented herein was 32.0 (0.5–101.1) IU/dl. This contrasted with a reported median (range) of 60.0 (5.0–219.0) IU/dl in a previous cross‐sectional study assessing the effect of heterozygous haemophilia carriership on bleeding symptoms from a survey of Dutch women who had been tested for carriership of haemophilia A or B before 2001. 4 This discrepancy may be explained by the fact that the carriers selected in the present study were symptomatic.
Although medications such as antifibrinolytics for HMB led to a reduction in bleeding, many WGH (8/14 treated for HMB) still experienced worse than normal bleeding during menstruation, making HMB the event with the lowest success in gaining haemostatic control. Similarly, a cross‐sectional study by Paroskie et al reported increased menstrual bleeding, although the haemophilia A carriers did not have a statistically significant increased duration of menstruation compared with non‐carriers and did not report use of antifibrinolytics for clinical management of HMB. In any case, factor deficiency among women and girls seems to have an impact on reproductive tract bleeding, with patients in this cohort experiencing excessive bleeding during menstruation and childbirth.
Several WGH included in this chart review experienced excessive bleeding during delivery or postpartum (44%), or miscarriage or stillbirth (20%). These findings are consistent with previous reports, although there is a current lack of data on the risk of miscarriage. 10 , 11 , 18 Factor concentrates and antifibrinolytics were also administered to WGH in preparation for or during delivery, or within 6 weeks postpartum. This is in accordance with guidance regarding haemostatic options for women wishing to preserve their fertility. 11 Women in this pregnancy subgroup also received transfusions during the postpartum period for bleeding; this was the only event of interest in this study during which transfusions were used. Newborns born to WGH can also be at increased risk of bleeding 10 ; 1 newborn in this chart review experienced bleeding complications at birth.
Treatments for women in this study were largely successful in addressing bleeding events and maintaining haemostasis during surgery. Several cross‐sectional studies examining bleeding in women report similar results. 4 , 6 , 8 , 19 A history of joint bleeds was reported in 19% (9/47) of patients in the current cohort, representing a similar proportion to that previously reported for females with mild FVIII deficiency (15%). 8 Previous research has demonstrated an increased bleeding tendency despite near‐normal levels of FVIII. 4 , 5 , 6 , 7 , 8
The women and girls described in this chart review experienced bleeds, despite being classified as having mild haemophilia. WGH present with symptoms related to reproductive health, as well as with bleeding events typically observed in males with haemophilia. For instance, in WGH with HMB, there is a bleeding event at regular intervals that should be addressed. The findings also highlight that monitoring of coagulation status and appropriate preparation are warranted in WGH undergoing surgery. 4 For these reasons, WGH may require specialty care for a range of clinical scenarios; patients should be treated in a setting where all required management options are available.
Although there is increasing recognition of both the clinical and psychological issues for WGH, 13 there are still numerous challenges in managing WGH. Haemophilia has been historically recognised as a male disease, 20 and WGH may also be less likely to seek care, potentially because of a lack of recognition of symptoms as abnormal and lack of awareness that WGH can warrant clinical treatment. 3 Bleeding symptoms are often under‐recognised owing to the overlap with bleeding symptoms in women who are not carriers (HMB, postpartum haemorrhage and post‐operative bleeding) and focus often lies on a more severe manifestation of haemophilia. 20 Finally, current treatment guidelines offer limited information on the management of WGH. 11
This study evaluates a range of bleeding events across different clinical scenarios in a real‐world setting, with data collected from experienced HTCs. However, the nature of the retrospective chart review and sampling bias are study limitations, as are the small number of sites included, potential for under‐ or over‐reporting of historical bleeding symptoms and limited or incomplete data in some cases (eg, impact of factor deficiency on the lives of patients, such as worries or concerns or missed days of work/school and information on resource use). Additionally, eligible patients were those who presented at selected HTCs with documentation of multiple visits and treatments and, therefore, may not represent the general WGH population, as additional patients who presented only for initial consultation were not included.
5. CONCLUSION
Results of this retrospective chart review, which represents a female population with mild haemophilia who presented at an HTC, demonstrate that WGH can have a bleeding phenotype that includes traumatic and joint bleeds, as well as the need for haemostasis management during certain clinical events. While the population presented in this study was limited to those women who presented to an HTC and required interventions for bleeding events, this chart review has allowed a view into medical information routinely collected in this population and brought awareness to the unresolved symptoms/bleeding events for some of these WGH. There are no set guidelines or specialised treatment centres for women; thus, patients are managed in a variety of ways. This is evident based on the results presented here and would apply to underdeveloped countries as well. Because prospective studies among WGH can have significant logistical challenges, such as identification of the appropriate patients and issues surrounding inclusion of women of childbearing age, results such as these from a chart review are valuable to highlight the importance of assessing the need for haemostasis management in WGH and can contribute to the design of future prospective studies evaluating optimal diagnosis, treatment and unmet needs in this group. Furthering clinical care for WGH is needed to reduce the psychosocial, emotional and economic impacts of a bleeding disorder diagnosis.
6. DATA SHARING STATEMENT
Qualified researchers may request access to patient level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan and dataset specifications. Patient‐level data will be anonymised, and study documents will be redacted to protect the privacy of our trial participants. Further details on Sanofi's data sharing criteria, eligible studies and process for requesting access can be found at: https://www.clinicalstudydatarequest.com/.
CONFLICT OF INTEREST
This research was funded by Sanofi. Sanofi and Sobi reviewed and provided feedback on the manuscript. The authors had full editorial control of the manuscript and provided their final approval of all content. AC: consultancy or honoraria/advisory committees for Bayer, Bioverativ, a Sanofi company, and Genentech. Her prior institution, Center For Inherited Blood Disorders, has received funding for research carried out in this work. RS Jr.: participated in advisory boards for uniQure, Sanofi, Genentech/Roche, Grifols, Kedrion, Octapharma, Novo Nordisk, Pfizer, Takeda, BioMarin, Spark and Catalyst; investigator‐initiated studies funded by Genentech, Grifols and Octapharma. NJ: employee of and hold equity interest in Sanofi. JT and ET: employee of Sanofi. MOO and RK: nothing to disclose.
ACKNOWLEDGEMENTS
This chart review was sponsored by Sanofi (Waltham, MA, USA). Medical writing and editing support were provided by Ashleigh Pulkoski‐Gross, PhD, CMPP, and Jennifer Alexander, MSc, MBA, CMPP, of JK Associates Inc., part of Fishawack Health (Conshohocken, PA, USA), and were funded by Sanofi.
[Correction added on 17 June 2021, after first online publication: The copyright line was changed.]
REFERENCES
- 1. Hermans C, Kulkarni R. Women with bleeding disorders. Haemophilia. 2018;24(Suppl 6):29‐36. [DOI] [PubMed] [Google Scholar]
- 2. Hirayama AB, Silva AKCD, Rocha JS, Roberti MDRF. Prevalence of symptoms in hemophilia carriers in comparison with the general population: a systematic review. Hematol Transfus Cell Ther. 2019;41:349‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. World Federation of Hemophilia . Carriers and women with hemophilia. http://www1.wfh.org/publication/files/pdf‐1471.pdf. Accessed November 2019.
- 4. Plug I, Mauser‐Bunschoten EP, Bröcker‐Vriends AHJT, et al. Bleeding in carriers of hemophilia. Blood. 2006;108:52‐56. [DOI] [PubMed] [Google Scholar]
- 5. Olsson A, Hellgren M, Berntorp E, Ljung R, Baghaei F. Clotting factor level is not a good predictor of bleeding in carriers of haemophilia A and B. Blood Coagul Fibrinolysis. 2014;25:471‐475. [DOI] [PubMed] [Google Scholar]
- 6. Paroskie A, Gailani D, DeBaun MR, Sidonio RF Jr. A cross‐sectional study of bleeding phenotype in haemophilia A carriers. Br J Haematol. 2015;170:223‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Paroskie A, Oso O, Almassi B, DeBaun MR, Sidonio RF Jr. Both hemophilia health care providers and hemophilia a carriers report that carriers have excessive bleeding. J Pediatr Hematol Oncol. 2014;36:e224‐e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sidonio RF, Mili FD, Li T, et al.; The Hemophilia Treatment Cancers Network . Females with FVIII and FIX deficiency have reduced joint range of motion. Am J Hematol. 2014;89:831‐836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(Suppl 6):1‐158. [DOI] [PubMed] [Google Scholar]
- 10. Kadir RA, Davies J, Winikoff R, et al. Pregnancy complications and obstetric care in women with inherited bleeding disorders. Haemophilia. 2013;19(Suppl 4):1‐10. [DOI] [PubMed] [Google Scholar]
- 11. Kulkarni R. Improving care and treatment options for women and girls with bleeding disorders. Eur J Haematol. 2015;95(Suppl 81):2‐10. [DOI] [PubMed] [Google Scholar]
- 12. Gilbert L, Paroskie A, Gailani D, Debaun MR, Sidonio RF. Haemophilia A carriers experience reduced health‐related quality of life. Haemophilia. 2015;21:761‐765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McLintock C. Women with bleeding disorders: clinical and psychological issues. Haemophilia. 2018;24(Suppl 6):22‐28. [DOI] [PubMed] [Google Scholar]
- 14. Govorov I, Ekelund L, Chaireti R, et al. Heavy menstrual bleeding and health‐associated quality of life in women with von Willebrand's disease. Exp Ther Med. 2016;11:1923‐1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Centers for Disease Control and Prevention . Community counts. https://www.cdc.gov/ncbddd/hemophilia/communitycounts/about.html. Accessed April 3, 2020.
- 16. American Thrombosis & Hemostasis Network . My Life, Our Future—Genotyping for progress in hemophilia. http://www.mylifeourfuture.org/. Accessed April 3, 2020.
- 17. World Federation of Hemophilia . Cornerstone initiative. https://www.wfh.org/en/our‐work‐reg‐national/cornerstone‐initiative. Accessed April 3, 2020.
- 18. Olsson A, Radulovic V, Wennerholm UB. Maternal and neonatal outcomes in carriers of haemophilia A and B: a Swedish Medical Birth Register study. Haemophilia. 2020;26:e14‐e17. [DOI] [PubMed] [Google Scholar]
- 19. Di Michele DM, Gibb C, Lefkowitz JM, Ni Q, Gerber LM, Ganguly A. Severe and moderate haemophilia A and B in US females. Haemophilia. 2014;20:e136‐e143. [DOI] [PubMed] [Google Scholar]
- 20. Staber J, Croteau SE, Davis J, Grabowski EF, Kouides P, Sidonio RF Jr. The spectrum of bleeding in women and girls with haemophilia B. Haemophilia. 2018;24:180‐185. [DOI] [PubMed] [Google Scholar]