

SUMMARY
Data-driven analyses are increasingly valued in modern medicine. We integrate quantitative proteomics and transcriptomics from over 1,000 post-mortem brains from six cohorts representing Alzheimer’s disease (AD), asymptomatic AD, progressive supranuclear palsy (PSP), and control patients from the Accelerating Medicines Partnership – Alzheimer’s Disease consortium. We define robust co-expression trajectories related to disease progression, including early neuronal, microglial, astrocyte, and immune response modules, and later mRNA splicing and mitochondrial modules. The majority of, but not all, modules are conserved at the transcriptomic level, including module C3, which is only observed in proteome networks and enriched in mitogen-activated protein kinase (MAPK) signaling. Genetic risk enriches in modules changing early in disease and indicates that AD and PSP have distinct causal biological drivers at the pathway level, despite aspects of similar pathology, including synaptic loss and glial inflammatory changes. The conserved, high-confidence proteomic changes enriched in genetic risk represent targets for drug discovery.
Graphical Abstract
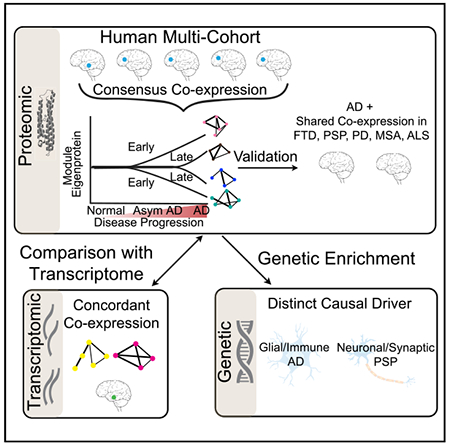
In Brief
Swarup et al. use a multi-omic, multi-cohort approach to identify robust early and late proteomic changes in AD and other neurodegenerative dementias and find that genetic risk is differentially enriched across disorders. Shared co-expression modules showing consistent molecular alterations at multi-omic levels are ripe for future investigation as drug targets.
INTRODUCTION
Dementia affects 9% of the population (Langa et al., 2017; McKhann et al., 2011) and exacts a large societal and financial burden (Alzheimer’s Association, 2019). The most common dementia is Alzheimer’s disease (AD), characterized pathologically by neurofibrillary tangles, consisting of filamentous hyperphosphorylated microtubule-associated protein tau (MAPT; tau), inclusions, and senile plaques, consisting of Aβ deposits (De Strooper and Karran, 2016; Goate et al., 1991; Lage et al., 2007; Schneider et al., 2009; Vinters, 2015). Several neurodegenerative conditions are related to AD due to shared alterations in proteostasis and pathological protein aggregates in brain including progressive supranuclear palsy (PSP), frontotemporal dementia (FTD), and Parkinson’s disease (PD) (Arendt et al., 2016; Cummings, 2003; Erkkinen et al., 2018; Kovacs, 2015). Although common and rare genetic variants in MAPT cause or increase the risk for AD, PSP (Coppola et al., 2012; Desikan et al., 2015), FTD, and PD, the risk for these disorders is heterogeneous and complex (Chang et al., 2017; Chen et al., 2018; Ferrari et al., 2014; Karch and Goate, 2015; Seelaar et al., 2011). So, despite advances in identifying genetic contributions to dementia disease risk, understanding how risk factors may converge on specific molecular pathways remains a critical gap in our knowledge (Bonham et al., 2018; Parikshak et al., 2015).
A powerful approach to provide a data-driven framework for connecting genetic risk to disease pathways is gene network analysis, which leverages gene co-expression to improve mechanistic models of pathophysiology (Parikshak et al., 2015). To accelerate this process, the National Institutes of Health (NIH) developed the Target Identification and Preclinical Validation Project of the Accelerating Medicines Partnership – Alzheimer’s Disease (AMP-AD) consortium (Hodes and Buckholtz, 2016) to integrate high-throughput genomic and molecular data from disease brain within a network-driven structure (Hodes and Buckholtz, 2016; Logsdon et al., 2019). Within this context, several large-scale RNA sequencing (RNA-seq) projects have been conducted (Allen et al., 2018; De Jager et al., 2018; Gaiteri et al., 2016; Logsdon et al., 2019; Mostafavi et al., 2018; Readhead et al., 2018; Zhang et al., 2013), identifying transcriptomic changes in the cerebral cortex from patients with AD and showing that genetic risk for AD, rather than being widely distributed, is enriched in specific transcriptomic networks. Complementary approaches based on proteomics quantified several thousand proteins and identified AD-related proteomic networks in post-mortem brain, but most have been limited to a single cohort (Johnson et al., 2018; McKenzie et al., 2017; Ping et al., 2018; Seyfried et al., 2017; Yu et al., 2018; Zhang et al., 2018).
Here, we took an integrative systems biology approach to identify neurodegenerative dementia-associated protein networks in post-mortem brain across multiple AD cohorts (Hodes and Buckholtz, 2016). To detect robust disease-associated changes, we used the same processing scheme to harmonize data across studies and applied a consensus network analysis approach to identify conserved co-expression modules (Langfelder and Horvath, 2008; Langfelder et al., 2013; Swarup et al., 2019). We subsequently characterized the temporal trajectories of distinct biological processes over disease progression by distinguishing early and late disease changes and identifying several co-expression modules correlated with neuropathology and cognitive decline. We validated these findings in an orthogonal dataset (Ping et al., 2018) and extended our initial findings across multiple neurodegenerative diseases, including FTD with TDP-43 (FTD-TDP), PSP with corticobasal degeneration (PSP-CBD), PD, PD with dementia (PD-D), multiple system atrophy (MSA), and amyotrophic lateral sclerosis (ALS). We found protein co-expression modules that were dysregulated in neurodegenerative dementias (AD, FTD-TDP, PSP-CBD, and PD-D), but not in related neurodegenerative diseases (PD, MSA, and ALS) where dementia is not the defining feature. We evaluated concordance of transcriptomic and proteomic signatures in a subset of samples, finding high correspondence overall, as well as proteome-specific changes. To understand the relationship of these core molecular processes with causal genetic drivers, we placed common variants associated with disease risk within co-expression modules to identify specific biological processes enriched in disease-specific risk variants.
RESULTS
Label-free high-throughput quantitative proteomics by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) was performed on five different study cohorts—the Baltimore Longitudinal Study of Aging (BLSA), Adult Changes of Thought (ACT), Mount Sinai Brain Bank (MSBB), Banner Sun Health Research Institute (Banner), and Mayo Clinic Brain Bank (Mayo; Figure 1)—totaling 798 samples representing AD, asymptomatic AD (AsymAD), PSP, and controls from prefrontal and temporal cortex (Figure 1). Detailed clinical and neuropathological scores were available for each cohort and were used in downstream analyses (Table S1). As a validation set, we generated quantitative proteomic data from University of Pennsylvania (UPenn) Brain Bank samples: 384 frontal cortical samples from patients with clinically diagnosed AD, FTD-TDP, PSP-CBD, PD, PD-D, MSA, and ALS (Figure 1). Using a label-free proteomics approach, we were able to identify approximately 65,000 peptides corresponding to approximately 5,000 proteins per sample. To circumvent the problem of missing protein abundance values across different datasets, we allowed for a maximum of 40% missing values. In total, 2,005 quantified proteins overlapped in all five datasets (prefrontal cortex for BLSA, ACT, MSBB, and Banner; temporal cortex for Mayo) and were used for our conservative downstream analysis.
Figure 1. Schematic Representation of the Various Analyses Performed in the Study.
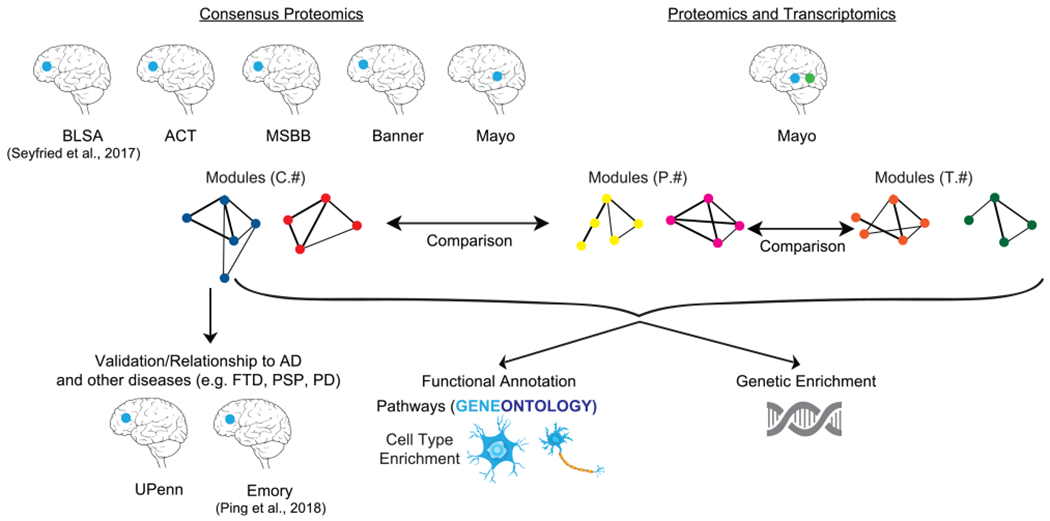
Consensus proteomics modules were generated from five different studies: the Baltimore Longitudinal Study of Aging (BLSA), Adult Changes of Thought (ACT), Mount Sinai Brain Bank (MSBB), Banner Sun Health Research Institute (Banner), and Mayo Clinic Brain Bank (Mayo). Furthermore, the Mayo dataset had matched samples where proteomics and transcriptomics data were available. Comparisons were then made among the consensus proteomics modules (labeled with “C”), Mayo-only proteomics modules (labeled with “P”), and Mayo-matched sample transcriptomics modules (labeled with “T”). Determining the relationship of consensus proteomic modules with other neurodegenerative diseases like frontotemporal dementia (FTD), progressive supranuclear palsy (PSP), and Parkinson’s disease (PD) was performed using completely separate datasets (UPenn and Emory). Functional annotation of the modules was performed using cell type and Gene Ontology. Common genetic enrichment analysis was used to understand the relationship of modules with causal genetic drivers. The blue dot indicates the location of proteomic brain samples. The green dot indicates location of transcriptomic brain samples.
Identification of Robust Disease-Relevant Protein Co-expression Signature
To find robust co-expression changes irrespective of brain banks and cortical regions, we employed a consensus-weighted gene co-expression network analysis (cWGCNA) across cohorts from the BLSA, ACT, MSBB, Banner, and Mayo brain banks (Figures 2A and S1A). In contrast to other analytical pipelines used for consensus network analyses (Johnson et al., 2020), the cWGCNA approach does not require batch regression for consensus module generation. We identified 10 consensus modules (labeled C1–C10) representing all four major brain cell types: neurons (C1 and C2 modules), microglia/endothelial cells (C10 module), astrocytes (C8 module), and oligodendrocytes (C5 module) (Figure 2B; Table S2). We identified two modules (C8 and C10) that were significantly associated with AD diagnosis (p < 0.05) in all five datasets, one module (C1) that was significantly correlated with diagnosis in four datasets, and three additional modules (C2, C3, and C4) that were significantly correlated with diagnosis (p < 0.05) in at least three of the five datasets (see Method Details) for a total of six AD-associated modules. Four consensus modules (C3, C4, C8, and C10) were upregulated in AD compared to controls, and two consensus modules (C1 and C2) were downregulated in AD (Figure 2B).
Figure 2. Consensus Proteomics Analyses.
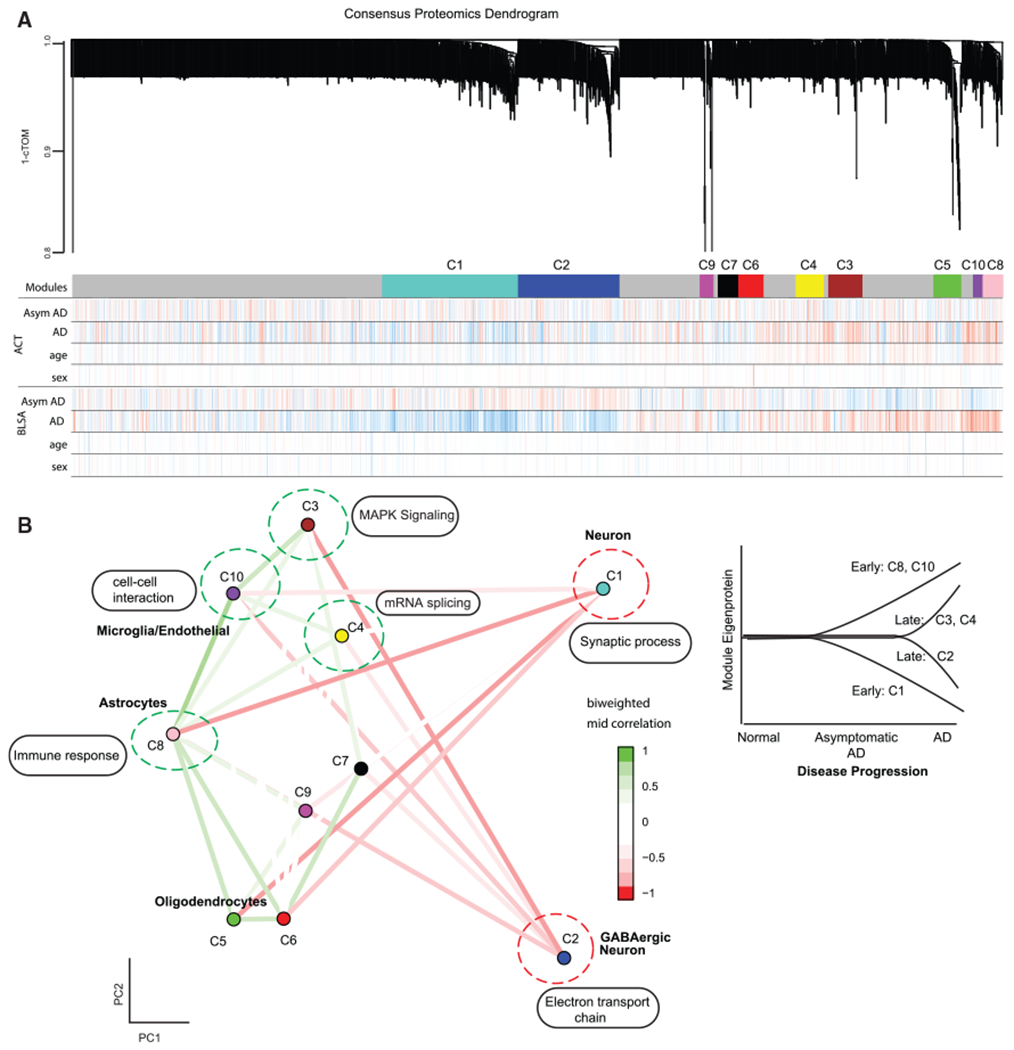
(A) Consensus proteomics dendrogram showing the proteomics modules from 2 of 5 different proteomics datasets (ACT and BLSA). Color bars below the modules show correlation of disease condition (AD, asymptomatic AD) and biological covariates (age, sex) with the expression of each protein. Red is positive correlation, and blue is anti-correlation.
(B) Multidimensional scaling plot on the left demonstrates relationship between consensus proteomics modules and clustering by cell-type relationship. Also shown are the major Gene Ontology enrichment terms for modules. Modules upregulated in AD (green dotted circle) and downregulated in AD (red dotted circle) are shown. On the right is a diagrammatic representation of the trajectory of module eigenprotein with the progression of Alzheimer’s disease, highlighting the early and late proteomic modules identified in the study.
cTOM, consensus topological overlap matrix; AD, Alzheimer’s disease; AsymAD, asymptomatic AD.
We leveraged three large cohorts of asymptomatic AD cases, where cognition is not affected but where subjects have sufficient neurofibrillary tangles and plaques for a pathological diagnosis (Hyman et al., 2012), providing a glimpse into early stages of the disease. We defined early AD molecular alterations as changes observed in AsymAD and AD, and late AD molecular alterations were defined as changes only seen in AD. Interestingly, one neuronal module (C1) was downregulated, and two glial modules (C8 and C10) were upregulated early. The modules altered late in disease course were not glial, but instead represented GABAergic neurons, mitogen-activated protein kinase (MAPK) signaling, and RNA splicing (C2, C3, and C4; Figure 2B). Table S3 summarizes characteristics of early and late modules associated with AD and their association with other neurodegenerative diseases. We also constructed an interactive graphical interface to allow exploration of individual proteins and their network relationships (https://labs.dgsom.ucla.edu/geschwind/files/view/html/ADpNET.html).
Identification of Early Changes in the AD Proteome
To identify potential drivers of disease pathology, we focused our attention on modules significantly correlated with AsymAD cases, representing the earliest disease phase. This highlighted module C1 (Figures 3A–3G and S1B–S1Q; Table S4) and the C8 and C10 modules, which were upregulated in AsymAD and AD cases (Figures 3H–3W and S2; Table S4). C1 represented genes highly expressed in neurons (hypergeometric test; p = 1.0e–23) and was enriched for synaptic processes, consistent with pre- and post-synaptic vesicle hub proteins such as HOMER1, DLG4, and SYT1 (Figure 3A). The hub gene SYT1, which mediates synaptic vesicle release, is reduced in human AD brain and has been shown to promote Aβ generation (Gautam et al., 2015; Öhrfelt et al., 2016; Yoo et al., 2001).
Figure 3. Early Proteomic Changes in AD.
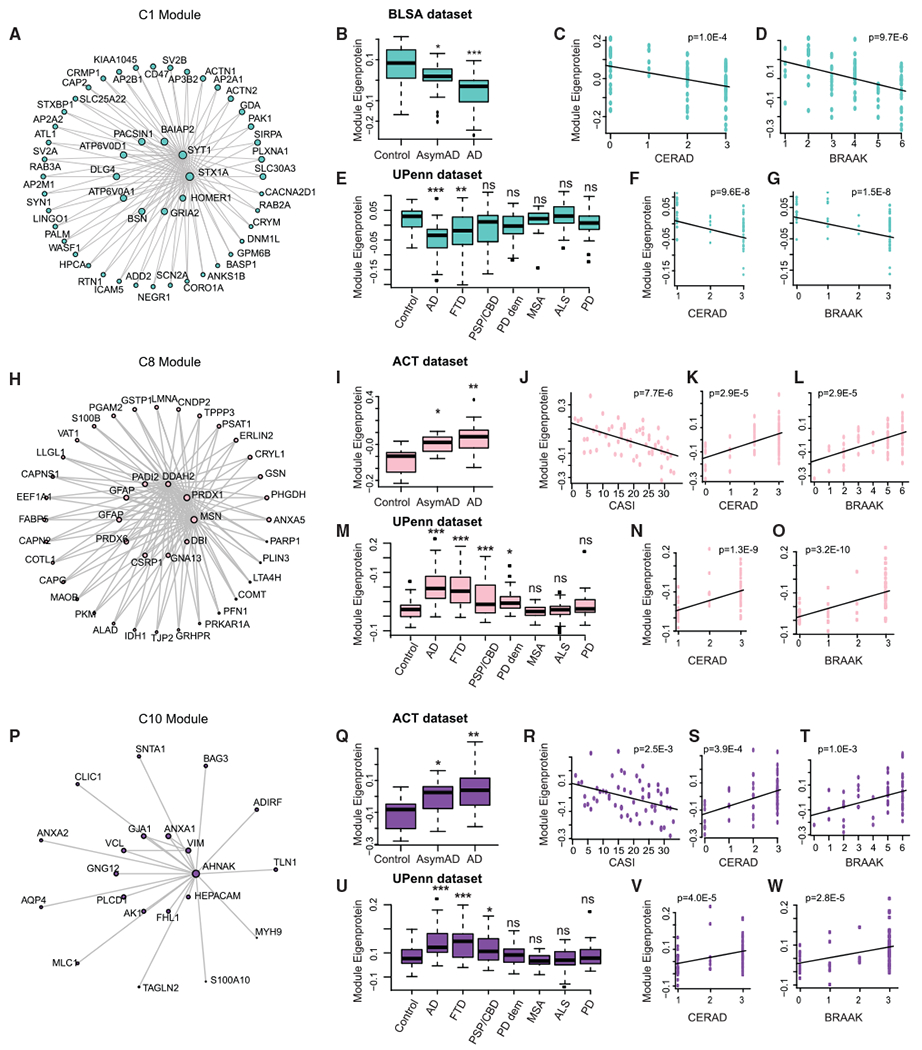
(A) Co-expression plot of the C1 module showing the hub proteins in the center.
(B–D) Plots showing C1 module eigenprotein trajectory with diagnosis (B), CERAD (C), and BRAAK (D) scores in BLSA dataset.
(E–G) Plots showing C1 module eigenprotein trajectory with diagnosis (E), CERAD (F), and BRAAK (G) scores in validation UPenn dataset.
(H) Co-expression plot of the C8 module showing the hub proteins in the center.
(I–L) Plots showing C8 module eigenprotein trajectory with diagnosis (I), CASI (J), CERAD (K), and BRAAK (L) scores in ACT dataset.
(M–O) Plots showing C8 module eigenprotein trajectory with diagnosis (M), CERAD (N), and BRAAK (O) scores in validation UPenn dataset.
(P) Co-expression plot of the C10 module showing the hub proteins in the center.
(Q–T) Plots showing C10 module eigenprotein trajectory with diagnosis (Q), CASI (R), CERAD (S), and BRAAK (T) scores in ACT dataset.
(U–W) Plots showing C10 module eigenprotein trajectory with diagnosis (U), CERAD (V), and BRAAK (W) scores in validation UPenn dataset.
For boxplots (B), (E), (I), (M), (Q), and (U), the center line indicates the median, box limits indicate the 25th and 75th percentiles, whiskers extend to the most extreme data point, which is no more than 1.5 times the interquartile range from the median, and outliers are represented by dots.
AD, Alzheimer’s disease; AsymAD, asymptomatic AD; FTD, frontotemporal dementia; PSP-CBD, progressive supranuclear palsy with corticobasal degeneration; PD, Parkinson’s disease; MSA, multiple systems atrophy; ALS, amyotrophic lateral sclerosis. *p < 0.05; **p < 0.01; ***p < 0.005; n.s., non-significant; p, p-value
In contrast, C8 represented genes highly expressed in astrocytes (hypergeometric test; p = 8.9e–7) and was enriched for immune response and cell-cell adhesion genes. One hub of the C8 module, MSN, is an essential protein involved in P2X7 receptor-mediated cleavage of amyloid precursor protein (APP) (Darmellah et al., 2012). Another hub protein—PRDX6, an antioxidant enzyme—is upregulated in astrocytes from patients with Lewy body dementia (Power et al., 2002) (Figure 3H). C10 was enriched in microglial and endothelial markers. One of its hubs, ANXA1, is a central player in the anti-inflammatory and neuroprotective role of microglia and the clearance of Aβ (McArthur et al., 2010; Park et al., 2017; Ries et al., 2016). Another hub, CLIC1, is a chloride channel expressed in microglia involved in mediating Aβ neurotoxicity (Milton et al., 2008; Skaper et al., 2013) (Figure 3P). We performed western blotting to validate that several C8 and C10 hub proteins (PADI2, GJA1, PLCD1, and HEPACAM) were increased in AD (Figure S3). This analysis clearly demonstrates that neuronal/synaptic gene downregulation and astroglial and microglial upregulation are consistent, early components of AD, consistent with previous analyses (Seyfried et al., 2017).
To identify which, if any, of these molecular traits were related to disease progression, we assessed their relationship to clinical-pathological variables. The C1 module eigenprotein was negatively correlated with amyloid plaques, Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) score (Mirra et al., 1991) (BLSA data, ρ = −0.40, p = 1.0e–4) and neurofibrillary tangles, and Braak score (Braak and Braak, 1991) (BLSA data, ρ = −0.45, p = 9.7e–6) across cases and controls (Figures 3C and 3D; Table S4). In contrast, the C8 module eigen-protein was positively correlated with CERAD score (ACT data, ρ = 0.52, p = 2.9e–5) and Braak score (ACT data, ρ = 0.52, p = 2.9e–5) and anti-correlated with cognitive status (Cognitive Abilities Screening Instrument [CASI] score; Teng et al., 1994) (ACT data, ρ = −0.55, p = 7.7e–6) (Figures 3J–3L; Table S4). A similar pattern was observed for the C10 module (Figures 3R–3T; Table S4) and similar trends were observed with the other study cohorts (Figures S1B–S1Q and S2; Table S4).
We next analyzed an independent dataset (UPenn; see Method Details), which also contained seven non-AD diseases for comparison. The observed trends for the C1, C8, and C10 modules, including correlations with CERAD and Braak scores, were recapitulated in AD (Figures 3E–3G, 3M–3O, and 3U–3W; Table S4). Notably, these early disease-associated modules were significantly altered only in conditions with prominent dementia (C1: FTD-TDP, PSP-CBD, and PD-D) (Figure 3E; Table S4) (C8: FTD-TDP, PSP-CBD, and PD-D) (Figure 3M; Table S4) (C10: FTD-TDP and PSP-CBD) (Figure 3U; Table S4), but not in other neurodegenerative diseases where dementia is not a primary feature (MSA, ALS, and PD). We validated our findings using data from a second independent study (Emory; Ping et al., 2018) that used multiplex isobaric tandem mass tags (TMTs) and offline fractionation for protein quantification that identified more than 11,000 proteins (Figures S2 and S3; Table S4). We also note that all prefrontal proteomic modules (F1–F10) have significant module preservation in the temporal cortex (Zsummary module preservation range 2.5 to 18.5; Table S5).
Identification of Late Changes in the AD Proteome
We next evaluated proteomic changes occurring in cases representing the later, symptomatic phases of AD, identifying one downregulated (C2) and two upregulated (C3 and C4) modules in AD, but not changing in AsymAD. C2, which consisted of mitochondrial electron transport chain subunits and ATPase subunits (Figures 4A–4G), was decreased with progression of disease as measured by CERAD and Braak scores (Figures 4B–4D; Table S4) and was enriched in GABAergic markers (Romanov et al., 2017). Upregulated module C3 (Figures 4H–4O), which was positively correlated with CERAD and Braak scores and anti-correlated with CASI scores (Figures 4I–4L; Table S4), was enriched with genes involved in MAPK signaling. Module C4 (Figures 4P–4V) was also positively correlated with CERAD and Braak scores (Figures 4Q–4S; Table S4), but was enriched for genes involved in mRNA splicing. Similar trends were observed for these modules with the other study cohorts (Figures 4E–4G, 4M–4O, 4T–4V, S4, and S5; Table S4).
Figure 4. Late Proteomic Changes in AD.
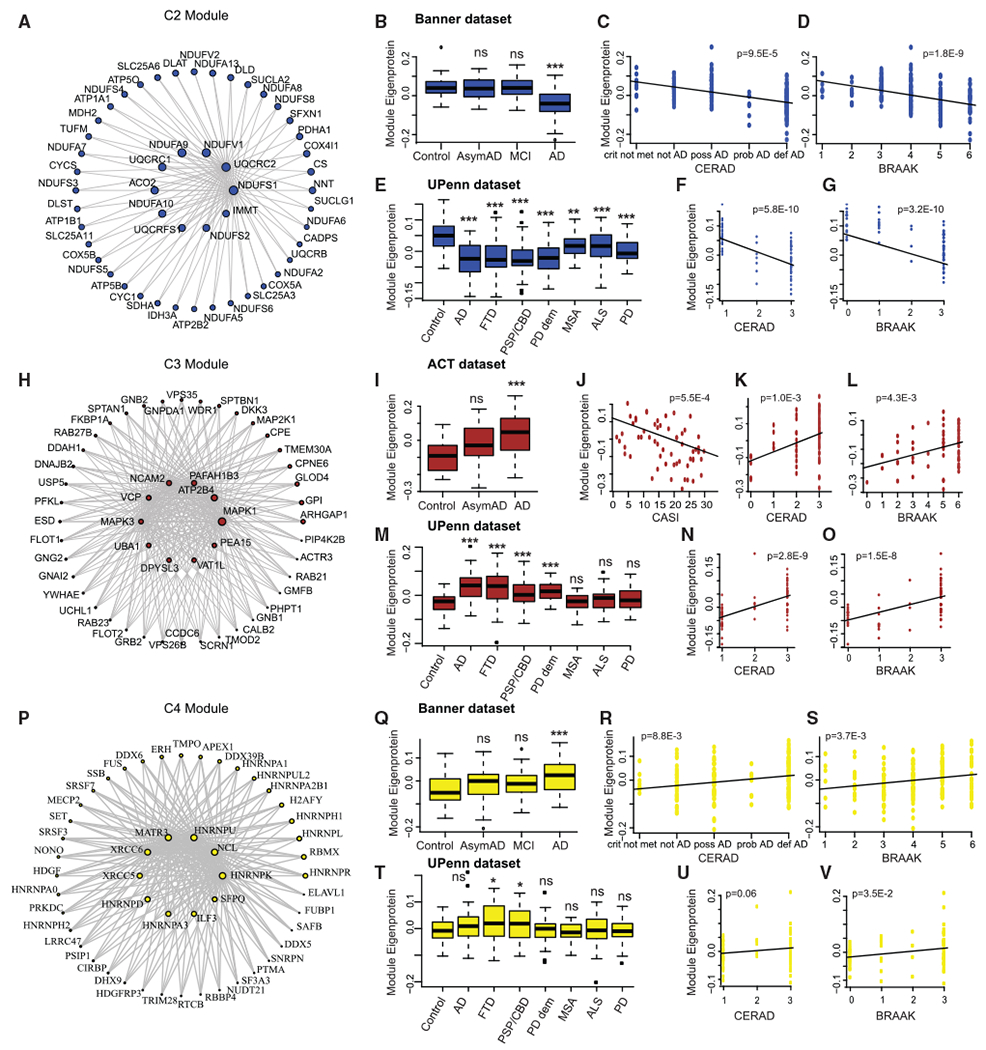
(A) Co-expression plot of the C2 module showing the hub proteins in the center.
(B–D) Plots showing C2 module eigenprotein trajectory with diagnosis (B), CERAD (C), and BRAAK (D) scores in Banner dataset.
(E–G) Plots showing C2 module eigenprotein trajectory with diagnosis (E), CERAD (F), and BRAAK (G) scores in validation UPenn dataset.
(H) Co-expression plot of the C3 module showing the hub proteins in the center.
(I–L) Plots showing C2 module eigenprotein trajectory with diagnosis (I), CASI (J), CERAD (K), and BRAAK (L) scores in ACT dataset.
(M–O) Plots showing C3 module eigenprotein trajectory with diagnosis (M), CERAD (N), and BRAAK (O) scores in validation UPenn dataset.
(P) Co-expression plot of the C4 module showing the hub proteins in the center.
(Q–S) Plots showing C4 module eigenprotein trajectory with diagnosis (Q), CERAD (R), and BRAAK (S) scores in Banner dataset.
(T–V) Plots showing C4 module eigenprotein trajectory with diagnosis (T), CERAD (U), and BRAAK (V) scores in validation UPenn dataset.
For boxplots (B), (E), (I), (M), (Q), and (T), the center line indicates the median, box limits indicate the 25th and 75th percentiles, whiskers extend to the most extreme data point which is no more than 1.5 times the interquartile range from the median, and outliers are represented by dots.
AD, Alzheimer’s disease; AsymAD, asymptomatic AD; FTD, frontotemporal dementia; PSP-CBD, progressive supranuclear palsy with corticobasal degeneration; PD, Parkinson’s disease; MSA, multiple systems atrophy; ALS, amyotrophic lateral sclerosis. *p < 0.05; **p < 0.01; ***p < 0.005; n.s., non-significant; p, p-value
See also Figures S4 and S5 and Tables S2, S3, and S4.
Comparison of the AD Proteome and Transcriptome
We reasoned that it would be important to understand how transcriptomic data were reflected at the level of proteomics collected from the same individuals in the same brain region, especially since little such comparative data are available in dementia. We leveraged 80 PSP, 80 AD, and 28 control cortical samples in the Mayo cohort in which proteomics and RNA-seq data were available from the same samples. We observed that for genes differentially expressed in PSP compared to AD (false discovery rate [FDR] < 0.1), the PSP-to-AD fold-change at the RNA level was significantly correlated with the fold-change at the protein level (β = 0.58, R2 = 0.56, p = 8.7e–165; Figure 5A). This suggests that slightly more than half of the variance in protein expression reflects gene expression, but an almost equivalent proportion of the signal is post-transcriptional, strongly supporting the utility of protein profiling and the complementarity of these two datasets.
Figure 5. Proteomics and Transcriptomic Analyses.
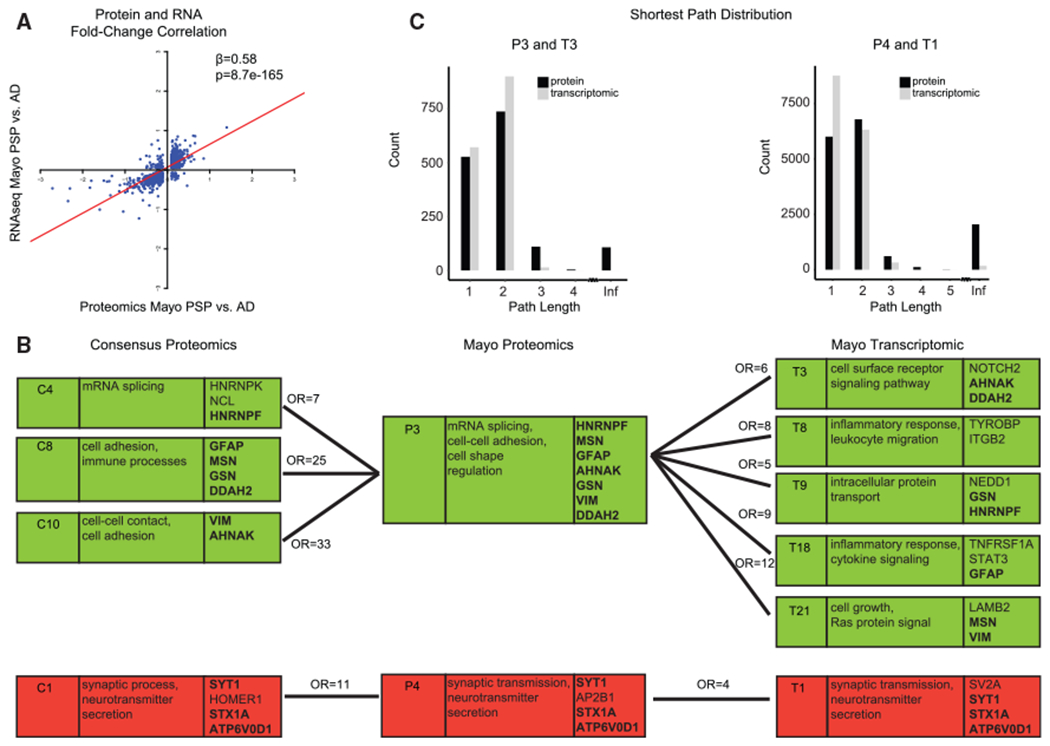
(A) For differentially expressed genes in PSP compared to AD (FDR < 0.1) in the Mayo dataset, the PSP-to-AD log2 fold-change at the RNA level correlated with the fold-change at the protein level.
(B) Relationship of consensus proteomics, Mayo proteomics, and Mayo transcriptomic modules. The first column is the module name, the second column includes Gene Ontology terms, and the third column includes hub genes of the module. Modules with significant hypergeometric overlap between consensus proteomics and Mayo proteomics and between Mayo proteomics and Mayo transcriptomic are shown. Numbers above connecting lines are the odds ratio of hypergeometric overlap. Bold gene names are hub genes shared across the consensus proteomics, Mayo proteomics, and Mayo transcriptomic modules. A green background denotes upregulation and a red bacgkround denotes downregulation in Alzheimer’s disease compared to controls or PSP.
(C) Distribution of shortest path between pairs of genes for P3 and T3 modules and for P4 and T1 modules. Infinite distance indicates unconnected gene pairs.
To delineate the conservation of biological processes at the proteome and transcriptome levels, we created separate co-expression networks for RNA (labeled T1–T21 modules from 11,140 transcripts) and protein (labeled P1–P15 modules from 3,849 proteins; Figure S6A) using only the Mayo cohort. As expected, all consensus proteomic modules overlapped with at least one “Mayo only” proteomic module (Figures 5B and S6B). Also per expectations, proteomic modules overall had lower connectivity than did the transcriptomic modules (Figure 5C; Table S6) and lower clustering coefficients (Table S6; p value range < 2.2e–16 to 7.2e–4), consistent with lower number of proteins compared with transcripts and subsequent higher connectivity within transcriptomic modules. The glial consensus modules upregulated early in AD, C8 (astrocyte) and C10 (microglia/endothelial cells), and the late upregulated module C4 (mRNA splicing) overlapped with the Mayo protein P3 module (C4-P3 hypergeometric overlap odds ratio (OR) = 7; C8-P3 hypergeometric overlap OR = 25; C10-P3 hypergeometric overlap OR = 33; Figure S6B). The Mayo P3 module was enriched for mRNA splicing and for genes that were markers for both astrocyte and microglial cell types, and it overlapped with multiple Mayo transcriptomic modules that were enriched for the same pathways and cell types: T3, T8, T9, T18, and T21 (Figures 5B and S6C; hypergeometric overlap, OR ranges 5–12). The consensus module downregulated in AD (C1) overlapped with the Mayo protein module P4 (OR = 11) and T1 transcriptomic module (OR = 4) (Figures 5B and S6C).
We next identified hub genes associated with disease altered at both the RNA and protein levels, which we reasoned would be worth highlighting as some may be candidate high-confidence therapeutic targets. Upregulated genes associated with processes associated with AD include HNRNPF (modules C4, P3, and T9; Figure 5B) (Geuens et al., 2016; Raj et al., 2018), MSN (modules C8, P3, and T21; Figure 5B) (Darmellah et al., 2012), GFAP (modules C8, P3, and T18; Figure 5B) (Dzamba et al., 2016; Efthymiou and Goate, 2017), DDAH2 (modules C8, P3, and T3; Figure 5B), GSN (modules C8, P3, and T9; Figure 5B), VIM (modules C10, P3, and T21; Figure 5B), and AHNAK (modules C10, P3, and T3; Figure 5B) (Sims et al., 2017). Those upregulated included synaptic proteins like SYT1 and STX1A, both involved in vesicle trafficking (Gautam et al., 2015; Smith et al., 2000), and ATP6V0D, involved in lysosomal acidification and linked to AD pathogenesis (Colacurcio and Nixon, 2016).
Non-overlapping Proteomic and Transcriptomic Modules
The electronic transport chain (C2) and MAPK (C3) late proteomic modules did not overlap with the transcriptomic modules, suggesting exclusive disease signatures identified via co-expression proteomic analyses. We found that a large percentage of genes from these non-overlapping proteomic modules were not assigned to any transcriptomic modules, indicating that the RNA of these unassigned genes, while expressed at moderate levels, did not have strong co-expression with genes in any transcriptomic module. Thus, we reasoned that these modules likely reflect post-transcriptional regulation, as they were not captured in the transcriptome (Hicke, 2001; Zhao et al., 2017). Consistent with this, we observe that the PSP-to-AD RNA-protein fold-change correlation was positive (β = 0.58) in modules captured in both RNA and protein, whereas the correlations were lower or negative for non-overlapping proteomic modules (β = −0.41 for C2 and β = 0.30 for C3). Non-overlapping proteomic modules represented genes with significantly lower RNA-to-protein correlation than average, consistent with data showing that MAPK pathway (C3) proteins show strong post-transcriptional (Sugiura et al., 2011; Whelan et al., 2012) and post-translational regulation (Lin et al., 2003).
Genetic Risk Is Enriched in Specific Proteomic Modules
The observed proteomic changes may be a cause or consequence of the disease. We next integrated genetic risk and genome-wide association studies (GWASs) to understand each module’s relationship to potential causal mechanisms (Gandal et al., 2018a; Parikshak et al., 2016). First, all early and late disease-associated modules showed significant correlation with APOE ε4 allele dosage (0, 1, or 2) in the expected direction. For example, modules upregulated in brains from patients diagnosed with AD (C8, C10, C3, and C4) showed positive correlation with APOE (Banner data, C8: ρ = 0.27, p = 1.7e–4; Banner data C10: ρ = 0.22, p = 2.4e–3; Banner data C3: ρ = 0.2, p = 5.8e–3; Mayo C4: ρ = 0.21, p = 3.1e–3) and the downregulated modules (C1 and C2) showed negative correlation with APOE (Banner data, C1: ρ = −0.19, p = 8.8e–3; Banner data, C2: ρ = −0.25, p = 5.2e–4), consistent with expectations (Table S4). Next, we used summary data from AD GWAS as input for Multi-marker Analysis of GenoMic Annotation (MAGMA) (de Leeuw et al., 2015), finding that common variants from AD GWAS (Lambert et al., 2013) were significantly enriched in genes within the consensus C8 (astrocyte) and C10 (microglia) modules (Figure 6A), in addition to the overlapping Mayo proteomics P3 module (Figure 6B) and Mayo transcriptomics T3 and T8 modules (Figure 6C). This is consistent with previous reports of AD candidate gene enrichment in glial modules (Johnson et al., 2020; Seyfried et al., 2017; Zhang et al., 2013). Combined with the early upregulation of C8 and C10 and their preservation in multiple datasets, this highlights the co-regulated set of genes in C8 and C10 as potential targets for disease modification.
Figure 6. Common Variant Enrichment.
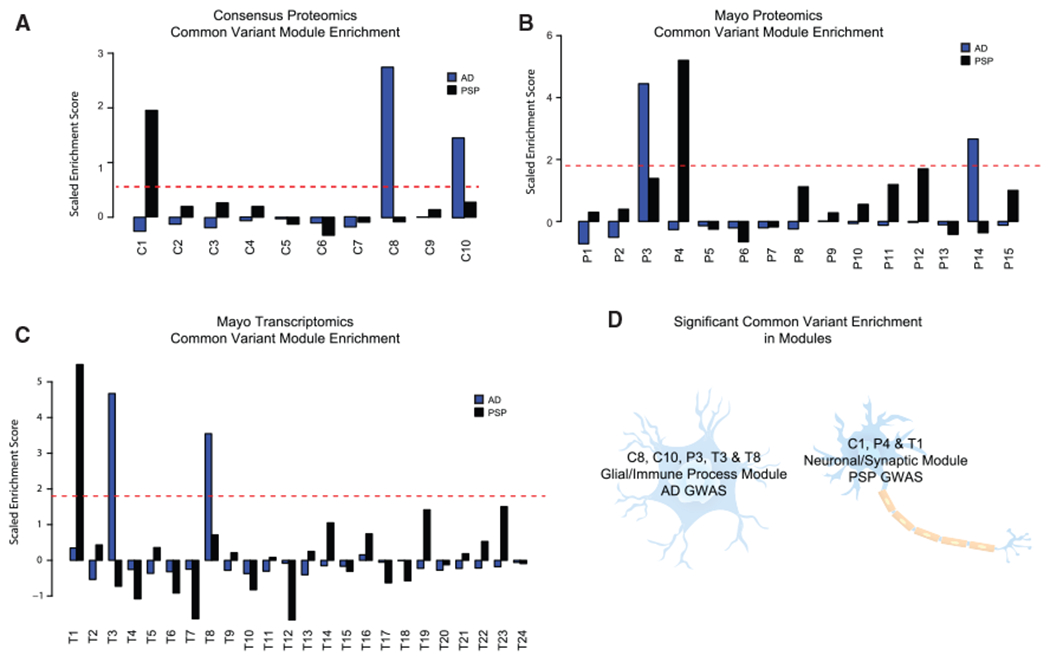
(A–C) Mean scaled enrichment of GWAS hits from AD (Lambert et al., 2013) and PSP (Höglinger et al., 2011) in (A) consensus proteomics modules, (B) Mayo proteomics modules, and (C) Mayo transcriptomic modules.
(D) Diagram showing enrichment of AD and PSP common variants from GWAS studies in significant consensus proteomics modules, Mayo proteomics modules, and Mayo transcriptomic modules, in addition to their cell-type enrichment.
In striking contrast, common variants from PSP GWAS (Höglinger et al., 2011) were significantly enriched in the early neuronal, C1 module (Figure 6A) and the overlapping Mayo proteomics P4 module (Figure 6B) and Mayo transcriptomic, T1, module (Figure 6C). These analyses illustrate two major points: (1) genetic risk enriches in modules with early changes in disease, consistent with the causal role of genetic risk variants; and (2) causal risk variants in these two tauopathies associated with dementia (AD and PSP) differ in terms of the specific biology and cell types that they causally impact (Figure 6D).
DISCUSSION
Quantifying changes in the brain proteome provides a direct means of understanding the molecular pathology in proteinopathies such as AD (Arnold et al., 2013; Forman et al., 2004; Ross and Poirier, 2004). Understanding the key drivers in a proteomic network that are enriched for causal genetic risk has direct implications for identifying druggable protein targets for the disease. By combining multiple cohorts of post-mortem human AD brains with label-free proteomics, we detected robust proteomic modules representing early and late changes associated with AD that were conserved in other neurodegenerative dementias and at the transcriptomic level. We found two specific consensus proteomic modules enriched for AD genetic risk and a distinct consensus module associated with PSP genetic risk variants. This analysis provides insights into the pathogenic processes related to AD onset and progression, many of which are conserved across neurodegenerative dementias. Despite these shared aspects of molecular pathology delineated by protein co-expression networks, we find that different dementias do show specificity with regards to enrichment for causal genetic risk, indicative of distinct inception points in these disorders.
One of the major challenges in using proteomics approaches is the limited scalability and reproducibility of proteomic studies to identify disease-specific alterations in large cohorts. While previous AD proteomic studies incorporated one or two datasets (Johnson et al., 2018; McKenzie et al., 2017; Ping et al., 2018; Seyfried et al., 2017; Yu et al., 2018; Zhang et al., 2018), we integrated data representing five different cohorts and used a bioinformatics approach to define robust disease-associated proteomic networks. While this work was in revision, a similarly large analysis of proteomic changes in AD using multiple cohorts from AMP-AD was published that highlighted multiple cell-type and molecular pathways that are altered in AD (Johnson et al., 2020), findings highly concordant with our current analysis. In contrast to Johnson et al., our cWGCNA approach bypassed the need for batch correction, which can lead to the removal or compression of the disease signal (Gandal etal., 2018b; Swarup et al., 2019), and we indeed observe larger signals in disease-associated module eigenproteins. Our work is complementary in that different pipelines and analytic approaches are used here, including consensus network analysis and an additional cohort for network construction, indicating that the major findings in both studies are robust to different analytical pipelines.
Our study is the largest to comprehensively compare RNA and protein co-expression in dementia. These early neuronal (C1) and glial (C8 and C10) consensus proteomic modules significantly overlapped with transcriptomic modules, suggesting these biological processes are altered at both the RNA and protein levels. Previous AD transcriptomic co-expression analyses also showed upregulation of inflammatory astrocytic and microglial signatures and downregulation of neuronal signatures (Mostafavi et al., 2018; Seyfried et al., 2017; Swarup et al., 2019; Wang et al., 2016). Our analysis also demonstrated the early disease-associated modules were correlated only with neurodegenerative conditions with dementia (FTD-TDP, PSP-CBD, and PD-D) and not in neurodegenerative diseases where dementia is not a primary feature (MSA, ALS, and PD).
Although these data highlight the role of neuronal loss (Braak and Braak, 1991; Braak et al., 2003; Davidson et al., 2007; De Strooper and Karran, 2016; Dickson et al., 2010; Hauw et al., 1994; Mackenzie et al., 2011) and astrocyte (González-Reyes et al., 2017; Perez-Nievas and Serrano-Pozo, 2018; Radford et al., 2015) and microglial (Bachiller et al., 2018; Fernández-Botrán et al., 2011; Hansen et al., 2018; Ishizawa and Dickson, 2001; Navarro et al., 2018; Radford et al., 2015) upregulation in the pathogenesis of all of these diseases, their time course and the enrichment of genetic risk in all three early disease-associated modules are consistent with the prediction that causal changes should occur at the earliest points in the disease course. However, the risk for different disorders was differentially distributed. Common partitioned heritability for PSP identified in GWASs was significantly enriched in the early C1 neuronal module, indicating that neuronal dysfunction is the primary causal driver in this disease. Synaptic loss is a common theme across all neurodegenerative diseases, but genetic enrichment in specific synaptic modules is not observed universally. In contrast, AD genetic signal from GWASs was enriched in the C8, astrocytic, and C10 microglia modules, which supports the emerging consensus of a glial-immune etiology in AD (Efthymiou and Goate, 2017; Heckmann et al., 2019; Heneka et al., 2015; Hong et al., 2016; Ising et al., 2019; Keren-Shaul et al., 2017; Liddelow et al., 2017; Nott et al., 2019; Wang et al., 2015). While some neuronal changes occur early in AD, as evidenced by the changes in C1, the causal elements reside in genes associated with glial cells.
As proteins represent druggable targets (Agora. Accelerating Medicines Partnership in Alzheimer’s Disease; Griffith et al., 2013; Hopkins and Groom, 2002; Oprea et al., 2018), we further examined the hubs of the C8 and C10 modules, which were early AD associated, had transcriptomic co-expression concordance, converged with other neurodegenerative dementias, and had AD causal genetic enrichment. For example, both GSN and MSN are preserved C8 hub genes linked to AD pathogenic mechanisms (Darmellah et al., 2012; Johnson et al., 2020; Seyfried et al., 2017). MSN and GSN are both considered druggable based on the Drug Gene Interaction database (Agora. Accelerating Medicines Partnership in Alzheimer’s Disease; Griffith et al., 2013). Other preserved C8 hub genes such as DDAH2 have not yet been linked to AD pathogenesis but are potentially druggable. In module C10, two hub genes, ANXA1 and GJA, have been previously associated with AD mechanisms (Kajiwara et al., 2018; McArthur et al., 2010).
In addition to these early disease-associated modules, we also identified late AD-associated modules (C2, C3, and C4), which may represent biological processes associated with chronic disease progression. When comparing late AD-associated consensus proteomic modules (C2, C3, and C4) to transcriptomic co-expression modules, the mRNA splicing C4 module was preserved in the transcriptomic network. Previous proteomic studies have identified RNA splicing (Golde et al., 1990; Love et al., 2015; Raj et al., 2018) signatures downregulated later in AD progression as well.
Our analyses did yield modules not observed in previous transcriptomic studies. Both C2 and C3, representing electron transport and MAPK pathways, respectively, did not overlap with transcriptomic modules. Accordingly, genes within these modules show lower protein-RNA correlation compared with the average observed across the genome. As late biological processes, these may be adaptive responses to AD pathology or propagate disease progression after AD pathology is established. Mitochondrial modules were associated with AD using transcriptomics and proteomics in previous studies (Johnson et al., 2018; Kim and Choi, 2015; Mostafavi et al., 2018; Munoz and Ammit, 2010; Ping et al., 2018; Shih et al., 2015). The mitochondrial C2 module was the only module shared across all neurodegenerative diseases, including those with and without dementia (AD, FTD TDP, PSP-CBD, PD-D, MSA, ALS, and PD). This highlights that dysfunctional energy metabolism, possibly due to impaired mitochondria trafficking or oxidative phosphorylation, is likely dysregulated in all of these neurodegenerative disorders at late stages (Briston and Hicks, 2018). C2 is not enriched in genetic risk and is dysregulated late in disease, suggesting that while it is shared across neurodegenerative disorders, it does not drive early disease pathophysiology.
Dysregulation of MAPK signaling was identified in a previous smaller, proteomic study (Seyfried et al., 2017) but has not been clearly identified in transcriptomic studies. MAPK signaling is implicated in many pathways dysregulated in dementia, including immune (Jones and Kounatidis, 2017; Lee and Kim, 2017; Zhu et al., 2002), oxidative stress (Munoz and Ammit, 2010; Shih et al., 2015), and tau hyperphosphorylation (Guise et al., 2001; Kim and Choi, 2010; Wang and Liu, 2008). The potential contribution of PEA-15, one of the C3 modules hubs, to AD pathogenesis has not been studied. Thus, in addition to identifying molecular disease processes previously associated with AD, proteomic co-expression analysis can identify less-established mechanisms.
Our consensus proteomic co-expression approach did not identify all processes associated with AD in previous proteomic and transcriptomic co-expression analyses (e.g., decreased cytoskeletal and microtubule signatures in AD) (Andreev et al., 2012; Seyfried et al., 2017; Zhang et al., 2018). Our study’s microtubule C5 module was not consistently downregulated in AD across cohorts. Our consensus proteomic modules also did not include cell cycle, chromatin modification, glucuronosyl-transferase, or axon guidance modules that were identified in several transcriptomic studies (Mostafavi et al., 2018; Wang et al., 2016; Zhang et al., 2013). This is perhaps, in part, a limitation of proteomic studies, where less soluble or very large membrane-associated proteins are more difficult to detect, and the conservative consensus approach, again supporting the complementarity of multi-omics approaches and the value of measuring distinct molecular species. Overall, our multi-omics, multi-cohort approach provides insights into early and late changes in AD that are shared with other neurodegenerative dementias but show disease specificity for genetic risk. Those modules with consistent molecular alterations at the genetic, transcriptomic, and proteomic levels are ripe for future investigation as drug targets.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Daniel H. Geschwind.
Material Availability
This study did not generate new unique reagents. Further information and requests for resources and reagents should be directed to the Lead Contact, Daniel H. Geschwind.
Data and Code Availability
All the proteomics data are available here: https://adknowledgeportal.synapse.org/Explore/Projects?Grant%20Number=U01AG046161. Links to data for each study can be found in the Key Resources Table. The accession number for the ACT data reported in this paper is https://www.synapse.org/ Synapse ID:syn5759376. The accession number for the MSBB data reported in this paper is https://www.synapse.org/ Synapse ID:syn3159438. The accession number for the Banner data reported in this paper is https://www.synapse.org/ Synapse ID:syn7170616. The accession number for the Mayo data reported in this paper is https://www.synapse.org/ Synapse ID:syn5550404. The accession number for the UPenn data reported in this paper is https://adknowledgeportal.synapse.org Synapse ID:syn21411742.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PLCD1 antibody | Abcam | Cat# ab134936; RRID:AB_2857853 |
| HEPACAM antibody | Abcam | Cat# ab130769; RRID:AB_2857854 |
| PADI2 antibody | Abcam | Cat# ab16478; RRID:AB_470249 |
| GAPDH antibody | Abcam | Cat# ab8245; RRID:AB_2107448 |
| GJA1 antibody | Sigma | Cat# C6219; RRID:AB_476857 |
| Biological Samples | ||
| Postmortem dorsolateral prefrontal and precuneus cortex brain tissue | Baltimore Longitudinal Study of Aging (BLSA) at Johns Hopkins University | https://www.hopkinsmedicine.org/research/labs/alzheimers-disease-research-center |
| Postmortem prefrontal cortex brain tissue | Adult Changes of Thought (ACT) at the University of Washington and Group Health Cooperative of Puget Sound | http://depts.washington.edu/mbwc/adrc/page/neuropathology |
| Postmortem prefrontal cortex brain tissue | Mount Sinai Brain Bank (MSBB) | https://icahn.mssm.edu/research/nih-brain-tissue-repository |
| Postmortem prefrontal cortex brain tissue | Banner Sun Health Research Institute (Banner) | https://www.bannerhealth.com/services/research/locations/sun-health-institute/programs/body-donation |
| Postmortem temporal cortex brain tissue | Mayo Clinic Brain Bank (Mayo) | https://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank |
| Postmortem prefrontal cortex brain tissue | University of Pennsylvania Brain Bank (UPenn) | https://www.med.upenn.edu/cndr/biosamples-brainbank.html |
| Postmortem dorsolateral prefrontal brain tissue | Emory University (Emory) | http://alzheimers.emory.edu/ |
| Deposited Data | ||
| Baltimore Longitudinal Study of Aging (BLSA) proteomic data | Seyfried et al., 2017 | https://doi.org/10.7303/syn3606086 |
| Adult Changes of Thought (ACT) proteomic data | This study | https://doi.org/10.7303/syn5759376 |
| Mount Sinai Brain Bank (MSBB) proteomic data | This study | https://repo-prod.prod.sagebase.org/repo/v1/doi/locate?id=syn3159438&type=ENTITY |
| Banner Sun Health Research Institute (Banner) proteomic data | This study | https://doi.org/10.7303/syn7170616 |
| Mayo Clinic Brain Bank (Mayo) proteomic and transcriptomic data | This study | https://doi.org/10.7303/syn5550404 |
| University of Pennsylvania (UPenn) proteomic data | This study | https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn21411742 |
| Emory proteomic data | Ping et al., 2018 | https://www.synapse.org/#!Synapse:syn10239444 |
| Cell type enrichment Markers | Zhang et al., 2016 | http://www.brainrnaseq.org/ |
| Alzheimer’s disease GWAS summary statistics | Lambert et al., 2013 | http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php |
| Progressive Supranuclear Palsy GWAS summary statistics | Höglinger et al., 2011 | https://www.niagads.org/datasets/ng00045 |
| Software and Algorithms | ||
| R | R Core Team | https://www.R-project.org/ |
| WGCNA R package | Langfelder and Horvath, 2008 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| Gene Ontology | http://geneontology.org/ | |
| igraph R package | Csardi and Nepusz, 2006 | https://igraph.org/r/ |
| MAGMA | de Leeuw et al., 2015 | https://ctg.cncr.nl/software/magma |
| Other | ||
| Interactive graphical interface for consensus proteomic network | https://coppolalab.ucla.edu/gclabapps/nb/browser?id=SwarupChangConsensusProteomics |
Code is available on GitHub: https://github.com/dhglab/Identification-of-conserved-proteomic-networks-in-neurodegenerative-dementia
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Samples
Postmortem human brain tissues were obtained from the Baltimore Longitudinal Study of Aging (BLSA) at Johns Hopkins University, Adult Changes of Thought (ACT) at the University of Washington and Group Health Cooperative of Puget Sound, Mount Sinai Brain Bank (MSBB) at Mount Sinai University, Banner Sun Health Research Institute (Banner), Mayo Clinic Brain Bank (Mayo) at the Mayo Clinic in Jacksonville, Florida, University of Pennsylvania Brain Bank (UPenn), and Emory University (Emory). All human samples were obtained under their respective institutional review board. For details about sample-level metadata including diagnosis, gender, neuropathological criteria, etc. please see Table S1. The BLSA samples consisted of 97 samples from the dorsolateral prefrontal cortex (BA9 area) and precuneus (parietal cortex, BA7) representing 15 controls, 15 AsymAD and 20 AD cases (Seyfried et al., 2017). The BLSA precuneus region was not used in our analyses. The ACT samples consisted of 65 samples from 12 controls, 14 AsymAD and 39 AD cases from prefrontal cortex area. The MSBB consisted of 251 prefrontal cortex samples from 78 controls, 28 AD possible, 13 AD probable and 132 AD definite cases based on clinical dementia rating (CDR) scores (Morris, 1993). The Banner samples were from prefrontal cortex of 30 controls, 28 mild cognitive impairment, 33 AsymAD and 98 confirmed AD cases. The Mayo samples were from temporal cortex consisting of 28 controls, 84 AD, and 84 PSP cases. The UPenn samples were from prefrontal cortex consisting of 43 controls, 42 AD, 58 ALS, 29 FTD-TDP, 22 MSA, 33 PD, 58 PD-dementia, and 48 PSP-CBD. Emory samples were previously published (Ping et al., 2018; Seyfried et al., 2017). This study was performed under the auspices of the UCLA Office of Human Research Protection, which determined that it was exempt because samples were anonymous pathological specimens (IRB# 15-001397).
METHOD DETAILS
Quantitative Proteomics
Label-free quantitative proteomics were performed at the Emory Proteomics Core, Emory University, USA. Detailed methods were published elsewhere (Seyfried et al., 2017). The label free quantitation (LFQ) algorithm in MaxQuant (Cox and Mann, 2008) was used for protein quantitation. The quantitation method only considered razor and unique peptides for protein level quantitation. The LFQ intensities were log2 transformed for downstream analyses.
Covariate correction
Protein LFQ (proteomics) and normalized FPKM values (RNA-seq) were assessed for effects from biological covariates (diagnosis, age, gender) and technical variables (batch, brain bank, etc.). We used a linear regression model accounting for biological and technical covariates depending upon the cohort to be analyzed. The final model used was implemented in R version 3.6.1 (R Core Team, 2019) as follows:
Consensus WGCNA
Consensus weighted co-expression network analysis (cWGCNA) was performed in R (R Core Team, 2019) using WGCNA package (Langfelder and Horvath, 2008). To identify consensus proteomics modules across different brain banks, we employed a signed cWGCNA approach by calculating component-wise values for topological overlap for individual brain banks. Biweighted mid-correlations were calculated for all pairs of genes, and then a signed similarity matrix was created. In the signed network, the similarity between genes reflects the sign of the correlation of their expression profiles. The signed similarity matrix was then raised to power β to emphasize strong correlations and reduce the emphasis of weak correlations on an exponential scale. The resulting adjacency matrix was then transformed into a topological overlap matrix (TOM). The consensus TOM (cTOM) was calculated by taking the component-wise minimum of TOMs in each dataset. Using 1 – cTOM as the distance measure, genes were hierarchically clustered (Parikshak et al., 2016). Module assignments were determined using a dynamic tree-cutting algorithm (cutreeHybrid, using the default parameters except deepSplit = 4, cutHeight = 0.999, minModuleSize = 20, dthresh = 0.07 and pamStage = FALSE). Network visualization was performed with the igraph package in R (Csardi and Nepusz, 2006).
Module eigenproteins were calculated as the first principal component of the coexpressed genes in the module (Langfelder and Horvath, 2007; Zhang and Horvath, 2005). The eigengene-based connectivity (kME) was used to represent the strength of a gene’s correlation with other gene module members. It is defined as the correlation between a gene’s expression and the module eigengene (Langfelder and Horvath, 2007; Zhang and Horvath, 2005). Hub genes of a module were considered genes with high kME.
QUANTIFICATION AND STATISTICAL ANALYSIS
Cell-type and Gene-ontology enrichment
For gene-set enrichment analysis of cell type (Miller et al., 2008; Romanov et al., 2017; Zhang et al., 2016) and gene ontology (Ashburner et al., 2000; Gene Ontology Consortium, 2019; Yu et al., 2012) we use the hypergeometric test, which is equivalent to a onesided Fisher’s exact test, to evaluate whether a gene set is enriched over background, providing a p value and enrichment value for gene set enrichment (Parikshak et al., 2016). An assumption with such a test is that the background set is similar to the gene set considered for enrichment in all factors other than pathway membership.
Module Eigenprotein Association with Disease
We determined if a module eigenprotein was significantly different from disease diagnosis (AD or AsymAD) versus control using the Wilcoxon rank sum test (Wilcoxon, 1945). A module was considered as late if it was significantly associated with AD compared to control in at least three of the five cohorts. A module was considered early if it was significantly associated with AsymAD and AD compared to control in at least one of three cohorts with AsymAD (Banner, ACT, BLSA) and significantly associated with AD in at least three of the five cohorts.
For the Mayo only proteomic module analyses, we compared AD to control and AD to PSP given the lower number of control available (28) compared to AD (84) and PSP (84). Comparing AD to control had reduced power versus comparing AD to PSP. To determine if a consensus proteomic module was associated with AD in the Mayo cohort, we only compared AD to control in order to be consistent with the above late module criteria in other cohorts, which also compared AD to controls.
Module eigenproteins were correlated with clinical score (CASI) and neuropathological scores (CERAD, Braak) using Pearson correlation (Pearson, 1931). We used the same method for clinical scores and neuropathological scores in UPenn and Emory datasets. For UPenn and Emory, we determined if the module eigenprotein was significantly different from disease diagnosis and control using linear regression.
Western blot validation
Brain homogenates (frontal cortex) in urea (50 μg) were mixed with Laemmli sample buffer and resolved by SDS-PAGE before an overnight wet transfer to nitrocellulose membranes (BioRad) as previously reported (Seyfried et al., 2017). Membranes were blocked with casein blocking buffer (Sigma B6429) and probed with primary polyclonal antibodies for Anti-PLCD1 antibody (Abcam #ab134936), anti-HEPACAM (Abcam #ab130769), anti-PADI2 antibody (Abcam #ab16478), anti-GAPDH antibody (Abcam #ab8245), and anti-Connexin-43 (Sigma #C6219) overnight at 4°C. Membranes were incubated with secondary antibodies conjugated to Alexa Fluor 680 (Invitrogen) or IRDye800 (Rockland) fluorophores for one hour at room temperature. Images were captured using an Odyssey Infrared Imaging System (Li-Cor Biosciences). Protein densitometry for relative quantification was performed using ImageJ open source software. For GJA1, the higher molecular weight band reflective of ubiquitination (Abreha et al., 2018) was analyzed separately from the lower molecular weight band. Densitometry was compared between AD and control using the Student’s t test (Student, 1908).
Differential expression analysis in PSP compared to AD
We used the set of differentially expressed genes in PSP compared to AD (FDR < 0.1) at the RNA and protein level. We correlated the fold-change in RNA level to the fold-change in protein level with linear regression and the Wald test to determine significance.
Comparison of the AD proteome in prefrontal and temporal cortex
We created a prefrontal consensus proteomic network with the four prefrontal cortex cohorts (BLSA, MSBB, Banner, ACT) using the same methodology and parameters when creating the consensus proteomic network with all five proteomic cohorts (BLSA, MSBB, Banner, ACT, Mayo) as described above. For each prefrontal cohort, we calculated prefrontal consensus module preservation using the Zsummary in the Mayo proteomic temporal cortex cohort (Langfelder et al., 2011). Zsummary greater than 2 indicates module preservation and Zsummary greater than 10 indicated strong module preservation.
Comparison of the AD proteome and transcriptome
We performed weighted co-expression network analysis (WGCNA) (Langfelder and Horvath, 2008) for only the Mayo proteomic and transcriptomic data resulting in a Mayo only proteomic network and transcriptomic network. Biweighted mid-correlations were calculated for all pairs of genes, and then a signed similarity matrix was created. The signed similarity matrix was then raised to power β to emphasize strong correlations and reduce the emphasis of weak correlations on an exponential scale. The resulting adjacency matrix was then transformed into a topological overlap matrix (TOM). Using 1 – TOM as the distance measure, genes were hierarchically clustered (Parikshak et al., 2016).
We identified consensus proteomic modules overlapping with Mayo proteomic modules, and Mayo proteomic modules overlapping with Mayo transcriptomic modules using the Fisher’s exact test (Fisher, 1922).
Module Connectivity in Proteomic versus Transcriptomic Modules
Hub genes had high eigengene-based connectivity (kME) (Langfelder and Horvath, 2007; Zhang and Horvath, 2005).
We compared intersecting genes from Mayo proteomic modules that had significant hypergeometric overlap with Mayo transcriptomic modules. To calculate the shortest path between all pairs of genes in a module, we first removed all edges with correlations from the adjacency matrix lower than the 75th percentile. The shortest path was calculated with the igraph package in R (Csardi and Nepusz, 2006). For pairs of genes that were connected, we determined if the mean shortest path in the Mayo proteomic module was significantly different than the mean shortest path in the Mayo transcriptomic modules using Student’s t test (Student, 1908). We calculated the clustering coefficient, which measures the connectivity among a gene’s neighbors, using the WGCNA R package (Langfelder and Horvath, 2008) and determined if it was significantly different in Mayo proteomic modules versus transcriptomic modules using Student’s t test (Student, 1908).
Genetic Risk GWAS Enrichment
We considered a module to be correlated with APOE ε4 allele if the correlation was significant in at least one of the three cohorts with APOE genotypes (Mayo, BLSA, Banner)
GWAS summary data were obtained from the International Genomics of Alzheimer’s Project (IGAP) for AD (Lambert et al., 2013) and for PSP(Höglinger et al., 2011). We used the MAGMA approach (de Leeuw et al., 2015) to assign each gene a score based on the best p value of a SNP in a given GWAS study within 20kB of the gene, and then set a p value cut-off at 0.05 to define the gene as included in the common variant set related to that study. We performed enrichment analysis with logistic regression controlling for gene length and other biases (Parikshak et al., 2016).
Supplementary Material
Highlights.
We distinguish robust early and late proteomic changes in AD in multiple cohorts
Changes in dementias are not preserved in other neurodegenerative diseases
Genetic risk is enriched in glial-immune modules for AD and synaptic for PSP
Protein expression variance is reflected both at RNA and post-transcriptional levels
ACKNOWLEDGMENTS
Support for this research was provided by funding from the National Institute of Neurological Disorders and Stroke (NINDS), United States (R25NS065723 Translational Neuroscience Training Grant, P30NS055077 NINDS Emory Neuroscience Core) and National Institute on Aging (NIA), United States (R01AG053960, R01AG057911, R01AG061800, RF1AG057470, RF1AG05747101, U01AG046161 Accelerating Medicine Partnership for Alzheimer’s disease, P50AG025688 Emory Alzheimer’s Disease Research Center).
The results published here are in whole or in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org/). BLSA data were generated from postmortem brain tissue collected through the National Institute on Aging’s Baltimore Longitudinal Study of Aging and provided by Dr. Levey from Emory University.
ACT data were generated from postmortem brain tissue collected through the Adult Changes in Thought study (E. Larsen), and The University of Washington ADRC (T. Montine), with the proteomics provided by Drs. Seyfried, Lah, and Levey from Emory University (NIA U01 AG046161; NIA P50 AG025688; NIA P30 NS055077).
MSBB data were provided by Dr. Levey from Emory University based on postmortem brain tissue collected through the Mount Sinai VA Medical Center Brain Bank provided by Dr. Eric Schadt from Mount Sinai School of Medicine.
Banner data were provided by Dr. Levey from Emory University. A portion of these data were generated from samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services, United States (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission, United States (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research, United States.
Mayo data were provided by Dr. Levey from Emory University based on postmortem brain tissue provided by the following sources: The Mayo Clinic Alzheimers Disease Genetic Studies, led by Dr. Nilufer Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimers Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by NIA grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949, NINDS grant R01 NS080820, CurePSP Foundation, United States, and support from Mayo Foundation, United States. Study data includes samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinsons Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimers Disease Core Center), the Arizona Department of Health Services, United States (contract 211002, Arizona Alzheimers Research Center), the Arizona Biomedical Research Commission, United States (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinsons Research, United States.
UPenn data were generated from postmortem brain tissue collected through the University of Pennsylvania and provided by Dr. Levey from Emory University. Emory data were generated from postmortem brain tissue collected through the Emory Alzheimers Disease Research Center Brain Bank and provided by Dr. Levey from Emory University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107807.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abreha MH, Dammer EB, Ping L, Zhang T, Duong DM, Gearing M, Lah JJ, Levey AI, and Seyfried NT (2018). Quantitative Analysis of the Brain Ubiquitylome in Alzheimer’s Disease. Proteomics 18, e1800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M, Wang X, Burgess JD, Watzlawik J, Serie DJ, Younkin CS, Nguyen T, Malphrus KG, Lincoln S, Carrasquillo MM, et al. (2018). Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement. 14, 352–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s Association (2019). 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 15, 321–387. [Google Scholar]
- Andreev VP, Petyuk VA, Brewer HM, Karpievitch YV, Xie F, Clarke J, Camp D, Smith RD, Lieberman AP, Albin RL, et al. (2012). Label-free quantitative LC-MS proteomics of Alzheimer’s disease and normally aged human brains. J. Proteome Res 11, 3053–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Stieler JT, and Holzer M (2016). Tau and tauopathies. Brain Res. Bull 126, 238–292. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Toledo JB, Appleby DH, Xie SX, Wang L-S, Baek Y, Wolk DA, Lee EB, Miller BL, Lee VM-Y, and Trojanowski JQ (2013). Comparative survey of the topographical distribution of signature molecular lesions in major neurodegenerative diseases. J. Comp. Neurol 521, 4339–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. ; The Gene Ontology Consortium (2000). Gene ontology: tool for the unification of biology. Nat. Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachiller S, Jiménez-Ferrer I, Paulus A, Yang Y, Swanberg M, Deierborg T, and Boza-Serrano A (2018). Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci 12, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonham LW, Karch CM, Fan CC, Tan C, Geier EG, Wang Y, Wen N, Broce IJ, Li Y, Barkovich MJ, et al. International FTD-Genomics Consortium (IFGC); International Parkinson’s Disease Genetics Consortium (IPDGC); International Genomics of Alzheimer’s Project (IGAP) (2018). CXCR4 involvement in neurodegenerative diseases. Transl. Psychiatry 8, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, and Braak E (2003). Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Briston T, and Hicks AR (2018). Mitochondrial dysfunction and neurodegenerative proteinopathies: mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans 46, 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, et al. ; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team (2017). A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JA, Chen Z, Won H, Huang AY, Lowe JK, Wojta K, Yokoyama JS, Bensimon G, Leigh PN, Payan C, et al. (2018). Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol. Neurodegener 13, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio DJ, and Nixon RA (2016). Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev 32, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppola G, Chinnathambi S, Lee JJ, Dombroski BA, Baker MC, Soto-Ortolaza AI, Lee SE, Klein E, Huang AY, Sears R, et al. ; Alzheimer’s Disease Genetics Consortium (2012). Evidence for a role of the rare p.A152T variant in MAPT in increasing the risk for FTD-spectrum and Alzheimer’s diseases. Hum. Mol. Genet 21, 3500–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Csardi G, and Nepusz T (2006). The igraph software package for complex network research. Int. J. Complex Syst 1695, 1–9. [Google Scholar]
- Cummings JL (2003). Toward a molecular neuropsychiatry of neurodegenerative diseases. Ann. Neurol 54, 147–154. [DOI] [PubMed] [Google Scholar]
- Darmellah A, Rayah A, Auger R, Cuif M-H, Prigent M,Arpin M, Alcover A, Delarasse C, and Kanellopoulos JM (2012). Ezrin/radixin/moesin are required for the purinergic P2X7 receptor (P2X7R)-dependent processing of the amyloid precursor protein. J. Biol. Chem 287, 34583–34595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson Y, Kelley T, Mackenzie IRA, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, and Mann DMA (2007). Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 113, 521–533. [DOI] [PubMed] [Google Scholar]
- De Jager PL, Yang H-S, and Bennett DA (2018). Deconstructing and targeting the genomic architecture of human neurodegeneration. Nat. Neurosci 21, 1310–1317. [DOI] [PubMed] [Google Scholar]
- de Leeuw CA, Mooij JM, Heskes T, and Posthuma D (2015). MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, and Karran E (2016). The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Schork AJ, Wang Y, Witoelar A, Sharma M, McEvoy LK, Holland D, Brewer JB, Chen C-H, Thompson WK, et al. ; ADNI, ADGC, GERAD, CHARGE and IPDGC Investigators (2015). Genetic overlap between Alzheimer’s disease and Parkinson’s disease at the MAPT locus. Mol. Psychiatry 20, 1588–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, and Josephs KA (2010). Neuropathology of variants of progressive supranuclear palsy. Curr. Opin. Neurol 23, 394–400. [DOI] [PubMed] [Google Scholar]
- Dzamba D, Harantova L, Butenko O, and Anderova M (2016). Glial Cells - The Key Elements of Alzheimer’s Disease. Curr. Alzheimer Res 13, 894–911. [DOI] [PubMed] [Google Scholar]
- Efthymiou AG, and Goate AM (2017). Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener 12, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkkinen MG, Kim M-O, and Geschwind MD (2018). Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol 10, a033118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Botrán R, Ahmed Z, Crespo FA, Gatenbee C, Gonzalez J, Dickson DW, and Litvan I (2011). Cytokine expression and microglial activation in progressive supranuclear palsy. Parkinsonism Relat. Disord 17, 683–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Hernandez DG, Nalls MA, Rohrer JD, Ramasamy A, Kwok JBJ, Dobson-Stone C, Brooks WS, Schofield PR, Halliday GM, et al. (2014). Frontotemporal dementia and its subtypes: a genome-wide association study. Lancet Neurol. 13, 686–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA (1922). On the interpretation of χ 2 from contingency tables, and the calculation of P. J. R. Stat. Soc 85, 87–94. [Google Scholar]
- Forman MS, Trojanowski JQ, and Lee VM-Y (2004). Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic break-throughs. Nat. Med 10, 1055–1063. [DOI] [PubMed] [Google Scholar]
- Gaiteri C, Mostafavi S, Honey CJ, De Jager PL, and Bennett DA (2016). Genetic variants in Alzheimer disease - molecular and brain network approaches. Nat. Rev. Neurol 12, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018a). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Haney JR, Parikshak NN, Leppa V, Ramaswami G, Hartl C, Schork AJ, Appadurai V, Buil A, Werge TM, et al. ; CommonMind Consortium; PsychENCODE Consortium; iPSYCH-BROAD Working Group (2018b). Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697.29439242 [Google Scholar]
- Gautam V, D’Avanzo C, Berezovska O, Tanzi RE, and Kovacs DM (2015). Synaptotagmins interact with APP and promote Aβ generation. Mol. Neurodegener 10, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium (2019). The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuens T, Bouhy D, and Timmerman V (2016). The hnRNP family: insights into their role in health and disease. Hum. Genet 135, 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. [DOI] [PubMed] [Google Scholar]
- Golde TE, Estus S, Usiak M, Younkin LH, and Younkin SG (1990). Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer’s disease using PCR. Neuron 4, 253–267. [DOI] [PubMed] [Google Scholar]
- González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, and Mora-Muñoz L (2017). Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci 10, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, Koval J, Das I, Callaway MB, Eldred JM, et al. (2013). DGIdb: mining the druggable genome. Nat. Methods 10, 1209–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guise S, Braguer D, Carles G, Delacourte A, and Briand C (2001). Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J. Neurosci. Res 63, 257–267. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J. Cell Biol 217, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, and Litvan I (1994). Preliminary NINDS neuropathologic criteria for Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). Neurology 44, 2015–2019. [DOI] [PubMed] [Google Scholar]
- Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, and Green DR (2019). LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 178, 536–551.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L (2001). Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol 2, 195–201. [DOI] [PubMed] [Google Scholar]
- Hodes RJ, and Buckholtz N (2016). Accelerating Medicines Partnership: Alzheimer’s Disease (AMP-AD) Knowledge Portal Aids Alzheimer’s Drug Discovery through Open Data Sharing. Expert Opin. Ther. Targets 20, 389–391. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Melhem NM, Dickson DW, Sleiman PMA, Wang L-S, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, et al. ; PSP Genetics Study Group (2011). Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet 43, 699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352,712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins AL, and Groom CR (2002). The druggable genome. Nat. Rev. Drug Discov 1, 727–730. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, et al. (2012). National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa K, and Dickson DW (2001). Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J. Neuropathol. Exp. Neurol 60, 647–657. [DOI] [PubMed] [Google Scholar]
- Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, Schwartz S, Albasset S, McManus RM, Tejera D, et al. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, Dammer EB, Duong DM, Yin L, Thambisetty M, Troncoso JC, Lah JJ, Levey AI, and Seyfried NT (2018). Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, et al. (2020). Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SV, and Kounatidis I (2017). Nuclear Factor-Kappa B and Alzheimer Disease, Unifying Genetic and Environmental Risk Factors from Cell to Humans. Front. Immunol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara Y, Wang E, Wang M, Sin WC, Brennand KJ, Schadt E, Naus CC, Buxbaum J, and Zhang B (2018). GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun 6, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, and Goate AM (2015). Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 77, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Kim EK, and Choi E-J (2010). Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405.20079433 [Google Scholar]
- Kim EK, and Choi E-J (2015). Compromised MAPK signaling in human diseases: an update. Arch. Toxicol 89, 867–882. [DOI] [PubMed] [Google Scholar]
- Kovacs GG (2015). Invited review: Neuropathology of tauopathies: principles and practice. Neuropathol. Appl. Neurobiol 41, 3–23. [DOI] [PubMed] [Google Scholar]
- Lage K, Karlberg EO, Sterling ZM, Olason PI, Pedersen AG, Rigina O, Hinsby AM, Tümer Z, Pociot F, Tommerup N, et al. (2007). A human phenome-interactome network of protein complexes implicated in genetic disorders. Nat. Biotechnol 25, 309–316. [DOI] [PubMed] [Google Scholar]
- Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. ; European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology (2013). Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langa KM, Larson EB, Crimmins EM, Faul JD, Levine DA, Kabeto MU, and Weir DR (2017). A Comparison of the Prevalence of Dementia in the United States in 2000 and 2012. JAMA Intern. Med 177, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2007). Eigengene networks for studying the relationships between co-expression modules. BMC Syst. Biol 1, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, and Horvath S (2011). Is my network module preserved and reproducible? PLoS Comput. Biol 7, e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Mischel PS, and Horvath S (2013). When is hub gene selection better than standard meta-analysis? PLoS ONE 8, e61505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, and Kim N-J (2017). Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung W-S, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-W, Chuang S-M, and Yang J-L (2003). ERK1/2 achieves sustained activation by stimulating MAPK phosphatase-1 degradation via the ubiquitin-proteasome pathway. J. Biol. Chem 278, 21534–21541. [DOI] [PubMed] [Google Scholar]
- Logsdon BA, Perumal TM, Swarup V, Wang M, Funk C, Gaiteri C, Allen M, Wang X, Dammer E, Srivastava G, et al. (2019). Meta-analysis of the human brain transcriptome identifies heterogeneity across human AD co-expression modules robust to sample collection and methodological approach. bioRxiv. 10.1101/510420. [DOI] [Google Scholar]
- Love JE, Hayden EJ, and Rohn TT (2015). Alternative Splicing in Alzheimer’s Disease. J. Parkinsons Dis. Alzheimers Dis 2, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IRA, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DMA, and Lee VMY (2011). A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 122, 111–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur S, Cristante E, Paterno M, Christian H, Roncaroli F, Gillies GE, and Solito E (2010). Annexin A1: a central player in the anti-inflammatory and neuroprotective role of microglia. J. Immunol 185, 6317–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie AT, Moyon S, Wang M, Katsyv I, Song W-M, Zhou X, Dammer EB, Duong DM, Aaker J, Zhao Y, et al. (2017). Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer’s disease. Mol. Neurodegener 12, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, et al. (2011). The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Oldham MC, and Geschwind DH (2008). A systems level analysis of transcriptional changes in Alzheimer’s disease and normal aging. J. Neurosci 28, 1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton RH, Abeti R, Averaimo S, DeBiasi S, Vitellaro L, Jiang L, Curmi PMG, Breit SN, Duchen MR, and Mazzanti M (2008). CLIC1 function is required for beta-amyloid-induced generation of reactive oxygen species by microglia. J. Neurosci 28, 11488–11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, and Berg L (1991). The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486. [DOI] [PubMed] [Google Scholar]
- Morris JC (1993). The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Gaiteri C, Sullivan SE, White CC, Tasaki S, Xu J, Taga M, Klein H-U, Patrick E, Komashko V, et al. (2018). A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci 21, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz L, and Ammit AJ (2010). Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology 58, 561–568. [DOI] [PubMed] [Google Scholar]
- Navarro V, Sanchez-Mejias E, Jimenez S, Muñoz-Castro C, Sanchez-Varo R, Davila JC, Vizuete M, Gutierrez A, and Vitorica J (2018). Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci 10, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M,Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366,1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öhrfelt A, Brinkmalm A, Dumurgier J, Brinkmalm G, Hansson O, Zetterberg H, Bouaziz-Amar E, Hugon J, Paquet C, and Blennow K (2016). The pre-synaptic vesicle protein synaptotagmin is a novel biomarker for Alzheimer’s disease. Alzheimers Res. Ther 8, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea TI, Bologa CG, Brunak S, Campbell A, Gan GN, Gaulton A, Gomez SM, Guha R, Hersey A, Holmes J, et al. (2018). Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov 17, 377. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, and Geschwind DH (2015). Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet 16, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. (2016). Genomewide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-C, Baik SH, Han S-H, Cho HJ, Choi H, Kim HJ, Choi H, Lee W, Kim DK, and Mook-Jung I (2017). Annexin A1 restores Aβ1-42 -induced blood-brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell 16, 149–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson ES (1931). The test of significance for the correlation coefficient. J. Am. Stat. Assoc 26, 128–134. [Google Scholar]
- Perez-Nievas BG, and Serrano-Pozo A (2018). Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping L, Duong DM, Yin L, Gearing M, Lah JJ, Levey AI, and Seyfried NT (2018). Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s Disease. Sci. Data 5, 180036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JHT, Shannon JM, Blumbergs PC, and Gai W-P (2002). Nonselenium glutathione peroxidase in human brain : elevated levels in Parkinson’s disease and dementia with lewy bodies. Am. J. Pathol 161, 885–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2019). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Radford RA, Morsch M, Rayner SL, Cole NJ, Pountney DL, and Chung RS (2015). The established and emerging roles of astrocytes and microglia in amyotrophic lateral sclerosis and frontotemporal dementia. Front. Cell. Neurosci 9, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S, Wang Y-C, Ng B, Gupta I, Haroutunian V, et al. (2018). Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet 50, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Readhead B, Haure-Mirande J-V, Funk CC, Richards MA, Shannon P, Haroutunian V, Sano M, Liang WS, Beckmann ND, Price ND, et al. (2018). Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 99, 64–82.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries M, Loiola R, Shah UN, Gentleman SM, Solito E, and Sastre M (2016). The anti-inflammatory Annexin A1 induces the clearance and degradation of the amyloid-β peptide. J. Neuroinflammation 13, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov RA, Zeisel A, Bakker J, Girach F, Hellysaz A, Tomer R, Alpár A, Mulder J, Clotman F, Keimpema E, et al. (2017). Molecular interrogation of hypothalamic organization reveals distinct dopamine neuronal subtypes. Nat. Neurosci 20, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, and Poirier MA (2004). Protein aggregation and neurodegenerative disease. Nat. Med 10 (Suppl), S10–S17. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Leurgans SE, and Bennett DA (2009). The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol 66, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelaar H, Rohrer JD, Pijnenburg YAL, Fox NC, and van Swieten JC (2011). Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J. Neurol. Neurosurg. Psychiatry 82, 476–486. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. (2017). A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 4, 60–72.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih R-H, Wang C-Y, and Yang C-M (2015). NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci 8,77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. ; ARUK Consortium; GERAD/PERADES, CHARGE, ADGC, EADI (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Facci L, and Giusti P (2013). Intracellular ion channel CLIC1: involvement in microglia-mediated β-amyloid peptide(1-42) neurotoxicity. Neurochem. Res 38, 1801–1808. [DOI] [PubMed] [Google Scholar]
- Smith SK, Anderson HA, Yu G, Robertson AG, Allen SJ, Tyler SJ, Naylor RL, Mason G, Wilcock GW, Roche PA, et al. (2000). Identification of syntaxin 1A as a novel binding protein for presenilin-1. Brain Res. Mol. Brain Res 78, 100–107. [DOI] [PubMed] [Google Scholar]
- Student. (1908). The probable error of a mean. Biometrika 6, 1–25. [Google Scholar]
- Sugiura R, Satoh R, Ishiwata S, Umeda N, and Kita A (2011). Role of RNA-Binding Proteins in MAPK Signal Transduction Pathway. J. Signal Transduct 2011, 109746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup V, Hinz FI, Rexach JE, Noguchi K-I, Toyoshiba H, Oda A, Hirai K, Sarkar A, Seyfried NT, Cheng C, et al. ; International Frontotemporal Dementia Genomics Consortium (2019). Identification of evolutionarily conserved gene networks mediating neurodegenerative dementia. Nat. Med 25, 152–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng EL, Hasegawa K, Homma A, Imai Y, Larson E, Graves A, Sugimoto K, Yamaguchi T, Sasaki H, Chiu D, et al. (1994). The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int. Psychogeriatr 6, 45–58, discussion 62. [DOI] [PubMed] [Google Scholar]
- Vinters HV (2015). Emerging concepts in Alzheimer’s disease. Annu. Rev. Pathol 10, 291–319. [DOI] [PubMed] [Google Scholar]
- Wang J-Z, and Liu F (2008). Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol 85,148–175. [DOI] [PubMed] [Google Scholar]
- Wang M, Roussos P, McKenzie A, Zhou X, Kajiwara Y, Brennand KJ, De Luca GC, Crary JF, Casaccia P, Buxbaum JD, et al. (2016). Integrative network analysis of nineteen brain regions identifies molecular signatures and networks underlying selective regional vulnerability to Alzheimer’s disease. Genome Med. 8, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan JT, Hollis SE, Cha DS, Asch AS, and Lee M-H (2012). Post-transcriptional regulation of the Ras-ERK/MAPK signaling pathway. J. Cell. Physiol 227, 1235–1241. [DOI] [PubMed] [Google Scholar]
- Wilcoxon F (1945). Individual comparisons by ranking methods. Biom. Bull 1, 80–83. [Google Scholar]
- Yoo BC, Cairns N, Fountoulakis M, and Lubec G (2001). Synaptosomal proteins, beta-soluble N-ethylmaleimide-sensitive factor attachment protein (beta-SNAP), gamma-SNAP and synaptotagmin I in brain of patients with Down syndrome and Alzheimer’s disease. Dement. Geriatr. Cogn. Disord 12, 219–225. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Petyuk VA, Gaiteri C, Mostafavi S, Young-Pearse T, Shah RC, Buchman AS, Schneider JA, Piehowski PD, Sontag RL, et al. (2018). Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann. Neurol 84, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005).A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol 4, 17. [DOI] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea L-G, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. (2013). Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Ma C, Gearing M, Wang PG, Chin L-S, and Li L (2018). Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer’s disease. Acta Neuropathol. Commun 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Roundtree IA, and He C (2017). Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol 18, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee H, Raina AK, Perry G, and Smith MA (2002). The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 11, 270–281. [DOI] [PubMed] [Google Scholar]
- https://agora.ampadportal.org/genes. 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the proteomics data are available here: https://adknowledgeportal.synapse.org/Explore/Projects?Grant%20Number=U01AG046161. Links to data for each study can be found in the Key Resources Table. The accession number for the ACT data reported in this paper is https://www.synapse.org/ Synapse ID:syn5759376. The accession number for the MSBB data reported in this paper is https://www.synapse.org/ Synapse ID:syn3159438. The accession number for the Banner data reported in this paper is https://www.synapse.org/ Synapse ID:syn7170616. The accession number for the Mayo data reported in this paper is https://www.synapse.org/ Synapse ID:syn5550404. The accession number for the UPenn data reported in this paper is https://adknowledgeportal.synapse.org Synapse ID:syn21411742.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PLCD1 antibody | Abcam | Cat# ab134936; RRID:AB_2857853 |
| HEPACAM antibody | Abcam | Cat# ab130769; RRID:AB_2857854 |
| PADI2 antibody | Abcam | Cat# ab16478; RRID:AB_470249 |
| GAPDH antibody | Abcam | Cat# ab8245; RRID:AB_2107448 |
| GJA1 antibody | Sigma | Cat# C6219; RRID:AB_476857 |
| Biological Samples | ||
| Postmortem dorsolateral prefrontal and precuneus cortex brain tissue | Baltimore Longitudinal Study of Aging (BLSA) at Johns Hopkins University | https://www.hopkinsmedicine.org/research/labs/alzheimers-disease-research-center |
| Postmortem prefrontal cortex brain tissue | Adult Changes of Thought (ACT) at the University of Washington and Group Health Cooperative of Puget Sound | http://depts.washington.edu/mbwc/adrc/page/neuropathology |
| Postmortem prefrontal cortex brain tissue | Mount Sinai Brain Bank (MSBB) | https://icahn.mssm.edu/research/nih-brain-tissue-repository |
| Postmortem prefrontal cortex brain tissue | Banner Sun Health Research Institute (Banner) | https://www.bannerhealth.com/services/research/locations/sun-health-institute/programs/body-donation |
| Postmortem temporal cortex brain tissue | Mayo Clinic Brain Bank (Mayo) | https://www.mayo.edu/research/departments-divisions/department-neuroscience-florida/brain-bank |
| Postmortem prefrontal cortex brain tissue | University of Pennsylvania Brain Bank (UPenn) | https://www.med.upenn.edu/cndr/biosamples-brainbank.html |
| Postmortem dorsolateral prefrontal brain tissue | Emory University (Emory) | http://alzheimers.emory.edu/ |
| Deposited Data | ||
| Baltimore Longitudinal Study of Aging (BLSA) proteomic data | Seyfried et al., 2017 | https://doi.org/10.7303/syn3606086 |
| Adult Changes of Thought (ACT) proteomic data | This study | https://doi.org/10.7303/syn5759376 |
| Mount Sinai Brain Bank (MSBB) proteomic data | This study | https://repo-prod.prod.sagebase.org/repo/v1/doi/locate?id=syn3159438&type=ENTITY |
| Banner Sun Health Research Institute (Banner) proteomic data | This study | https://doi.org/10.7303/syn7170616 |
| Mayo Clinic Brain Bank (Mayo) proteomic and transcriptomic data | This study | https://doi.org/10.7303/syn5550404 |
| University of Pennsylvania (UPenn) proteomic data | This study | https://adknowledgeportal.synapse.org/Explore/Studies?Study=syn21411742 |
| Emory proteomic data | Ping et al., 2018 | https://www.synapse.org/#!Synapse:syn10239444 |
| Cell type enrichment Markers | Zhang et al., 2016 | http://www.brainrnaseq.org/ |
| Alzheimer’s disease GWAS summary statistics | Lambert et al., 2013 | http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php |
| Progressive Supranuclear Palsy GWAS summary statistics | Höglinger et al., 2011 | https://www.niagads.org/datasets/ng00045 |
| Software and Algorithms | ||
| R | R Core Team | https://www.R-project.org/ |
| WGCNA R package | Langfelder and Horvath, 2008 | https://cran.r-project.org/web/packages/WGCNA/index.html |
| Gene Ontology | http://geneontology.org/ | |
| igraph R package | Csardi and Nepusz, 2006 | https://igraph.org/r/ |
| MAGMA | de Leeuw et al., 2015 | https://ctg.cncr.nl/software/magma |
| Other | ||
| Interactive graphical interface for consensus proteomic network | https://coppolalab.ucla.edu/gclabapps/nb/browser?id=SwarupChangConsensusProteomics |
Code is available on GitHub: https://github.com/dhglab/Identification-of-conserved-proteomic-networks-in-neurodegenerative-dementia