

Abstract
Scope
Combining different “omics” data types in a single, integrated analysis may better characterize the effects of diet on human health.
Methods and results
The performance of two data integration tools, similarity network fusion tool (SNFtool) and Data Integration Analysis for Biomarker discovery using Latent variable approaches for “Omics” (DIABLO; MixOmics), in discriminating responses to diet and metabolic phenotypes is investigated by combining transcriptomics and metabolomics datasets from three human intervention studies: a postprandial crossover study testing dairy foods (n = 7; study 1), a postprandial challenge study comparing obese and non‐obese subjects (n = 13; study 2); and an 8‐week parallel intervention study that assessed three diets with variable lipid content on fasting parameters (n = 39; study 3). In study 1, combining datasets using SNF or DIABLO significantly improve sample classification. For studies 2 and 3, the value of SNF integration depends on the dietary groups being compared, while DIABLO discriminates samples well but does not perform better than transcriptomic data alone.
Conclusion
The integration of associated “omics” datasets can help clarify the subtle signals observed in nutritional interventions. The performance of each integration tool is differently influenced by study design, size of the datasets, and sample size.
Keywords: classification, data integration, Data Integration Analysis for Biomarker discovery using Latent variable approaches for “Omics”, nutritional intervention, Similarity Network Fusion tool
“Omics” technologies like metabolomics or transcriptomics can give different snapshots of the molecular changes that are associated with diet. However, combining such “omics” data, may offer a deeper understanding of the holistic effects of nutrition on biological processes. The authors explore, in three independent diet‐related studies, how two different data integration tools can help to identify diet‐related molecular signatures.
1. Introduction
Nutrigenomics approaches are increasingly applied in human nutritional sciences to comprehensively model the complex mechanisms that link diet to health. Large multi‐omics datasets have been generated in nutritional studies, including whole‐genome gene expression (transcriptome) and the profiling of small molecules detectable in biofluids (metabolome). To date, analysis of data is usually performed separately for each “omics” layer.[ 1 , 2 ] However, the combination of “omics” datasets, may be key to understand how the system functions as a whole as each dataset contributes to a larger, common biological system.[ 3 , 4 , 5 ] This integration may identify related changes in gene expression and metabolite flux that would be difficult to detect when analyzed independently. Indeed, integrating transcriptomic and metabolomic data from human blood samples has already revealed novel insights into the molecular mechanisms of clinical traits underlying normal physiology and disease by highlighting the cross‐talk between biological layers at the pathway level.[ 3 ]
In human nutrition intervention studies, data integration techniques could also advance understanding of the metabolic changes attributable to diet or food compounds. Such signals of metabolic change are often subtle and difficult to elucidate against a complex background of biological and environmental variation. Several studies have successfully used data integration tools to identify groups of biomarkers that are associated with diet‐related outcomes, for example the modulation of insulin sensitivity induced by caloric restriction intervention[ 6 ] or change in body weight.[ 7 ] However, data integration of nutritional “omics” datasets remains underexploited.
Different methods have been developed for integrating “omics” layers. This study evaluates two recently developed methods for integrating “omics” data that can be used to group samples and identify biomarkers, themes that are relevant to nutrition and disease‐focused studies. The first method is the similarity network fusion tool (SNF),[ 8 ] a correlation‐based, unsupervised tool that previously performed well in detecting subtle signatures in datasets compared to alternative unsupervised methods.[ 9 ] SNF uses correlation matrices, generated separately for the related “omics” datasets, to create a “fused”, integrated network that models the relationship between individual samples. The second method is the Data Integration Analysis for Biomarker discovery using Latent variable approaches for “Omics” studies (DIABLO),[ 10 , 11 ] a supervised, multivariate method (component‐based) that maximizes the discrimination between predefined sample groups while associating each pair of “omics” datasets.[ 10 , 12 , 13 , 14 ] Therefore DIABLO can identify a set of correlated variables (such as genes or metabolites) that could discriminate different responses to diet.
This study evaluates the performance of the SNF and DIABLO methods for the integration of nutritional “omics” data in three independent nutrition studies. The studies represent some of the common types of approaches that are used in nutritional studies, including crossover and parallel design, postprandial tests (to assess the immediate response to food intake), and fasting measurements (to assess the longer‐term consequences of diet on metabolism).
2. Experimental Section
2.1. Study Designs
SNF and DIABLO data integration methods were applied in an exploratory analysis of data obtained from three clinical human studies in which the effects of diet on transcriptomics and metabolomics or lipidomic signatures were evaluated in blood samples (Figure 1 ). Study 1 (the F3 study), a randomized controlled crossover design, assessed the dynamic postprandial effects of two dairy products (800 g yogurt or acidified milk) on the whole blood transcriptome (Illumina RNAseq) and serum metabolome (untargeted UHPLC/Q‐TOF‐MS analysis) of seven healthy young men.[ 15 , 16 , 17 ] Study 2 (the MECHE study) also used postprandial testing (response at 4h post‐challenge) to evaluate the transcriptome (human whole‐genome GeneChip microarray, in PBMCs) and serum lipid profile (Orbitrap LC‐MS, identification of a selection of lipids) of thirteen men with different metabolic phenotypes (obese/non‐obese) to an oral lipid tolerance test (OLTT).[ 18 , 19 , 20 , 21 ] Study 3 (the MARIS study) used a randomized controlled parallel study design to evaluate an 8 week dietary intervention, testing three dietary patterns: a Western‐type diet high in saturated fatty acids (SFA diet), a Western‐type diet high in MUFA from olive oil (MUFA diet), and a Mediterranean‐type diet (MED diet) with equivalent MUFA amounts to the MUFA diet.[ 22 ] The transcriptome (human whole‐genome GeneChip microarray, in PBMCs) and serum metabolome (NMR, identification of a selection of metabolites from various classes) were assessed in 39 overweight or obese men and women using fasting samples collected after a 2 week run‐in period (SFA diet) and after the dietary interventions.[ 23 , 24 ] All three studies included in this analysis were completed in accordance with the ethical standards of the responsible committee on human experimentation and with the guidelines laid down in the 1975 Helsinki Declaration, as revised in 1983. The studies are registered at ClinicalTrials.gov (study 1: NCT02230345, study 2: NCT01172951, and study 3: NCT00405197). A full description of study designs is given in Sections S1‐S3, Supporting Information.
Figure 1.
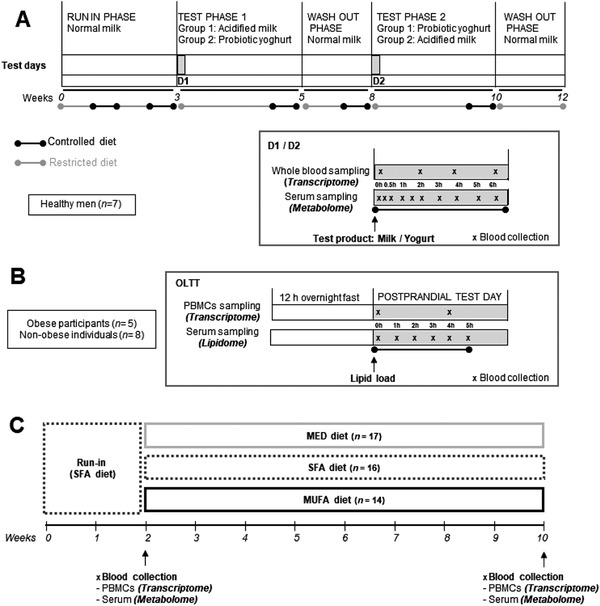
Overview of study designs used for data integration. A) Study 1 used a randomized, controlled, crossover study design to test the postprandial and short‐term effects of acidified milk and probiotic yogurt.[ 15 , 31 On dairy test days (D1 and D2) blood samples were collected fasting and postprandially for transcriptome and untargeted metabolome profiling (n = 7 subjects). Dietary restriction applied 3 days before the postprandial tests and diet was controlled throughout the study. B) Study 2 evaluated the response of obese (n = 5) and non‐obese (n = 8) participants to a standard metabolic challenge comprised of a lipid overload that was completed after an overnight fast. The postprandial response to the challenge was evaluated by blood sampling over 5 h for lipid profiling and transcriptome analysis.[ 20 ] C) Study 3 used a parallel, controlled study design to evaluate the effect of different dietary patterns on the fasting metabolic profile and transcriptome. After a 2 week run‐in phase of SFA diet, participants were randomly assigned to test for 8 weeks either a MED diet (n = 17), SFA diet (n = 16), or MUFA diet (n = 14).[ 22 ] D1/D2, dairy test; MED, Mediterranean; SFA, saturated fatty acid.
2.2. Preparation of Data
Each dataset was preprocessed to filter artifacts and low‐level signal according to the original study protocols (see Sections S1‐S3, Supporting Information). Additional filtering of the data with baseline measures deducted was applied to refine data integration performance [ 8 ] ( Section S4, Supporting Information). Total filtered features retained for data integration analysis were: study 1, n = 5043 genes, n = 1100 metabolites (untargeted, not identified); study 2, n = 1040 genes, n = 42 lipids (identified and quantified); study 3, n = 1012 genes, n = 129 metabolites (identified and quantified). The datasets were then processed according to a common analysis pipeline (Figure S1, Supporting Information) in the R environment (R v 3.5.3; R Foundation for Statistical Computing, Vienna, Austria).
2.3. SNF Analysis
The SNF analysis protocol was based on the methods described by Wang et al.[ 8 ] using the R package “SNFtool” (v 2.3.0) (Section S4, Supporting Information). SNF first calculates dissimilarity distances between samples for each separate dataset to create affinity/similarity matrices that can be interpreted as networks. Each individual “network” is then iteratively modified by SNF, finally converging on a unique, integrated network. Transcriptome, metabolome/lipidome and integrated SNF networks were visualized by extracting the strongest connections between the samples. SNF integrated models were validated using bootstrapping tests that compared the model classification to models using randomized data (p < 0.05) (see Section S4, Supporting Information). Classification performance of the models was evaluated by the classification error rate (CER) (ranging from 0 to 1), which was estimated by an internal (M‐fold) cross‐validation analogous to that of the mixOmics “perf” function. CERs for the integrated and non‐integrated models were compared using linear mixed‐effects models. Post hoc pairwise comparisons were estimated, where indicated (p < 0.05), using marginal means (emmeans)[ 25 ] (significant effects considered where p adj < 0.05).
2.4. PLS‐DA and DIABLO Analyses
For each study, separate partial least squares discriminant analysis (PLS‐DA) models for transcriptomics and metabolomics datasets were created using mixOmics (v 6.6.2).[ 26 ] Classification performance of the models was evaluated by the CERs using the same cross‐validation test as described for SNF. Goodness‐of‐fit and predictability were verified with R2 and Q2 parameters.
The integration of the two “omics” datasets using DIABLO applied the workflow proposed by Rohart et al.[ 10 ] and Singh et al.[ 11 ] using mixOmics. For each study, a DIABLO model was built with settings to maximize the separation between treatment groups while accounting for correlation between the “omics” datasets. Validity of the DIABLO models was assessed by a permutation test that compared the CERs of the original DIABLO models to CERs of models built with randomly permuted samples (significance where p < 0.05). Relative “weights” of each dataset in the final integrated model were compared to evaluate the relative importance of each dataset.[ 10 ] Finally, CERs of DIABLO models were compared to those of PLS‐DA models using the same method as defined for SNF to evaluate whether the integration of the two “omics” datasets improved classification performance.
A multilevel decomposition was applied to PLS‐DA and DIABLO analyses for study 1 to account for the crossover design.[ 27 , 28 ] A detailed description of the PLS‐DA and DIABLO workflows is given in Section S4, Supporting Information and model settings are presented in Table S1, Supporting Information.
2.5. Comparison of SNF and DIABLO
For each study, the CERs of the SNF and DIABLO models could be directly compared using paired, two sample t‐tests (p < 0.05), as identical test and training sets were used in M‐fold cross‐validations. When both methods showed good performance (validated CER < 0.05), further evaluation of the features (genes and metabolites/lipids) selected by the models was carried out including network and enrichment analyses. The top 5% most important features for each integrated models were extracted and the strongest connections between selected features (ρ < −0.9 and ρ > 0.9, Spearman's correlation) were represented in networks. Over‐representation analysis (ORA) using Fisher's exact test was used to assess the functional relevance of the top 5% most important genes selected by integrated models. Full details are found in Section S4, Supporting Information.
3. Results
3.1. SNF and DIABLO Performed Well to Discriminate the Two Postprandial Dairy Tests (Study 1)
Study 1 sought to characterize the postprandial response to acidified milk and yogurt. The SNF models generated for the transcriptome or metabolome data alone (respectively shown in the form of affinity matrices in Figure 2A,B) did not completely distinguish the responses to the dairy products, despite the observation of some grouping of samples especially for the metabolome. However, the integration of the datasets using the SNF tool revealed two distinct clusters in the integrated SNF affinity matrix, comprised only of samples representing the postprandial response to either acidified milk or yogurt (Figure 2C). The grouping of the samples in the SNF model were statistically validated by bootstrapping permutation tests (p = 0.0002). In line with the strength of the SNF model in separating the samples, the model performed well in prediction tests, with a statistically significantly lower CER than either model created with the separate datasets (Table 1 ).
Figure 2.
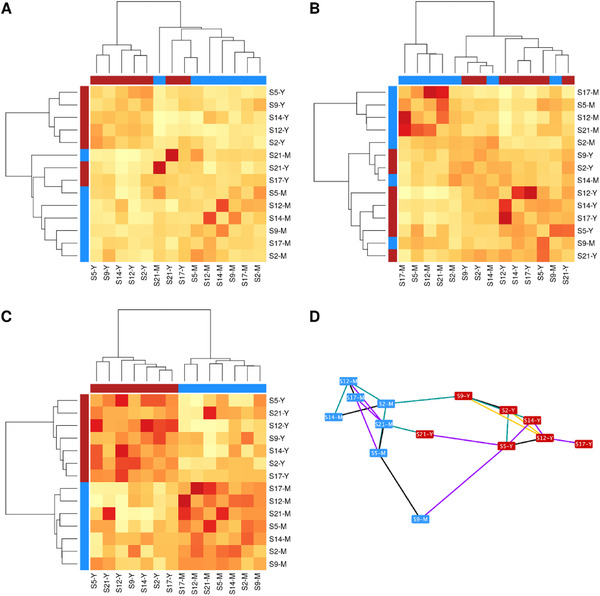
Visualization of models constructed with SNF for study 1. Affinity matrices for net iAUC show metabolite, gene, and SNF integrated models (respectively, A–C); SNF network showing the top 30% connections between samples using Cytoscape with edge weighted spring embedded layout in panel D. Samples are labeled by subject (S) number and intervention type milk (M, n = 7) or yogurt (Y, n = 7), with colors to indicate test meal: M (blue), Y (red). Diagonal of heatmaps shows median similarity across all samples. Network connections are colored according to whether the connection was identified in the top 30% connections for networks created with the metabolome (turquoise), transcriptome (purple), in both metabolome and transcriptome separate networks (black) or only with the datasets combined (SNF model) (yellow). iAUC, net incremental area under curve; SNF, similarity network fusion.
Table 1.
Classification error rates (CER) for SNF models (separate and integrated models) and for PLS‐DA and DIABLO models presented for all studies.CER is validated by M‐fold cross‐validation tests (respectively, 7‐, 5‐, and 10‐fold for studies 1, 2, and 3)
| Study 1 CER ± SEM | Study 2 CER ± SEM | Study 3 CER ± SEM | ||||
|---|---|---|---|---|---|---|
| Milk versus yogurt | Non‐obese versus obese | All diets | SFA diet versus MED diet | SFA diet versus MUFA diet | MUFA diet versus MED diet | |
| SNF analysis | ||||||
| Metabolome/lipidome model | 0.21a ± 0.004 | 0.15a ± 0.003 | 0.41a ± 0.005 | 0.13b ± 0.003 | 0.29a ± 0.006 | 0.30a ± 0.006 |
| Transcriptome model | 0.14b ± 0 | 0.03c ± 0.006 | 0.38b ± 0.004 | 0.19a ± 0.005 | 0.21b ± 0.004 | 0.33b ± 0.004 |
| SNF integrated model | 0.02c ± 0 | 0.08b ± 0.002 | 0.30c ± 0.005 | 0.08c ± 0.002 | 0.23b ± 0.005 | 0.32b ± 0.004 |
| DIABLO analysis | ||||||
| Metabolome/lipidome (PLS‐DA) | 0.14a ± 0 # | 0.13a ± 0.007 # | 0.27a ± 0.003 # | 0.11a ± 0.004 # | 0.18a ± 0.002 # | 0.19a ± 0.004 # |
| Transcriptome (PLS‐DA) | 0.14a ± 0 | 0b ± 0 # | 0.06c ± 0.002 # | 0.07b ± 0.003 # | 0.01b ± 0.003 # | 0.07b ± 0.005 # |
| DIABLO model | 0.03b ± 0.007 # | 0b ± 0 # | 0.08b ± 0.003 # | 0.06b ± 0.004 # | 0.02b ± 0.003 # | 0.08b ± 0.005 # |
Different letters (a–c) indicate significant differences between CERs for comparisons between models for each study (as assessed by linear mixed‐effect models with post hoc pairwise comparisons, p adj < 0.05). #indicates a difference comparing equivalent models created by the SNF tool and DIABLO (as assessed by paired t‐test, p < 0.05).CER, classification error rate; DIABLO, data integration analysis for biomarker discovery using latent variable approaches for “omics” studies; MED, Mediterranean; PLS‐DA, partial least squares discriminant analysis; SFA, saturated fatty acid; SNF, similarity network fusion.
A network representing the most important connections between samples in the SNF model shows spatial separation of milk and yogurt postprandial samples (Figure 2D). The network connections were generally specific to the metabolome or the transcriptome rather than both, while some connections were only found when both datasets are combined.
The multivariate analyses of each datasets separately (PLS‐DA) and successfully discriminated the postprandial dairy responses, as shown by the score plots (Figure S2, Supporting Information; R2 and Q2 values in Table S2, Supporting Information). The integration using DIABLO resulted in a valid model (permutation test, p = 2.2×10–16, Figure S3, Supporting Information) and performed better in classifying the samples than when the datasets were analyzed separately (Table 1). A strong association was observed between the first components of the two data types (Pearson correlation, ρ = 0.86, p = 8.0×10–5) and correspondingly the relative contribution (“weights”) of the metabolome and transcriptome datasets in the integrated DIABLO model were not significantly different (Table S3, Supporting Information).
The integration of study 1 datasets with DIABLO and SNF both resulted in models showing similar performance in the prediction of type of dairy consumed (CER < 0.05), although the CER was slightly better for SNF (Table 1). Comparison of the top 5% most important metabolites and genes selected by each integrated model revealed some similarities despite the different strategies used to construct the models. Of these features, 25% were present in both models, with more common genes (27%) than common metabolites (16%). The similarity between the models was greater when considering only the most correlated genes and metabolites (e.g., those with |ρ| > 0.90), as shown in the gene‐metabolite networks (Figure 3 ); 41% of all selected features were found in both networks. Pathway analysis did not reveal a significant enrichment among the genes identified as discriminant for either model.
Figure 3.
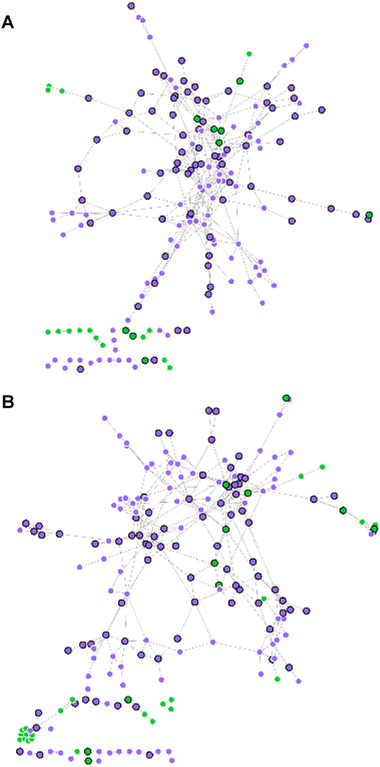
Network plots for the top 5% most important genes and metabolites for differentiating blood samples taken after milk intake or yogurt intake in study 1, selected by A) SNF and B) DIABLO. Connections between nodes (metabolites, green; genes, purple) are shown for the strongest associations (ρ < −0.90 or ρ > 0.90, Spearman's correlation) (SNF, n = 199 nodes; DIABLO, n = 209 nodes). Nodes present in both networks are highlighted by a black outline and a larger size (metabolites, n = 12; genes, n = 80). DIABLO, data integration analysis for biomarker discovery using latent variable approaches for “Omics” studies; PLS‐DA, partial least squares discriminant analysis; SNF, similarity network fusion.
3.2. Using Postprandial Transcriptome and Lipid Data to Separate Responses to an OLTT (Study 2)
The MECHE metabolic challenge study investigated the metabolic impact of an OLTT in the postprandial state, focusing on the differences in this response between obese and non‐obese individuals. The affinity matrix for the transcriptome alone grouped together the five OLTT responses of the obese subjects correctly while the lipidome misclassified one obese individual (Figure S4A,B, Supporting Information). Integration of transcriptome and lipidome datasets for MECHE using SNF resulted in a valid model (p = 0.003) that broadly grouped the samples into OLTT responses of obese and non‐obese subjects, with one misclassification of a response from an obese individual (BMI 34.4 kg m−2), which was grouped with the responses from non‐obese participants (Figure S4C, Supporting Information). The CER of the integrated model was thus a little lower than the separate lipidome model though higher than the transcriptome alone (Table 1). The network derived from the SNF model (Figure S4, Supporting Information) shows the BMI groups were well separated when only visualizing the strongest associations between samples, with the lipidome showing particular importance in defining connections within the non‐obese while associations within the obese group were captured by the integrated datasets.
PLS‐DA analysis of the transcriptome dataset alone correctly classified all phenotypic responses to the OLTT in all M‐fold tests, while the lipidome PLS‐DA model showed significantly weaker predictive performance (Table 1). PLS‐DA performance parameters R2 and Q2 values are presented in Table S2, Supporting Information. The integrated DIABLO model also correctly predicted all phenotypic responses to the OLTT (permutation tests, p = 2.2×10–16, Figure S3, Supporting Information) (Table 1). Despite a strong association between the first components of the two data types (Pearson correlation, ρ = 0.77, p = 0.002), comparison of the relative “weights” of the datasets in the final DIABLO model showed that the transcriptome dataset was given significantly more importance than the lipidome dataset (Table S3, Supporting Information). The spatial separation of the samples and their groupings for each model are shown in the score plots (Figure S5, Supporting Information).
For this study, the integration of datasets using DIABLO resulted in better classification of the phenotype groups, with lower CER for DIABLO than SNF (Table 1). Comparisons of the top features selected for each model were not completed due to the high CER obtained for the SNF model, which implies that extracted features based on this model would not characterize the groups well.
3.3. Data Integration for the Long‐Term Dietary Intervention (Study 3)
MARIS was an 8 week dietary fat and dietary quality modification intervention study. When the three dietary interventions were included in the SNF analysis, regardless of whether metabolome, transcriptome, or integrated datasets were used, affinity matrices (Figures S6A, S7A, and S8A, Supporting Information) and CERs (Table 1) did not show good classification of samples. Conversely, when only two diets were included per analysis, sample clustering was improved for the metabolome and transcriptome data (Figures S6B–D and S7B–D, Supporting Information). Moreover, the integration of the datasets using SNF enabled clustering that separated the SFA diet group from that of the MED diet group (p = 0.0005). However, other comparisons using SNF integrated datasets were not significant (MUFA vs MED: p = 0.07; MUFA vs SFA, p = 0.06) (Figure S8B–D, Supporting Information). The comparison between MED and SFA diets also showed the best spatial separation in the sample networks (Figure S9, Supporting Information), with only one sample that was misplaced in both groups. The metabolome was important in defining the similarities within each group (Figure S9A, Supporting Information, turquoise connections).
PLS‐DA analyses of study 3 showed that the transcriptome performed significantly better in classifying the samples than the metabolome for all dietary comparisons (Table 1; score plots available in Figure S10, Supporting Information, R2 and Q2 values in Table S2, Supporting Information). Furthermore, the DIABLO integration of the datasets did not significantly improve the CER compared to the transcriptome alone for any of the comparisons although the integrated models were validated (p < 0.0001 for all permuted tests, Figure S3C–F, Supporting Information). The relative importance of the transcriptomics dataset was also shown by the significantly greater weight of the transcriptome dataset in the DIABLO model as compared to the metabolome dataset (Table S3, Supporting Information). In addition, the “omics” datasets were not strongly associated except for the SFA v. MED model (Pearson correlations: all diets ρ = 0.50, p = 0.001, MUFA vs SFA ρ = 0.56, p = 0.001, MUFA vs MED ρ = 0.58, p = 0.002, SFA vs MED ρ = 0.81, p < 0.0001), confirming the different contribution of each dataset to the final model. The lowest CER was observed for the SFA versus MUFA comparison using the transcriptome PLS‐DA model (Table 1).
For study 3, the integration of datasets using DIABLO resulted in lower CER than the integration using SNF, for all comparisons of the three diets (Table 1). As for study 2, due to the high CERs obtained for the SNF models, comparisons of the top features selected for each model were not completed.
4. Discussion
In our data integration analyses of three different nutritional studies, we show that the integration of related “omics” datasets, by DIABLO or SNF, can help to differentiate responses to diet. The inherent differences of using a supervised (e.g., DIABLO) or unsupervised (e.g., SNF) data integration approach have implications for their utility for nutritional studies. As expected, DIABLO performed well in extracting common discriminating signals of diet or metabolic phenotype, even where the dietary effects were small. Conversely, SNF performed well in classifying samples where the dietary effects were more marked but also enabled the detection of outliers or novel groups.
4.1. DIABLO could Discriminate Sample Groups for all Three Study Designs
DIABLO applies a powerful supervised method that discriminates predefined groups and thus was expected to be relevant for nutritional datasets as the effects of diet can be subtle. One feature of DIABLO is its ability to handle “omics” datasets that are “unbalanced” (i.e., one dataset carries more discriminatory information than the other(s)). Specifically, the capacity to attribute a “weight” to datasets is critical in allowing DIABLO to combine datasets while maintaining the importance of the discriminatory features. In both studies 2 and 3, the transcriptome was assigned a significantly greater “weight” in the models than the metabolome. Consequently, the integration of “omics” datasets using DIABLO was very similar to the separate analysis using the transcriptome only and did not further improve sample classification. The lower importance of the lipidome/metabolome in the DIABLO model for studies 2 and 3 could be explained by the reduced number of compounds. In contrast, the untargeted approach used in study 1 captured the broad spectrum of metabolites that could respond to diet similarly to the approach used for the transcriptome.
In the current analyses, DIABLO models were built using a “full weighted design matrix,”[ 10 ] that maximizes the separation between samples groups while taking into account the correlation between “omics” datasets (design matrix parameter set to 0.1). However, by using a “full design matrix” instead (design matrix parameter set to 1), the model would maximize the correlation between “omics” datasets, prioritizing the association between features of the metabolome with features of the transcriptome. Thus, although the integration of data with DIABLO did not improve sample classification in study 2 and 3 compared to transcriptome data alone, DIABLO remains useful in offering a method to associate features of the metabolome and transcriptome that similarly discriminate diet.
4.2. SNF Showed Potential to Reveal Novel Sample Groups
In contrast to DIABLO, SNF models are unsupervised and thus do not use any a priori information to separate sample groupings. For this reason, the use of a classification measure (i.e., CER) as a performance criterion to compare the two methods might be expected to favor DIABLO. Nevertheless, SNF performed slightly better than DIABLO in study 1.
Interestingly, while DIABLO gives an estimate of the overall contribution of each dataset (by the relative weights), the SNF networks provide a deeper insight into the importance of each dataset by specifying the dataset(s) used to build each connection between samples. The lack of connections defined by both individual datasets (e.g., black connections in Figure 2D), confirmed the value of integrating the datasets to model the two postprandial dairy responses.
The SNF model for the postprandial metabolic challenge in study 2 failed to improve classification compared to the transcriptome alone. One explanation for the weaker performance for the SNF in this study is suggested in the characteristics of the obese outlier for the SNF model, who was the oldest participant in the study. This participant may have a metabolic response profile distinct from both non‐obese and younger obese participants; indeed the broad descriptor “obesity” may comprise multiple subtypes.[ 29 , 30 ] A potential advantage of the SNF tool for nutritional studies is the ability to separate samples by undefined factors (for example environmental or biological factors). This could lead to the identification of new metabolic subgroups, but it would require either a stronger signal or a larger cohort than used in study 2 to clarify the groupings.
The results from study 3 suggest two important aspects of study design that can affect the performance of the SNF models: the number of dietary groups studied and the use of fasting blood samples to assess long‐term effects of diet. It was noteworthy that a significant improvement was observed for the integrated SFA versus MED SNF model, whereas the evaluation of all three diets together suggested no clear discrimination. The nutritional content of the SFA and MED diets were also the most differing of the three diets, testifying the tendency of SNF to identify the strongest signals in the data. The discrimination of more than two dietary intervention groups by SNF using fasting, long‐term data was challenging in this study. The effect of the diet might be more visible if the inter‐variation between participants was reduced (e.g., crossover study design). However, the study diet was well controlled (all food was provided to participants) and, the SFA and MED diet could be differentiated by SNF. Moreover, the strong performance of the DIABLO models for all comparisons of study 3 show the value of the tool in giving weight to the features that discriminate the intervention groups, even if the diets are relatively similar or if the long‐term, physiological effects of diet are being assessed.
4.3. Strengths and Limitations
We assessed two different data integration tools that hold promise for nutritional datasets using three independent, distinct dietary studies. The use of the same M‐fold validation test with identical samples per fold allowed a direct comparison of the CERs for the tools in each study although it is acknowledged that the methods were not designed with the same purpose. We chose to include a filtering step in the analysis pipeline to limit the noise in the data as advocated in the previous analysis of Tini et al.,[ 9 ] and this was an essential step in the preparation of our data due to limited sample size in our studies. In larger nutritional datasets, models could be explored using filtered and unfiltered data to confirm the utility of the filtering step for clarifying the dietary signals. Although the results were validated internally by the M‐fold validation test, an external validation of the results would be useful to further confirm the robustness of these models. While the number of participants in our studies was a limitation of our work, the strength of the models in the internal validation tests despite this limitation underlines the potential of these tools for integrating dietary datasets.
Another consideration in our study was the differences in the choice of “omic” platform and the resulting variation in number of features per dataset. These differences most likely explain why transcriptomic datasets carried most of the information when integrating the datasets in studies 2 and 3. Moreover, such differences, by influencing the performance of the integration tools, might limit the direct comparison of the data integration for different study designs. However, given that these studies reflected the variation of techniques used across existing nutritional studies it was considered relevant to apply data integration approaches to studies with different “omic” data types. It is also noteworthy that the choice of omics data as well as the omics platform selected could affect the success of data integration. While we evaluated commonly available blood “omics” datasets, depending on the research question, the use of other “omics” approaches (e.g., proteomics) may be considered more relevant.
The potential to use SNF and DIABLO to support the biological interpretation of the combined “omics” signal was explored for study 1 in which both models performed well in classifying the samples. Different features were found to be important in defining the models, which may reflect the inherent differences in the methods, although very discriminatory features were found for both models. Discriminatory genes did not represent an enriched metabolic pathway in either model though the limited identification of the untargeted metabolic dataset prevented an integrated pathway analysis of the two datasets.
4.4. Conclusions and Perspectives
The application of data integration methods to combine related “omics” datasets may help to discriminate responses to different diets and identify related biological signals that are regulated by diet. SNF and DIABLO data integration methods seem to offer different advantages for the analysis of human nutrition intervention/challenge datasets. Generally, DIABLO performed well in our relatively small datasets to identify the features that can differentiate diets or metabolic phenotypes. However, given the complex responses of humans to diet, SNF may be relevant for the identification and the investigation of new metabolic phenotypes.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
K.J.B.‐P., G.P., M.H., C.C.J.R.M., A.F., N.V., F.P.P., L.A.A., H.M.R., M.I., and G.V., designed the research (project initiation, project conception, development of overall research plan, and study oversight); K.J.B.‐P., G.P., M.H., C.C.J.R.M., and N.V., conducted the research (hands‐on conduct of the experiments and data collection); K.J.B.‐P., G.P., M.H., and C.C.J.R.M., analyzed data or performed statistical analysis; K.J.B.‐P., G.P., M.H., C.C.J.R.M., H.M.R., and G.V., wrote the paper, K.J.B.‐P., G.P., M.H., C.C.J.R.M., N.V., L.A.A., H.M.R., L.B., M.I., and G.V., critically reviewed the manuscript, K.J.B.‐P had primary responsibility for final content. All authors read and approved the final manuscript.
Supporting information
Supporting information
Acknowledgements
Study 1 received funding from the Joint Programming Healthy Diet for a Healthy Life (JPI HDHL) funded EU Food Biomarkers Alliance “FOODBAll” (14/JP‐HDHL/B3076). Study 2 was funded by the National Children's Research Centre, Crumlin, Ireland (Grant number: B/11/1), by Science Foundation Ireland Joint Programmes Initiative (14/JP‐HDHL/B3076) FoodBAll (The Food Biomarkers Alliance), and through “JINGO” 2007–2013 (07FHRIUCD1). Dr. Nadine Zangger and Dr. Mauro Delorenzi of the Bioinformatics Core Facility (Swiss Institute of Bioinformatics, Lausanne) are thanked for their support in the analysis of the raw transcriptome data from study 1. In addition, the authors thank Prof. François P. Pralong for his role as PI for study 1. The authors would like to thank Flavia Marzetta (Vital‐IT group, SIB) for critical reading of the manuscript and helpful suggestions.
Burton‐Pimentel K. J., Pimentel G., Hughes M., Michielsen C. C., Fatima A., Vionnet N., Afman L. A., Roche H. M., Brennan L., Ibberson M., Vergères G., Discriminating Dietary Responses by Combining Transcriptomics and Metabolomics Data in Nutrition Intervention Studies. Mol. Nutr. Food Res. 2021, 65, 2000647. 10.1002/mnfr.202000647
Data Availability Statement
Transcriptomic data for the 3 studies are available at:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE98645
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?accv = GSE56609
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE30509
Other data reported is available from the corresponding author upon reasonable request.
References
- 1. van Ommen B., Stierum R., Curr. Opin. Biotechnol. 2002, 13, 517. [DOI] [PubMed] [Google Scholar]
- 2. Wittwer J., Rubio‐Aliaga I., Hoeft B., Bendik I., Weber P., Daniel H., Mol. Nutr. Food Res. 2011, 55, 341. [DOI] [PubMed] [Google Scholar]
- 3. Bartel J., Krumsiek J., Schramm K., Adamski J., Gieger C., Herder C., Carstensen M., Peters A., Rathmann W., Roden M., Strauch K., Suhre K., Kastenmuller G., Prokisch H., Theis F. J., PLoS Genet. 2015, 11, e1005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang S., Chaudhary K., Garmire L. X., Front. Genet. 2017, 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nath A. P., Ritchie S. C., Byars S. G., Fearnley L. G., Havulinna A. S., Joensuu A., Kangas A. J., Soininen P., Wennerstrom A., Milani L., Metspalu A., Mannisto S., Wurtz P., Kettunen J., Raitoharju E., Kahonen M., Juonala M., Palotie A., Ala‐Korpela M., Ripatti S., Lehtimaki T., Abraham G., Raitakari O., Salomaa V., Perola M., Inouye M., Genome Biol. 2017, 18, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dao M. C., Sokolovska N., Brazeilles R., Affeldt S., Pelloux V., Prifti E., Chilloux J., Verger E. O., Kayser B. D., Aron‐Wisnewsky J., Ichou F., Pujos‐Guillot E., Hoyles L., Juste C., Dore J., Dumas M. E., Rizkalla S. W., Holmes B. A., Zucker J. D., Clement K., Consortium M. I.‐O., Front. Physiol. 2018, 9, 1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wahl S., Vogt S., Stuckler F., Krumsiek J., Bartel J., Kacprowski T., Schramm K., Carstensen M., Rathmann W., Roden M., Jourdan C., Kangas A. J., Soininen P., Ala‐Korpela M., Nothlings U., Boeing H., Theis F. J., Meisinger C., Waldenberger M., Suhre K., Homuth G., Gieger C., Kastenmuller G., Illig T., Linseisen J., Peters A., Prokisch H., Herder C., Thorand B., Grallert H., BMC Med. 2015, 13, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang B., Mezlini A. M., Demir F., Fiume M., Tu Z., Brudno M., Haibe‐Kains B., Goldenberg A., Nat. Methods 2014, 11, 333. [DOI] [PubMed] [Google Scholar]
- 9. Tini G., Marchetti L., Priami C., Scott‐Boyer M. P., Briefings Bioinf. 2019, 19, 1269. [DOI] [PubMed] [Google Scholar]
- 10. Rohart F., Gautier B., Singh A., Le Cao K. A., PLoS Comput. Biol. 2017, 13, e1005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh A., Shannon C. P., Gautier B., Rohart F., Vacher M., Tebbutt S. J., Le Cao K. A., Bioinformatics 2019, 35, 3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bouvier‐Muller J., Allain C., Enjalbert F., Farizon Y., Portes D., Foucras G., Rupp R., J. Dairy Sci. 2018, 101, 2248. [DOI] [PubMed] [Google Scholar]
- 13. Romdhane S., Devers‐Lamrani M., Beguet J., Bertrand C., Calvayrac C., Salvia M. V., Jrad A. B., Dayan F. E., Spor A., Barthelmebs L., Martin‐Laurent F., Sci. Total Environ. 2019, 651, 241. [DOI] [PubMed] [Google Scholar]
- 14. Jiang R., Zhao C., Gao B., Xu J., Song W., Shi P., Int. J. Biochem. Cell Biol. 2018, 102, 1. [DOI] [PubMed] [Google Scholar]
- 15. Burton K. J., Rosikiewicz M., Pimentel G., Butikofer U., von Ah U., Voirol M. J., Croxatto A., Aeby S., Drai J., McTernan P. G., Greub G., Pralong F. P., Vergeres G., Vionnet N., Br. J. Nutr. 2017, 117, 1312. [DOI] [PubMed] [Google Scholar]
- 16. Burton K. J., Pimentel G., Zangger N., Vionnet N., Drai J., McTernan P. G., Pralong F. P., Delorenzi M., Vergeres G., PLoS One 2018, 13, e0192947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pimentel G., Burton K. J., von Ah U., Butikofer U., Pralong F. P., Vionnet N., Portmann R., Vergeres G., J. Nutr. 2018, 148, 851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ryan M. F., O'Grada C. M., Morris C., Segurado R., Walsh M. C., Gibney E. R., Brennan L., Roche H. M., Gibney M. J., Am. J. Clin. Nutr. 2013, 97, 261. [DOI] [PubMed] [Google Scholar]
- 19. Matone A., O'Grada C. M., Dillon E. T., Morris C., Ryan M. F., Walsh M., Gibney E. R., Brennan L., Gibney M. J., Morine M. J., Roche H. M., Mol. Nutr. Food Res. 2015, 59, 2279. [DOI] [PubMed] [Google Scholar]
- 20. Morris C., O'Grada C. M., Ryan M. F., Gibney M. J., Roche H. M., Gibney E. R., Brennan L., Lipids Health Dis. 2015, 14, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fatima A., Connaughton R. M., Weiser A., Murphy A. M., O'Grada C., Ryan M., Brennan L., O'Gaora P., Roche H. M., Mol. Nutr. Food Res. 2018, 62, 1700388. [DOI] [PubMed] [Google Scholar]
- 22. Bos M. B., de Vries J. H., Feskens E. J., van Dijk S. J., Hoelen D. W., Siebelink E., Heijligenberg R., de Groot L. C., Nutr., Metab. Cardiovasc. Dis. 2010, 20, 591. [DOI] [PubMed] [Google Scholar]
- 23. Michielsen C. C. J. R., Hangelbroek R. W. J., Feskens E. J. M., Afman L. A., Mol. Nutr. Food Res. 2019, 63, 1801095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Dijk S. J., Feskens E. J., Bos M. B., de Groot L. C., de Vries J. H., Muller M., Afman L. A., J. Nutr. 2012, 142, 1219. [DOI] [PubMed] [Google Scholar]
- 25. Lenth R., emmeans: Estimated Marginal Means, aka Least‐Squares Means, R package version 1.3.3. 2019. [Google Scholar]
- 26. Le Cao K. A., Rohart F., Gonzalez I., Dejean S., Gautier B., Bartolo F., contributions from Monget, P. , Coquery J., Yao F. Z., Liquet B., mixOmics: Omics Data Integration Project, R package version 6.1.1. 2016. [Google Scholar]
- 27. Westerhuis J. A., van Velzen E. J., Hoefsloot H. C., Smilde A. K., Metabolomics 2010, 6, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liquet B., Le Cao K. A., Hocini H., Thiebaut R., BMC Bioinformatics 2012, 13, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Field A. E., C. A. Camargo, Jr. , Ogino S., JAMA, J. Am. Med. Assoc. 2013, 310, 2147. [DOI] [PubMed] [Google Scholar]
- 30. Achilike I., Hazuda H. P., Fowler S. P., Aung K., Lorenzo C., 2015, 39, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burton K. J., Krüger R., Scherz V., Münger L. H., Picone G., Vionnet N., Bertelli C., Greub G., Capozzi F., Vergères G., Nutrients 2020, 12, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Data Availability Statement
Transcriptomic data for the 3 studies are available at:
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE98645
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?accv = GSE56609
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE30509
Other data reported is available from the corresponding author upon reasonable request.