

Abstract
Activation of innate immune signaling in the tumor microenvironment is central to a successful anti-tumor immune response, and it is in large part mediated by cytosolic double-stranded DNA sensing. Here, Carozza et al. (2020b) report potent and selective inhibitors of ENPP1, a negative regulator of innate immune signaling, which are shown to potentiate anti-tumor immune responses.
The ability to therapeutically harness the immune system against cancer has transformed the field of oncology. While many of the successful immune therapies primarily rely on overcoming immune checkpoints that prevent the full activation of the adaptive immune system, there is ample evidence that advanced tumors can also dampen innate immune signaling leading to an immune suppressive tumor microenvironment (TME). Sensing of cytosolic double-stranded DNA (dsDNA) by the cGAS-STING pathway has emerged as a key mechanism to activate pro-inflammatory innate immune responses in the TME, primarily through the induction of type I interferon (IFN) signaling (Ablasser and Chen, 2019). Yet, evidence suggests that tumors employ negative feedback mechanisms to suppress cGAS-STING signaling and instead co-opt its chronic activation to spread to distant organs (Bakhoum et al., 2018).
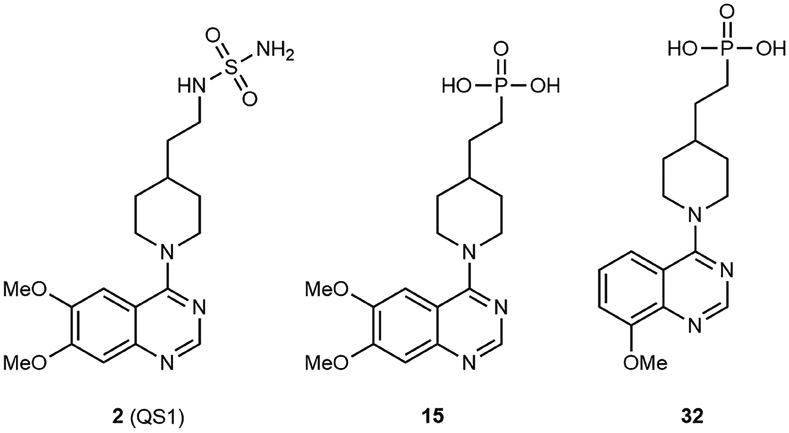
Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1) is an ATPase that plays a critical role in phosphate balance central to bone mineralization via the generation of pyrophosphate (Onyedibe et al., 2019). Recent discoveries have shown that ENPP1 is also responsible for the hydrolysis of 2′,3′-cGAMP (Carozza et al., 2020a; Li et al., 2014) and is expressed in the TME in cancers where it blunts cGAMP-mediated activation of the immune response. Yet, to date, the small number of ENPP1 inhibitors that have been developed are unlikely to be suitable for in vivo experiments (Onyedibe et al., 2019). Among the most suitable compounds are quinazoline derivatives such as compound 2 (QS1; Figure 1) (Patel et al., 2009). In this issue of Cell Chemical Biology, Carozza et al. (2020b) describe the discovery of potent and selective tools and possible drug leads that derive from compound 2. This report describes the origin of compounds used in a previous publication (Carozza et al., 2020a) and discloses additional in vivo data to further link ENPP1 to immune evasion in cancers.
Figure 1.
Small Molecule Inhibitors of ENPP1 Hydrolysis of 2′,3′-cGAMP
In order to provide ENPP1 inhibitors suitable for in vivo experiments, Carozza et al. (2020b) critically evaluated the conditions of ENPP1 enzymatic assays. Assays used to discover and characterize ENPP1 inhibitors have typically been performed under basic conditions where ENPP1 has greater enzymatic activity. The authors developed a biochemical assay with recombinant murine ENPP1 at pH of 7.4 and discovered that known ENPP1 inhibitors, such as compound 2, are significantly less potent at physiological conditions.
Since ENPP1 is a metalloenzyme that requires two catalytic zinc atoms in the active site, the authors proposed that a sulfamoylate-zinc interaction contributes to ENPP1 inhibition by QS1. This would account for the pH sensitivity of inhibition. They surmised that a phosphonic acid zinc-binding group that remains anionic at pH 7.4 would be more effective at a lower pH. Indeed, they demonstrated multiple examples of phosphonic acid such as compound 15 with <50 nM K1s at both pH 9.0 and 7.4. The phosphonic acids are also cellularly impermeable, which is suitable for ENPP1, an extracellularly disposed membrane-bound protein. This is an important reminder to consider carefully how well in vitro assay conditions translate to the relevant physiological conditions. The authors further demonstrated the relevance of their assay conditions by inhibiting soluble ENPP1 in mouse and human plasma.
The authors then solved an X-ray costructure of compound 15 bound to the nuclease-like domain of murine ENPP1 that demonstrates the phosphonate-Zn interaction. In fact, the phosphonate achieved interactions with both Zn atoms as well as asparagine 259 in a nearly identical manner to the phosphate of AMP, the product of ATP and 2′,3′-cGAMP hydrolysis by ENPP1. The quinazoline of 15 bound in the nucleotide binding pocket where bases of the ENPP1 substrates also bound. However, the quinazoline filled the pocket more fully, accounting for the increased affinity of the quinazoline and quinoline ENPP1 inhibitors over AMP.
The authors prepared several analogs to establish structure-activity relationships (SARs) for murine ENPP1 inhibition at pH 7.4. They demonstrated that the phosphonic acids are superior to a set of zinc-binding group-matched pairs beyond aminosulfonamides. They further explored substitution on the quinazoline and discovered that certain 8-alkoxy quinazolines, such as compound 32, provided potency below the limit of their assay (K1 < 2 nM). It is interesting, however, that much of the SAR is not explained by the binding pose of compound 15 in ENPP1, suggesting that there are multiple possible conformations of the quinazolines that can result in potent inhibition. A recent publication of compound 1 and related compounds bound to human ENPP1 (Dennis et al., 2020) is evidence that multiple binding modes are possible. Dennis et al. observed a different binding mode for the quinazoline of compound 1 than that observed for the quinazoline compound 15 in this report, and a direct interaction of the aminosulfonamide with either of the catalytic zincs was not observed.
Developing potent and specific ENPP1 inhibitors has the potential to restore pro-inflammatory signaling and type I IFN in the TME, which would potentiate the effects of a multitude of anti-cancer therapeutic modalities that rely on IFN signaling. This includes radiation therapy, as shown by Carozza et al., as well as other DNA-damaging chemotherapeutics. Given the centrality of cGAS-STING signaling in tumor response to immunotherapy, inhibition of ENPP1 has the potential to potentiate the effect of immune checkpoint blockade; however, this has yet to be shown.
Over the past decade there has been a concerted effort to therapeutically activate STING in the TME through the development of small molecule agonists. Whereas inhibition of ENPP1 would achieve a similar outcome, it is likely to exhibit very different kinetics. STING agonists have been delivered primarily through intratumoral administration where rapid clearance is a critical limitation. Furthermore, dose-limiting toxicities from systemic IFN activation are likely to narrow the therapeutic window. Conversely, ENPP1 inhibition acts at much smaller scales within the TME, which would likely limit systemic toxicity. Furthermore, sustained release of tumor-derived cGAMP would enable consistent activation of host STING in a manner conducive to a sustained anti-tumor effect.
Given the cGAMP-independent functions of ENPP1, it will be important to pay close attention to the side-effect profile of ENPP1 inhibitors as these progress to first-in-human testing. Patients with ENPP1 mutations exhibit a constellation of symptoms including generalized arterial calcification and hypophosphatemic rickets secondary to dysregulation of pyrophosphate metabolism (Lorenz-Depiereux et al., 2010). Furthermore, ENPP1 polymorphisms have been linked to the onset of type 2 diabetes mellitus (Sortica et al., 2015), likely due to direct interaction between the ENPP1 cytoplasmic domain with the insulin receptor. While it is unlikely that short-term treatment of patients with cancer would yield symptoms reminiscent of lifelong ENPP1 loss of function, data from human genetic studies provide an excellent framework to carefully assess any potential side effects arising from this emerging promising class of inhibitors.
ACKNOWLEDGMENTS
S.F.B. is supported by the Office of the Director, National Institutes of Health under Award Number DP5OD026395 High-Risk High-Reward Program, the NCI Breast Cancer SPORE (P50CA247749), the Burroughs Wellcome Fund Career Award for Medical Scientists, the Parker Institute for Immunotherapy at MSKCC, the Josie Robertson Foundation, and the MSKCC core grant P30CA008748.
Footnotes
DECLARATION OF INTERESTS
D.C. is an employee and holds shares of Volastra Therapeutics Inc. S.F.B. holds a patent related to some of the work described targeting CIN and the cGAS-STING pathway in advanced cancer. He owns equity in, receives compensation from, and serves as a consultant on the Scientific Advisory Board and Board of Directors of Volastra Therapeutics Inc. He has also consulted for Sanofi and received sponsored travel from the Prostate Cancer Foundation and both travel and compensation from Cancer Research UK.
REFERENCES
- Ablasser A, and Chen ZJ (2019). cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carozza JA, Böhnert V, Nguyen KC, Skariah G, Shaw KE, Brown JA, Rafat M, von Eyben R, Graves EE, Glenn JS, et al. (2020a). Extracellular cGAMP is a cancer-cell-produced immunotransmitter involved in radiation-induced anticancer immunity. Nat. Can 1,184–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carozza JA, Brown JA, Böhnert V, Fernandez D, AlSaif Y, Mardjuki RE, Smith M, and Li L (2020b). Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem. Biol 27, this issue, 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis ML, Newman J, Dolezal O, Hattarki M, Surjadi RN, Nuttall SD, Pham T, Nebl T, Camerino M, Khoo PS, et al. (2020). Crystal structures of human ENPP1 in apo and bound forms. Acta Crystallogr. D Struct. Biol 76, 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Yin Q, Kuss P, Maliga Z, Millán JL, Wu H, and Mitchison TJ (2014). Hydrolysis of 2‘3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat. Chem. Biol 10, 1043–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, and Strom TM (2010). Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am. J. Hum. Genet 86, 267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onyedibe KI, Wang M, and Sintim HO (2019). ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors-A STING in the Tale of ENPP1. Molecules 24, 4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SD, Habeski WM, Cheng AC, de la Cruz E, Loh C, and Kablaoui NM (2009). Quinazolin-4-piperidin-4-methyl sulfamide PC-1 inhibitors: alleviating hERG interactions through structure based design. Bioorg. Med. Chem. Lett 19, 3339–3343. [DOI] [PubMed] [Google Scholar]
- Sortica DA, Buffon MP, Souza BM, Nicoletto BB, Santer A, Assmann TS, Crispim D, and Canani LH (2015). Association between the ENPP1 K121Q polymorphism and risk of diabetic kidney disease: a systematic review and meta-analysis. PLoS One 10, e0118416. [DOI] [PMC free article] [PubMed] [Google Scholar]