

Abstract
Neutrophils are innate immune cells that play important roles in many physiological and pathological processes, including immune defense and cancer metastasis. In addition to the release of proinflammatory cytokines, chemokines, and cytoplasmic granules containing digestive proteins, in recent years, neutrophils have been observed to release neutrophil extracellular traps (NETs) that consist of extracellular DNA associated with antimicrobial proteins, such as histones and myeloperoxidase. These NETs are increasingly being recognized as an important mechanism of neutrophil host defense and function. This chapter will summarize the current literature on the known processes of NET formation and describe in detail an immunofluorescence approach that can be employed to visualize and quantify NETs in vitro.
Keywords: NETs, Extracellular DNA, Activation, Myeloperoxidase, Sytox, PicoGreen, Citrullination
1. Introduction
Neutrophils are the body’s first line of defense against pathogens. They rapidly migrate to sites of infection by following gradients of chemoattractants such as endogenous interleukin (IL)-8 or bacterial N-formyl peptides where they can neutralize pathogens through several mechanisms. Neutrophils can eliminate pathogens by phagocytosis, release of destructive reactive oxygen species (ROS), and secretion of chemokines and cytokines that can propagate inflammation and recruit and activate additional immune cells [1, 2]. Furthermore, neutrophils can exocytose cytoplasmic granules containing degradative enzymes and antimicrobial proteins that can neutralize pathogens extracellularly; this process is termed degranulation [1, 2]. More recently, a novel mode of extracellular killing has been observed: the release of neutrophil extracellular traps (NETs), which are DNA webs, decorated with antimicrobial proteins and enzymes, can entrap and neutralize pathogens close to the neutrophil [3, 4]. In addition to the antimicrobial effect of NETs, recent studies have also reported their involvement in the propagation of inflammation and in noninfectious disease settings such as autoimmunity, thrombosis, and cancer metastasis [2, 5–7]. Thus, a better understanding of the triggers and mechanisms of NET formation and their downstream effects may be relevant to many biomedical fields.
1.1. Composition of NETs and Their Formation
NET formation is commonly induced in vitro by chemicals such as phorbol myristate acetate (PMA) or calcium ionophores (A23187). PMA-induced NET release requires ROS production mediated by NADPH oxidase, while A23187-induced NET release occurs independent of NADPH oxidase activity and instead relies on mitochondrial-derived ROS [8]. Likewise, physiologically, E. coli lipopolysaccharide (LPS)-induced NETs require NADPH oxidase activity, while S. aureus and Leishmania induce NETs independent of NADPH oxidase activity [9, 10]. Using PMA as the classic inducer of NETs, the intracellular processes involved in NET release have been investigated in detail. It has been observed that around 2 h poststimulation, there is breakdown of the nuclear envelop, which allows enzymes such as myeloperoxidase (MPO) and neutrophil elastase (NE) to migrate from cytoplasmic granules into the nucleus where they help facilitate chromatin decondensation [4]. There is also activation of protein arginine deiminase (PAD)4, which citrullinates histones 3 and 4 to aid chromatin decondensation. Around 4 h poststimulation, neutrophils burst to release NETs consisting of nuclear DNA and associated antimicrobial enzymes and proteins [4]. Recent proteomic studies have revealed that depending upon the stimulus, different proteins will be associated with the NET structures and their posttranslational modifications may also differ [11, 12]. Thus, NETs induced under different conditions may have different biological effects. Typically, during NET release, neutrophil membrane integrity is compromised and neutrophil death results, leading to the process being named “suicidal NETosis”. However, other more rapid pathways have also been reported, including “vital” NET release, where neutrophil viability and function are not compromised after NET release [7, 13, 14], and the release of NETs that are composed of mitochondrial DNA [14–16].
1.2. Visualizing NETs
NETs are commonly visualized ex vivo or in vitro by fluorescence microscopy after staining with a DNA-binding dye and/or immunofluorescence using antibodies against MPO, NE, and/or citrullinated histones that are specific to NET formation. Several groups have also examined NETs by electron microscopy, reporting that NETs are made up of 15–26 nm wide filaments with 25–60 nm globular domains [3, 17]. NET release can also be visualized in real time using intravital microscopy or live-cell imaging [18–21]. In culture, NETs typically appear spread out and filamentous; however, in vivo, NETs appear round and aggregated, likely to indicate spatial restrictions of the tissue.
1.3. Quantifying NETs
In addition to qualitatively visualizing NETs, NET release can also be quantified. The most common methods include quantification of the area covered by extracellular DNA after staining, [22] fluorescence spectroscopy of the culture media after adding DNA-binding dyes, or a modified ELISA against MPO-DNA or NE-DNA complexes characteristic of NETs [23]. More recently, a new method of quantification using multispectral imaging flow cytometry was reported [24] and algorithms for automated quantification of fluorescence images have been made available [25, 26]. However, for all of these methods, in particular, when detecting NETs in vivo, care should be taken to conclude the formation of NETs as opposed to ETs derived from other innate immune cells since monocyte and macrophage-derived ETs have also been reported to be associated with MPO and elastase that were first thought to be neutrophil-specific [27, 28 ]. Quantification of extracellular DNA using DNA-binding dyes may also be confounded by cell-free DNA. Thus, the appropriate controls must be included to rule out cell death, such as necrosis, which would lead to the release of cell-free DNA.
2. Materials
2.1. Equipment
Sodium heparin-coated vacutainers.
50 ml Conical tubes.
Plastic 1.5 ml Pasteur pipettes.
12-Well flat-bottom culture plates.
24-Well flat-bottom culture plates.
Black flat bottom 96-well plates.
Large volume centrifuge.
Centrifuge.
Tissue Culture Hood.
Tissue Culture Incubator.
Inverted confocal fluorescence microscope.
Fluorescence plate reader (excitation: ~480 nm; emission: ~520 nm).
2.2. Reagents
Hank’s balanced salt solution (HBSS) without Ca2+ and Mg2+ (see Note 1).
12% Dextran solution: v/v, diluted with HBSS without Ca2+ and Mg2+.
Histopaque® 1077 and 1119.
1.8% saline solution: 1.8 g NaCl in 1 L distilled water and sterile filter.
OptiMEM medium.
RPMI1640 medium.
Fetal bovine serum (FBS).
Goat serum.
Phosphate-buffered saline (PBS).
0.05% PBS-T: Tween-20 in PBS v/v.
4% paraformaldehyde w/v (PFA) (see Note 2).
Blocking solution for staining: 5% goat serum v/v in PBS.
Phorbol myristate acetate (PMA) as a positive control for NET release.
SYTOX® Green Nucleic Acid Stain.
Quant-iT™ PicoGreen® dsDNA Kit.
Antibody reactive against human neutrophil elastase.
Antibody reactive against citrullinated histone 3.
Antibody reactive against myeloperoxidase.
Secondary antibody solution: 1:500 v/v Alexa Fluor® 564-labelled antibody in blocking solution.
3. Methods
The methods described below outline the process for (1) human neutrophil isolation from peripheral blood; (2) neutrophil culture; (3) immunostaining of NETs; and (4) spectroscopic quantification of NETs.
3.1. Isolating Human Neutrophils from the Peripheral Blood
Collect blood by venepuncture into heparin-coated vacutainers. Proceed with the subsequent steps using sterile reagents in a tissue culture hood.
Pool blood into a 50 ml conical tube and add dextran to form a 1.2% solution. For example, for 30 ml of whole blood, add 3 ml of 12% dextran.
Invert 5–10× gently to mix and leave at room temperature for 20 min for red blood cells (RBCs) to settle.
While RBCs are settling, set up 15 ml conical tubes with 3 ml of Histopaque® 1119 at the bottom and carefully overlay this with 3 ml of Histopaque® 1077. If the interface is prepared properly, there should be a sharply demarcated line between the two Histopaque® layers. If not, discard and prepare again.
After RBCs have settled, the top layer of plasma should be transferred to the top of the Histopaque® layers using plastic Pasteur pipettes.
Centrifuge the Histopaque/Plasma layers at 450 × g for 25 min without brakes at room temperature to separate out the neutrophils from mononuclear cells (see Fig. 1).
Carefully suction off the top three layers and dispose before collecting the neutrophil layer into a new 50 ml tube.
Add HBSS (with no Ca2+ or Mg2+) up to 50 ml and centrifuge at 450 × g for 10 min at room temperature with brakes.
Remove the HBSS and resuspend the cell pellet.
To remove contaminating RBCs, resuspend the cell pellet, add 25 ml of sterile distilled water to the tube, invert to mix, and immediately add 25 ml of sterile 1.8% saline solution to restore tonicity.
Centrifuge at 450 × g for 10 min at room temperature to pellet the neutrophils.
Remove the supernatant and resuspend the neutrophil pellet in OptiMEM media supplemented with 2% FBS (see Note 3).
Cells can be counted, checked for purity by flow cytometry for neutrophil markers such as CD16b, and plated for experiments.
Fig. 1.
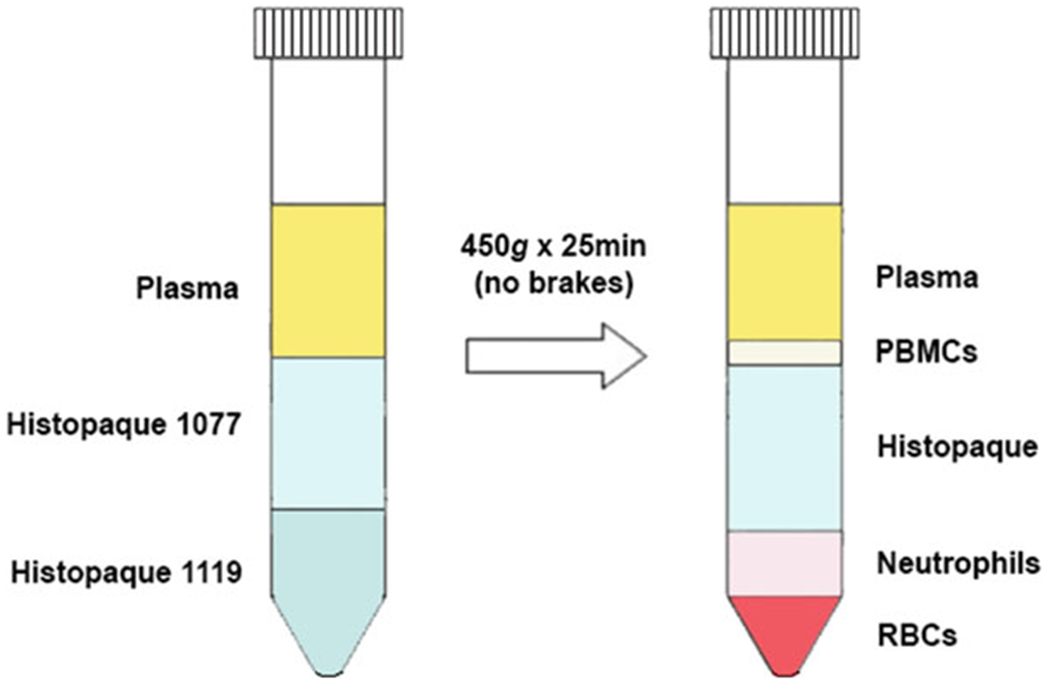
Isolation of neutrophils using Histopaque®. Neutrophils can be separated from peripheral blood mononuclear cells (PBMCs) and red blood cells (RBCs) by centrifuging blood plasma on layers of Histopaque® 1077 and 1119
3.2. Preparation of Neutrophil Cultures
Neutrophils are seeded into sterile culture plates for NET visualization and quantification experiments. For visualization experiments, 2 × 105 neutrophils are seeded per well of a 24-well plate, and for quantification, 1 × 106 neutrophils are seeded per well of a 12-well plate.
The stimulus of interest is added to neutrophils. As a positive control, 50 nM PMA should be added to the cells.
Culture neutrophils in 37 °C/5% CO2 for an optimized time-point before analysis (see Note 4).
3.3. Immunostaining of NETs
Prepare a 167 nM SYTOX® Green solution just before use by diluting 1 μl of the dye in 30 μl HBSS and then using 1 μl of this for every ml of HBSS. Store in the dark until ready for use. SYTOX® Green is a cell membrane-impermeable DNA-binding dye.
After the optimized treatment timepoint, take the plate of neutrophils out of the incubator, aspirate the medium, and gently wash (see Note 5) the wells with 1 ml of HBSS twice.
Gently add 0.5 ml of SYTOX® Green solution to each well for 15 min at room temperature in the dark.
Aspirate the dye and gently wash twice with HBSS. From this point onward, perform all incubations in the dark.
Gently add 0.5 ml of 4% PFA to each well for 10 min at room temperature in the dark.
Aspirate PFA and wash twice with HBSS.
Block each well with 0.5 ml of blocking solution for 1 h at room temperature in the dark.
Prepare 0.5 ml of primary antibody solution for each well (1:100 citrullinated histone 3 or MPO or NE in blocking solution). Be sure to include an isotype-matched IgG control (see Note 6).
Incubate overnight at 4 °C and wash twice with PBS-T.
Add 0.5 ml of secondary antibody solution to each well.
Incubate for 1 h at room temperature in the dark.
After washing twice with PBS-T, NETs are ready to be viewed using an inverted confocal fluorescence microscope.
3.4. Semi-Quantification of NETs
Collect the neutrophil conditioned media after treatment for the optimized timepoint (Subheading 3.2).
Centrifuge the conditioned media at 2000 × g for 5 min at 4 °C and collect the supernatant (see Note 7).
While samples are being prepared, the Quant-iT™ PicoGreen® kit can be thawed and 1 × TE buffer is made according to the manufacturer’s instructions using sterile, distilled, DNase-free water.
After the supplied 100 μg/ml phage lambda DNA standard has been thawed and thoroughly vortexed, the standard curve can be prepared following the manufacturer’s instructions in nucleic acid- and DNAse-free Eppendorf tubes using TE buffer. The standard curve typically ranges from 1 ng/ml to 1 μg/ml, but can be diluted down to 25 pg/ml with accuracy.
In black flat-bottom 96-well plates, prepare different dilutions of each conditioned media using TE buffer to determine the optimal dilution required for downstream analysis. From our experience, culture of 1 × 106 neutrophils in 1 ml of media with 50 nM PMA for 4 h requires a 1:4 dilution of the conditioned media prior to analysis for the sample fluorescence to fit within the fluorescence levels of the standard curve. (Once the optimized dilution has been determined, samples can be diluted in each well, i.e., First pipette 75 μl of TE buffer into all wells, followed by 25 μl of each sample).
Standards should be analyzed in duplicate, and each sample should be analyzed in triplicate.
Once all standards and samples have been pipetted into the plate, prepare the PicoGreen® dye by diluting the provided dye stock in TE buffer 200-fold in a plastic container. This solution is susceptible to photodegradation, and so prepare just before use and store in the dark.
Vortex the dye solution thoroughly and pipette 10 μl of the dye to the side of each well. The dye droplet will fall and mix with the sample over time.
After the dye has been added to all wells, ensure that the dye droplet has mixed with each sample by tapping the plate side to side gently. From this point onward, the plate is light-sensitive and should be covered.
Incubate the plate at room temperature for 5 min and then read using a fluorescent plate reader (excitation: 480 nm; emission: 520 nm).
Using the fluorescence levels from the standard curve, the total amount of extracellular DNA in each conditioned media can be calculated (see Note 8).
Acknowledgments
Funding: Supported by grant R01AI121183 (to VMA) from The National Institute of Allergy and Infectious Diseases (NIAID), the National Institutes of Health (NIH).
Footnotes
Conflicts of interest: The authors have no conflicts of interest.
HBSS without calcium and magnesium is used throughout the experiment to minimize neutrophil activation during the purification and staining steps.
Care should be taken when handling PFA due to known toxicity.
Neutrophils appear to form NETs spontaneously when exposed to serum-free conditions, and so media supplemented with 2% FBS are used throughout the experiments.
It is well-established that PMA induces optimal NET formation after 3–4 h of stimulation, but a time course study may be needed for the stimulus of interest to ensure analysis at the right timepoint.
To minimize disruptions to the NET structures, wash plates gently by pipetting gently down the side of each well and aspirating from the wall of the well.
If dual staining with both citrullinated histone 3 and MPO or elastase antibodies is desired, DAPI or Hoechst 33342 will need to be used as the DNA stain.
At this point, the supernatant can be stored at −80 °C until analysis.
Make sure that any dilution factors prior to analysis are taken into account.
References
- 1.Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13(3):159–175. 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- 2.Nathan C (2006) Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 6(3):173–182. 10.1038/nri1785 [DOI] [PubMed] [Google Scholar]
- 3.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535. 10.1126/science.1092385 [DOI] [PubMed] [Google Scholar]
- 4.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176(2):231–241. 10.1083/jcb.200606027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mayadas TN, Cullere X, Lowell CA (2014) The multifaceted functions of neutrophils. Annu Rev Pathol 9:181–218. 10.1146/annurev-pathol-020712-164023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tong M, Abrahams VM (2019) Neutrophils in preterm birth: friend or foe? Placenta 495(1):60–63. 10.1016/j.placenta.2019.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yipp BG, Kubes P (2013) NETosis: how vital is it? Blood 122(16):2784–2794. 10.1182/blood-2013-04-457671 [DOI] [PubMed] [Google Scholar]
- 8.Douda DN, Khan MA, Grasemann H, Palaniyar N (2015) SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci U S A 112(9):2817–2822. 10.1073/pnas.1414055112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185(12):7413–7425. 10.4049/jimmunol.1000675 [DOI] [PubMed] [Google Scholar]
- 10.Rochael NC, Guimaraes-Costa AB, Nascimento MT, DeSouza-Vieira TS, Oliveira MP, Garcia e Souza LF, Oliveira MF, Saraiva EM (2015) Classical ROS-dependent and early/rapid ROS-independent release of neutrophil extracellular traps triggered by Leishmania parasites. Sci Rep 5:18302. 10.1038/srep18302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, Wright HL (2019) Caught in a trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front Immunol 10:423. 10.3389/fimmu.2019.00423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, Migliorini P (2019) Neutrophil extracellular traps (NET) induced by different stimuli: a comparative proteomic analysis. PLoS One 14(7):e0218946. 10.1371/journal.pone.0218946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong M, Potter JA, Mor G, Abrahams VM (2019) Lipopolysaccharide-stimulated human fetal membranes induce neutrophil activation and release of vital neutrophil extracellular traps. J Immunol 203(2):500–510. 10.4049/jimmunol.1900262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU (2009) Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16(11):1438–1444. 10.1038/cdd.2009.96 [DOI] [PubMed] [Google Scholar]
- 15.McIlroy DJ, Jarnicki AG, Au GG, Lott N, Smith DW, Hansbro PM, Balogh ZJ (2014) Mitochondrial DNA neutrophil extracellular traps are formed after trauma and subsequent surgery. J Crit Care 29(6):1133 e1131–1133 e1135. 10.1016/j.jcrc.2014.07.013 [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Li T, Chen S, Gu Y, Ye S (2015) Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol 67(12):3190–3200. 10.1002/art.39296 [DOI] [PubMed] [Google Scholar]
- 17.Onouchi T, Shiogama K, Mizutani Y, Takaki T, Tsutsumi Y (2016) Visualization of neutrophil extracellular traps and fibrin meshwork in human Fibrinopurulent inflammatory lesions: III Correlative light and electron microscopic study. Acta Histochem Cytochem 49(5):141–147. 10.1267/ahc.16028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury CA, Zhang K, Conly J, Kubes P (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18(9):1386–1393. 10.1038/nm.2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Buhr N, von Kockritz-Blickwede M (2016) How neutrophil extracellular traps become visible. J Immunol Res 2016:4604713. 10.1155/2016/4604713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13(4):463–469. 10.1038/nm1565 [DOI] [PubMed] [Google Scholar]
- 21.Barr FD, Ochsenbauer C, Wira CR, Rodriguez-Garcia M (2018) Neutrophil extracellular traps prevent HIV infection in the female genital tract. Mucosal Immunol 11(5):1420–1428. 10.1038/s41385-018-0045-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez AS, Bardoel BW, Harbort CJ, Zychlinsky A (2014) Induction and quantification of neutrophil extracellular traps. Methods Mol Biol 1124:307–318. 10.1007/978-1-62703-845-4_20 [DOI] [PubMed] [Google Scholar]
- 23.Sil P, Yoo DG, Floyd M, Gingerich A, Rada B (2016) High throughput measurement of extracellular DNA release and quantitative NET formation in human neutrophils in vitro. J Vis Exp 112:52779. 10.3791/52779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao W, Fogg DK, Kaplan MJ (2015) A novel image-based quantitative method for the characterization of NETosis. J Immunol Methods 423:104–110. 10.1016/j.jim.2015.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mohanty T, Sorensen OE, Nordenfelt P (2017) NETQUANT: automated quantification of neutrophil extracellular traps. Front Immunol 8:1999. 10.3389/fimmu.2017.01999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Breda SV, Vokalova L, Neugebauer C, Rossi SW, Hahn S, Hasler P (2019) Computational methodologies for the in vitro and in situ quantification of neutrophil extracellular traps. Front Immunol 10:1562. 10.3389/fimmu.2019.01562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Granger V, Faille D, Marani V, Noel B, Gallais Y, Szely N, Flament H, Pallardy M, Chollet-Martin S, de Chaisemartin L (2017) Human blood monocytes are able to form extracellular traps. J Leukoc Biol 102(3):775–781. 10.1189/jlb.3MA0916-411R [DOI] [PubMed] [Google Scholar]
- 28.Doster RS, Rogers LM, Gaddy JA, Aronoff DM (2018) Macrophage extracellular traps: a scoping review. J Innate Immun 10(1):3–13. 10.1159/000480373 [DOI] [PMC free article] [PubMed] [Google Scholar]