

Abstract
Acid ceramidase (murine gene code: Asah1) (50 kDa) belongs to N-terminal nucleophile hydrolase family. This enzyme is located in the lysosome, which mediates conversion of ceramide (CER) into sphingosine and free fatty acids at acidic pH. CER plays an important role in intracellular sphingolipid metabolism and its increase causes inflammation. The mammalian target of rapamycin complex 1 (mTORC1) signaling on late endosomes (LE)/lysosomes may control cargo selection, membrane biogenesis and exosome secretion, which may be fine controlled by lysosomal sphingolipids such as CER. This lysosomal-CER-mTOR signaling may be a crucial molecular mechanism responsible for development of arterial medial calcification (AMC). Torin-1 (5mg/kg/day), an mTOR inhibitor significantly decreased aortic medial calcification accompanied with decreased expression of osteogenic markers like osteopontin and RUNX2 and upregulation of SM22-α in mice receiving high dose of Vitamin D (500,000 IU/kg/day). Asah1fl/fl/SMCre mice had markedly increased co-localization of mTORC1 with Lamp-1(lysosome marker) and decreased co-localization of VPS16 (a multivesicular bodies (MVBs) marker) with Lamp-1, suggesting mTOR activation caused reduced MVBs interaction with lysosomes. Torin-1 significantly reduced the co-localization of mTOR vs Lamp-1, increased lysosome-MVBs interaction which was associated with reduced accumulation of CD63 and annexin 2 (exosome markers) in the coronary arterial wall of mice. Using CASMCs, Pi-stimulation significantly increased p-mTOR expression in Asah1fl/fl/SMCre CASMCs as compared to WT/WT cells, associated with increased calcium deposition and mineralization. Torin-1 blocked Pi-induced calcium deposition and mineralization. siRNA mTOR and Torin-1 significantly reduce co-localization of mTORC1 with Lamp-1, increased VPS16 vs Lamp-1 co-localization in Pi-stimulated CASMCs, associated with decreased exosome release. Functionally, Torin-1 significantly reduce arterial stiffening as shown by restoration from increased pulse wave velocity and decreased elastin breaks. These results suggest that lysosomal CER-mTOR signaling may play a critical role for the control of lysosome-MVB interaction, exosome secretion and arterial stiffening during AMC.
Keywords: Vascular calcification, mTOR, exosome, sphingolipids, arterial stiffness
Introduction
Vascular calcification (VC) involved formation and accumulation of calcium phosphate salts within the vascular wall that has been associated with aging, chronic kidney disease (CKD), diabetes mellitus, and atherosclerosis1. In response to various physiological stimuli or a mineral imbalance, vascular smooth muscle cells (VSMCs) secrete small, specialized membrane-bound bodies termed matrix vesicles (MVs) which act to nucleate calcium/phosphate (Ca/Pi) crystals in the form of hydroxyapatite. These MVs or exosomes (<200 nm) form the first nidus for mineralization2. Literature reported that enhanced exosome excretion may trigger smooth muscle cell (SMC) phenotype changes3–5, the apatite nucleation6, and calcifying nidus formation7, ultimately leading to extracellular matrix (ECM) mineralization8–11. Recently, it has been reported that bioactive sphingolipids and ceramide (CER) are associated with cytoskeletal rearrangements in synthetic VSMCs lead to multivesicular bodies (MVBs) trafficking and elevated exosome secretion in VC9,12,6.
Given that sphingolipids are involved in cardiovascular pathophysiology and biomineralization. Acid ceramidase (murine gene code: Asah1) (50 kDa) belongs to N-terminal nucleophile hydrolase family. Deficiency of AC activity lead to Farber disease in humans13. Case reports have mentioned some manifestations of this disease including enlargement and calcification of axillary lymph nodes14. This enzyme is located in the lysosome, which mediates conversion of CER into sphingosine and free fatty acids at acidic pH15. CER plays an important role in intracellular sphingolipid metabolism and its increase causes inflammation16. In human femoral arterial SMCs, it was reported that CER enhanced the Ox-LDL-induced VSMC mineralization via p38 MAPK signaling pathway, both CER and Ox-LDL stimulated phosphorylation of p38 MAPK. However, inhibition of p38 MAPK blocked this mineralization process17. In Ox-LDL stimulated human VSMCs, a significantly reduced calcium deposition was observed with PDTC, a NF-κB inhibitor. However, this effect of PDTC on calcification was reversed by C2-ceramide treatment, indicating that NF-κB regulates VC in a ceramide-dependent manner18. Recently, a study identified serum- and glucocorticoid-inducible kinase 1 (SGK1) which is up-regulated in VSMCs during pro-calcifying conditions and act as downstream of ASM/CER mediated calcification effects. The study demonstrated that knockdown of SGK1 suppresses the pro-calcific effects of ASM/CER treatment in VSMCs. Hence, concluded that ASM/ceramide via promoting SGK1-dependent osteo-/chondrogenic transdifferentiation of VSMCs contributes to the development of VC during hyperphosphatemia19. Kapustin et al reported that neutral sphingomyelinase 2 (N-SMase2) inhibition reduces mineralization in response to osteogenic medium in human coronary artery SMC9. Recently, our group demonstrated that SMC specific lysosomal acid ceramidase (Ac) and acid sphingomyelinase (ASM) controlled CER metabolism play an important role in the development and progression of arterial medial calcification (AMC)20,21. These studies demonstrated that either deletion of lysosomal Asah1 gene or over‐expression of Smpd1 gene specifically in SMCs cause reduction of lysosome-MVB interaction and consequent prolongation of MVBs fate resulting in enhanced SMCs-derived exosome/small extracellular vesicle release, SMC phenotypic transition initiating AMC. However, the finer molecular mechanisms how lysosomal-sphingolipid metabolism regulates exosome excretion from MVBs in SMCs during the development and progression AMC is still unknown.
Recent studies showed that the mammalian target of rapamycin complex (mTORC) signaling on late endosomes (LE)/lysosomes may control cargo selection, membrane biogenesis and lysosome trafficking in lysosomal degradation pathways22, which is fine controlled by sphingolipids, ceramide and sphingosine-1-phosphate (S1P)23. This sphingolipid-regulated mTORC signaling has been reported to be involved in VC24, which may be a crucial molecular mechanism responsible for the development of calcification of atheromatous plaques and even in arterial medial layer due to lysosome dysfunction that we observed recently25–27. In fro/fro (N-SMase2−/−) fibroblasts, study anticipated that N-SMase2 and CER are the important mediators in the regulation of hyaluronan synthesis, via microdomains and the Akt/mTOR pathway28. An in vitro study demonstrated that ceramide-1-phosphate (C1P) causes phosphorylation of mTORC1 thereby regulating macrophage proliferation. mTORC1 signaling on late endosomes (LE)/lysosomes seems to control vesicular formation and trafficking in lysosomal degradation pathways22. In VSMCs and mice model, miR-30b was shown to markedly inhibit activation of the mTOR signaling pathway which attenuated the VC during hyperphosphatemia in CKD29. Another study in rat cultured VSMCs demonstrated that high Pi -induced expression of Akt /mTOR pathway are involved in VSMC calcification through the activation of Cbfα130. Exosomes isolated from high glucose-induced human umbilical vein endothelial cell (HUVEC) promoted VSMCs calcification involving Notch3 protein mediated mTOR signaling pathway31. In this regard, rapamycin was reported to decrease VC by inhibiting mTOR pathway under in vitro and in vivo24. Recent studies have indicated that inhibition of lysosomal and endosomal function also promoted osteogenic transformation of VSMCs leading to VC32. Based on these observations, we hypothesized that lysosomal sphingolipids may contribute to mTOR activation on lysosomes and blunt their fusion to MVBs involved in exosome excretion from SMCs during AMC.
Material and Methods
Animal Studies
SMC-specific Asah1 gene knockout (Asah1fl/fl/SMCre) mice (N-Acylsphingosine Amidohydrolase 1 (Asah1) were employed for this study, characterized in our previous study20. Male C57BL/6 J wild-type and Asah1fl/fl/SMCre mice of 12–14 weeks were used. All protocols and experiments were carried out in compliance with appropriate guidelines and regulations by Institutional Animal Care and Use Committee of Virginia Commonwealth University. Animals were thereafter categorized into 8 groups, for each mouse strain (Asah1fl/fl/SMCre and WT/WT) subcutaneously injected active vitamin D (Vit D) (500,000 IU/Kg/bw/day) or matched vehicle (5% v/v ethanol) for 3–4 days to develop AMC33,34. Following 16–17 days of post injection period, animals were sedated with 2% isoflurane through a nose cone. Started 3 days prior to Vit D treatment, intraperitoneally injection of Torin-1 (a pharmacological inhibitor of mTORC1) (5 mg/kg BW)35,36 were continued for two weeks (n = 5–6 per group). The Vit D solution was prepared as described previously20,33. Blood samples were collected, and plasma was isolated, stored at −80 °C. Mice were sacrificed, heart and aorta were retrieved, with a portion of each preserved in 10% buffered formalin for histopathological observations and immunostaining. The remaining portion of heart and aorta were frozen in liquid nitrogen and stored at −80 °C to perform dual fluorescence staining and confocal microscopy.
Calcium staining, mineralization and immunohistochemistry
Alizarin Red S staining was performed33 for detection of AMC. Different washes of alcohol and xylene followed by distilled water was used for deparaffinization of samples. The arterial sections were stained by 1% Alizarin Red S solution for 5 min followed by incubation with acetone for 20 sec and another 20 sec with acetone-xylene. The reddish color indicated positively stained area. Mineralization was observed by Von Kossa staining as previously described37 and following manufacturer’s protocol (ab150687, Abcam, USA). To perform the histological examinations, paraffin sections were made using fixed mouse heart and aortas, where tissues were dehydrated and embedded in paraffin and cut into 5 μm sections. Immunohistochemical examinations were performed as stated earlier38 or according to the manufacturer’s protocol for CHEMICON IHC Select HRP/DAB Kit (EMD Millipore, MA)33. Briefly, after antigen recovery with citrate buffer, 3% H2O2 was used to quench endogenous peroxidase activity. The tissue was blocked by using 2.5% horse serum for 1 hour at RT followed by incubation with primary antibodies against SM22-α (1:1500), RUNX2 (1:300), OSP (1:100), CD63 (1:50) and AnX2 (1:50) overnight at 4 °C and subsequently with biotinylated secondary antibodies and a streptavidin peroxidase complex (PK-7800, Vector Laboratories, Burlingame, CA, USA). The tissue slides were treated in a sequential manner in conformity with manufacturer’s protocol. Finally, the tissues were counterstained with hematoxylin, dehydrated and mounted using DPX. The area percentage of the positive staining was measured using Image Pro Plus 6.0 software33,39.
Confocal Microscopy.
Co-localization of the MVB protein marker VPS16 against mTORC1 and lysosomal protein marker Lamp-1, indicating mTOR activation and MVB-lysosome interaction in SMCs was evaluated by indirect immunofluorescent staining. Primary antibodies VPS16 (1:300), mTORC1 (1:100) and Lamp-1 (1:200) were used to incubate acetone fixed frozen aortic tissue slides overnight at 4 °C. The aortic tissue slides were incubated with Alexa-488 or Alexa-555-labeled secondary antibodies for 1 hour at RT to perform the double immunofluorescent staining. Finally, slides were mounted by DAPI-mounting solution, and then analyzed by confocal laser scanning microscope (Fluoview FV1000, Olympus, Japan). As mentioned previously39,40 the Image Pro Plus 6.0 software was used for analysis of images (Media Cybernetics, Bethesda, MD), which measured and represented co-localization as the Pearson Correlation Coefficient (PCC)33.
Primary culture of mouse CASMCs and Alizarin Red S staining
Isolated mouse CASMCs as described previously41 were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco), supplemented with 10% FBS (Gibco) and 1% penicillin‐streptomycin (Gibco) in humidified 100% air and 5% CO2 mixture at 37°C. Confluent cells were treated with phosphate (Pi) (3 mmol/L)20, and osteogenic medium was changed every two days for 2 weeks to develop in vitro calcification model42. Briefly, CASMCs cells were incubated with 1 mL of 1% Alizarin Red S stain for 5 minutes, washed with diH2O and visualized using a phase microscope. CASMCs were treated with Torin-1 (a pharmacological inhibitor of mTORC1) (100 nm)43 and incubated for 24 hours. Besides, CASMCs were also treated with mouse specific Silencer® Select siRNA mTOR (Assay ID s80903, Thermoscientific, USA) and Silencer® Select Negative siRNA. Manufactures protocol was followed during siRNA transfection process [sc-29528 (siRNA Transfection Medium: sc-36868, siRNA Transfection Reagent: sc-29528)].
Western blot analysis
Briefly, equal amount of protein was resolved on 4–20% precast polyacrylamide gels (Bio-Rad, USA) and transferred to nitrocellulose using Trans-Blot turbo transfer system (Bio-Rad, USA). The membrane was blocked and incubated with primary antibodies rabbit polyclonal to p-mTOR (Ser2448) (1:1000 Cell Signaling, USA) and rabbit anti-vinculin (1:1000, Thermoscientific, USA) at 4 °C overnight followed by incubation with donkey anti-rabbit-HRP IgG (1:5000, Santa Cruz Biotechnology, Dallas, TX, USA) for 1 hour at room temperature. The chemiluminescence technique using LI-COR Odyssey Fc was then used for identification of bands and the band intensity of target proteins were normalized to vinculin and determined by Image J software version 1.44p (NIH, Bethesda, MD, USA)44.
Nanoparticle Tracking Analysis (NTA)
The exosome size and concentration in the cell culture media was evaluated by Nanoparticle Tracking Analysis (NTA) using the light scattering mode of the NanoSight LM10 (Nano Sight Ltd., Enigma Business Park, Amesbury, United Kingdom). The detached cells or debris were removed from cell culture supernatant by centrifugation at 300 g at 4 oC for 10 min. Further filtered through 0.22 μm filters to remove apoptotic bodies and microvesicles, and finally ultracentrifuged at 100,000 × g for 90 min at 4°C (Beckman 70.1 T1 ultracentrifuge rotator)45. In order to carry out the NTA, the exosome pellet was diluted in PBS. Each diluted sample was captured in 5 frames (30 s each) with background level 10, camera level 12, and shutter speed 30 respectively. The captured video was evaluated with NTA software (Version 3.2 Build 16). The PRISM software (GraphPad, San Diego, CA, USA) was used to plot an average particle size distribution graph9.
Non-Invasive, in Vivo Measurement of Aortic Pulse Wave Velocity (PWV)
Arterial stiffness was measured by Pulse Wave Velocity (PWV) using a high-resolution Doppler ultrasound instrument (Vevo2100, Visual-Sonics, Toronto, ON, Canada)46. Briefly, anesthetization of animals was managed by 2% isoflurane laid on a temperature-controlled platform where the body temperature of mice was maintained at 37 °C to eliminate the movement artifacts. Furthermore, parameters like electrocardiogram (ECG), heart rate (HR), and respiratory rate were observed. The isoflurane concentration was adjusted to 1.5% to maintain the heart rate (HR) of about 450 bpm. Flow wave Doppler measurement of aortic outflow was performed by a 6 mm, 30 MHz pulsed Doppler probe with a pulse repetition frequency of 40 kHz. Aortic outflow profiles were taken longitudinally at proximal and distal locations to the heart. Similar heart rate was observed in both proximal and distal point measurements. PWV was measured at both the proximal and distal locations from the peak of the ECG R wave to the foot of the flow waves for 5 replicates in each mouse. PWV (mm/ms) is based on TT(Dt) and the distance between the measurement sites (Dd), where TT (Transit time) is the difference between the time of arrival at proximal and distal points46.
Elastin Staining by Verhoeff’s Van Gieson stain
The elastic fibers in aortic tissue sections were stained using Verhoeff’s Van Gieson staining kit following manufacturer’s instructions (Abcam, Cambridge, MA, USA).
Statistical analysis.
All of the values are expressed as mean ± SEM. Significant differences among multiple groups were examined by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. P < 0.05 was considered statistically significant.
Results
mTOR signaling inhibition reduces lysosomal-CER related arterial stiffness in Asah1fl/fl/SMCre mice.
Arterial stiffness measured by non-invasive ultrasonography and assessed by in vivo aortic pulse-wave velocity (PWV), was significantly increased due to SMC specific Asah1 gene deletion in Asah1fl/fl/SMCre mice under control conditions compared with wild type littermates (WT/WT) as shown (Fig. 1A). The summarized data are depicted in the bar graph (Fig.1C). Besides, Vit D treatment significantly increased arterial stiffness in Asah1fl/fl/SMCre mice compared with littermates (WT/WT). Moreover, Torin-1 (a pharmacological inhibitor of mTORC1) treatment significantly decreased PWV in all the above-mentioned genotypes of mice during AMC under Vit D‐treated conditions. The arterial stiffness change was more noticeable in Asah1fl/fl/SMCre mice with or without progression of AMC. These results indicate that lysosomal-CER may contribute to arterial stiffness mediated via mTOR pathway during development and progression of AMC.
Figure 1.
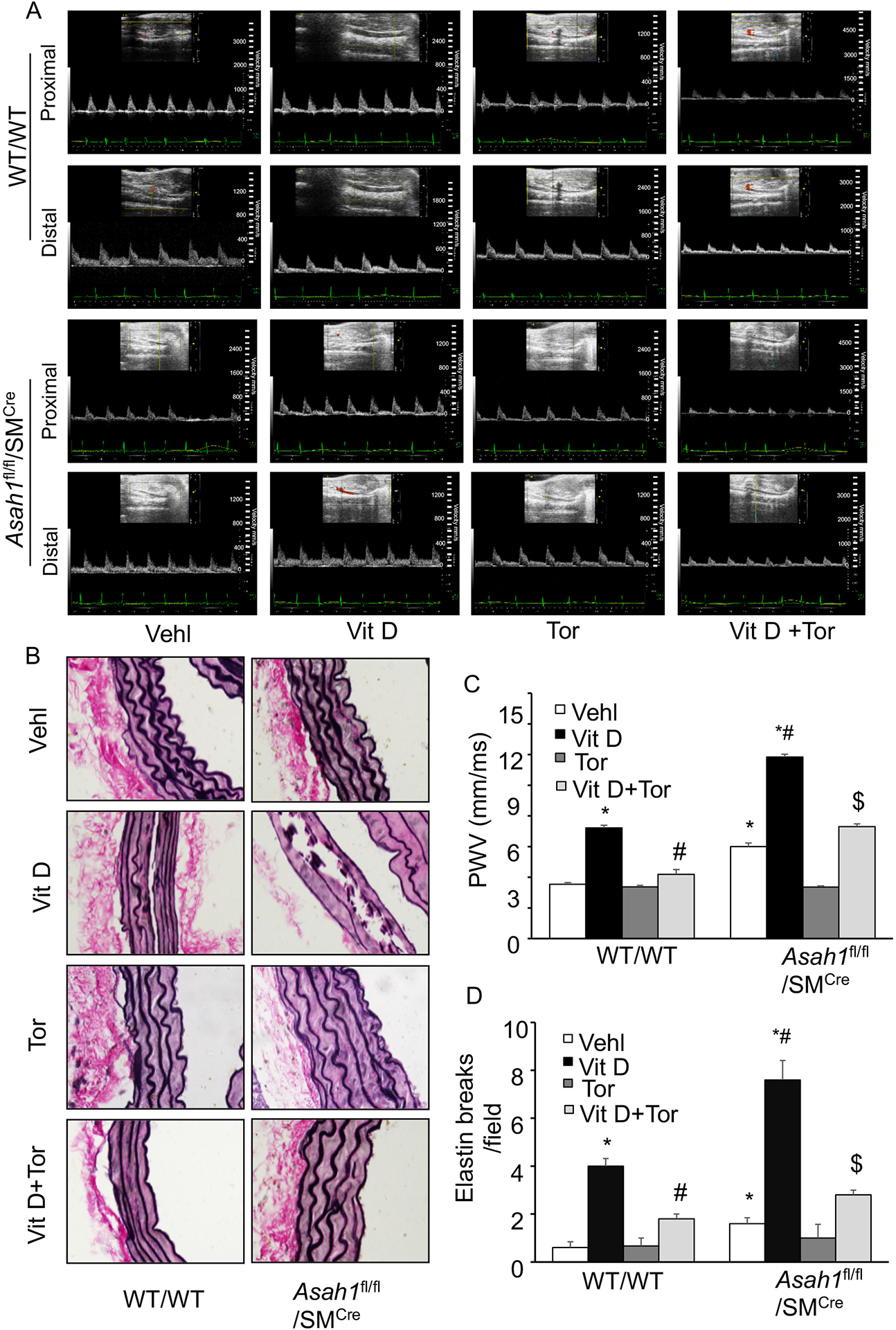
Effect of SMC‐specific Asah1 deletion on arterial stiffness in Vit D‐treated SMC specific Asah1 KO mice. (A) Representative Doppler images showed Doppler velocity signals. (B) Representative images of aortic sections showed damage to elastic fibers. (C, D) Summarized bar graph showing the effect of Torin-1 on the PWV and elastin breaks. n=5, SMC: smooth muscle cell; Vehl: vehicle; Vit D: vitamin D; Tor: Torin-1, ‘n’ is mouse number. *P < 0.05 vs WT/WT Vehl; #P < 0.05 vs WT/WT Vit D group; $P< 0.05 vs. Asah1fl/fl/SMCre Vit D group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown as mean ± SEM of values.
Further, we observed the effect of SMC specific Asah1 gene deletion on the structural changes of aortic media such as elastin degradation during arterial stiffness in Asah1fl/fl/SMCre mice. Elastin staining by Verhoff’s Van Geison stain was determined on histological aortic sections. Due to SMC specific Asah1 gene deletion, integrity of the elastic laminae in the aortic media seems to be compromised, and treatment with Vit D enhanced thinning and increased breaks of elastin as shown in Fig. 1B, distortion of junctions in between inner and outermost layers, and solid plates formation or sheaths disorganization. Fig. 1D shows the summarized data in the bar graph. These histological changes were easily found in the calcified aortas of Asah1fl/fl/SMCre mice as compared to WT/WT, and Torin-1 treatment efficiently averted these histopathological changes in the aortic media during AMC, as shown by significant decrease in PWV and elastic breaks, suggesting that mTOR signaling may be crucial pathway in lysosomal-CER related arterial stiffness.
mTOR signaling inhibition suppresses lysosomal-CER related AMC in Asah1fl/fl/SMCre mice.
Firstly, we observed p-mTOR expression in the aorta of WT/WT and Asah1fl/fl/SMCre mice, and found that p-mTOR expression was significantly increased in Vit D treated Asah1fl/fl/SMCre mice compared with littermates (WT/WT) as shown in Fig. 2A, 2B. Besides, we explored the role of mTOR signaling inhibition in lysosomal-CER related AMC. Consistent with our previous findings, (Fig. 2C, 2D) showed calcium/mineral deposition in the arterial media, as assessed by Alizarin Red S and Von Kossa staining was significantly increased in Vit D treated Asah1fl/fl/SMCre mice compared with littermates (WT/WT), whereas Torin-1 treatment significantly ameliorated the calcium/mineral deposition in these mice. Importantly, decrease in calcium/mineral deposition was observed in all the genotypes of mice, however much more prominent changes were observed in mice with SMC specific Asah1 gene deletion. The summarized data are shown in the bar graph (Fig. 2E, 2F).This AMC was accompanied with phenotypic switch of arterial medial SMCs, because OSP, RUNX2 immunostaining was markedly increased and SM22-α was decreased in the aortic medial wall of Asah1fl/fl/SMCre mice compared with wild type (Fig. 3A, 3B, 3C). However, Torin-1 treatment reversed these changes in the phenotype of aortic medial SMCs as shown by significantly decreased immunostaining of OSP, RUNX2 associated with increased SM22-α. The bar shows summarized data in (Fig. 3D, 3E, 3F). These results indicate that SMC specific Asah1 gene deletion which perturbates the sphingolipid rheostat in arterial medial SMC’s lead to phenotype change in SMCs mediated via mTOR activation during the development of AMC.
Figure 2.
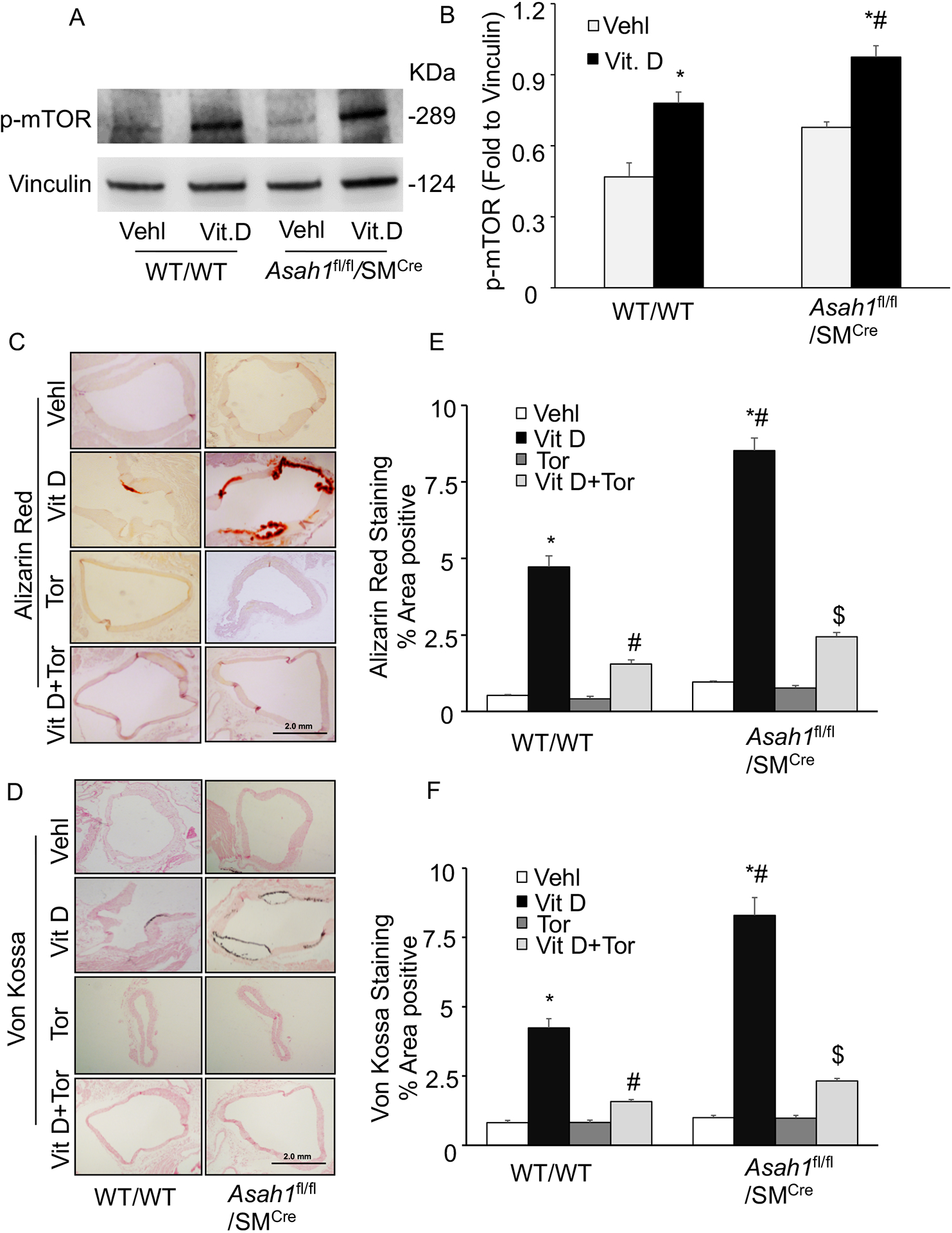
Effect of mTOR-inhibition on AMC in the Asah1fl/fl/SMCre mice. (A, B) Western blot analysis of enhanced expression of p-mTOR (Ser2448) in Vit D-treated Asah1fl/fl/SMCre mice. n=4. Representative photomicrographs showed (C) calcium deposition by Alizarin Red S staining (red color) and (D) mineral deposition by Von Kossa staining (black color). (E, F) Summarized bar graph showed Torin-1 significantly decreased calcium deposition and mineralization in aortic medial wall. n=5, SMC: smooth muscle cell; Vehl: vehicle; Vit D: vitamin D; Tor: Torin-1. ‘n’ is mouse number. *P < 0.05 vs WT/WT Vehl; #P < 0.05 vs WT/WT Vit D group; $P< 0.05 vs. Asah1fl/fl/SMCre Vit D group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown as mean ± SEM of values.
Figure 3.
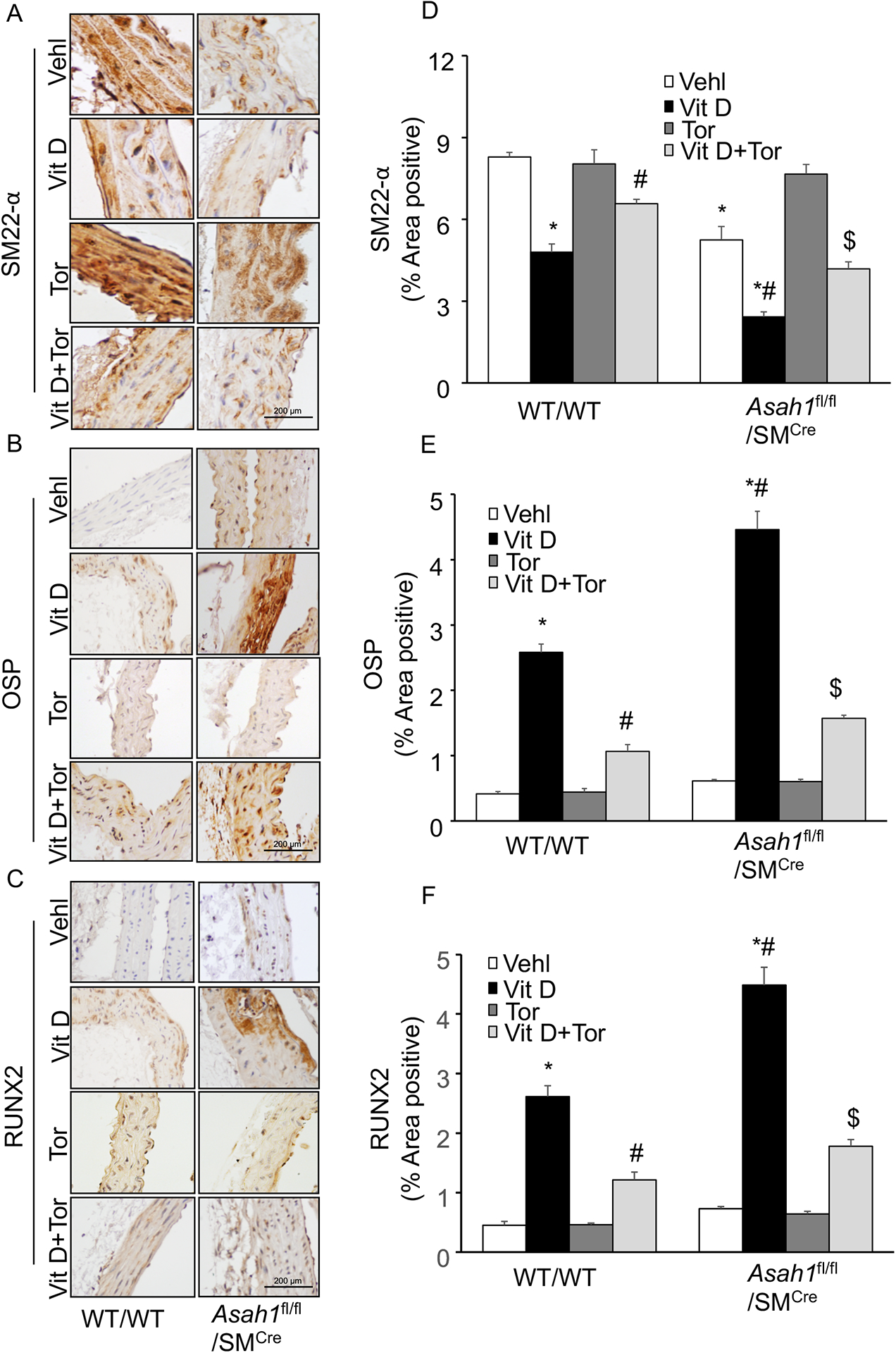
mTOR-inhibition prevented phenotype switch in the Asah1fl/fl/SMCre mice. Representative images from the aortic sections depicts changes in the expression of (A) SM22‐α (brown stain) (B) OSP and (C) RUNX2 in the aortic media of Asah1fl/fl/SMCre mice. Summarized bar graph showed Torin-1 significantly increased (D) SM22‐α and decreased (E) OSP (F) RUNX2 in aortic medial wall of mice receiving Vit D injections. n=5, SMC: smooth muscle cell; Vehl: vehicle; Vit D: vitamin D; Tor: Torin-1; OSP, osteopontin; RUNX2, runt‐related transcription factor 2, ‘n’ is mouse number. *P < 0.05 vs WT/WT Vehl; #P < 0.05 vs WT/WT Vit D group; $P< 0.05 vs. Asah1fl/fl/SMCre Vit D group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown as mean ± SEM of values.
mTOR activation reduced lysosome-MVBs interactions in lysosomal-CER related AMC in Asah1fl/fl/SMCre mice.
Next, we explored whether SMC specific Asah1 gene deletion in the aortic medial SMCs lead to mTOR activation indicated by co-localization of mTOR with lysosome as shown in Fig. 4A. Here, we observed significantly increased co-localization of mTORC1 (green) with Lamp‐1 (lysosome marker, red) in the aortic medial SMCs in Asah1fl/fl/SMCre mice (larger yellow spots) compared with littermates (WT/WT) (fewer yellow spots) under control conditions which was enhanced by Vit D treatment as summarized in the bar diagram (Fig. 4C). Furthermore, we found that mTOR activation in aortic medial SMCs is associated with markedly decreased co-localization of VPS16 (a multivesicular bodies (MVBs) marker, green) with Lamp‐1 (lysosome marker, red) in Asah1fl/fl/SMCre mice (fewer yellow spots)compared with wild type littermates (larger yellow spots) with or without Vit D treatment, indicating reduced MVBs interaction with lysosomes (Fig. 4B). However, Torin-1 treatment markedly decreased co-localization of mTORC1 (green) with Lamp‐1 (lysosome marker, red) associated with significantly increased co-localization of VPS16 (MVBs marker, green) with Lamp‐1 (lysosome marker, red) in aortic media with or without Vit D treatment in all these mice as depicted in the bar graph (Fig. 4D), suggesting that mTOR signaling pathway may be implicated in the lysosome‐MVBs interaction in lysosomal-CER related AMC.
Figure 4.
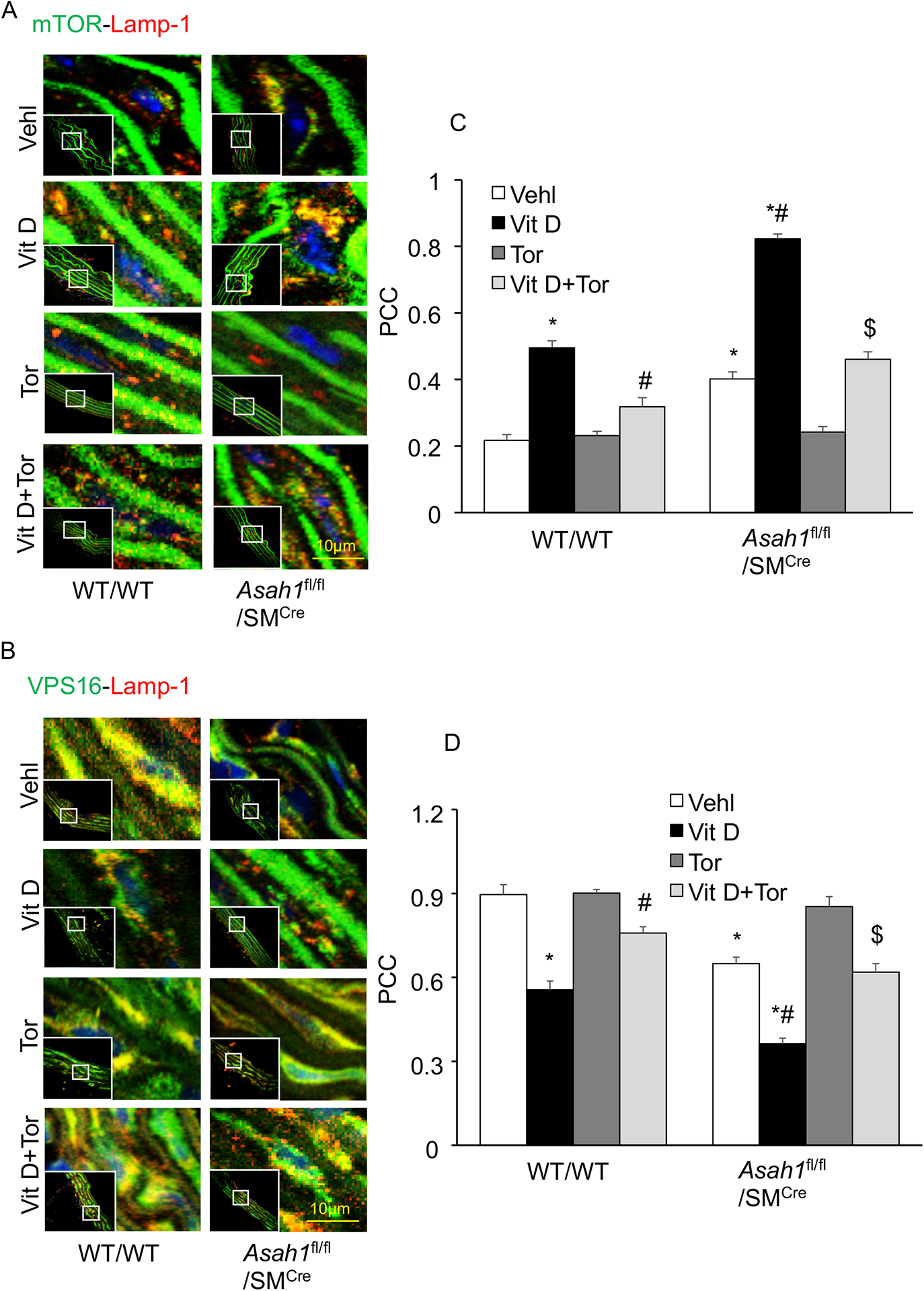
mTOR activation and lysosome-MVB interaction in the Asah1fl/fl/SMCre mice. Representative confocal images showed (A) co-localization of mTORC1 (green) and Lamp-1(red) in aortic medial wall (B) co-localization of VPS16 (green) and Lamp-1(red) and, (C) Bar graph shows Torin-1 significantly decreased the co-localization of mTORC1/Lamp-1 which was enhanced due SMC Asah1 gene deletion in Asah1fl/fl/SMCre mice. (D) Bar graph shows Torin-1 significantly increased the co-localization of VPS16/Lamp-1 which was reduced due SMC Asah1 gene deletion in Asah1fl/fl/SMCre mice. n=5–6, SMC: smooth muscle cell; Vehl: vehicle; Vit D: vitamin D; Tor: Torin-1, PCC: Pearson correlation coefficient, ‘n’ is mouse number. *P < 0.05 vs WT/WT Vehl; #P < 0.05 vs WT/WT Vit D group; $P< 0.05 vs. Asah1fl/fl/SMCre Vit D group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown as mean ± SEM of values.
mTOR signaling inhibition decreased exosome markers in lysosomal-CER related AMC in Asah1fl/fl/SMCre mice.
Literature cites that exosomes or microvesicles plays a crucial role in the tissue calcification process. Next, we sought to determine whether exosomes during calcification are ameliorated via the mTOR signaling pathway, using Torin-1 as mTOR inhibitor. In consistent to our previous results we found that SMC specific Asah1 gene deletion markedly increased the accumulation of exosome markers such as CD63 and annexin 2 (AnX2) in the coronary arterial wall of Asah1fl/fl/SMCre mice compared to their littermates (WT/WT) as shown in (Fig.5A, 5B). However, mTOR inhibition by Torin-1 significantly reduced accumulation of CD63 and AnX2 in the coronary arterial wall of these mice as depicted in the bar graph (Fig.5C, 5D). These results indicate that exosome increase in lysosomal-CER related AMC is mediated via the mTOR signaling pathway.
Figure 5.
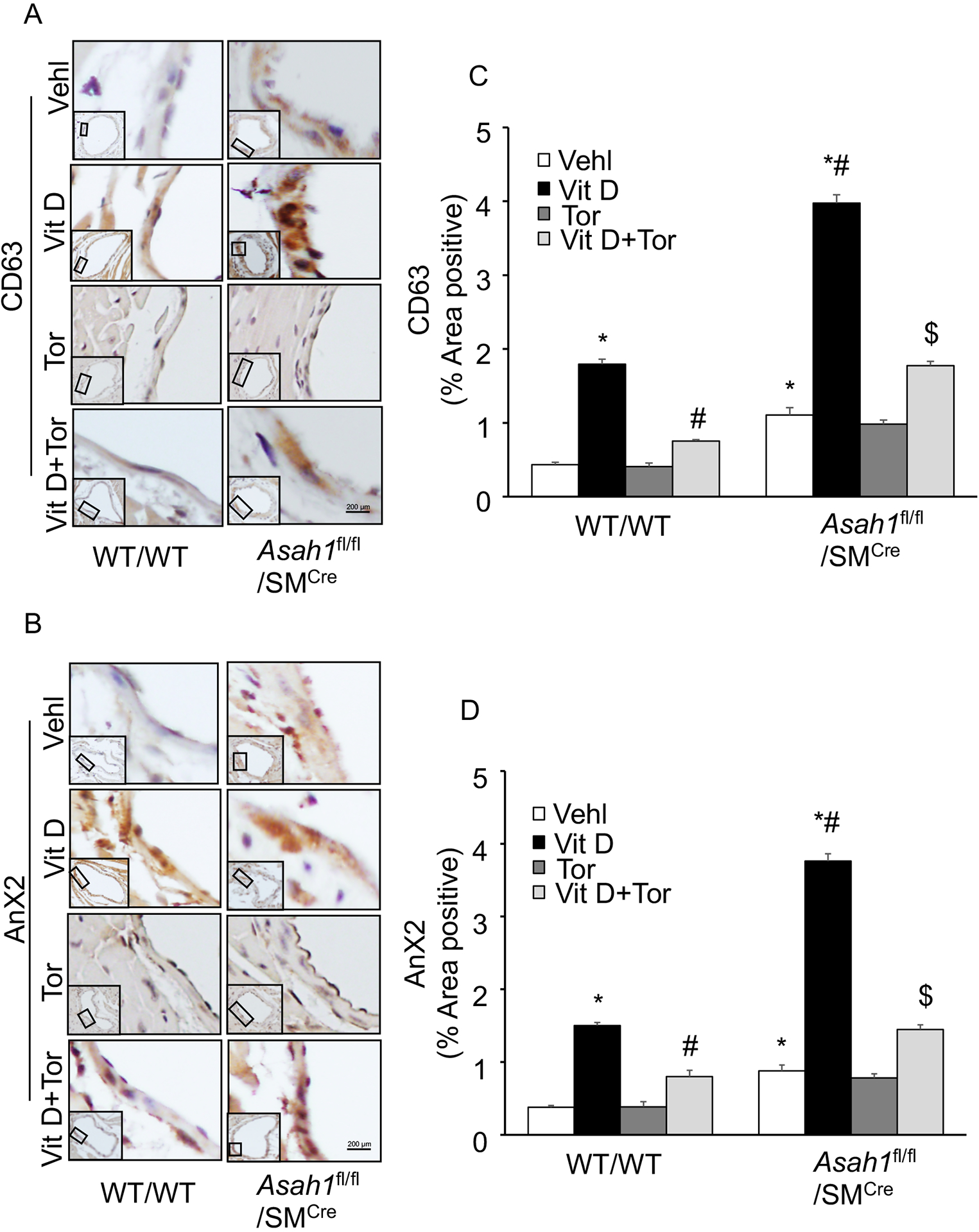
Effect of mTOR inhibition on exosome markers in the Asah1fl/fl/SMCre mice. Representative immunohistochemical images depicts that exosome makers (A) CD63 (brown stain) and (B) AnX2 staining in the coronary arterial wall. (C, D) The summarized bar graph showing area positive staining percentage demonstrates that Torin-1 significantly decreased expression of CD63 and AnX2 in the coronary arterial wall of mice receiving Vit D injection. n=5, SMC: smooth muscle cell; Vehl: vehicle; Vit D: vitamin D; Tor: Torin-1, ‘n’ is mouse number. *P < 0.05 vs WT/WT Vehl; #P < 0.05 vs WT/WT Vit D group; $P< 0.05 vs. Asah1fl/fl/SMCre Vit D group by one-way one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown as mean ± SEM of values.
mTOR activation mediates Pi-induced lysosomal-CER related calcification in CASMCs in vitro.
To test whether lysosomal-CER related calcification is mediated via mTOR signaling in Pi treated CASMCs in vitro. Firstly, we demonstrated that p-mTOR (Ser2448), a 289 kD cytoplasm protein is enriched in CASMCs by western blot analysis, and treatment of CASMCs isolated from Asah1fl/fl/SMCre mice with high Pi significantly increased p-mTOR (Ser2448) expression as compared to WT/WT CASMCs (Fig 6A,6B).
Figure 6.
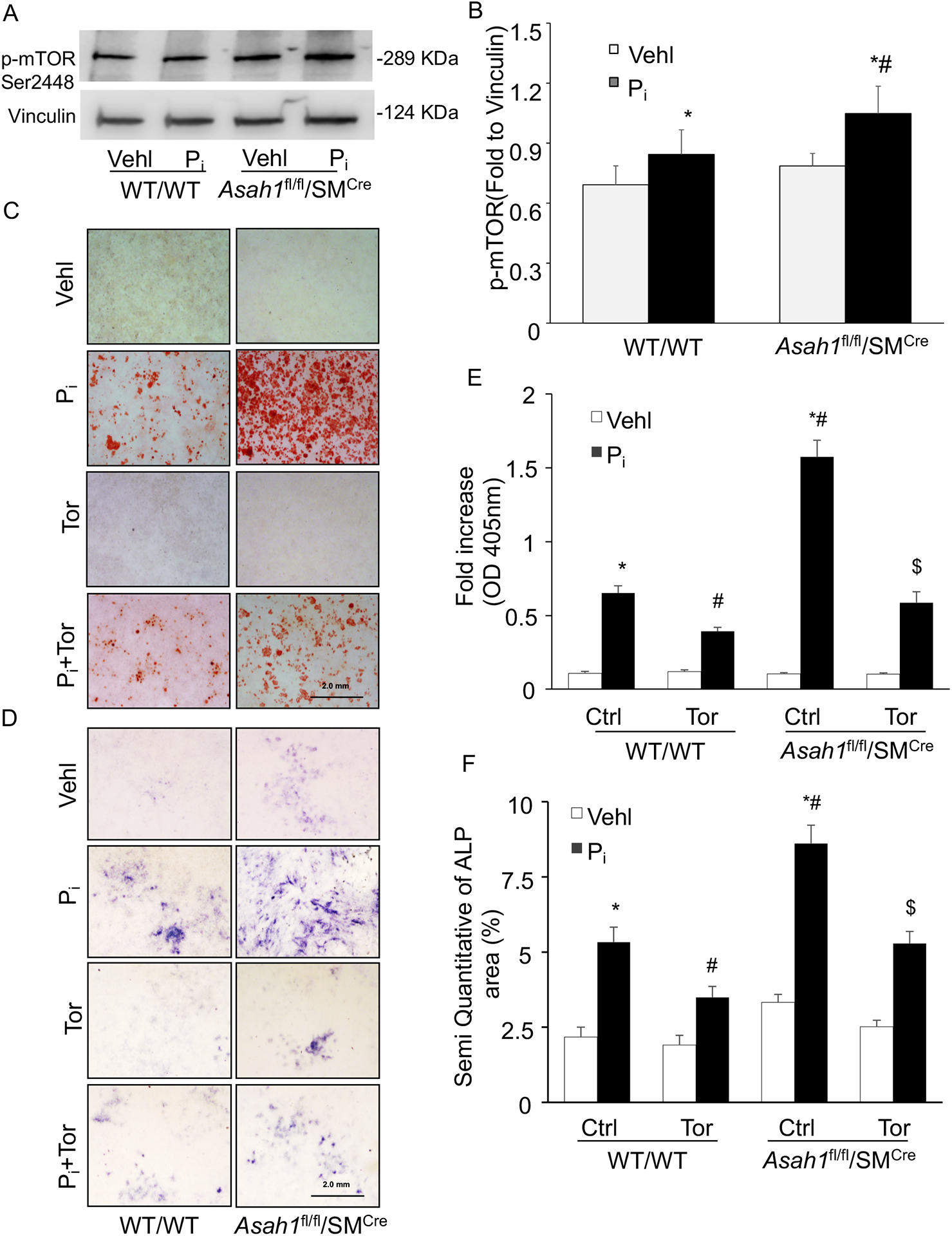
mTOR activation and calcification in the Pi-treated Asah1fl/fl/SMCre CASMCs. (A, B) Western blot analysis of enhanced expression of p-mTOR (Ser2448) in Pi-treated Asah1fl/fl/SMCre CASMCs. (C) Representative images showed calcium deposition by Alizarin Red S staining (red color) and (D) mineral deposition by alkaline phosphatase (blue color) staining. (E, F) Summarized bar graph showed Torin-1 significantly decreased Pi-induced calcium deposition and mineralization in Asah1fl/fl/SMCre CASMCs as compared to WT/WT cells. n=5, Pi: Phosphate; Tor: Torin-1, ‘n’ is experimental repeats. *P< 0.05 vs. WT/WT Vehl; #P< 0.05 vs. WT/WT Pi; $ P< 0.05 vs. Asah1fl/fl/SMCre Pi group by one-way ANOVA followed by Holm-Sidak’s test. Data are shown mean ± SEM of values.
Moreover, we observed that Torin-1 treatment significantly reduced Pi-induced calcium and mineral deposition, as determined by alizarin red and alkaline phosphatase (ALP, blue color) both in Asah1fl/fl/SMCre and WT/WT CASMCs as shown in Fig. 6C and 6D. The summarized data in the bar graph depicts that changes were more prominent with Ac gene deletion in Pi-treated Asah1fl/fl/SMCre CASMCs as compared to WT/WT cells (Fig. 6E, 6F). These data confirm that lysosomal-CER-mTOR signaling via contributes to the development of VSMC calcification.
Regulation of lysosome-MVBs interaction by lysosomal-CER-mTOR signaling pathway in Pi-induced CASMCs in vitro.
Using confocal microscopy, we observed marked increase in the co-localization of mTORC1 (green) with Lamp‐1 (lysosome marker, red), a lysosome targeting of mTORC1, in the CASMCs isolated from Asah1fl/fl/SMCre mice (larger yellow spots) compared with WT/WT (fewer yellows dots) under vehicle conditions and was enhanced during Pi -stimulation in the Asah1fl/fl/SMCre CASMCs as shown in Fig. 7 and Fig. 8. However, siRNA against mTOR and Torin-1 significantly decreased co-localization of mTORC1 (green) with Lamp‐1 (lysosome marker, red) both in Asah1fl/fl/SMCre and WT/WT CASMCs as shown in Fig. 7A, 8A. The summarized data is shown in the bar graph (Fig. 7C, 8C). Reduced lysosome-MVB interaction as indicated by markedly decreased co-localization of VPS16 (MVB marker, green) with Lamp‐1 (lysosome marker, red), was observed in Asah1fl/fl/SMCre CASMCs (fewer yellows dots) as compared to WT/WT cells (larger yellow spots) with or without Pi treatment (Fig. 7B and 8B), which was reversed by siRNA mTOR and Torin-1 treatment as shown in the bar graph (Fig. 7D and 8D). These results suggest that lysosomal-CER-mTOR signaling pathway may contribute to lysosome-MVBs interaction in CASMCs during calcification.
Figure 7.
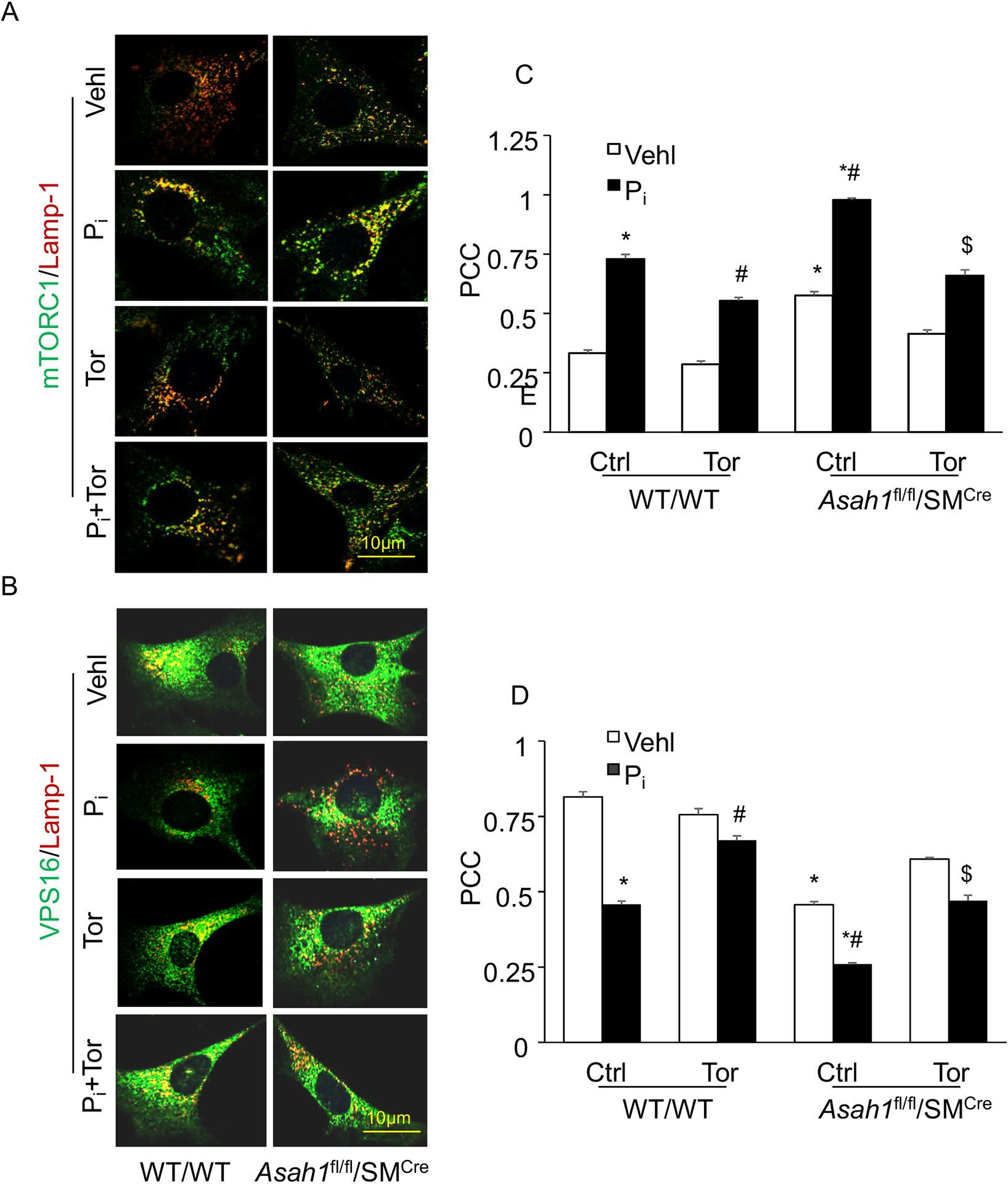
Pharmacological inhibition of mTOR signaling abrogates lysosome-MVBs interaction in Pi-treated Asah1fl/fl/SMCre CASMCs. Representative confocal images showed (A) co-localization of mTORC1 (green) and Lamp-1(red), indicative of mTOR activation. (B) co-localization of VPS16 (green) and Lamp-1(red), indicative of lysosome-MVBs interaction (C, D) Bar graph shows significant increase in the co-localization of mTORC1/Lamp-1 associated with decreased co-localization of VPS16/Lamp-1 in Pi-treated Asah1fl/fl/SMCre CASMCs as compared WT/WT cells (fewer yellows dots) whereas Torin-1 reversed this trend in these cells. n=5, Pi: Phosphate; Tor: Torin-1, ‘n’ is experimental repeats. *P< 0.05 vs. WT/WT Vehl; #P< 0.05 vs. WT/WT Pi; $P< 0.05 vs. Asah1fl/fl/SMCre Pi group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown mean ± SEM of values.
Figure 8.
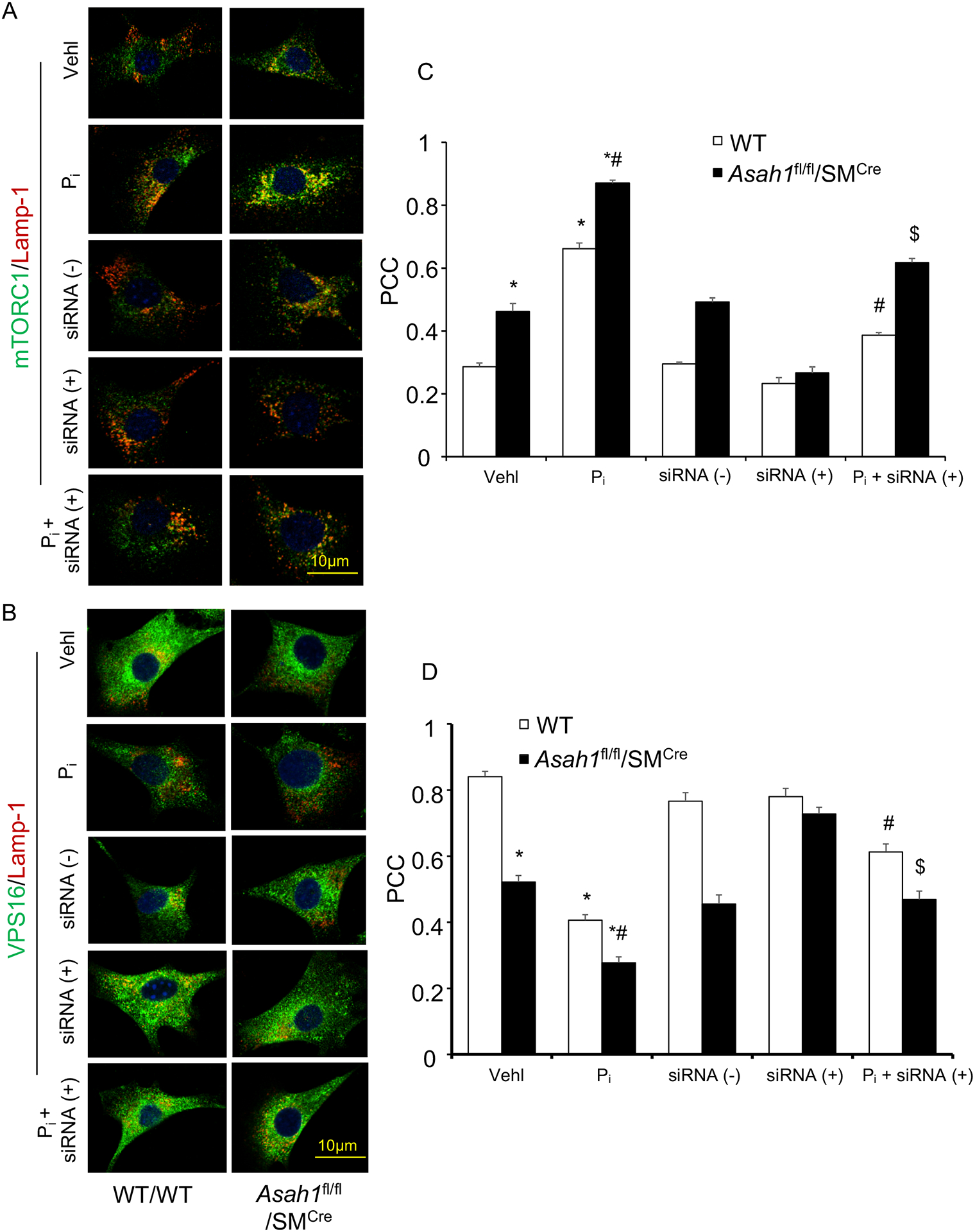
siRNA mTOR inhibition abrogates lysosome-MVBs interaction in Pi-treated Asah1fl/fl/SMCre CASMCs. Representative confocal images showed (A) co-localization of mTORC1 (green) and Lamp-1(red), indicative of mTOR activation. (B) co-localization of VPS16 (green) and Lamp-1(red), indicative of lysosome-MVBs interaction (C, D) Bar graph shows significant increase in the co-localization of mTORC1/Lamp-1 associated with decreased co-localization of VPS16/Lamp-1 in Pi-treated Asah1fl/fl/SMCre CASMCs as compared WT/WT cells (fewer yellows dots) whereas siRNA mTOR reversed this trend in these cells. n=3–4, Pi: Phosphate; PCC: Pearson correlation coefficient, ‘n’ is experimental repeats. * P < 0.05 vs. WT/WT Vehl; #P < 0.05 vs. WT/WT Pi group; $P < 0.05 vs. Asah1fl/fl/SMCre Pi group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown mean ± SEM of values.
Role of lysosomal-CER- mTOR signaling in the exosome secretion in Pi-induced CASMCs in vitro.
Using a nanoparticle tracking analysis system, we found that with or without Pi -stimulation, excretion of exosomes (<200 nm) were found to be significantly increased in CASMCs of Asah1fl/fl/SMCre CASMCs compared to WT/WT, which was enhanced by Pi stimulation, as depicted by representative 3-D histograms in Fig. 9A and 9B. However, siRNA mTOR and Torin-1 significantly decreased the excretion of exosomes with or without Pi - stimulation in Asah1fl/fl/SMcre CASMCs. The summarized data in the bar graph depicted vesicle counts of <200 nm which shows a significant decrease upon prior treatment of siRNA mTOR and Torin-1 in Pi -stimulated Asah1fl/fl/SMCre and WT/WT CASMCs (Fig. 9C and 9D). These results suggest that lysosomal CER -mTOR signaling regulates exosome release from CASMCs which may trigger calcification nidus formation.
Figure 9.
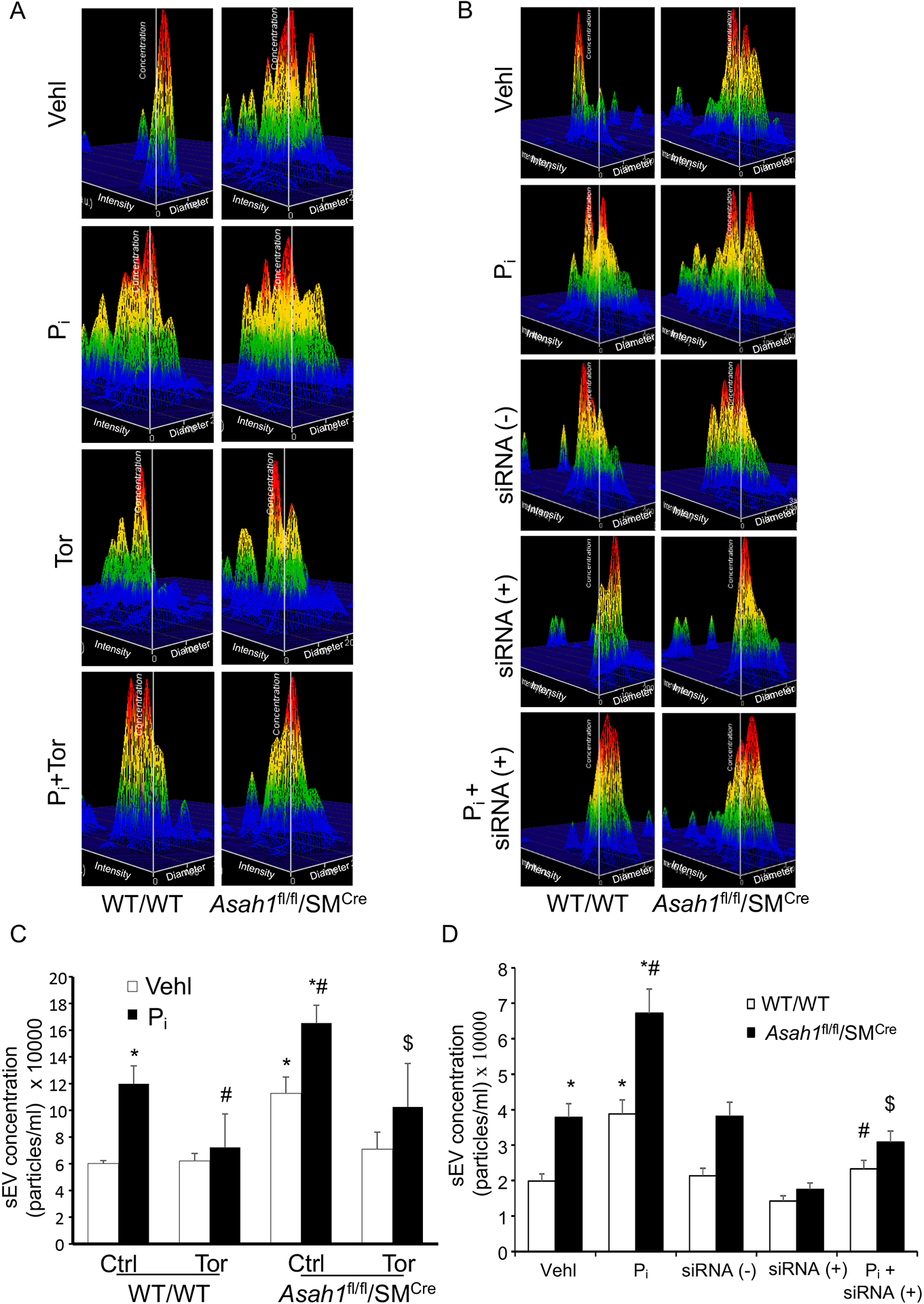
siRNA and pharmacological mTOR inhibition reduces exosome secretion in Pi-treated Asah1fl/fl/SMCre CASMCs. (A, B) Representative 3-D histograms of secretion of exosomes (<200 nm) from CASMCs. (C, D) Bar graph show increased exosome release from Asah1fl/fl/SMcre CASMCs with or without Pi treatment as compared to WT/WT cells which was significantly decreased by siRNA mTOR and Torin-1. n=5–6, Pi: Phosphate; Tor: Torin-1, ‘n’ is experimental repeats. *P< 0.05 vs. WT/WT Vehl; #P< 0.05 vs. WT/WT Pi; $P< 0.05 vs. Asah1fl/fl/SMCre Pi group by one-way ANOVA with Holm-Sidak’s test post-hoc analysis. Data are shown mean ± SEM of values.
Discussion
The present study first time revealed that lysosomal-CER‐mTOR signaling is critical for the control of lysosome fusion to MVBs, exosome secretion, osteogenic shift and arterial stiffening in AMC. Asah1 gene deletion that encodes for a lysosomal Ac to increase ceramide demonstrated that lysosomal-CER‐mTOR signaling may be one of the mechanistic pathways contributing to the development and progression of AMC. Non‐invasive imaging technique showed that PWV, which is correlated to arterial stiffness is significantly increased upon SMC specific Asah1 gene deletion in Asah1fl/fl/SMCre mice as compared to their littermates which was further enhanced during hypercalcemia induced by high doses of Vit D. Histopathological analysis revealed that damage to elastin fibers in the aortic media of Asah1fl/fl/SMCre mice, associated with distorted junctions and formation of solid plates or sheaths, resulting in damaged arterial architecture. However, Torin-1, a pharmacological inhibitor of mTOR efficiently averted these histopathological changes in the aortic media during AMC, as shown by significant decrease in PWV and elastic breaks. Moreover, pretreatment of these mice with Torin‐1 significantly decreased the Vit D-induced AMC and expression of osteogenic markers like osteopontin and RUNX2 associated with increased levels of SM22-α. Furthermore, we observed a significant increase in the co-localization of mTORC1 with Lamp‐1(lysosome marker) in Asah1fl/fl/SMCre mice suggesting mTOR activation which was accompanied with decreased co-localization of VPS16 (MVBs marker) with Lamp‐1 indicating reduced MVBs interaction with lysosomes in these mice. However, mTOR inhibition by Torin‐1 showed marked decrease in the co-localization of mTOR vs Lamp‐1 associated with increased VPS16 vs Lamp‐1 interaction. Torin‐1 was also found to reduce accumulation of CD63 and annexin 2 (AnX2) (exosome markers) in the coronary arterial wall of Asah1fl/fl/SMCre mice compared to wild type littermates. In CASMCs under in vitro conditions, we validated our in vivo findings. Firstly, we observed that Pi-stimulation significantly increased expression of p-mTOR in WT CASMCs which was further enhanced due to Ac gene deletion in Pi-treated Asah1fl/fl/SMCre CASMC, indicating that CER contributes to activation of mTOR. Torin-1, an mTOR inhibitor markedly decreased the Pi-induced calcium and mineral deposition as shown by the presence of decreased Alizarin Red-stained nodules and ALP staining both in Asah1fl/fl/SMCre cells and WT/WT cells. In addition, we found that siRNA mTOR and Torin-1 significantly decreased co-localization of mTORC1 with Lamp-1, but increased co-localization of VPS16 with Lamp-1 in these cells, signifying that activation of mTOR signaling inhibits lysosome fusion with MVBs. We further found that Ac gene deletion led to increased Pi-induced excretion of exosomes (<200), which was blocked by siRNA mTOR and Torin‐1 suggesting the role of lysosomal-CER-mTOR signaling in lysosome interactions with MVBs and exosome secretion. This lysosomal-CER-mTOR signaling in lysosome-MVB interactions, exosome secretion and arterial stiffening may represent a novel molecular mechanism involved in the development of AMC.
Earlier we found that increased lysosomal CER due to Ac gene deletion in SMCs markedly increased calcification in the arterial media of mice as well as isolated CASMCs from these mice. Here in the present study we postulate that mTOR signaling may be a mechanistic pathway through which lysosomal-sphingolipid pathway contribute to the AMC. However, the proper molecular mechanisms involved in AMC are still unknown. An important, change seen during the development and progression of AMC is stiffening of the large elastic arteries. Studies have shown increase in large elastic artery stiffness contributes to cardio vascular disease (CVD) risk and is associated with pathophysiological conditions like hypertension, left ventricular hypertrophy, stroke, sub-endocardial ischemia and cardiac fibrosis47. Literature cites that arterial stiffness increase during AMC which is hallmark of aging process48,49. Sphingolipid such as ceramide regulate actin cytoskeleton dynamics in VSMCs which is crucial for mechanosensing in these cells during aging process50,51. In the current study, we determined whether lysosomal-CER-mTOR signaling contributes to arterial stiffness. We found that Ac gene deletion in arterial SMCs increased PWV, measure of arterial stiffness52 which was further increased due to Vit D treatment, and blocking mTOR signaling using Torin‐1, a pharmacological inhibitor of mTORC1, significantly decreased PWV in these mice model of AMC. In the mice and rabbits fed a western diet blockade of glycosphingolipid synthesis can lower arterial stiffness during the development of atherosclerosis53. Recently, our laboratory demonstrated that SMC specific overexpression of Smpd1 gene that encodes for acid sphingomyelinase to increase CER in arterial media of Smpd1trg/SMCre mice observed significantly increased arterial stiffness in these mice indicating lysosomal-CER contributes to arterial stiffness20,21. In human ELN in a bacterial artificial chromosome (hBAC) hBAC-mNULL mice, mTOR inhibition by Rapalog reduces the aberrant mechanosignaling and aortic stiffening54. In another study, increased mTOR activation was observed in the large arteries of the old mice which was associated with arterial stiffness in these arteries55. Altogether, all these findings support our data which shows that sphingolipid-mTOR pathway contribute to arterial stiffness.
Literature reports that AMC is an actively regulated process controlled by osteogenic differentiation of SMCs and matrix mineralization56,57. Recent reports from our laboratory described that lysosomal-sphingolipids may be important mediators of VC20,21. But the exact molecular mechanism how lysosomal sphingolipids contribute to AMC is still poorly understood. A study revealed that the Lysosome Associated Protein Transmembrane 4B (LAPTM4B) interacts with CER, and is also involved in the 4F2 cell-surface antigen heavy chain (4F2hc)-mediated regulation of mTORC signaling58. The findings of this study showed decreased phosphorylation of the mTORC1 substrate S6K in LAPTM4B knockout cells indicating that LAPTM4B promotes mTORC activation. Further, the study also reported that Asah1 gene deletion which can increase CER levels increased levels of phospho-S6K indicating mTORC activation, however silencing of Smpd1 gene which will block CER formation from sphingomyelin decreased the levels of phospho-S6K, suggesting that lysosomal-CER is associated with mTOR activation58. In this context, we observed Asah1 gene deletion in the arterial SMCs led to mTOR activation and increased AMC both in vitro and in vivo while as blocking mTOR signaling by Torin-1 reduced AMC under these conditions. Under in vitro and in vivo conditions, inhibition of the mTOR activation signaling pathway by miR-30b was shown to markedly attenuated the VC in hyperphosphatemia of chronic kidney disease (CKD)29. In rat cultured VSMCs high Pi induced expression of Akt /mTOR pathway result in VSMC calcification mediated via Cbfα1 actiavtion30.VSMCS treated with exosomes isolated from high glucose-induced human umbilical vein endothelial cell promoted calcification involving Notch3 protein mediated through mTOR signaling pathway31. In Human aortic vascular smooth muscle cells (HAVSMCs), bone marrow mesenchymal stem cell (BMSC) derived exosomes inhibited high phosphorus-induced calcification via modifying miRNA profiles. The study further revealed that inhibition of calcification by BMSC derived exosomes involves mTOR, MAPK, and Wnt signaling pathway using functional miRNA-gene regulatory network59. Also, rapamycin, a mTOR inhibitor was reported to decrease VC by inhibiting mTOR pathway both in vitro and in vivo24. In patients with end-stage renal disease, it was found that inflammation induced-osteogenic differentiation in vascular tissues contributed to VC and also enhanced expression of bone formation biomarkers, which was regulated by the activation of the mTORC1 pathway. These findings support our hypothesis and suggest that lysosomal-CER pathway may contribute to mTOR activation which may be involved in calcification process during the development of AMC.
Arterial SMCs undergo phenotypic changes which simulates osteogenesis5,60. During the AMC, medial VSMCs displayed osteogenic phenotype which mainly contributes to VC61–63. In human VSMCs, C2-ceramide treatment result in Ox-LDL-induced VC through TLR4/NF-κB/CER signaling18. In human femoral arterial SMCs, increased N-SMase activity and CER levels increased Ox‐LDL‐induced matrix mineralization in these cultured SMCs17. Recently, our group reported that SMC specific lysosomal Asah1 or Smpd1 gene expression had remarkable effects on SMC marker (SM22-α) and phenotypic switch markers such OSP and RUNX2 expression suggesting that lysosomal sphingolipid‐CER pathway contribute to the phenotype change in arterial medial SMCs undergoing AMC20,21. PDMP, a ceramide analogue, via inhibition of mTORC1 activity reduced osteoblastic proliferation and pre-osteoblastic cell differentiation by translocation of mTORC1 from late endosome/ Lysosome to the endoplasmic reticulum64. In the present study, we found that pretreatment of SMC specific Asah1 KO mice with Torin-1, a pharmacological mTOR inhibitor, markedly decreased the expression of osteogenic markers like osteopontin and RUNX2 associated with increased levels of contractile marker SM22-α in Vit D-induced mice model. During aging process, VSMCs phenotypic transformation is closely linked with increased arterial stiffening and blood pressure65. The mTOR signaling pathway plays a crucial role in VSMC proliferation, hypertension and atherosclerosis66,67 and hence, has been identified as a therapeutic target67,68. Furthermore, several studies have shown that the mTOR signalling pathway is involved in VSMC dedifferentiation69–71. However, the regulation of the mTOR/p70S6K signalling pathway and its effects on the phenotypic switching of VSMCs in arterial medial calcification is still poorly understood. In VSMCs, homocysteine-induced proliferation, migration, and phenotypic transformation was reduced by overexpression of miR-145 mediated via decreased PI3K, Akt, and mTOR expression72. Recently, a study showed that rapamycin treatment markedly declined the kaempferol-induced osteogenetic activity associated with decreased RUNX2 and osterix expression involving mTOR, suggesting contribution of mTOR-RUNX2/osterix signaling in osteogenic effect under in vitro and in vivo73. In another study, sirolimus (also known as rapamycin) diminished advanced glycation end product (AGE)-induced phenotypic transition of VSMCs indicated by increased expression OSP, ALP and decreased expression of α-SMA via inhibition of the ILK/mTOR/p70S6K signaling pathway74. VSMC calcification has been observed to be a manifestation of cellular senescence, and replicative senescence of these cells enhances age-associated medial artery calcification75,76. Several studies reported that replicative senescence of VSMCs was significantly inhibited via inhibition of mTOR-signaling pathway77,78. In in CaCl2‐induced aortic aneurysm rat model, pretreatment with rapamycin decreased the OSP expression in aortic SMCs indicating phenotypic switch of aortic SMCs to contractile one via mTOR activation suggesting a crucial role of mTOR-signaling pathway in phenotypic transition of SMCs79. Taken together, these data support our hypothesis that the mTOR-signaling is associated with the osteogenic effect of lysosomal-sphingolipids in SMC specific Asah1 gene KO mice.
Imbalanced mineral metabolism in SMCs enhanced secretion of calcifying exosomes via sphingolipid/CER pathway causing arterial calcification9,80. These SMC‐derived vesicles were having increased levels of exosomes markers such as CD63, AnX2 and ALP80. Several studies demonstrated that both intimal and medial calcifications is accompanied with increased exosomes (size of 40–100 or to 140 nm in size) in the vascular interstitial space2,9,12. Evidences showed that augmented exosome secretion may elicit SMC phenotype changes3,5 contributing to extracellular matrix (ECM) mineralization9,11. It is already reported that sphingolipid pathway plays a curial role in the EV production in different cells both in vitro and in vivo81,82. In our previous study, we demonstrated that deletion of lysosomal Asah1 gene specifically in SMCs caused reduction of lysosome-MVB interaction and enhanced SMCs-derived exosome release, SMC phenotypic transition during AMC20. PDMP, a ceramide analogue, allows normal lysosomal functioning or activation, and their fusion with MVBs via inhibition of mTORC1 activity64. In the present study, we investigated whether inhibition of mTOR-signaling pathway block lysosomal-CER mediated exosome secretion which act as a triggering mechanism in the AMC. Firstly, we observed enrichment of activated p-mTOR in Pi-stimulated CASMCs which was increased due to Ac gene deletion in these cells.
We also observed markedly increased co-localization of mTORC1 with lysosome marker (Lamp-1) in Asah1 gene KO CASMCs as compared WT/WT under vehicle conditions indicating mTOR activation which was associated with decreased co-localization of MVB marker (VPS16) with lysosome marker (Lamp-1) suggesting less lysosomal fusion with MVBs. However, mTOR inhibition at genetic and pharmacological level significantly decreased co-localization of mTORC1 with lysosome marker (Lamp-1), but increased co-localization of MVB marker (VPS16) with lysosome marker (Lamp-1) both in WT/WT and Asah1fl/fl/SMCre CASMCs signifying that inhibition of mTOR signaling promotes lysosome fusion with MVBs. Furthermore, Nanoparticle tracking analysis demonstrated that mTOR inhibition in these CASMCs under osteogenic conditions decreased the excretion of exosome (<200 nm in size), suggesting the role of lysosomal-CER-mTOR signaling in lysosome interactions with MVBs/ exosome secretion. mTOR inhibition was also found to reduce the accumulation of CD63 and AnX2 (exosome markers) in the coronary arterial wall of Vit D-treated mice. High Pi-induced calcification in HAVSMCs was alleviated with treatment of BMSC-derived exosomes (CD63 and CD81 as exosome markers) through modifying miRNA profiles involving mTOR, MAPK, and Wnt signaling pathway59. Cardiac progenitor cell-derived exosomes (CD63 and CD81 as exosome markers) promoted H9C2 cell growth via activation of Akt/mTOR pathway in a time-dependent manner suggesting that PI3K/AKT/mTOR pathway may have important regulatory function in cardioprotection83. In cervical intraepithelial neoplasia patients, a non-significant difference of PI3k/Akt/mTOR gene expression was observed in cervical cancer tissue and the exosomes extracted from vaginal secretion but were significantly increased corresponding adjacent control tissues suggesting that PI3k/Akt/mTOR signaling pathway mediated by exosomes may provide candidate diagnostic biomarkers or potential therapeutic targets84. Fibroblast-derived exosomes mediate mobilization of autocrine Wnt10b-mTOR pathway to promote axonal regeneration, a crucial step towards healing the injured central nervous system85. Together, all these studies conclude that lysosomal-CER in association with mTOR activation enhance exosome release during calcification process as shown in Fig. 10. In this context, our findings indicate that lysosomal Ac associated CER-mTOR signaling may regulate lysosome fusion with MVBs, exosome secretion and arterial stiffening which may represent a novel molecular mechanism involved in the development of AMC.
Figure 10.
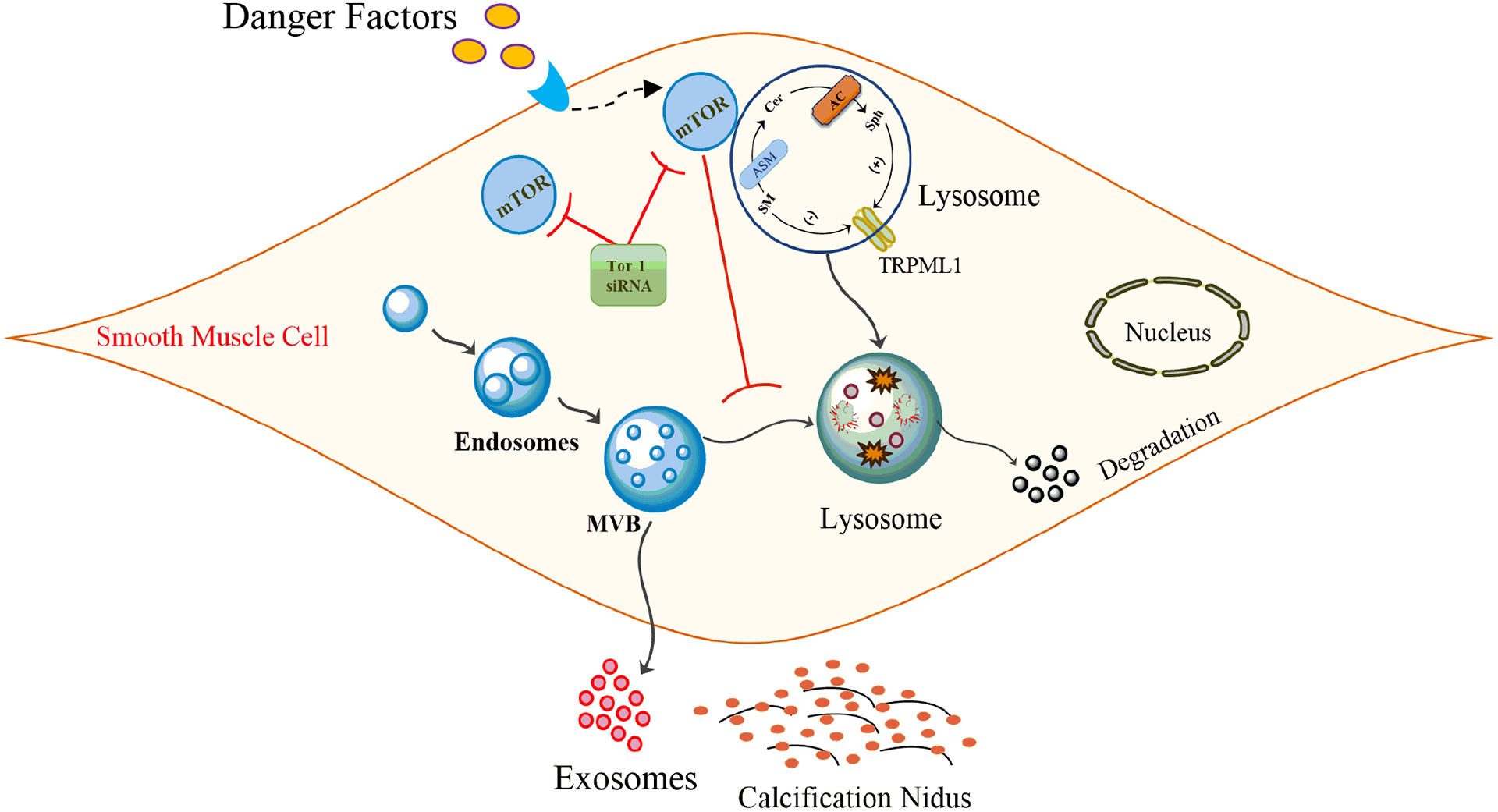
Schematic model indicates that lysosomal Ac associated CER-mTOR signaling may regulate lysosome fusion with MVBs and exosome secretion which may represent a novel molecular mechanism involved in the development of AMC. Ac: Acid ceramidase; SMCs: Smooth muscle cells; AMC: Arterial medial calcification.
Acknowledgment
This study was supported by grants from the National Institutes of Health (HL122937, HL057244 and HL075316).
Nonstandard Abbreviations:
- AC
Acid Ceramidase
- AMC
Arterial Medial Calcification
- AnX2
Annexin-II
- ASM
Acid Sphingomyelinase
- CASMCs
Coronary Artery Smooth Muscle Cells
- CER
Ceramide
- ILV
Intraluminal Vesicles
- Lamp-1
Lysosome Associated Membrane Protein-1
- mTORC1
Mammalian Target of Rapamycin Complex 1
- MVBs
Multivesicular Bodies
- OSP
Osteopontin
- RUNX2
Runt-related transcription factor 2
- SM22-α
Smooth Muscle 22α
- SMC
Smooth Muscle Cells
- VC
Vascular Calcification
- Vit D
Vitamin D
- VPS16
Vacuolar Protein Sorting-associated Protein 16
- VSMCs
Vascular Smooth Muscle Cells
Footnotes
Conflict of Interest Statement
The authors declare no competing financial and non-financial interests.
Data Availability Statement
All data generated or analyzed during this study are included in this article.
References
- 1.Shanahan CM, Crouthamel MH, Kapustin A & Giachelli CM Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res 109, 697–711, doi: 10.1161/CIRCRESAHA.110.234914 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kapustin AN & Shanahan CM Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J Physiol 594, 2905–2914, doi: 10.1113/JP271340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin ME, Chen T, Leaf EM, Speer MY & Giachelli CM Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am J Pathol 185, 1958–1969, doi: 10.1016/j.ajpath.2015.03.020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith ER Vascular Calcification in Uremia: New-Age Concepts about an Old-Age Problem. Methods Mol Biol 1397, 175–208, doi: 10.1007/978-1-4939-3353-2_13 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Steitz SA et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 89, 1147–1154, doi: 10.1161/hh2401.101070 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Kapustin AN & Shanahan CM Calcium regulation of vascular smooth muscle cell-derived matrix vesicles. Trends Cardiovasc Med 22, 133–137, doi: 10.1016/j.tcm.2012.07.009 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Nakahara T et al. Coronary Artery Calcification: From Mechanism to Molecular Imaging. JACC Cardiovasc Imaging 10, 582–593, doi: 10.1016/j.jcmg.2017.03.005 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Goettsch C et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. The Journal of clinical investigation 126, 1323–1336, doi: 10.1172/JCI80851 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kapustin AN et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116, 1312–1323, doi: 10.1161/CIRCRESAHA.116.305012 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Krohn JB, Hutcheson JD, Martinez-Martinez E & Aikawa E Extracellular vesicles in cardiovascular calcification: expanding current paradigms. The Journal of physiology 594, 2895–2903, doi: 10.1113/JP271338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krohn JB et al. Discoidin Domain Receptor-1 Regulates Calcific Extracellular Vesicle Release in Vascular Smooth Muscle Cell Fibrocalcific Response via Transforming Growth Factor-beta Signaling. Arterioscler Thromb Vasc Biol 36, 525–533, doi: 10.1161/ATVBAHA.115.307009 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapustin AN et al. Prothrombin Loading of Vascular Smooth Muscle Cell-Derived Exosomes Regulates Coagulation and Calcification. Arterioscler Thromb Vasc Biol 37, e22–e32, doi: 10.1161/ATVBAHA.116.308886 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Sugita M, Dulaney JT & Moser HW Ceramidase deficiency in Farber’s disease (lipogranulomatosis). Science 178, 1100–1102, doi: 10.1126/science.178.4065.1100 (1972). [DOI] [PubMed] [Google Scholar]
- 14.Mondal RK, Nandi M, Datta S & Hira M Disseminated lipogranulomatosis. Indian Pediatr 46, 175–177 (2009). [PubMed] [Google Scholar]
- 15.Mao C & Obeid LM Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta 1781, 424–434, doi: 10.1016/j.bbalip.2008.06.002 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haus JM et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58, 337–343, doi: 10.2337/db08-1228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liao L et al. Ceramide mediates Ox-LDL-induced human vascular smooth muscle cell calcification via p38 mitogen-activated protein kinase signaling. PLoS One 8, e82379, doi: 10.1371/journal.pone.0082379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y et al. TLR4/NF-kappaB/Ceramide signaling contributes to Ox-LDL-induced calcification of human vascular smooth muscle cells. Eur J Pharmacol 794, 45–51, doi: 10.1016/j.ejphar.2016.11.029 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Luong TTD et al. Acid sphingomyelinase promotes SGK1-dependent vascular calcification. Clin Sci (Lond) 135, 515–534, doi: 10.1042/CS20201122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhat OM et al. Arterial Medial Calcification through Enhanced small Extracellular Vesicle Release in Smooth Muscle-Specific Asah1 Gene Knockout Mice. Sci Rep 10, 1645, doi: 10.1038/s41598-020-58568-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhat OM, Yuan X, Cain C, Salloum FN & Li PL Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: A ceramide-mediated vasculopathy. J Cell Mol Med 24, 539–553, doi: 10.1111/jcmm.14761 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber LA & Teis D Lysosomal signaling in control of degradation pathways. Curr Opin Cell Biol 39, 8–14, doi: 10.1016/j.ceb.2016.01.006 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Taniguchi M et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem 287, 39898–39910, doi: 10.1074/jbc.M112.416552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao Y et al. Mammalian target of rapamycin signaling inhibition ameliorates vascular calcification via Klotho upregulation. Kidney Int 88, 711–721, doi: 10.1038/ki.2015.160 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Bao J, Li G, Yuan X, Li PL & Gulbins E Contribution of p62 to Phenotype Transition of Coronary Arterial Myocytes with Defective Autophagy. Cell Physiol Biochem 41, 555–568, doi: 10.1159/000457877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bao JX et al. Implication of CD38 gene in autophagic degradation of collagen I in mouse coronary arterial myocytes. Front Biosci (Landmark Ed) 22, 558–569, doi: 10.2741/4502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y et al. Defective autophagosome trafficking contributes to impaired autophagic flux in coronary arterial myocytes lacking CD38 gene. Cardiovasc Res 102, 68–78, doi: 10.1093/cvr/cvu011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin J, Berdyshev E, Poirer C, Schwartz NB & Dawson G Neutral sphingomyelinase 2 deficiency increases hyaluronan synthesis by up-regulation of Hyaluronan synthase 2 through decreased ceramide production and activation of Akt. The Journal of biological chemistry 287, 13620–13632, doi: 10.1074/jbc.M111.304857 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu TH et al. Restoration of microRNA-30b expression alleviates vascular calcification through the mTOR signaling pathway and autophagy. J Cell Physiol 234, 14306–14318, doi: 10.1002/jcp.28130 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Yu Y, Zhang HX & Wu XL [The expression of Akt/mTOR in VSMC calcification induced by high phosphate and its regulation of Cbfalpha1]. Zhonghua Yi Xue Za Zhi 98, 1446–1451, doi: 10.3760/cma.j.issn.0376-2491.2018.18.016 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Lin X et al. Exosomal Notch3 from high glucose-stimulated endothelial cells regulates vascular smooth muscle cells calcification/aging. Life Sci 232, 116582, doi: 10.1016/j.lfs.2019.116582 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Cai Y, Wang XL, Flores AM, Lin T & Guzman RJ Inhibition of endo-lysosomal function exacerbates vascular calcification. Sci Rep 8, 3377, doi: 10.1038/s41598-017-17540-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schieder M, Rotzer K, Bruggemann A, Biel M & Wahl-Schott C Planar patch clamp approach to characterize ionic currents from intact lysosomes. Sci Signal 3, pl3, doi: 10.1126/scisignal.3151pl3 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Price PA, Buckley JR & Williamson MK The amino bisphosphonate ibandronate prevents vitamin D toxicity and inhibits vitamin D-induced calcification of arteries, cartilage, lungs and kidneys in rats. J Nutr 131, 2910–2915, doi: 10.1093/jn/131.11.2910 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Vogel KR, Ainslie GR, Jansen EE, Salomons GS & Gibson KM Torin 1 partially corrects vigabatrin-induced mitochondrial increase in mouse. Ann Clin Transl Neurol 2, 699–706, doi: 10.1002/acn3.200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gurley JM, Griesel BA & Olson AL Increased Skeletal Muscle GLUT4 Expression in Obese Mice After Voluntary Wheel Running Exercise Is Posttranscriptional. Diabetes 65, 2911–2919, doi: 10.2337/db16-0305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoenderop JG, van der Kemp AW, Urben CM, Strugnell SA & Bindels RJ Effects of vitamin D compounds on renal and intestinal Ca2+ transport proteins in 25-hydroxyvitamin D3–1alpha-hydroxylase knockout mice. Kidney Int 66, 1082–1089, doi: 10.1111/j.1523-1755.2004.00858.x (2004). [DOI] [PubMed] [Google Scholar]
- 38.Bhat OM et al. Interleukin-18-induced atherosclerosis involves CD36 and NF-kappaB crosstalk in Apo E−/− mice. J Cardiol 66, 28–35, doi: 10.1016/j.jjcc.2014.10.012 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Boini KM et al. Implication of CD38 gene in podocyte epithelial-to-mesenchymal transition and glomerular sclerosis. J Cell Mol Med 16, 1674–1685, doi: 10.1111/j.1582-4934.2011.01462.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang C et al. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 60, 154–162, doi: 10.1161/HYPERTENSIONAHA.111.189688 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li X et al. Control of autophagy maturation by acid sphingomyelinase in mouse coronary arterial smooth muscle cells: protective role in atherosclerosis. J Mol Med (Berl) 92, 473–485, doi: 10.1007/s00109-014-1120-y (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu D et al. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1 mediated pathway. J Cell Mol Med 19, 165–174, doi: 10.1111/jcmm.12373 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chandrika G, Natesh K, Ranade D, Chugh A & Shastry P Mammalian target of rapamycin inhibitors, temsirolimus and torin 1, attenuate stemness-associated properties and expression of mesenchymal markers promoted by phorbol-myristate-acetate and oncostatin-M in glioblastoma cells. Tumour Biol 39, 1010428317695921, doi: 10.1177/1010428317695921 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Yuan X et al. Inhibitory effects of growth differentiation factor 11 on autophagy deficiency-induced dedifferentiation of arterial smooth muscle cells. Am J Physiol Heart Circ Physiol 316, H345–H356, doi: 10.1152/ajpheart.00342.2018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan X, Bhat OM, Lohner H, Zhang Y & Li PL Endothelial acid ceramidase in exosome-mediated release of NLRP3 inflammasome products during hyperglycemia: Evidence from endothelium-specific deletion of Asah1 gene. Biochim Biophys Acta Mol Cell Biol Lipids 1864, 158532, doi: 10.1016/j.bbalip.2019.158532 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tong X et al. Pro-atherogenic role of smooth muscle Nox4-based NADPH oxidase. J Mol Cell Cardiol 92, 30–40, doi: 10.1016/j.yjmcc.2016.01.020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quinn U, Tomlinson LA & Cockcroft JR Arterial stiffness. JRSM Cardiovasc Dis 1, doi: 10.1258/cvd.2012.012024 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shroff RC & Shanahan CM The vascular biology of calcification. Semin Dial 20, 103–109, doi: 10.1111/j.1525-139X.2007.00255.x (2007). [DOI] [PubMed] [Google Scholar]
- 49.Mackey RH, Venkitachalam L & Sutton-Tyrrell K Calcifications, arterial stiffness and atherosclerosis. Adv Cardiol 44, 234–244, doi: 10.1159/000096744 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Qiu H et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 107, 615–619, doi: 10.1161/CIRCRESAHA.110.221846 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zeidan YH, Jenkins RW & Hannun YA Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J Cell Biol 181, 335–350, doi: 10.1083/jcb.200705060 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hartley CJ, Taffet GE, Michael LH, Pham TT & Entman ML Noninvasive determination of pulse-wave velocity in mice. Am J Physiol 273, H494–500, doi: 10.1152/ajpheart.1997.273.1.H494 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Chatterjee S et al. Inhibition of glycosphingolipid synthesis ameliorates atherosclerosis and arterial stiffness in apolipoprotein E−/− mice and rabbits fed a high-fat and -cholesterol diet. Circulation 129, 2403–2413, doi: 10.1161/CIRCULATIONAHA.113.007559 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiao Y et al. mTOR (Mechanistic Target of Rapamycin) Inhibition Decreases Mechanosignaling, Collagen Accumulation, and Stiffening of the Thoracic Aorta in Elastin-Deficient Mice. Arterioscler Thromb Vasc Biol 37, 1657–1666, doi: 10.1161/ATVBAHA.117.309653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lesniewski LA et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 16, 17–26, doi: 10.1111/acel.12524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reynolds JL et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol 15, 2857–2867, doi: 10.1097/01.ASN.0000141960.01035.28 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Shroff RC et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol 21, 103–112, doi: 10.1681/ASN.2009060640 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou K et al. A Ceramide-Regulated Element in the Late Endosomal Protein LAPTM4B Controls Amino Acid Transporter Interaction. ACS Cent Sci 4, 548–558, doi: 10.1021/acscentsci.7b00582 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo Y et al. Bone marrow mesenchymal stem cell-derived exosomes alleviate high phosphorus-induced vascular smooth muscle cells calcification by modifying microRNA profiles. Funct Integr Genomics 19, 633–643, doi: 10.1007/s10142-019-00669-0 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Giachelli CM, Liaw L, Murry CE, Schwartz SM & Almeida M Osteopontin expression in cardiovascular diseases. Ann N Y Acad Sci 760, 109–126, doi: 10.1111/j.1749-6632.1995.tb44624.x (1995). [DOI] [PubMed] [Google Scholar]
- 61.Tanimura A, McGregor DH & Anderson HC Matrix vesicles in atherosclerotic calcification. Proc Soc Exp Biol Med 172, 173–177, doi: 10.3181/00379727-172-41542 (1983). [DOI] [PubMed] [Google Scholar]
- 62.Pai A, Leaf EM, El-Abbadi M & Giachelli CM Elastin degradation and vascular smooth muscle cell phenotype change precede cell loss and arterial medial calcification in a uremic mouse model of chronic kidney disease. Am J Pathol 178, 764–773, doi: 10.1016/j.ajpath.2010.10.006 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El-Abbadi MM et al. Phosphate feeding induces arterial medial calcification in uremic mice: role of serum phosphorus, fibroblast growth factor-23, and osteopontin. Kidney Int 75, 1297–1307, doi: 10.1038/ki.2009.83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ode T et al. PDMP, a ceramide analogue, acts as an inhibitor of mTORC1 by inducing its translocation from lysosome to endoplasmic reticulum. Exp Cell Res 350, 103–114, doi: 10.1016/j.yexcr.2016.11.011 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Lu QB et al. Nesfatin-1 functions as a switch for phenotype transformation and proliferation of VSMCs in hypertensive vascular remodeling. Biochim Biophys Acta Mol Basis Dis 1864, 2154–2168, doi: 10.1016/j.bbadis.2018.04.002 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Rzucidlo EM Signaling pathways regulating vascular smooth muscle cell differentiation. Vascular 17 Suppl 1, S15–20, doi: 10.2310/6670.2008.00089 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Chong ZZ, Shang YC & Maiese K Cardiovascular disease and mTOR signaling. Trends Cardiovasc Med 21, 151–155, doi: 10.1016/j.tcm.2012.04.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurdi A, De Meyer GR & Martinet W Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br J Clin Pharmacol 82, 1267–1279, doi: 10.1111/bcp.12820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martin KA et al. The mTOR/p70 S6K1 pathway regulates vascular smooth muscle cell differentiation. Am J Physiol Cell Physiol 286, C507–517, doi: 10.1152/ajpcell.00201.2003 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Ding M et al. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler Thromb Vasc Biol 31, 1403–1410, doi: 10.1161/ATVBAHA.110.216804 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pan S et al. Folic acid inhibits dedifferentiation of PDGF-BB-induced vascular smooth muscle cells by suppressing mTOR/P70S6K signaling. Am J Transl Res 9, 1307–1316 (2017). [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang M et al. MiR-145 alleviates Hcy-induced VSMC proliferation, migration, and phenotypic switch through repression of the PI3K/Akt/mTOR pathway. Histochem Cell Biol 153, 357–366, doi: 10.1007/s00418-020-01847-z (2020). [DOI] [PubMed] [Google Scholar]
- 73.Zhao J et al. Kaempferol promotes bone formation in part via the mTOR signaling pathway. Mol Med Rep 20, 5197–5207, doi: 10.3892/mmr.2019.10747 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Z et al. Effect of sirolimus on arteriosclerosis induced by advanced glycation end products via inhibition of the ILK/mTOR pathway in kidney transplantation recipients. Eur J Pharmacol 813, 1–9, doi: 10.1016/j.ejphar.2017.06.038 (2017). [DOI] [PubMed] [Google Scholar]
- 75.Nakano-Kurimoto R et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol 297, H1673–1684, doi: 10.1152/ajpheart.00455.2009 (2009). [DOI] [PubMed] [Google Scholar]
- 76.Iijima K [Bone and calcium update; diagnosis and therapy of bone metabolism disease update. Regulatory Mechanism of Mammalian Sirtuin SIRT1 in Vascular calcification: impact of vascular smooth muscle cell senescence]. Clin Calcium 21, 53–60, doi:CliCa111218031810 (2011). [PubMed] [Google Scholar]
- 77.Zhan JK et al. Adiponectin attenuates the osteoblastic differentiation of vascular smooth muscle cells through the AMPK/mTOR pathway. Exp Cell Res 323, 352–358, doi: 10.1016/j.yexcr.2014.02.016 (2014). [DOI] [PubMed] [Google Scholar]
- 78.Zhan JK et al. The protective effect of GLP-1 analogue in arterial calcification through attenuating osteoblastic differentiation of human VSMCs. Int J Cardiol 189, 188–193, doi: 10.1016/j.ijcard.2015.04.086 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Cao J et al. Rapamycin inhibits CaCl2-induced thoracic aortic aneurysm formation in rats through mTOR-mediated suppression of proinflammatory mediators. Mol Med Rep 16, 1911–1919, doi: 10.3892/mmr.2017.6844 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trajkovic K et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247, doi: 10.1126/science.1153124 (2008). [DOI] [PubMed] [Google Scholar]
- 81.Serban KA et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep 6, 31596, doi: 10.1038/srep31596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bianco F et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J 28, 1043–1054, doi: 10.1038/emboj.2009.45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li S et al. Cardiac progenitor cellderived exosomes promote H9C2 cell growth via Akt/mTOR activation. Int J Mol Med 42, 1517–1525, doi: 10.3892/ijmm.2018.3699 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang W et al. The exosome-mediated PI3k/Akt/mTOR signaling pathway in cervical cancer. Int J Clin Exp Pathol 12, 2474–2484 (2019). [PMC free article] [PubMed] [Google Scholar]
- 85.Tassew NG et al. Exosomes Mediate Mobilization of Autocrine Wnt10b to Promote Axonal Regeneration in the Injured CNS. Cell Rep 20, 99–111, doi: 10.1016/j.celrep.2017.06.009 (2017). [DOI] [PubMed] [Google Scholar]