

Abstract
The eIF2α kinase general control non-depressible 2 integrates translation initiation rates to amino acid availability. General control non-depressible 2 also regulates translation initiation during synaptic plasticity and GCN2−/− mice show improved memory compared with wild-type mice with a reduced threshold for triggering late long-term potentiation. This property suggests that inhibiting general control non-depressible 2 function might represent a therapeutic avenue for improving memory. We screened for general control non-depressible 2 inhibitors using a small library of known kinase inhibitors and ATP-analogs and identified three compounds – indirubin-3′-monoxime, SP600125 and a SyK inhibitor with activity against general control non-depressible 2. All three compounds inhibit the ability of general control non-depressible 2 to phosphorylate eIF2α in vitro as well as in vivo following UV-treatment of mouse embryonic fibroblasts. Using computer-assisted modeling, we modeled the binding of the inhibitors in the ATP-binding site of general control non-depressible 2. This work provides the molecular basis for undertaking structure–activity relationships of these compounds in order to develop specific inhibitors of general control non-depressible 2.
Keywords: eIF2α phosphorylation, GCN2, general control non-derepressible-2, kinase inhibitors, translation
Eukaryotic initiation factor 2 is a trimeric factor that plays a central role in the control of translation initiation upon various stresses (1). In growing cells, this factor recruits Met-tRNAiMet and GTP to form a ternary complex (TC) that then associates with the 40S ribosome. Once an appropriate initiation codon is found on the mRNA, GTP is hydrolyzed and eIF2•GDP is released and recycled to eIF2•GTP by the guanine nucleotide exchange factor, eIF2B (2). Various cellular stresses induce the phosphorylation of the α subunit of eIF2, which in turn confers high-affinity binding to eIF2B and renders eIF2 a competitive inhibitor of eIF2B – effectively decreasing levels of the TC (1,3). Since eIF2α is in excess of eIF2B, small changes in phospho-eIF2α levels can significantly inhibit translation.
Four different kinases are known to phosphorylate eIF2α; each being activated in response to specific stresses and environmental stimuli (1). These are as follows: (i) the protein kinase RNA-dependent (PKR) which is activated by double stranded RNA, as occurs during infection with RNA viruses (4), (ii) the PKR-like endoplasmic reticulum (ER) kinase (PERK), an ER-associated kinase that is activated following accumulation of unfolded proteins in the ER (5), (iii) the heme-regulated inhibitor (HRI), a kinase which phosphorylates eIF2α upon heme deprivation (6) and, (iv) the general control non-derepressible-2 (GCN2) kinase known to induce eIF2α phosphorylation under amino acid starvation (2,7). eIF2α is phosphorylated under a variety of stresses, with different kinases implicated in different events. For example, heat shock or arsenite treatment of erythroid cells activates HRI (8), whereas arsenite treatment stimulates eIF2α phosphorylation in a PKR-dependent manner (9,10). Deprivation of glucose (11) or serum (12), proteasome inhibition (13) and UV irradiation (14) have been shown to induce eIF2α phosphorylation through activation of GCN2. Although eIF2α phosphorylation causes a near-global reduction in protein synthesis activity, it plays a secondary role in activating translation of a subset of mRNAs that contain multiple open reading frames upstream (uORFs) of the major initiation codon. This mechanism was elegantly described in yeast for GCN4, a transcription factor that activates genes in the amino acid biosynthesis pathway (2). The same type of translational control has been described for some mammalian transcripts, with genes activated under stress appearing to regulate cell survival (1,15).
GCN2 is conserved from yeast to mammals and monitors the concentration of amino acids in the cell by binding to uncharged tRNAs via its histidyl-tRNA synthetase (HisRS)-like domain (Figure 1A). This triggers conformational changes that lead to autophosphorylation on two threonines followed by dimerization, and subsequent binding to, and phosphorylation of, eIF2α (2). Recently, GCN2-mediated eIF2α phosphorylation has been implicated in the control of memory and synaptic plasticity (16,17). GCN2-null mice, or mice heterozygous for the non-phosphorylable eIF2α(S51A) mutant, show reduced threshold to trigger late long-term potentiation (L-LTP) in the hippocampal region, a phenotype associated with improved memory (16,17). This raises the interesting possibility that reducing eIF2α phosphorylation in the brain using small molecules could improve long-term memory.
Figure 1:
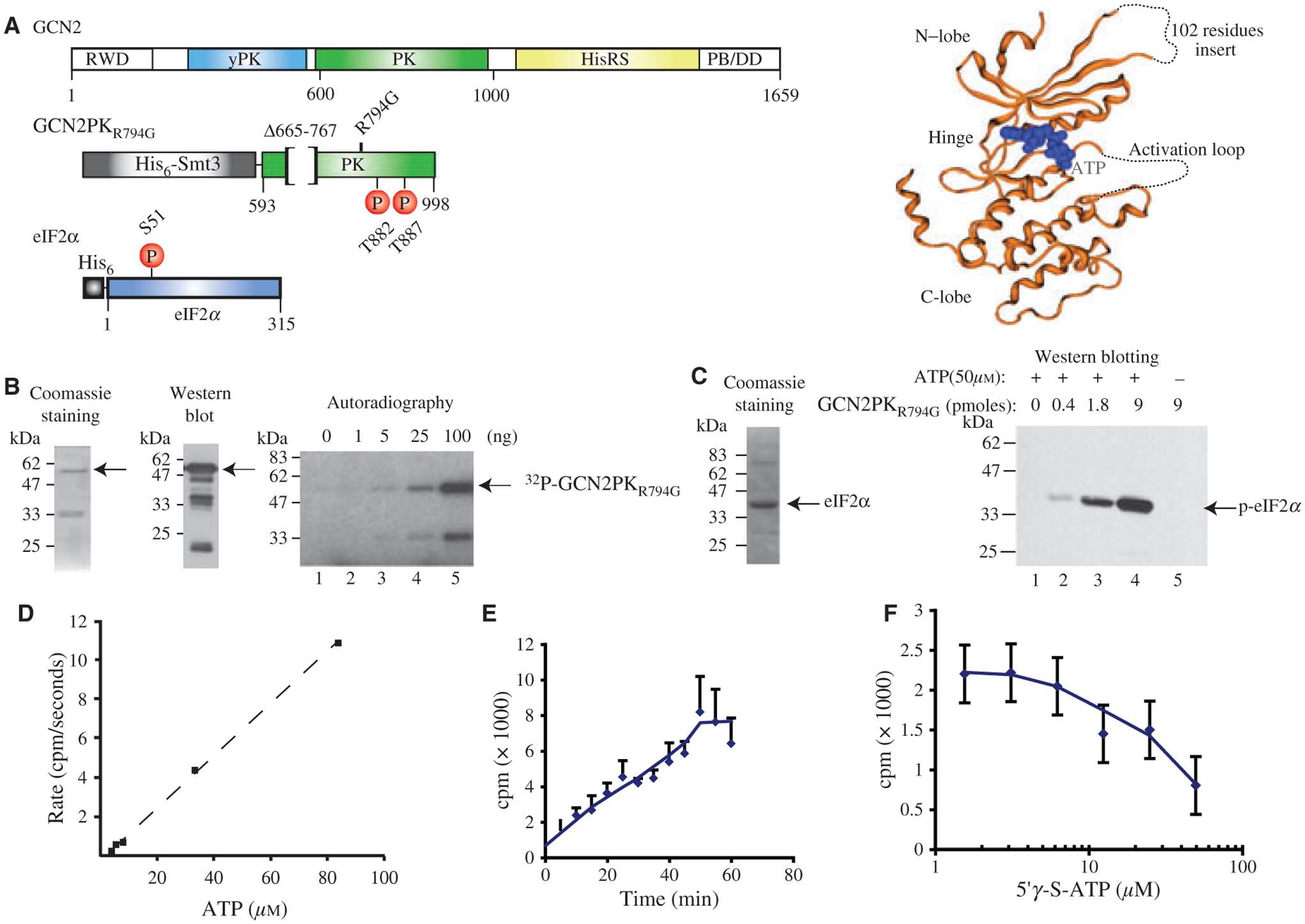
Proteins and assays used in this study. (A) Right panel: Schematic diagram of recombinant GCN2 and eIF2α proteins. Top panel: Yeast GCN2 consists of 5 domains: RWD: RING finger-containing, WD-repeat-containing, and yeast DEAD (DEXD)-like proteins; yPK: pseudo-protein kinase domain; PK: protein kinase domain; HisRS: histidine tRNA synthetase-like binding domain; PB/DD: ribosome binding and dimerization domain. Middle panel: GCN2 protein kinase domain used in this study (18). The protein contains a His6-Smt3 tag for purification, is missing residues 665–767 to improve protein stability during purification and has a R794G mutation that renders the kinase domain constitutively active (24). The two sites of auto-phosphorylation (Thr882 and Thr887) leading to activation of the enzyme are indicated. Lower panel: Mouse eIF2α bears a His6 tag at its N-terminus for purification and the phosphorylation site (S51) is indicated. Left Panel: Crystallographic structure of the kinase domain of yeast GCN2 bound to Mg•ATP. The GCN2 kinase is shown in orange ribbon drawing, the Mg•ATP is shown in blue. The 102-residues missing loop absent from the purified kinase protein used herein as well as the disordered activation loop are represented by a dotted line as determined previously (18). (B) Purity and activity of the GCN2 kinase domain used in this study. Left panel shows a Coomassie-stained polyacrylamide gel of His6-tagged-purifed GCN2 kinase domain. The middle panel shows a Western blot analysis of GCN2PKR794G using an anti-His6-tag antibody. The right panel shows the auto-phopshorylation activity of GCN2PKR794G. The indicated amount of purified GCN2PKR794G was incubated for 30 min in the presence of γ32P-ATP as described in the Methods and Materials. SDS-sample buffer was then added to each reaction, the samples boiled, separated by SDS-PAGE, dried and exposed to film (Kodak X-OMAT). (C) Yeast GCN2 can phosphorylate mouse eIF2α. The left panel shows a Coomassie-stained SDS-PAGE of purified recombinant eIF2α. The right panel is a Western blot illustrating detection of eIF2α phosphorylation by GCN2PKR794G. Fifteen nmoles of eIF2α was incubated with the indicated amounts of GCN2PKR794G for 30 min at 30 °C. eIF2α phosphorylation was then detected by Western blotting. (D) Autophosphorylation of GCN2PKR794G is linear over a large range of ATP concentration. Nine pmoles of GCN2PKR794G was incubated with increasing amounts of γ-32P-ATP for 30 min at 30 °C followed by quenching with 500 uM of unlabeled ATP. The amount of radiolabel incorporated was determined by filtration through nitrocellulose filters and the amount of radioactivity retained determined by Cerenkov counting. (E) Kinase activity of GCN2PKR794G is linear for 50 min. Kinase reactions were performed in the presence of 50 μM γ-32P-ATP for the indicated periods of time and quantitated as described in (D). (F) The ATP analog 5′γ-S-ATP inhibits GCN2PKR794G kinase activity. Kinase reactions were performed and quantitated as in (D).
Herein, we describe the identification of small-molecule inhibitors of GCN2 and show that they can prevent the phosphorylation of eIF2α upon UV irradiation in mouse embryonic fibroblasts (MEFs). Based on the crystallographic structure of GCN2, we used computer-assisted modeling to dock the molecules into the active site. These studies provide an understanding into the binding GCN2 inhibitors and provide a starting point from which more potent and specific derivatives can be developed.
Methods and Materials
Expression vectors
The expression vector pET26b-GCN2PKR794GΔ665–767 ((henceforth refered to as GCN2PKR794G)) was a kind gift from Dr. S.K. Burley (SGX Pharmaceuticals, San Diego, CA, USA) (18). The protein encoded from this vector contains a single amino acid substitution that constitutively activates the kinase and a 665–767 amino acid deletion to remove a proteolytically sensitive loop that allows stable bacterial expression of the protein. The mouse eIF2α gene was amplified by PCR from a mouse cDNA library using the following oligonucleotides: 5′-ACAGCCGCATATGCCGGGGCTAAGTTGTAGA TTTT-3′ (NdeI site underlined) and 5′-CCGGGATCCTTAATCTT CAGCTTTGGCTTCCATT-3′ (BamHI site underlined) and cloned into the NdeI/BamHI restriction sites of pET-15b, to generate pET-15b/eIF2α.
Compounds
The Screen-well™ kinase inhibitor library was purchased from Biomol International and a kinase focussed-library containing 1000 compounds initially from ChemDiv was a kind gift from Dr. D.Y. Thomas (McGill University, Montreal, Canada). This library was selected from a 5000 kinase-targeted compound library to obtain the maximum diversity, as defined by Trepalin et al. (19). The ChemDiv original library was designed by pharmacophore search and docking using ChemDiv Chemosoft™ software and other commercially available softwares like Accelrys (Accelrys Software Inc., San Diego, CA, USA), MOE (Chemical Computing Group, Montreal, Canada) and Daylight (Chemical information system Inc. Aliso Viejo, CA, USA). The 1000-compounds library used here contained 430 unique heterocyclic chemotypes. The resupply of staurosporine, SP600125, indirubin-3′-monoxime, 3-(1-Methyl-1H-indol-3-yl-methylene)-2-oxo-2,3-dihydro-1H-indole-5-sulfonamide (refered herein as SyK inhibitor [SyKI]) and analogs were from Calbiochem (San Diego, CA, USA). Adenosine 5′-O-(3-thiotriphosphate) (5′γ-S-ATP) was purchased from Sigma (St. Louis, MO, USA). The indirubin-3′-monoxime analogs were obtained ‘de novo’ from Calbiochem (San Diego, CA, USA) and were selected according to their structural similarity to the initial hit.
Protein expression and purification
Escherichia coli BL21(DE3)/pLysS were transformed with the plasmids pET-26b-GCN2PKR794G and pET-15b/eIF2α. After selection of single colony transformants, one liter cultures were grown at 37 °C to an OD600 of 0.6. Protein expression was induced for 4 h at 30 °C by adding 0.5 mM isopropyl β-d-thiogalactopyranoside and cells were harvested by centrifugation at 2300 g for 15 min, followed by one freeze-thaw cycle. The pellet was resuspended in buffer A (50 mM Tris–HCl [pH 8.0], 50 mM NaCl, 1 mM EDTA, 5% glycerol and 5 mM DTT) supplemented with 5 mM imidazole. Protein purifications were performed using Ni++-NTA agarose (Qiagen, Valencia, CA, USA) (20).
In vitro kinase assays
The GCN2PKR794G autophosphorylation assay was performed essentially as described (21). In brief, 9 pmoles (0.5 μg) of GCN2PKR794G was incubated with 50 μM of γ32P-ATP (or otherwise indicated) in the presence of kinase buffer (20 mM Tris–HCl [pH 7.6], 10 mM MgCl2, 100 mM NaCl and 0.25 mg/mL bovine serum albumin) in a 10 μL reaction. The reaction was stopped by the addition of 1× SDS-sample buffer, boiled and analyzed on an SDS-polyacrylamide gel. The gel was dried and exposed to Kodak X-OMAT X-ray film. The eIF2α kinase reactions were performed by incubating 9 pmoles (0.5 μg) of GCN2PKR794G with 14.9 pmoles (0.5 μg) of eIF2α and 50 uM ATP in kinase buffer. The extent of eIF2α phosphorylation was determined by Western blotting using a phospho-specific eIF2α antibody (Biosource, Camarillo, CA, USA). A pan-eIF2α antibody (Abcam, Cambridge, MA, USA) was used to quantitate the amount of material loaded on the gel. For screening of compounds, we performed an autophosphorylation assay as described above, except that the reaction was stopped by the addition of 500 uM of cold ATP and filtered through a 96-well Multiscreen HTS™ Immobilon-P filter (Millipore). The filter was preblocked with 0.1 M sodium pyrophosphate beforehand. Following filtration, the plate was washed three times with kinase buffer supplemented with 0.1 M sodium pyrophosphate, the filters were then punched out of the plate, transferred to a scintillation vial and the bound radioactivity determined by Cerenkov counting.
UV irradiation of MEFS
UV-C treatments were performed on GCN2+/+ or GCN2−/− MEFs, graciously obtained from Dr. Nahum Sonenberg (McGill University) (22). For these experiments, 2.5 × 105 cells were plated per well of a 6-well plate. The following day, the cells were washed twice with phosphate-buffered saline (PBS), irradiated with 300 J/cm2 UV-C (254 nm; UV Stratalinker 1800, Stratagene, La Jolla, CA, USA) a layer of PBS covering the cells, and then put back at 37 °C for 30 min in the presence of DMEM with 10% serum. After incubation, the cells were scraped from the plate and collected by centrifugation at 1000 g for 10 min. The cell pellet was resuspended in lysis buffer (10 mM Tris–HCl [pH 8.0], 1.5 mM MgCl2, 5 mM KCl, 0.5% NP-40 (Sigma), 2 mM DTT, 1 mM phenylmethylsulphonyl fluoride, 10 mM pyrophosphate, 10 mM β-glycerophosphate, 50 mM NaF, 1 μg/mL pepstatin, 1 μg/mL, leupeptin, 1 μg/mL aprotonin), incubated on ice for 5 min, followed by one freeze-thaw cycle and a centrifugation at 5000 g for 8 min at 4 °C. Thirty micrograms of proteins from the supernatant was then loaded on an SDS-polyacrylamide gel and the extent of eIF2α phosphorylation determined by Western blotting. For compound testing, cells were pretreated for 30 min at the indicated concentrations, washed twice with PBS supplemented with compounds, irradiated, and incubated again at 37 °C for 30 min in DMEM in the presence of compounds before lysis and Western blotting analysis. The amount of material loaded on the gel was determined by probing the membranes with pan-eIF2α or anti-tubulin antibodies (Sigma).
In silico modeling of GCN2 active site movement
Structural motion in the GCN2 binding pocket was examined by superposing the apo (PDB code 1ZYC) and ATP bound (PDB code 1ZYD) wild-type structures based on the binding pocket residues. The proteins were aligned and superposed using the automatic sequence and structure alignment tool (pro_Align) in the MOE software package (MOE software (version 2007.09). The default alignment settings were used to obtain an initial rough superposition of the bound and apo forms, that was further refined using the pro_Consensus tool to focus the superposition on the active site residues. The resulting alignment was used to compare the binding pocket in the apo and ATP bound forms. Van der Waals (VDW) molecular interaction surfaces were computed and drawn around the active site pockets to further illustrate changes in the binding pocket shape upon ligand binding, and to aid in determining clashes between the binding pocket and potential ligands. A 2D interaction diagram of the ATP interaction with GCN2 was computed using the default settings of the ligand interaction application in the MOE software. The resulting diagram was used to examine key ligand–protein interactions.
In silico compound docking
The knowledge-based docking of the GCN2 inhibitors was first initiated by performing a search on the PDB database for kinase structures similar in sequence to GCN2 which contained bound ligands that were also similar to the identified compounds. The PDB was searched using two criteria: a BLAST E-value of <10 with the GCN2 sequence and a MACCS similarity of >0.8 between the ligand in the PDB and the GCN2 hits. A Blast expect value (the E-value) describes the number of hits one can ‘expect’ to see by chance when searching a database of a particular size. A MACCS key tanimoto (MDL Information Systems, Inc, San Leandro, CA, USA) similarity of 0.8 was chosen to ensure high similarity between the ligands under study and the ligands retrieved from the PDB search. All structures having sequences with E-values < 10 (23) and MACSS tanimoto similarities of >0.8 were kept. This retrieved many unrelated structutres, which were then filtered manually based on 3D structural similarity with the GCN2 protein structure. The models of the hits bound to GCN2 hits were generated by aligning and superposing each representative PDB structure onto the apo and ATP bound GCN2 structures (1ZYC and 1ZYD, respectively). The alignments were refined to emphasize superposition of the hinge-region residues. The coordinates of the superposed ligands from the related PDB complexes were then used as starting points to produce refined docked poses in the GCN2 pocket. Where the ligands from the PDB complex are identical to the GCN2 hits, no further modifications were required. A final refinement of the poses was performed using the LigX module in MOE to minimize each ligand in the fields of the fixed GCN2 receptor pocket. The minimizations were performed using the MMFF94s forcefield in MOE, with a 0.1 kcal/mol Å2 root-mean-squared gradient termination criterion. The ligand poses produced by the above procedure were examined before and after the minimizations to gain insights into possible binding modes of these ligands in GCN2.
Results
Autophosphorylation of GCN2 as an assay to identify GCN2 inhibitors
Although the isolated protein kinase (PK) domain of GCN2 is inert in vitro, the activity can be rescued by a single mutation in the catalytic domain (R794G) that renders it constitutively active (GCN2PKR794G) (24). It constitutes a very useful tool for the screening of small-molecules inhibitors of GCN2 since it bypasses the requirement for activation by uncharged tRNA. The limitation of using this mutant, however, stems from the fact that it is about 75-fold more active than the wild-type protein in vitro (18) and, hence, it is difficult to quantitatively transpose an effect observed with an inhibitor on GCN2PKR794G to the full-length wild-type GCN2 protein. We nonetheless used this mutant for the chemical inhibitors screen because of its ease in allowing detection kinase activity. GCN2PKR794G (Figure 1A, middle panel) was expressed and purified as described (18) with the exception that the plasmid-derived Smt3 tag was not removed. After purification, full-length protein was detected by Coomassie staining and Western blotting (Figure 1B). To verify that the purified GCN2PKR794G was active, we performed in vitro auto-phosphorylation assays (Figure 2B; right panel). We observed the incorporation of 32P-label into full-length GCN2PKR794G as well as into a polypeptide that migrated at ~33kDa (Figure 2B; right panel). We do not know the origins of the 33 kDa polypeptide, but suspect that it could be a co-purifying bacterial protein that is non-specifically phosphorylated by GCN2PKR794G. We do not believe that it represents an endogenous bacterial protein capable of auto-phosphorylation since we failed to observed this product when kinase reactions were performed with Ni++-purified bacterial lysates lacking GCN2PKR794G (data not shown). These results indicate that recombinant GCN2PKR794G is active in our autophosphorylation reaction. To determine if purified GCN2PKR794G could phosphorylate mouse eIF2α, we performed in vitro kinase reactions. Incubation of GCN2PKR794G with purified eIF2α resulted in a dose-dependent phosphorylation of eIF2α (Figure 1C; right panel). Omission of GCN2 (lane 1) or ATP (lane 5) did not phosphorylate eIF2α indicating that the reaction is GCN2- and ATP-dependent.
Figure 2:
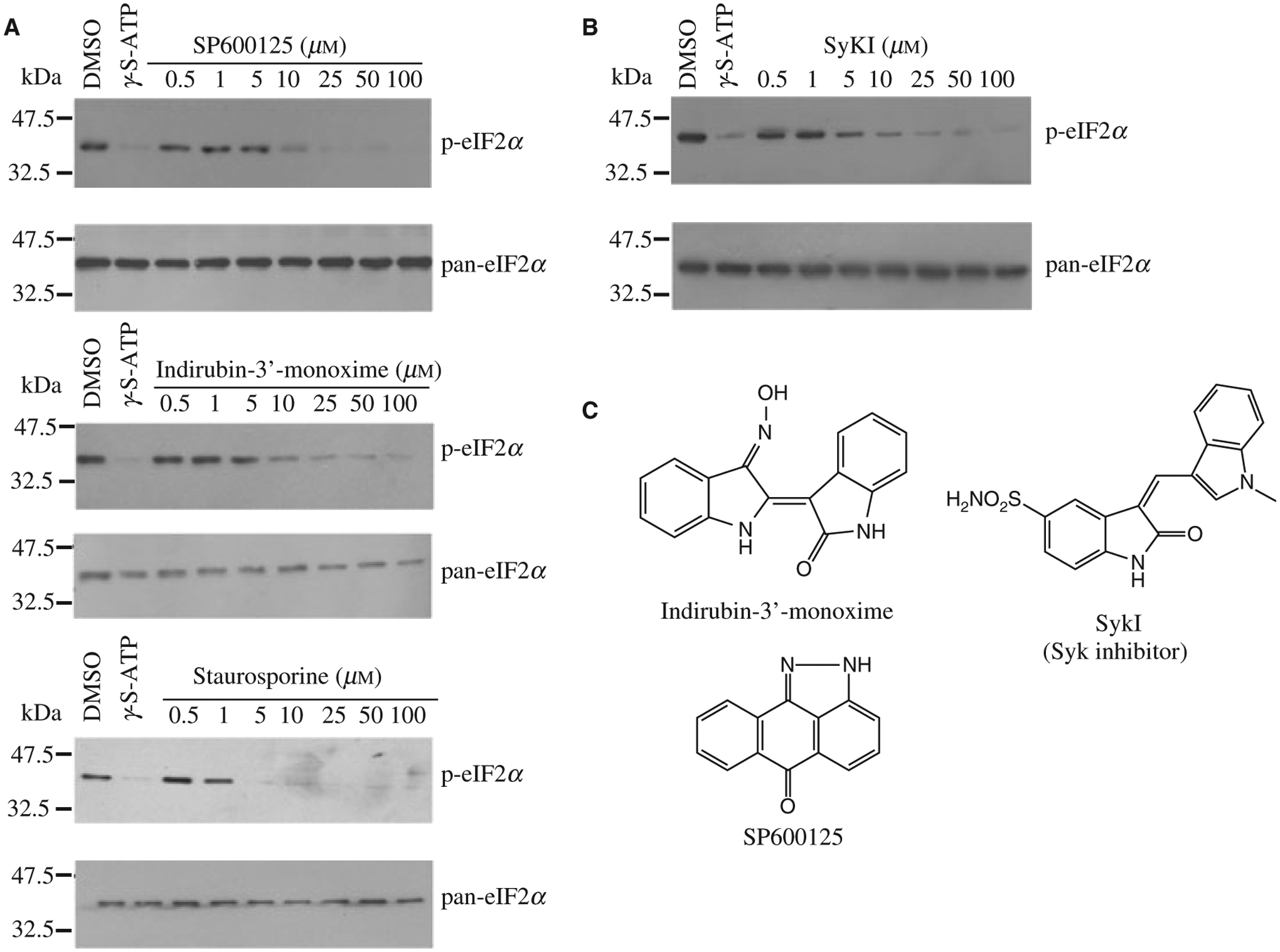
Inhibition of GCN2PKR794G-mediated eIF2α phosphorylation. (A) Indirubin-3′-monoxime, SP600125 and staurosporine block GCN2PKR794G-mediated phosphorylation of eIF2α. Following kinase reactions, the extent of eIF2α phosphorylation was determined by Western blotting using anti-phospho-eIF2α antibodies. Pan-eIF2α antibody was used to control for the sample loadings. The ATP competitive inhibitor, 5′γ-S-ATP, was used as positive control in these reactions. (B) Inhibition of GCN2PKR794G-mediated eIF2α phosphorylation by the SYK inhibitor, SyKI. Kinase reactions were analyzed as described in (A). (C) Schematic diagram of indirubin-3′-monoxime, SP600125, and SyKI.
To determine the concentration of labeled ATP that could be used in our assay and still maintain linear incorporation into GCN2PKR794G, we titrated γ32P-ATP into kinase reactions and found that incorporation was linear with γ32P-ATP concentrations up to 80 μM. Kinetic analysis of the reaction revealed that the kinase reactions remained linear for approximately 50 min, with a slight plateau occurring between 50 and 60 min (Figure 1E). The non-hydrolyzable ATP-analog, 5′γ-S-ATP was able to block phosphorylation of GCN2PKR794G in a dose-dependent manner (Figure 1F), and was used as a positive control in our screens.
Identification and characterization of GCN2PKR794G inhibitors
To screen for inhibitors of GCN2PKR794G we accessed two small chemical libraries; one containing 80 known kinase inhibitors and the other one consisting of a focussed kinase inhibitor library of 1000 compounds (See Methods and Materials). Each compound was tested in duplicate at 100 μM in the autophosphorylation assay described above and those that were able to cause a 70% reduction in GCN2PKR794G kinase activity were considered ‘hits’. These compounds were then titrated in the GCN2PKR794G autophosphorylation assay and the IC50’s determined (Table 1).
Table 1:
Hits identified in the GCN2 autophosphorylation assay
| Compound | Known target | IC50 (μM) |
|---|---|---|
| Staurosporine | Wide array of targets | 2.7 ± 0.2 |
| Damnacanthal | P56lck tyrosine kinase | 24.7 ± 2.7 |
| Hypericin | PKC | 46.8 ± 5.6 |
| 2-Aminopurine | p58 PITSLRE beta1 | 23.2 ± 3.9 |
| GW5074 | cRaf | 26.5 ± 4.1 |
| Rottlerin | PKC delta | 19.8 ± 2.1 |
| SP600125 | JNK | 24.8 ± 1.9 |
| Indirubin-3′-monoxime | GSK3 beta, CDK5/P25, CDK2 | 26.1 ± 3.6 |
| Kenpaullone | GSK3 beta | 11.2 ± 1.1 |
| Apigenin | CK | 22.9 ± 2.3 |
The IC50 with the standard deviation were calculated using the Sigmastat software (Aspire Software International, Ashburn, VA, USA).
As a confirmatory assay, we tested the ability of the ‘hits’ to block GCN2PKR794G mediated phosphorylation of eIF2α. In vitro kinase assays were performed in the presence of increasing concentrations of compounds and we found that only indirubin-3′-monoxime, SP600125 and staurosporine were able to prevent eIF2α phosphorylation in vitro (Figure 2A). Due to staurosporine’s promiscuity (25), we did not pursue further characterization of this compound. We then tested 14 different analogs of indirubin-3′-monoxime and found one of these, which we call SyKI, showed activity similar to indirubin-3′-monoxime (Figure 2B). This compound has been previously identified as a spleen tyrosine kinase (SYK) inhibitor (26) (Figure 2C).
Assessing activity of GCN2 inhibitors in vivo
We next sought to test the ability of indirubin-3′-monoxime, SP600125 and SyKI to prevent eIF2α phosphorylation upon GCN2 activation in cells. eIF2α is rapidly phosphorylated in a GCN2- dependent manner in cells exposed to UV-irradiation (Figure 3A, compare lanes 5–8 to 1–4) (14). Exposure of cells to UV-C caused phosphorylation of eIF2α (Figure 3B, compare lane 2 to 1). Treatment of cells with compounds blunted UV-C-induced eIF2α phosphorylation in a dose-dependent manner (Figure 3B, compare lanes 3–9 to 2). SP600125 appeared to be the most potent compound showing an IC50 of ~2 μM compared with an IC50 of 20 μM for indirubin-3′-monoxime and SyKI (Figure 3B). To ensure that the inhibition of eIF2α phosphorylation was due to a blockade of GCN2 activation, we subjected GCN2+/+ and GCN2−/− MEFs to UV-C irradiation in the presence of 20 μM compounds and probed for phosphorylation of eIF2α (Figure 3C). The results indicate that following treatment of GCN2−/− MEFs with UV-C in the presence of compounds, the levels of eIF2α phosphorylation remain unchanged indicating that the compounds are targeting a GCN2-dependent process in vivo.
Figure 3:
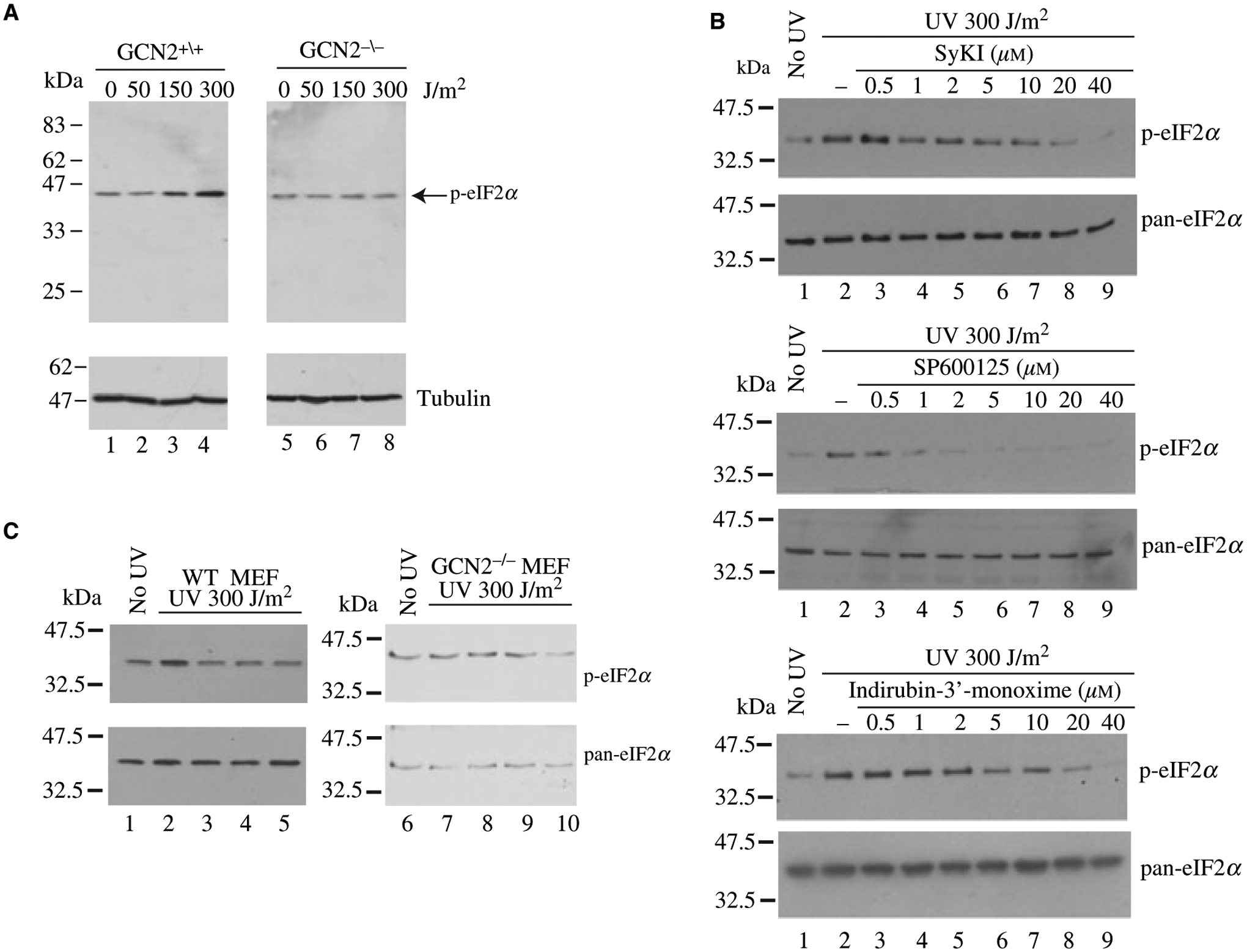
Inhibition of GCN2 prevents UV-induced eIF2α phosphorylation in vivo. (A) UV irradiation induces eIF2α phosphorylation in a GCN2-dependent manner. WT (lanes 1–4) or GCN2−/− (lanes 5–8) MEFs were irradiated with increasing doses of UV-C, followed by a 30 min incubation at 37 °C. Cells were then harvested and the levels of eIF2α phosphorylation determined by Western blotting. (B) Inhibition of UV-induced eIF2α phosphorylation by GCN2 inhibitors. WT MEFs were irradiated (lanes 2–9) with 300 J/cm2 UV-C in the absence (lane 2) or presence (lanes 3–9) of GCN2 inhibitors. The extent of eIF2α phosphorylation was verified by Western blotting. (C) GCN2 phosphorylation does not affect the basal phosphorylation status of eIF2α in GCN2−/− MEFs. WT (lanes 1–5) or GCN2−/− (lanes 6–10) MEFs were irradiated (lanes 2–5 and 7–10) with UV-C in the absence (lanes 2 and 7) or presence of 20 μM SyKI (lanes 3 and 8), 20 μM SP600125 (lanes 4 and 9), and 20 μM Indirubin-3′-monoxime (lanes 5 and 10). The extent of eIF2α phosphorylation was determined by Western blotting.
Analysis of the ATP-binding to GCN2
To gain a better understanding of ligand binding to GCN2, molecular modeling studies were performed. The recently determined crystal structures of wild-type and mutant forms of GCN2 (PDB codes 1ZXE, 1ZYC, 1ZYD, 1ZY5 and 1ZY4) (18) show that GCN2 exhibits a bi-lobate structure similar to that of other kinases, with an alpha-helix-rich C-terminal lobe connected via an ATP binding ‘hinge-region’ to beta-sheet rich N-terminal lobe. The structural similarity of GCN2 to other kinases such as CDK2, Src, Akt2, cAPK and Hck has been reported (18). We first analysed the structure of apo GCN2 or bound to ATP to examine the changes that occur in the protein upon ATP binding. This was a necessary prelude to enable us to propose binding modes for the small molecule inhibitors of GCN2. As observed by Padyana et al. (18), comparison of both GCN2 conformations, the apo and ATP-bound structures, shows that the ATP binding cleft is significantly closed in the apo structure – even more so than the binding pocket of cAPK (PDB code 1ATP), one of the most closed kinase structures known (27). Due to the closed nature of the binding pocket of GCN2 in the apo form, conformational rearrangement of pocket residue side-chains must occur before ATP binding is possible.
In Figure 4A, the superimposed binding site residues of the apo structure (red) and the ATP-bound structure (green) show that the hinge region residue conformations remain relatively unchanged upon binding, while the C-lobe and N-lobe both move outwards to open up the binding pocket, with the N-lobe showing more substantial backbone motion than the C-lobe. A top-view of molecular interaction surfaces drawn around the superimposed apo and ATP bound GNC2 structures (Figure 4B) shows that ATP would clash with the binding pocket of the apo structure, specifically with ASP-853. To accommodate ATP in the binding pocket, ASP-853 must rotate towards, and form an interaction with, LYS-628. This conformational change opens up the pocket around the hinge region and allows ATP to bind. Furthermore, The ASP-853 residue is quite sterically restricted in this environment, and can adopt essentially one of only two possible conformations; the conformation exemplified by the apo structure, and the conformation exemplified by the ATP bound structure. The important interactions between ATP and the GCN2 binding pocket are summarized in Figure 5C. They include the canonical kinase H-bonds to the hinge region residues GLU-789 and CYS-791, and a water-mediated interaction between the ATP, ASP-853 residue and the ‘gatekeeper’ residue MET-788.
Figure 4:
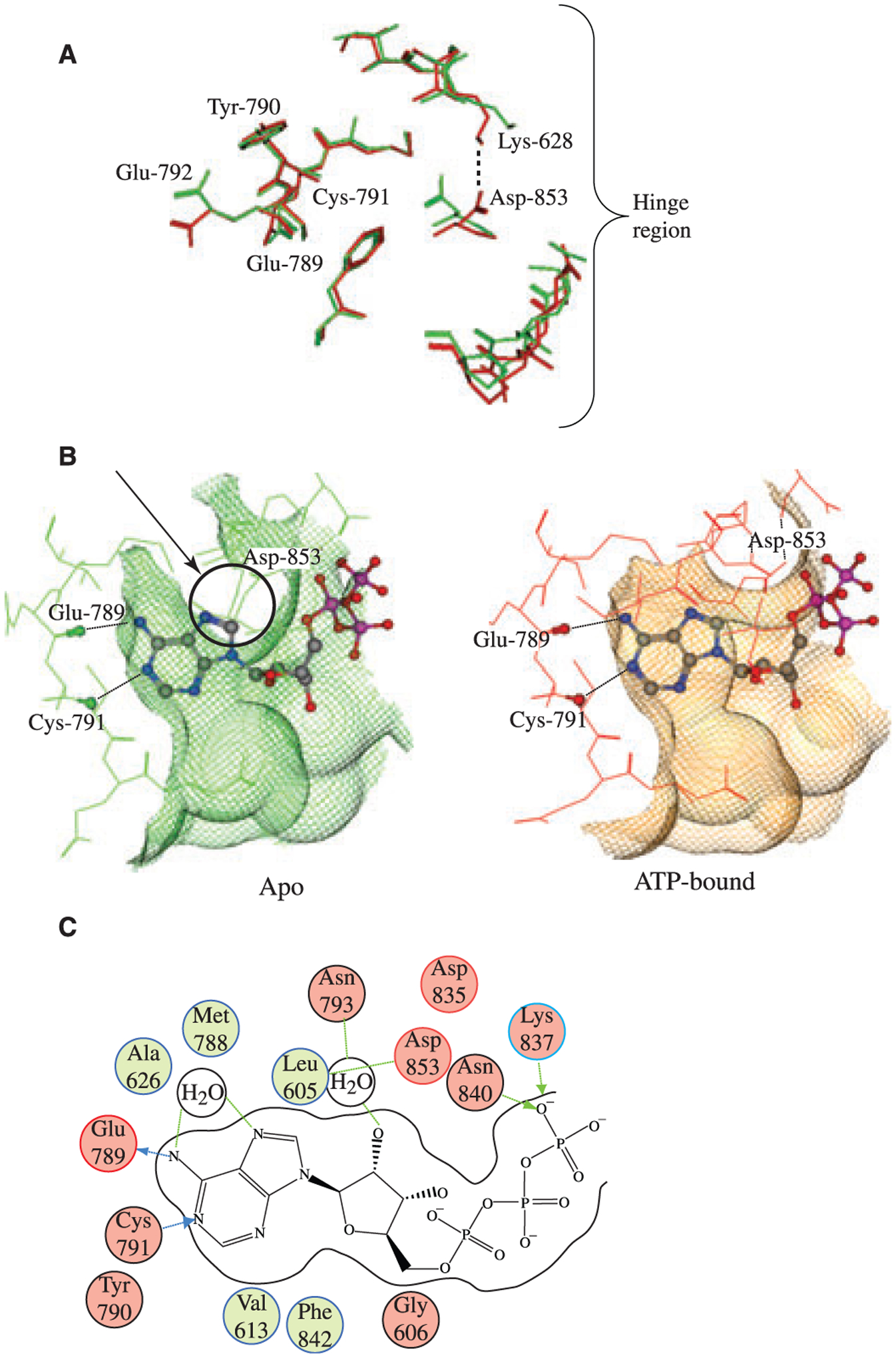
Analysis of the GCN2 ATP binding pocket. (A) Comparison of apo (green) and ATP bound (red) GCN2 structures. The most striking feature of pocket motion upon binding is the rotation of ASP-853 towards LYS-628, which opens up the binding pocket in the hinge region and allows ATP to bind. (B) Top view of apo (red) and ATP bound (green) GCN2 superimposed with bound ATP. In apo GCN2 the ASP-853 residue is in a conformation that would sterically clash with ATP. In order to bind ATP, the ASP-853 residue (indicated by an arrow and circled) must rotate out of position in order to open up the pocket around the hinge region, as demonstrated by the ATP bound structure (green). The conformation of the ASP-853 residue in the bound GCN2 structure may be partially stabilized by ionic/H-bond interaction with LYS-628; upon binding, LYS-628 rotates away from its conformation in the apo GCN2 structure and adopts a conformation more suitable for interaction with ASP-853. (C) 2D diagram of ATP interactions with GCN2. The canonical ligand H-bonds to the hinge region are made with the backbone atoms of GLU-789 and CYS-791. The diagram also highlights the water-mediated H-bonds between the purine ring on ATP and the ASP-853 and MET-788 residues backbone. This water molecule is conserved in both the apo and the bound GCN2 structures.
Figure 5:
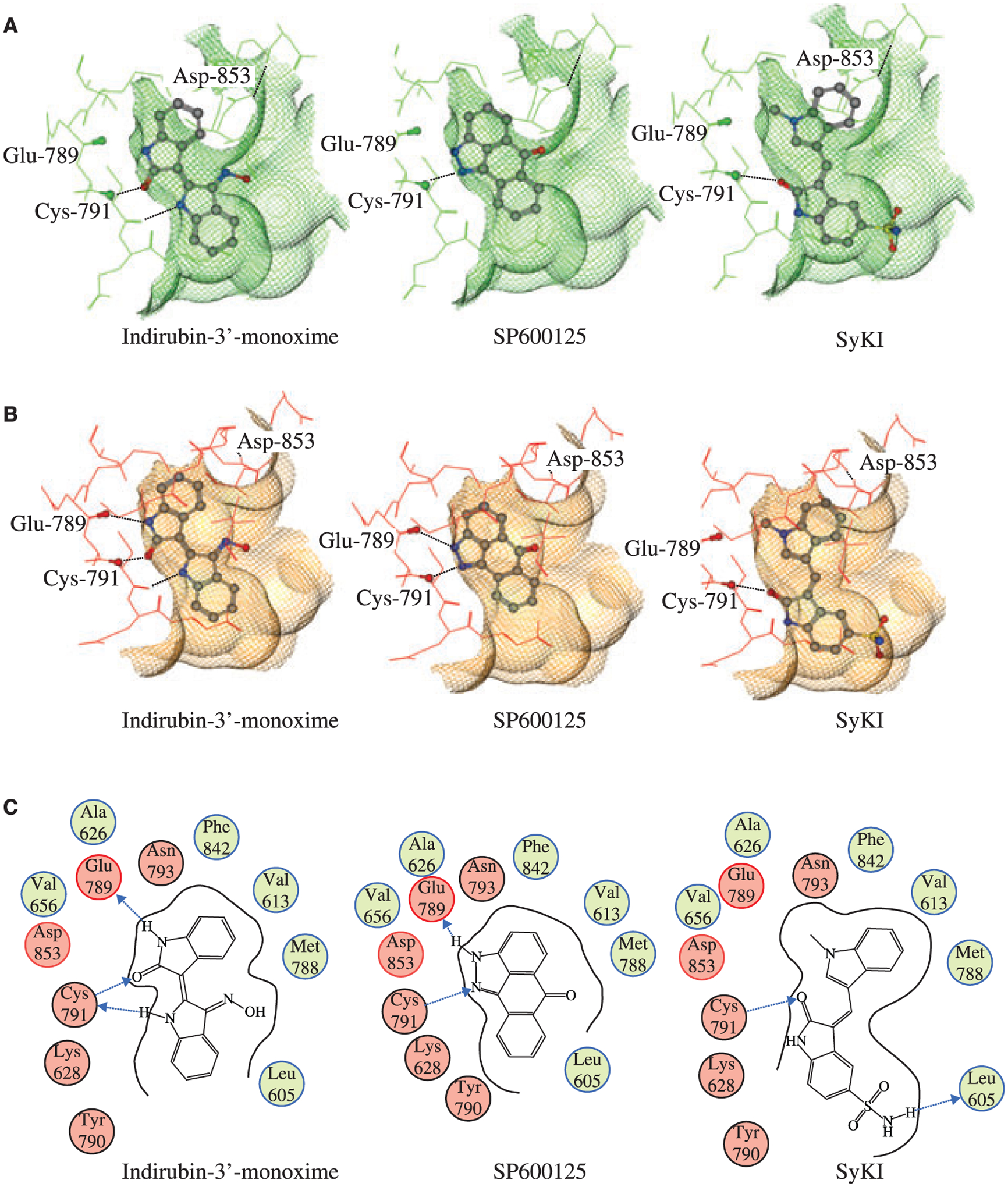
Modeling of indirubin-3′-monoxime, SP600125, and SyKI into the ATP-binding pocket of GCN2. (A) Ligand poses in the apo form of GCN2. (B) Ligand poses in the ATP bound form of GCN2. (C) 2D diagram of indirubin-3′-monoxime, SP600125, and SyKI interacting with GCN2.
Small molecule docking to the ATP-binding site of GCN2
Since indirubin-3′-monoxime, SP600125, and SyKI identified herein are known kinase inhibitors, a similar search to what has been previously described (28) was performed on the PDB database for kinase structures similar in sequence to GCN2 containing bound ligands similar to the compounds described herein. For each of the compounds the ‘best’ representative PDB complex was chosen based on structural similarity of the binding pocket in the hinge region. In the case of SP600125 and indirubin-3′-monoxime, but not that of SyKI, an exact match was found: the structure of indirubin-3′-monoxime in complex with GSK-3β (PDB 1Q41) (29) and that of SP600125 in complex with JNK3 (PDB 1PMV) (30). Docked poses were generated as described in Methods and Materials and they are given for both the apo GCN2 structure and the ATP bound GCN2 structure (Figure 5). Van der Waals interaction surfaces drawn as a grid around the pockets allow assessment of the binding poses by visual inspection. The models generated show that indirubin-3′-monoxime as well as SP600125 are able to make hydrogen bonding to the same residues in GCN2 involved in the interaction with ATP, namely, GLU-789 and CYS-791. Also, it can be seen that indirubin-3′-monoxime, but not SP600125, exhibit significant VDW clashes with the apo GCN2 binding pocket indicating that, as with ATP, the binding of indirubin-3′-monoxime would require, at a minimum, a conformational relocation of ASP-853 before binding would be possible. Since no relevant structures were retrieved of SyKI bound to a kinase, we used the docking of indirubin-3′-monoxime to GCN2 as a starting point, since this compound shares common structural features with SyKI. However, in this case, clashes between the GCN2 pocket and SyKI appeared. Interestingly, the crystal structure of the PIM1 kinase bound to PLX-K031 (PDB 1YXX, (31)), an indirubin-3′-monoxime analog, shows that the orientation of the molecule is inverted compared that of indirubin-3′-monoxime bound to GSK-3Beta (29). We thus used this structure to generate a starting orientation of SyKI that is rotated 180° from the proposed indirubin-3′-monoxime pose. The proposed rotation of the molecule produced a pose with both a good fit into the GCN2 binding pocket as well as formation of a hydrogen bond with CYS-791. It should be noted that, like Indirubin-3′-monoxime, the SyKI would clash with the apo form of the GCN2 pocket.
Discussion
One of the key molecular mechanisms activated during cellular stress is eIF2α phosphorylation which leads to a rapid inhibition of protein synthesis (1). This mechanism is considered to be an efficient protective mechanism to prevent the translational machinery from continuing to utilize vital resources in situations where these are limiting or during viral infection. Despite the obvious advantages of inducing eIF2α phosphorylation to protect cells under stress, recent findings uncovered that preventing GCN2-mdiated eIF2α phosphorylation in the brain improves long-term memory in mice (32). We describe herein the identification of three small-molecule inhibitors of the eIF2α kinase GCN2 that inhibit eIF2α phosphorylation in vitro and in vivo. SP600125 was originally identified from a screen for Jun N-terminal kinase inhibitor and found to target proteins of that family (33). Indirubine-3′-monoxime is an inhibitor of CDK family kinases and later found to also target GSK3β (34). SyKI was obtained from a screen for inhibitors of SYK (26). SP600125 and indirubin-3′-monoxime have been shown to bind to the ATP-binding pocket of their respective target. SyKI is likely to bind to the same domain since it is a nucleotide analog, although this remains to be formally demonstrated. Although we did not pursue the characterization of staurosporine as an inhibitor of GCN2, we note that the eIF2α kinases, PKR and HRI have been previously shown to be resistant to this inhibitor suggesting intrinsic differences in the structure of the ATP-binding domain of PKR and HRI compared with GCN2 (35).
During the course of a study aimed at characterizing UV-induced eIF2α phosphorylation, Deng et al. (14) used SP600125 to verify the involvement of the JNK kinase on eIF2α phosphorylation (since this compound was initially identified as a JNK inhibitor). In the study by Deng et al. (14), SP600125 did not prevent the phosphorylation of eIF2α following UV irradiation. However, in our hands, SP600125 was capable of blocking GCN2-mediated phosphorylation of eIF2α (Figure 3). One major difference between our study and that of Deng et al. (14) is that the latter used NIH3T3 cells whereas we used MEFs in our study. To assess if the observed differences could be due to the cell lines used, we UV irradiated NIH3T3 or MEF in the presence or absence of SP600125 and determined the extent of eIF2α phosphorylation. As observed by Deng et al. (14), we found that in NIH3T3 cells, SP600125 does not prevent eIF2α phosphorylation compared with MEFs (F.R., data not shown).
The mechanism of activation of GCN2 by UV-light remains unclear, but might involve the accumulation of cleaved tRNA by the RNAse angiogenin following cellular stresses (36). During amino acid starvation, the (HisRS)-like domain of GCN2 binds to uncharged tRNAs and activates the kinase domain through a proposed two-step mechanism (18). When inactive, the kinase domain of GCN2 remains closed by non-covalent interactions between specific residues that prevent ATP binding (18). Activation of GCN2 induces a movement of the hinge region to allow a productive ATP-binding conformation followed by the autophosphorylation of GCN2 and eIF2α binding and phosphorylation. Based on our modeling results, we propose that, like ATP, Indirubin-3′-monoxime and SyKI can only interact with GCN2 when it is activated because the closed conformation of the kinase domain in the inactivated state could not accommodate the compounds. Since it can fit in both GCN2 conformations, SP600125 would however be able to freely bind GCN2 and prevent its activation. It is noteworthy that SP600125 is more potent at preventing eIF2α phosphorylation in cells than the two other compounds and could happen if SP600125 bound both GCN2 conformations.
Bain et al. have tested the potency of a number of previously identified kinase inhibitors against a large panel of kinases (37). Included in their analysis were indirubin-3′-monoxime and SP600125, but not SyKI. Both indirubin-3′-monoxime and SP600125 inhibited a number of other kinases indicating cross-specificity and underscores the need to craft these compounds into more specific inhibitors of GCN2. Our in silico analysis of these compounds complexed to GCN2 represents a first-step in this direction. Analysis of the computer-generated models for the GCN2-inhibitor complexes revealed that all three compounds are well-accommodated in the ATP-binding site. The ATP-bound form of the protein was initially chosen for the docking studies, mainly because the binding pocket in the apo structure of GCN2 is quite closed and cannot accommodate ATP. Thus, most ligands would not dock to the closed form in any reasonable fashion. However, since some of the active kinase inhibitors were relatively small molecules, docking to the apo structure was performed in order to see if it was possible for any of these molecules to fit into the closed form of the binding pocket (based on their docked pose in the open form). We note that in the case of indirubin-3′-monoxime and SyKI, a small rearrangement of the ATP-binding pocket of GCN2 is required for the proper binding of the molecules. This is somewhat expected, since as previously discussed, the GCN2 ATP-binding pocket must undergo conformational rearrangement in order to bind ATP. Interestingly, compound SP600125 seems able to fit into the apo GCN2 binding pocket without VDW clashes, suggesting that unlike ATP and the other ligands discussed here, SP600125 may be able to bind to the GCN2 pocket without any conformational rearrangements. In the ATP-bound form of GCN2, the posed ligands indicated that the compounds do not form any VDW clashes with the GCN2 pocket, suggesting that these ligands (at least in these poses) could be accommodated by the same GCN2 pocket conformation that binds ATP. Further structure–activity relationship exploration around these scaffolds would be necessary to clearly establish the reliability of the predicted poses. However, there is some experimental evidence supporting our modeling results from the fact that similar scaffolds bind to kinases in similar fashions. This is referred to as ‘knowledge-based’ prediction of kinase inhibitor binding modes as outlined in (27). In sum, our work provides the molecular basis for undertaking more detailed and intensive structure–activity relationships of these compounds to develop more specific and potent inhibitors of GCN2.
Acknowledgments
This work was supported by a CIHR Team Grant (#CTP-79858) on the Molecular Basis of Translational Control of Memory Formation, and by NIH grant RO1 GM061262 (SKB).
References
- 1.Wek RC, Jiang HY, Anthony TG (2006) Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans;34: 7–11. [DOI] [PubMed] [Google Scholar]
- 2.Hinnebusch AG (2000) Mechanism and regulation of methionyl-tRNA binding to ribosomes. In: Sonenberg N, Hershey JWB, Mathews MB, editors. Mechanism and Regulation of Methionyl-TRNA Binding to Ribosomes. Cold Spring Harbor, NY: Cold Spring harbor Laboratory Press; p. 184–243. [Google Scholar]
- 3.Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG (2001) Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol;21:5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barber GN (2005) The dsRNA-dependent protein kinase, PKR and cell death. Cell Death Differ;12:563–570. [DOI] [PubMed] [Google Scholar]
- 5.Wek RC, Cavener DR (2007) Translational control and the unfolded protein response. Antioxid Redox Signal;9:2357–2371. [DOI] [PubMed] [Google Scholar]
- 6.Chen JJ (2007) Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood;109: 2693–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, Jefferson LS, Cavener DR (2002) The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol;22:6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu L, Han AP, Chen JJ (2001) Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol Cell Biol;21:7971–7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ito T, Yang M, May WS (1999) RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J Biol Chem;274:15427–15432. [DOI] [PubMed] [Google Scholar]
- 10.Patel CV, Handy I, Goldsmith T, Patel RC (2000) PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem;275:37993–37998. [DOI] [PubMed] [Google Scholar]
- 11.Yang R, Wek SA, Wek RC (2000) Glucose limitation induces GCN4 translation by activation of Gcn2 protein kinase. Mol Cell Biol;20:2706–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berlanga JJ, Santoyo J, De Haro C (1999) Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2alpha kinase. Eur J Biochem/FEBS;265:754–762. [DOI] [PubMed] [Google Scholar]
- 13.Jiang HY, Wek RC (2005) Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J Biol Chem;280:14189–14202. [DOI] [PubMed] [Google Scholar]
- 14.Deng J, Harding HP, Raught B, Gingras AC, Berlanga JJ, Scheuner D, Kaufman RJ, Ron D, Sonenberg NI (2002) Activation of GCN2 in UV-irradiated cells inhibits translation. Curr Biol;12:1279–1286. [DOI] [PubMed] [Google Scholar]
- 15.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev;12:982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, Bidinosti M (2007) eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell;129:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M et al. (2005) Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature;436:1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padyana AK, Qiu H, Roll-Mecak A, Hinnebusch AG, Burley SK (2005) Structural basis for autoinhibition and mutational activation of eukaryotic initiation factor 2alpha protein kinase GCN2. J Biol Chem;280:29289–29299. [DOI] [PubMed] [Google Scholar]
- 19.Trepalin SV, Gerasimenko VA, Kozyukov AV, Savchuk NP, Ivaschenko AA (2002) New diversity calculations algorithms used for compound selection. J Chem Inf Comput Sci;42:249–258. [DOI] [PubMed] [Google Scholar]
- 20.Cencic R, Robert F, Pelletier J (2007) Identifying small molecule inhibitors of eukaryotic translation initiation. Methods Enzymol; 431:269–302. [DOI] [PubMed] [Google Scholar]
- 21.Zhu S, Sobolev AY, Wek RC (1996) Histidyl-tRNA synthetase-related sequences in GCN2 protein kinase regulate in vitro phosphorylation of eIF-2. J Biol Chem;271:24989–24994. [DOI] [PubMed] [Google Scholar]
- 22.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell;6:1099–1108. [DOI] [PubMed] [Google Scholar]
- 23.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol;215:403–410. [DOI] [PubMed] [Google Scholar]
- 24.Qiu H, Hu C, Dong J, Hinnebusch AG (2002) Mutations that bypass tRNA binding activate the intrinsically defective kinase domain in GCN2. Genes Dev;16:1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Brian CA, Ward NE (1990) Staurosporine: a prototype of a novel class of inhibitors of tumor cell invasion? J Natl Cancer Inst;82:1734–1735. [DOI] [PubMed] [Google Scholar]
- 26.Lai JY, Cox PJ, Patel R, Sadiq S, Aldous DJ, Thurairatnam S et al. (2003) Potent small molecule inhibitors of spleen tyrosine kinase (Syk). Bioorg Med Chem Lett;13:3111–3114. [DOI] [PubMed] [Google Scholar]
- 27.Zheng J, Knighton DR, ten Eyck LF, Karlsson R, Xuong N, Taylor SS, Sowadski JM (1993) Crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MgATP and peptide inhibitor. Biochemistry;32:2154–2161. [DOI] [PubMed] [Google Scholar]
- 28.Ghose AK, Herbertz T, Pippin DA, Salvino JM, Mallamo JP (2008) Knowledge based prediction of ligand binding modes and rational inhibitor design for kinase drug discovery. J Med Chem;51:5149–5171. [DOI] [PubMed] [Google Scholar]
- 29.Bertrand JA, Thieffine S, Vulpetti A, Cristiani C, Valsasina B, Knapp S, Kalisz HM, Flocco M (2003) Structural characterization of the GSK-3beta active site using selective and non-selective ATP-mimetic inhibitors. J Mol Biol;333:393–407. [DOI] [PubMed] [Google Scholar]
- 30.Scapin G, Patel SB, Lisnock J, Becker JW, LoGrasso PV (2003) The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem Biol;10:705–712. [DOI] [PubMed] [Google Scholar]
- 31.Kumar A, Mandiyan V, Suzuki Y, Zhang C, Rice J, Tsai J, Artis DR, Ibrahim P, Bremer R (2005) Crystal structures of proto-oncogene kinase Pim1: a target of aberrant somatic hyper-mutations in diffuse large cell lymphoma. J Mol Biol;348:183–193. [DOI] [PubMed] [Google Scholar]
- 32.Costa-Mattioli M, Sonenberg N (2008) Translational control of gene expression: a molecular switch for memory storage. Prog Brain Res;169:81–95. [DOI] [PubMed] [Google Scholar]
- 33.Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW (2001) SP600125, an anthrax-pyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci USA;98:13681–13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, Eisen-brand G, Meijer L (2001) Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors?. J Biol Chem;276:251–260. [DOI] [PubMed] [Google Scholar]
- 35.Martin de la Vega C, Garcia A, Martin ME, Alcazar A, Marin O, Quevedo C, Salinas M (1999) Resistance of initiation factor 2 (eIF-2alpha) kinases to staurosporine: an approach for assaying the kinases in crude extracts. Cell Signal;11:399–404. [DOI] [PubMed] [Google Scholar]
- 36.Yamasaki S, Ivanov P, Hu GF, Anderson P (2009) Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol;185:35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bain J, McLauchlan H, Elliott M, Cohen P (2003) The specificities of protein kinase inhibitors: an update. The Biochemical journal;371:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]