

Abstract
Failure to regenerate myocardium after injury is a major cause of mortality and morbidity in adult humans. Direct differentiation of human induced pluripotent stem cells (iPSCs) into cardiomyocytes provides an invaluable resource to pursue cell-based cardiac regeneration. Beyond the potential for clinical therapies, iPSC technology also enables the generation of cardiomyocytes, which recapitulates patient-specific phenotypes, thus presenting a powerful in-vitro cell-based model to understand disease pathology and guide precision medicine. Here we describe protocols for reprogramming human dermal fibroblasts and blood cells into iPSCs by the non-integrative Sendai virus system and for the monolayer differentiation of iPSCs to cardiomyocytes using chemically defined media.
Keywords: Heart regeneration, Induced pluripotent stem cells (iPSCs), Cardiomyocytes, Patient-specific, Reprogramming, Differentiation, Sendai virus, Chemically defined, Monolayer differentiation
1. Introduction
Induced pluripotent stem cells (iPSCs) provide an unprecedented opportunity for understanding heart physiology and diseases, cardiovascular regeneration, and drug discovery[1-6]. In 2006, Takahashi and Yamanaka et al. revolutionarily transformed mouse fibroblasts to embryonic stem cell-like pluripotent cells using four transcription factors: OCT4 (POU5F1), SOX2, KLF4, and MYC[7]. In 2007, two independent groups Takahashi et al. and Yu et al. successfully generated human induced pluripotent cells from fibroblasts[8,9]. These iPSCs resemble embryonic stem cells (ESCs) in that they possess the potential to undergo indefinite self-renewal and have the ability to differentiate into any specialized cell lineage[10]. Most importantly, iPSCs circumvent the ethical concerns and immunogenicity of human ESCs, thus becoming an attractive alternative for autologous tissue repair and regeneration[1,11]. Advances in Yamananka’s original method have reduced the possibility of genome instability and DNA aberration[12-17]. Delivery methods have evolved from integrative viral vectors (retrovirus and lentivirus) to non-integrative systems, including episomal DNA vectors, synthetic mRNA, protein, and Sendai virus (SeV)[16-18,15,19]. With progress in the iPSC field, generation of iPSCs will be safe and efficient for clinical applications.
The principle of differentiating iPSCs into cardiomyocytes is to recapitulate key steps in embryonic cardiac development, leading from the formation of mesodermal cells to functional cardiomyocytes[20,21]. These steps involve stage-specific activation and inhibition of specific signaling pathways with defined growth factors. Stimulation of activin/Nodal, BMP (Bone Morphogenetic Protein), Wnt (wingless/INT protein) and FGF (Fibroblast Growth Factor) signaling promotes cardiac mesoderm formation[22,23]. Subsequent inhibition of Wnt, BMP, and TGF3β induces cardiac specification[24,23]. In the most recent development of monolayer differentiation techniques, the combination of growth factors was replaced by small molecules that modulate only WNT signaling[25,26]. The purity of cardiomyocytes from these protocols was enhanced by using glucose-deprived culture media containing abundant lactate, which favors the survival of cardiomyocytes over other cardiac cell lineages[27]. These chemically defined protocols have largely reduced cost and labor while increasing consistency for large-scale de novo production of cardiomyocytes.
In this chapter, we describe a detailed protocol to reprogram human iPS cells from dermal fibroblasts and blood cells by using a non-integrative Sendai (SeV) virus encoding the four Yamanaka factors (OCT4, SOX2, KLF4 and c-MYC). We also lay out a thorough procedure for differentiation of iPSCs into high-purity functional cardiomyocytes using a chemically defined monolayer method.
2. Materials
2.1. For Fibroblast transduction:
- Reagents:
- Sendai virus (encoding OSKM factors)
- Inactivated MEF cells (feeder cells)
MEF Media: 90% DMEM; 10% Heat Inactivated Fetal Bovine Serum; 1% L-glutamine;1% Antibiotic-Antimyotic Solution.
ES Media: 80% Knockout DMEM (or Knockout DMEM/F12); 20% Knockout Serum Replacer; 1% L-glutamine; 1% Non-essential Amino Acids; 1% Antibiotic-Antimyotic Solution; 10 ng/mL bFGF.
2.2. For Blood cell transduction:
- Reagents:
- Sendai virus (encoding OSKM factors)
- Inactivated MEF cells (feeder cells)
- Polybrene
Expansion Media: QBSF base medium; 1% Antibiotic-Antimyotic Solution; 10 μg/mL Ascorbic Acid; 10 ng/mL IL3; 2 U/mL EPO; 40 ng/mL IGF1; 1 μM Dexamethasone; 50 ng/mL SCF.
MEF Media: 8.7%IMDM base medium; 10% Heat Inactivated Fetal Bovine Serum ; 1% L-Glutamine; 1% Antibiotic-Antimyotic Solution; 1% Non-essential Amino Acids; 1.8 μl/ml 55mM 2-Mercaptoethanol (2-ME).
MEF Media with Cytokines: 8.7%IMDM base medium; 10% Heat Inactivated Fetal Bovine Serum; 1% L-Glutamine (Glutamax); 1% Antibiotic-Antimyotic Solution; 1% Non-essential Amino Acids; 1.8 μl/mL 55mM 2-ME; 10 μg/mL Ascorbic Acid; 10 ng/mL IL3; 2 U/mL EPO; 40 ng/mL IGF1; 50 ng/mL SCF; 1 μM Dexamethasone; 10 ng/mL bFGF.
ES Media: See section 2.1.
2.3. Checking transgene by RT-PCR
- Primers (human):
SeV Forward GGA TCA CTA GGT GAT ATC GAG C Reverse ACC AGA CAA GAG TTT AAG AGA TAT GTA TC SOX2 Forward ATG CAC CGC TAC GAC GTG AGC GC Reverse AAT GTA TCG AAG GTG CTC AA KLF4 Forward TTC CTG CAT GCC AGA GGA GCC C Reverse AAT GTA TCG AAG GTG CTC AA c-MYC Forward TAA CTG ACT AGC AGG CTT GTC G Reverse TCC ACA TAC AGT CCT GGA TGA TGA TG OCT3/4 Forward CCC GAA AGA GAA AGC GAA CCA G Reverse AAT GTA TCG AAG GTG CTC AA Program: 94 for 5min; 30-35 cycles of: 95 for 30 seconds, 55 for 30 seconds, 72 for 30 seconds. Followed by 72 for 10mins, 4 °C forever.
2.4. Materials for the Maintenance of human iPSCs in a feeder-free system
mTESR 1 Kit (or other feeder-free hESC culture media); Matrigel hESC - qualified Matrix; ReLeSR; DMEM/F-12; ESC-qualified FBS; 70% Ethanol (EtOH)
DMSO.
2.5. Cardiomyocyte Differentiation Media and Reagents
2.5.1. Reagents
Matrigel-Growth Factor Reduced (GFR)
ROCK inhibitor (Y-27632)
PBS
ReLeSR dissociation reagent
STOP media (DMEM/FBS 1:1 mix)
TrypLE Express Enzyme (1x)
DNase
2.5.2. Media
RPMI1640 B-27 minus Insulin: RPMI1640; 2% B-27 supplement minus insulin; 1.5% Antibiotic-Antimycotic.
RPMI1640 B-27 with Insulin: RPMI1640; 2% B-27 Supplement; 1.5% Antibiotic-Antimycotic.
CM Enrichment Media: RPMI 1640 w/o Glucose; 2% B-27 supplement; Sodium-D-Lactate (final concentration 5mM); 1.5% Antibiotic-Antimycotic.
3. Methods
3.1. Fibroblast Transduction
Workflow see Figure 1.
Figure 1. Workflow of reprogramming human dermal fibroblast cells into iPSCs.

D=Day.
A. Fibroblast preparation
- Day -5:
- Coat a 10 cm culture dish with 1% gelatin and allow it to sit for 1 hr at room temperature.
- Thaw fibroblast cells: Remove 1 vial of MEF from liquid nitrogen and quick thaw in 37 °C of water bath. Transfer to a 15-mL conical tube and add 2 mL of MEF media dropwise. Pipet up and down to mix. Spin at 200x g for 5 min at room temperature.
- Remove supernatant and resuspend pellet in 10 mL of fresh MEF media.
- Plate cells onto a gelatin-coated 10-cm dish and incubate at 37 °C 5% CO2 until confluent.
- Day -2:
- Remove MEF media and rinse with 3 mL of PBS. Add 2 mL of TrypLE and incubate at 37 °C 5% CO2 for approximately 2-3 min until cells have lifted from the bottom of the dish.
- Add 2 mL of MEF media to stop reaction and transfer to 15cc conical tube.
- Add 1 mL of MEF media to the plate to rinse the remaining cells from the dish and add to the conical tube.
- Spin cells at 200x g for 5 min at RT. Decant supernatant and add fresh MEF media.
- Plate cells onto 2 wells of a gelatin-coated 6-well plate at a density to achieve approximately 5x105 (80-90% confluency) per well after 48 hr (Figure 2a).
Figure 2. Reprogramming of human dermal fibroblast cells into iPSCs.
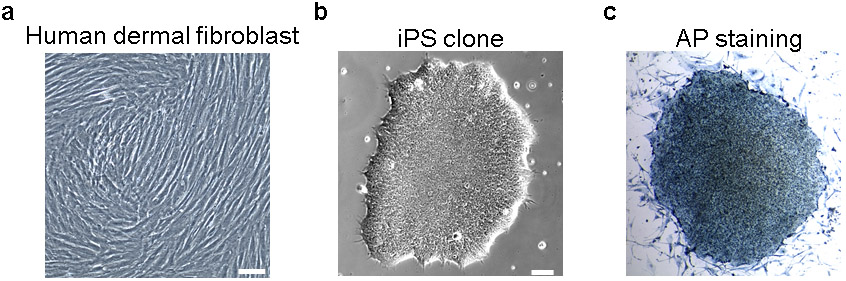
(a) Freshly isolated human dermal fibroblast cells in vitro. (b) Representative image of human iPS clone derived from fibroblast cells by using Sendai virus encoding the four Yamanaka factors. (c) Characterization of iPS clone shows positive alkaline phosphatase (AP) staining. Scale bars, 100 μm.
B. Transduction
- Day 0: Perform transduction
- Warm 2 mL of MEF media.
- Add Sendai virus at a MOI of 3 to 2 mL of prewarmed MEF media (See NOTE 1).
- Thoroughly mix by gently pipetting up and down.
- Within 5 min of mixing, aspirate MEF media from fibroblast cells. Add 1 mL of the virus mixture to each well. Incubate overnight at 37 °C 5% CO2.
- Day 6: Prepare feeder dishes
- Prepare 3 x 10 cm gelatin-coated MEF dishes for each cell line that has been transduced. Coat dishes with 1% gelatin for 1 hr. Plate 0.9-1x106 of inactivated MEFs per dish. Incubate until ready for use the next day.
- Day 7: Plate transduced cells onto 10 cm feeder plates
- Remove cells by adding 500 μL of TrypLE.
- Incubate at room temperature for 1-3 min until the cells have rounded.
- Add 1 mL of MEF media. Collect the cells and transfer to a 15 mL conical tube.
- Rinse wells with an additional 1 mL of MEF media. Spin cells at 200 x g for 5 mins.
- Count the cells and plate at a density of 5x104, 1x105, and 2x105 on the 3 feeder dishes containing inactivated MEFs. Add 8-10 mL of MEF media. Incubate the plates at 37°C, 5% CO2 overnight. (See NOTE 4).
- Day 8 to 28: Feeding and monitoring cells
- Change to ES media 24 hr after plating onto feeders.
- Replace media every other day.
- Start observing cells for colony formation on day 8. It may take up to 4 weeks before the clones will begin to appear.
C. Clone Expansion
Picking clones
Once the clones have reached the appropriate size, seed a 12-well plate with inactivated MEF cells 1-2 days prior to picking the clones.
Identify several clones that are large enough to passage and mark them using either a cell marker or felt-tip pen.
Cut one clone into several smaller pieces using a finely pulled glass pipet with a sealed tip. Transfer all pieces into 1 well of the 12-well feeder plate containing 1 mL of ES media.
Proceed to transfer the remaining marked colonies in the same manner, allowing for 1 clone per well.
Number each well of the 12-well plate to identify individual clones.
Incubate plate for 48 hr before changing media to allow the pieces to attach. Continue with media changes every day.
Continue to passage, expand, and maintain the newly named clones using standard culture procedures for ES cells (Figure 2b).
D. Confirmation of vector-free iPS clones:
After 10 passages, check iPS clones for removal of the Sendai reprogramming vectors by performing RT-PCR.
If c-MYC is still present after 10 passages, you can enhance removal by incubating the cultures at 38-39 °C for 5 days. (c-MYC contains a temperature-sensitivity mutation, whereas the other genes do not.) Repeat RT-PCR.
Continue confirmation assays by performing AP staining (Figure 2c), qRT-PCR, and immunocytochemistry (Figure 3) with pluripotency markers.
Figure 3. Human iPSCs express pluripotency markers.

Human iPSCs show positive staining for the nuclear marker Oct-4 (green, top) and the cytoplasmic marker TRA-1-60 (red, bottom) determined by immunofluorescent microscopy. Scale bars, 100 μm.
3.2. Blood Cell Transduction
A. PBMC Isolation and Expansion
Day-14: Isolation
Mix blood 1:1 with warm PBS.
Gently layer over 10 mL of warm Histopaque.
Spin at 300-400x g for 30 consecutive min.
Use a sterile pipet to collect the buffy coat at the center of the interfaces.
Increase the volume by adding 5 mL of sterile PBS.
Spin at 300x g for 10 min.
- If necessary, perform RBC lysis.
- - Add 5 mL of RBC lysis buffer and incubate RT for 10 min. Fill with PBS and spin 300x g for 10 min.
Remove the supernatant and resuspend in 10 mL of sterile PBS. Spin 300x g 10 min.
Repeat the above wash step. Discard supernatant, resuspend in 5 mL of PBS, and count the cells.
Transfer 1-2 x 106 cells to a conical tube and spin at 300x g for 10 min.
Resuspend in 2 mL of EM media and transfer to 1 well of a 12-well plate. Incubate for 3 days.
Freeze remaining cells at ~2x106 using 90% FBS and 10% DMSO.
B. PBMC Expansion:
- Day -11: Expansion
- Transfer the cells into a sterile 15 mL conical tube.
- Wash the well once with 1 mL of QBSF base media to collect any adherent cells and add to the conical tube.
- Spin cells at 300x g for 10 min.
- Resuspend in 2 mL of EM and plate the cell suspension in 1 well of fresh 12-well plate.
- Day -8 and day -4:
- Repeat procedure from day -11.
C. Transduction:
- Day 0: Transduction
- Count the cells, transfer 2.5-5.0 x 105 to a conical tube, and spin at 300x g for 10 min.
- Warm 1 mL of EM.
- Add Sendai virus at a MOI of 3 to 2 mL of prewarmed MEF media (See NOTE 1).
- Add 4 μg/mL of polybrene.
- Within 5 min, decant supernatant from spun cells and resuspend in approximately 300 μL of virus mixture, mix gently.
- Plate each line onto 1 well of 24-well plate. Incubate overnight at 37 °C 5% CO2.
- Day 1: Replace media
- 24 hr after transduction, collect the cells in a 15-ml conical tube and spin at 300x g for 10 min.
- Resuspend the cells in 500 μL EM media and replate in a 24-well plate.
- Incubate until Day 3 (See NOTE 7).
- Day 3: Replace media
- Collect the cells and spin at 300x g for 10 min.
- Count the cells and plate 2 x105 onto 10-cm feeder plate using 10 mL of MEF media containing cytokines and 10 ng/ml bFGF. Incubate for 48 hr.
- Day 5: Replace and change media
- Aspirate the media and replace it with 10 mL of MEF media containing 10 ng/mL bFGF (NO CYTOKINES).
- Incubate for 48 hr.
- Day 7: Replace and change media
- Aspirate the media and replace it with 10 mL of media made up of 50% MEF media containing bFGF and 50% ESC media.
- Incubate for 48 hr.
- Day 9: Change media
- Replace the media with 10 mL of ESC media.
- Continue to replace the media daily, watching for the development of clones.
- Day 15+: Clone formation
- Clones should be reaching appropriate sizes for transfer. To help avoid differentiation, try to transfer clones close to 3 weeks or later post-transduction.
D. Clone Expansion:
Picking clones:
See section 3.1C.
E. Confirmation of vector-free iPS clones:
See section 3.1D.
3.3. Maintenance of human iPSCs
A. Prepare Matrigel plates
Prepare Matrigel (0.5 mg for 6 mL of DMEM/F-12) by thawing Matrigel aliquot(s) on ice.
Dilute the Matrigel into the appropriate amount of ice cold DMEM/F-12 according to the protein concentration on manufacturer’s protocol.
Distribute the diluted Matrigel into the appropriate number of wells for passaging iPS cells. 2 mL for each well of a 6-well plate.
-
Let the plates sit for a minimum of 1 hr under the hood prior to use.
(Wrap plates in Parafilm and store at 4 °C for up to 2 weeks. Bring to RT prior to use.)
B. Passaging of iPSCs
(1 well of a 6-well plate at optimal density of 70-80%; cells should be split every 5-7 days.)
Aspirate the growth media from the currently growing undifferentiated iPSCs and rinse with PBS.
Add 1 mL of room temperature ReLeSR to each well of a 6-well plate.
Aspirate off the ReLeSR and incubate at 37 °C for 5 min.
After incubation, gently tap the plate and add 1 mL of the culture media.
Gently pipet up and down 5x with a P1000 pipet to break colonies into small clumps (See NOTE 8)
Transfer the cell suspension to a 1.5-mL microcentrifuge tube.
Pipet up and down gently 3-5x to ensure that cells are in small clusters. (5-10 cells/cluster).
Obtain new Matrigel coated plate(s). Aspirate off the Matrigel. Pipet the cell suspension up and down 1-2 times gently, taking care not to break up cell aggregates. Evenly divide the cell suspension into the new well(s) (See NOTE 9).
Add culture media so that each well has a total volume of 2-3 mL (See NOTE 10).
Move the plate quickly back and forth and side to side to disperse the cells evenly across the well surface. Place in a 37 °C, 5% CO2 incubator.
Change the media daily and check for contamination under an inverted phase contrast microscope.
3.4. Human cardiomyocyte differentiation
Workflow see Figure 4.
Figure 4. Workflow of iPSC differentiation to cardiomyocytes using a chemically defined method.
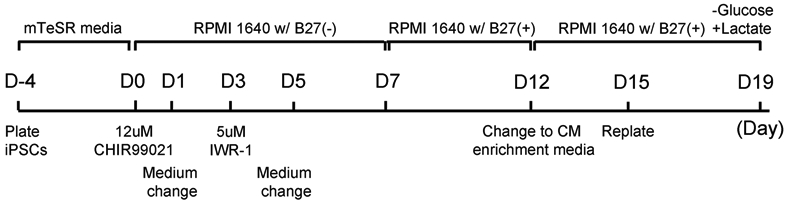
D=Day.
A. Matrigel plate preparation
Prepare the Matrigel as done in Section 3.3.
B. iPS passaging
DAY -4:
Refer to section 3.3B.
Evenly distribute the cell suspension into Matrigel-coated wells/plates to achieve 80-90% confluency within 4 days (See NOTE 11).
Add culture media containing 10 μM of ROCK inhibitor to bring the final volume for each well of a 6-well plate to 2 mL (1 mL for one 12-well plate).
Move the plate quickly back and forth and front to back 5x to ensure even distribution of cells around the wells.
Incubate at 37°C 5% CO2.
Replace the media with fresh culture media on the following day.
Continue with daily media changes until Day 0, when cells should be ~70-80% confluent.
C. Differentiation
DAY 0: (CHIR99021) Cells should be at least 80% confluent.
- Aspirate the culture media and replace with RPMI 1640 w/ B-27 (−) insulin media.
- 2 mL for each well of 12-well plate
- 4 mL for each well of 6-well plate
Add 12 μM of CHIR99021 (10mM CHIR99021 stock).
Incubate for 24 hr.
DAY 1: (Media change)
Aspirate off media containing the CHIR99021 and replace with fresh RPMI 1640 w/ B-27 (−).
Incubate for 48 hr.
DAY 3: (IWR-1)
Change the media on all wells to fresh RPMI1640 w/ B-27 (−).
Add 5 μM of IWR-1 (10 mM IWR-1 stock).
Incubate for 48 hr.
DAY 5: (Media change)
Remove the IWR-1 media from all wells and add fresh RPMI 1640 w/ B-27 (−).
DAYS 7 and 10: (Media change with Insulin)
Remove RPMI 1640 w/ B-27 (−) and add fresh RPMI 1640 w/ B-27 supplement media. You should be able to see cells starting to contract between days 8 & 9 (See NOTE 12).
DAY 12: (Media change with lactate for CM enrichment)
Remove the RPMI 1640 w/ B-27 supplement and replace it with CM enrichment media.
-
Continue to culture in with CM enrichment media until the day of dissociation
(See NOTE 12).
DAY 15: (iPSC-CM dissociation)
D. CM Dissociation:
- Matrigel Plate Preparation
- Prepare Matrigel as done in Section 3.3.
- Distribute the diluted Matrigel into the number of wells needed for passaging cells.
- Let the plate sit for a minimum of 1 hr under the hood prior to use.
- Dissociation
- Wash the cells with PBS.
- Add 1 mL of TrypLE to each well and incubate for 5-7 min at 37 °C.
- After 5-7 mins, gently pipette up and down ~10x to dislodge and break up cell clumps.
- Incubate the plate for an additional 5-7 min.
- Add 1 mL of stop media containing 10 μg/ml of DNase and gently pipette up and down until all cells are dislodged from the bottom of the well and have dissolved into the cell suspension without visible clumps.
- Pass the cells through a 70-micron cell strainer inserted into a sterile 15-ml or 50-ml conical tube.
- Add an additional 1 mL of stop media to the well to rinse and filter.
- Spin at 600 x g for 8 min.
- Aspirate off the supernatant and resuspend the pellet by gently tapping bottom of tube.
- Add 1 mL of CM enrichment media.
- Count the cells.
- Plate the cells on the desired cell culture plates needed for experiments.
- Recommended densities:
- 24-well plate with 13 mm coverslips: 1-2x 105/well;
- 6-well plate: 0.8-1x 106/well.
- Confirm differentiation efficiency by flow cytometry or immunocytochemistry (Figure 5).
Figure 5. iPSC derived cardiomyocytes at Day 16 of differentiation.

More than 90% of the cells show positive staining for the cardiomyocyte marker α-actinin. iPSC derived cardiomyocytes exhibit sarcomere structure by immunofluorescent microscopy at high magnification (60x). Scale bars, 100 μm (brightfield); 20 μm.
Acknowledgement
This work was supported by the Richard King Mellon Foundation Institute for Pediatric Research (UPMC Children’s Hospital of Pittsburgh), by a Transatlantic Network of Excellence grant by Foundation Leducq (15CVD03), Children’s Cardiomyopathy Foundation, NIH grant R01HL106302 (to B.K.), and AHA Career Development Awards (to Lu H.). We thank Dan Roden and Kevin Bersell for generously sharing a human iPS cell line CiPS001-13 (Vanderbilt University). We thank Ashok Srinivasan for his critical reading of the chapter.
Footnotes
Cytotoxicity may be seen 24-48 hr post-transduction, which can affect up to 50% of the cells. This indicates high virus uptake.
High cell density before Day 5 may be seen. Do not plate onto feeders prior to Day 7.
This is to determine the correct plating density. Once it has been determined, 3 plates may not be necessary for future transductions.
During clone formation, if the feeder layer becomes overly spent and is thinning, add fresh feeders to the 10 cm plate.
After 3+ weeks post-transduction, if the feeder layer appears thick, gently peel away the areas where possible and replace with fresh feeders.
You may see drastic cell death (>60%) 24-48 hr post transduction. Large aggregated cells may be seen.
Differentiated colonies will remain attached.
The typical split-ratio is ~1:40 if passaging from a well that is approximately 70-80% confluent.
10 μM of ROCK inhibitor may be added to each well to ensure survival and reduce spontaneous differentiation.
The typical split ratio is ~1:30 if passaging from a well that is approximately 70-80% confluent.
Differentiation between Days 7-10 and 12-15: additional media changes with noted media maybe required if media turns yellow prior to indicated media change days.
References
- 1.Shi Y, Inoue H, Wu JC, Yamanaka S (2017) Induced pluripotent stem cell technology: a decade of progress. Nature Reviews Drug Discovery 16:115–130. doi: 10.1038/nrd.2016.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L (2011) Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471:225–229. doi: 10.1038/nature09747 [DOI] [PubMed] [Google Scholar]
- 3.Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang Y-S, Schaniel C, Lee D-F, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR (2010) Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465:808–812. doi: 10.1038/nature09005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kehat I, Kenyagin-Karsenti D, Snir M, Segev H, Amit M, Gepstein A, Livne E, Binah O, Itskovitz-Eldor J, Gepstein L (2001) Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. The Journal of clinical investigation 108:407–414. doi: 10.1172/JCI12131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL (2010) Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Research 4:107–116. doi: 10.1016/J.SCR.2009.11.004 [DOI] [PubMed] [Google Scholar]
- 6.Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, Mich-Basso J, Lis A, Hassan N, London B, Bett GC, Tobita K, Rasmusson RL, Yang L (2014) Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovasc Res 104 (2):258–269. doi: 10.1093/cvr/cvu205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126 (4):663–676. doi: 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- 8.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. doi: 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 9.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318 (5858):1917–1920. doi: 10.1126/science.1151526 [DOI] [PubMed] [Google Scholar]
- 10.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM (1998) Embryonic stem cell lines derived from human blastocysts. Science (New York, NY) 282:1145–1147. doi: 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- 11.van Laake LW, Passier R, Doevendans PA, Mummery CL (2008) Human Embryonic Stem Cell–Derived Cardiomyocytes and Cardiac Repair in Rodents. Circulation Research 102:1008–1010. doi: 10.1161/CIRCRESAHA.108.175505 [DOI] [PubMed] [Google Scholar]
- 12.Sánchez Alvarado A, Yamanaka S (2014) Rethinking differentiation: stem cells, regeneration, and plasticity. Cell 157:110–119. doi: 10.1016/j.cell.2014.02.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giorgetti A, Montserrat N, Aasen T, Gonzalez F, Rodríguez-Pizà I, Vassena R, Raya A, Boué S, Barrero MJ, Corbella BA, Torrabadella M, Veiga A, Izpisua Belmonte JC (2009) Generation of induced pluripotent stem cells from human cord blood using OCT4 and SOX2. Cell stem cell 5:353–357. doi: 10.1016/j.stem.2009.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K (2009) Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458:771–775. doi: 10.1038/nature07864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.FUSAKI N, BAN H, NISHIYAMA A, SAEKI K, HASEGAWA M (2009) Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proceedings of the Japan Academy, Series B 85:348–362. doi: 10.2183/pjab.85.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, Thomson JA (2009) Human induced pluripotent stem cells free of vector and transgene sequences. Science (New York, NY) 324:797–801. doi: 10.1126/science.1172482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warren L, Manos PD, Ahfeldt T, Loh Y-H, Li H, Lau F, Ebina W, Mandal PK, Smith ZD, Meissner A, Daley GQ, Brack AS, Collins JJ, Cowan C, Schlaeger TM, Rossi DJ (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell stem cell 7:618–630. doi: 10.1016/j.stem.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Kim CH, Moon JI, Chung YG, Chang MY, Han BS, Ko S, Yang E, Cha KY, Lanza R, Kim KS (2009) Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell 4 (6):472–476. doi: 10.1016/j.stem.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ban H, Nishishita N, Fusaki N, Tabata T, Saeki K, Shikamura M, Takada N, Inoue M, Hasegawa M, Kawamata S, Nishikawa S-I (2011) Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proceedings of the National Academy of Sciences of the United States of America 108:14234–14239. doi: 10.1073/pnas.1103509108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson EN (2006) Gene regulatory networks in the evolution and development of the heart. Science (New York, NY) 313:1922–1927. doi: 10.1126/science.1132292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abu-Issa R, Kirby ML (2007) Heart Field: From Mesoderm to Heart Tube. Annual Review of Cell and Developmental Biology 23:45–68. doi: 10.1146/annurev.cellbio.23.090506.123331 [DOI] [PubMed] [Google Scholar]
- 22.Johansson BM, Wiles MV (1995) Evidence for involvement of activin A and bone morphogenetic protein 4 in mammalian mesoderm and hematopoietic development. Mol Cell Biol 15 (1):141–151. doi: 10.1128/mcb.15.1.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G (2011) Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 8 (2):228–240. doi: 10.1016/j.stem.2010.12.008 [DOI] [PubMed] [Google Scholar]
- 24.Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE (2007) Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A 104 (23):9685–9690. doi: 10.1073/pnas.0702859104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, Raval KK, Zhang J, Kamp TJ, Palecek SP (2012) Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proceedings of the National Academy of Sciences of the United States of America 109:E1848–1857. doi: 10.1073/pnas.1200250109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC (2014) Chemically defined generation of human cardiomyocytes. Nat Methods 11 (8):855–860. doi: 10.1038/nmeth.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tohyama S, Hattori F, Sano M, Hishiki T, Nagahata Y, Matsuura T, Hashimoto H, Suzuki T, Yamashita H, Satoh Y, Egashira T, Seki T, Muraoka N, Yamakawa H, Ohgino Y, Tanaka T, Yoichi M, Yuasa S, Murata M, Suematsu M, Fukuda K (2013) Distinct metabolic flow enables large-scale purification of mouse and human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell 12 (1):127–137. doi: 10.1016/j.stem.2012.09.013 [DOI] [PubMed] [Google Scholar]