

Abstract
Sigma (σ) receptors represent attractive targets for the development of potential agents for the treatment of several disorders, including Alzheimer's disease and neuropathic pain. In the search for multitarget small molecules (MSMs) against such disorders, we have re-discovered chromenones as new affine σ1/σ2 ligands. 6-(4-(Piperidin-1-yl)butoxy)-4H-chromen-4-one (7), a previously identified MSM with potent dual-target activities against acetylcholinesterase and monoamine oxidase B, also exhibited σ1/σ2 affinity. 6-(3-(Azepan-1-yl)propoxy)-4H-chromen-4-one (20) showed a Ki value for σ1 of 27.2 nM (selectivity (σ1/σ2) = 28), combining the desired σ1 receptor affinity with a dual inhibitory capacity against both acetyl- and butyrylcholinesterase. 6-((5-Morpholinopentyl)oxy)-4H-chromen-4-one (12) was almost equipotent to S1RA, an established σ1 receptor antagonist.
Chromenone derivatives were identified as σ1 and σ2 receptor ligands (12, 20) and multitarget small molecules (7).
Sigma (σ) receptors represent a unique receptor class involved in a number of biological processes and classified into two subtypes, sigma-1 (σ1) and sigma-2 (σ2). Both receptors have been implicated in several neurological and neurodegenerative disorders, such as Alzheimer's disease (AD), as well as cancer.1,2 Although σ1/σ2 receptors represent structurally different proteins, a number of pharmacologically active compounds possess affinity towards both. Accordingly, receptor-subtype selectivity becomes a crucial aspect in the discovery of σ receptor targeting drugs,1–3 the design of molecular hybrids,4 and the development of compounds for radioligand-based molecular imaging techniques.5
The σ1 receptor is a ligand-regulated molecular chaperone involved in Ca2+ signalling at the endoplasmic reticulum (ER), the management of diverse types of ion channels, and the modulation of ER and mitochondrial (dis)functions.6 The σ1 receptors are known to play key roles in the central nervous system modulating cognitive capacities such as memory and learning.7 These receptors are addressed by trophic factors and by some anti-amnesic drugs acting as σ1 receptor agonists by increasing cholinergic and glutamatergic activity.1,6,8 Antagonists of the σ1 receptor are valuable pharmacological tools to manage neuropathic pain.9 They decrease the capsaicin-induced mechanical allodynia, while σ1 receptor agonists have the opposite effect, which is currently being used for the functional analysis to assign the agonistic or antagonistic profile of ligands.10,11
With respect to structure–activity relationships and σ1/2 agonist/antagonist capacity, a large number of ligands bearing a diverse array of structural motifs have been identified.1 Among them, piperidines,12 such as the antipsychotic haloperidol (Fig. 1) or (+)-MR200,13 ethylenediamine derivatives such as BD1008 (Fig. 1),14 piperazines, such as CM156,15 pyrimidines, such as Lan-0101,16 as well as aminoalkyl-substituted 1,3-dioxanes17 and aminoalkyloxy-substituted isoxazoles18 typically act as σ1 receptor antagonists. Most, but not all,19 of such ligands share a common structural feature, a basic nitrogen, that is pivotal for their interaction with the σ1 receptor. S1RA (Fig. 1),20,21 a highly selective σ1 receptor antagonist, reached phase II clinical trials for the treatment of neuropathic pain.22
Fig. 1. Known σ1/2 receptor modulators, general structures for (aminoalkoxy)chromones and selective receptor ligands I (R3 = Ar, n = 2–9)24 and representatives of chemotype II, i.e. the multitarget small molecule 7 and the potent σ1/2 receptor modulator 12.

Surprisingly in spite of being a privileged motif in medicinal chemistry,23 chromones have been scarcely investigated as σ1/2 receptor modulators. In fact, to the best of our knowledge, only one report of Erickson and co-workers on (aminoalkoxy)chromones (I) (Fig. 1) with potent (16–100 nM) binding affinities has been published.24
In search for multitarget small molecules (MSMs)25 and prompted by the previously described 6-(4-(piperidin-1-yl)butoxy)-4H-chromen-4-one (7),26 a chromenone of type II (Fig. 1) showing potent dual-target activity towards human acetylcholinesterase (hAChE) and human monoamine oxidase B (hMAO-B) (Table S1, ESI†), we initiated the σ1/2 receptor binding analysis of a recently synthesized chromenone library, designed as a combination of different linker lengths and different terminal amine components.26 We discovered in this study that (i) the MSM 7 exhibited moderate dual σ1/2 affinity, (ii) 6-(3-(azepan-1-yl)propoxy)-4H-chromen-4-one (20) combined the desired σ1 receptor affinity with a dual inhibitory capacity against both cholinesterases, and (iii) 6-((5-morpholinopentyl)oxy)-4H-chromen-4-one (12) was almost equipotent to S1RA,21 an established σ1 receptor antagonist.
By using a validated radioligand assay (see ESI,† Table S2), we evaluated the chromenone library of compounds 1–22 (ref. 26) as ligands of the human σ1 (hσ1) and rat σ2 (rσ2) receptor. Haloperidol, SA4503 (cutamesine), a synthetic σ1 receptor agonist, and S1RA were included as reference compounds. The results are shown in Table 1. A structure–activity-relationship analysis of the σ1/2 receptor binding affinity was possible in view of the multitude combinations of the terminal amine motifs and the lengths of the linker (Table 1). These observations could be summarized as follows. The majority of investigated chromenones were potent and selective σ1 receptor ligands, with affinity values ranging from Ki = 19.6 nM (12) to Ki = 12.1 μM (16). An increased linker length caused, by trend, an improved σ1 receptor affinity and pentamethylene compounds with n = 4, such as 4, 12, 15, and 18 were the most potent in each corresponding subseries.
Affinity values (Ki, nM) of selected chromenones at the human σ (hσ1) and rat σ2 (rσ2) receptors.
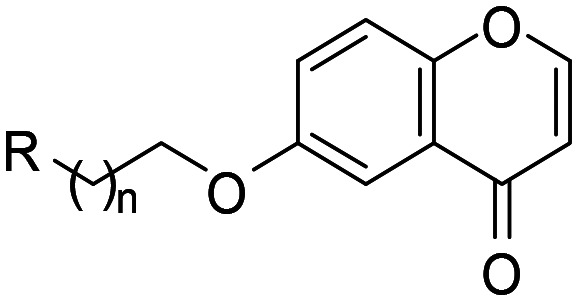
| |||||
|---|---|---|---|---|---|
| Compd | R | n | hσ1 | rσ2 | Selectivity hσ1//rσ2 |
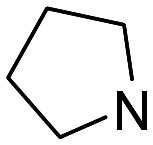
| 1 |
|
1 | 2870 | 12 500 | 4.4 |
| 2 | 2 | 499 | 23 900 | 48 | |
| 3 | 3 | 227 | 3110 | 14 | |
| 4 | 4 | 87.0 | 1450 | 17 | |
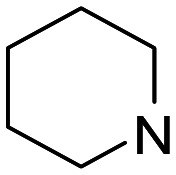
| 5 |
|
1 | 825 ± 146 | 9070 | 11 |
| 6 | 2 | 65.5 ± 4.5 | 2890 | 44 | |
| 7 | 3 | 309 ± 45 | 1970 ± 1400 | 6.4 | |
| 8 | 4 | 213 | 490 | 2.3 | |
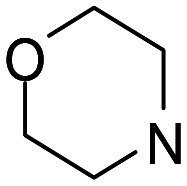
| 9 |
|
1 | >10 000 | >10 000 | n.d. |
| 10 | 2 | 245 | 22 500 | 92 | |
| 11 | 3 | 23.9 ± 9.7 | 14 600 | 610 | |
| 12 | 4 | 19.6 ± 3.2 | 2630 ± 1200 | 130 | |
| 13 |
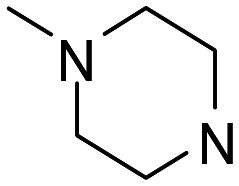
|
1 | 604 | 14 200 | 24 |
| 14 | 2 | 809 | 17 100 | 21 | |
| 15 | 4 | 82.5 | 4100 | 50 | |
| 16 |
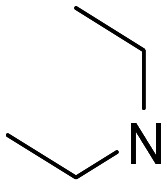
|
1 | 12 100 | 30 000 | 2.5 |
| 17 | 2 | 1220 | 8360 | 6.9 | |
| 18 | 4 | 218 | 2500 | 11 | |
| 19 |
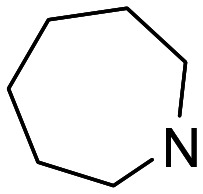
|
1 | 113 ± 28 | 1620 | 14 |
| 20 | 2 | 27.2 ± 6.8 | 750 ± 250 | 28 | |
| 21 |
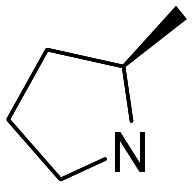
|
3 | 104 | 1090 | 10 |
| 22 | 4 | 134 | 583 | 4.4 | |
| Haloperidol | 3.53 ± 0.47 | 20.5 ± 1.0 | 5.8 | ||
| SA4503 | 3.77 ± 0.11 | n.d.a | n.d. | ||
| S1RA b | 17.0 | 9300 | 550 | ||
Not determined.
Data from ref. 21.
Hence, in the first subset (ligands 1–4), the affinity gradually increased with the linker length, from n = 1 to 4. Chromenone 4 (n = 4) was the most potent σ1 receptor ligand containing a pyrrolidine as basic center (Ki = 87.0 nM, selectivity ratio (sel.) (σ2/σ1) = 17) and showed modest hAChE (IC50 = 7.20 μM) with no remarkable MAO inhibition (Table S1, ESI†). An attractive MSM was previously identified in this subseries.26 This compound (3; hAChE: IC50 = 3.74 μM; hMAO-B: IC50 = 19.6 μM) has now been discovered as a σ1 receptor ligand with good affinity (Ki = 227 nM). The incorporation of a (R)-2-methyl group in the pyrrolidine ring of ligands 3 and 4 led to the enantiomerically pure chromenones 21 and 22, with n = 3 and 4, respectively. This simple modification afforded either a 2.2 more (21) or 1.5-fold less (22) potent σ1 receptor agent (Table 1).
The most potent σ1 receptor ligand within the piperidine subset (ligands 5–8) was 6 (Ki = 65.5 nM; sel. (σ2/σ1) = 44), showing very modest hAChE (IC50 = 15.9 μM), but no noteworthy MAO inhibition. Compound 7 combines pronounced hAChE and hMAO-B inhibition (Table 2; IC50 = 5.58 μM and 7.20 μM, respectively) with σ1 receptor (Ki = 309 nM) and modest σ2 receptor (Ki = 1.97 μM) affinity.
Inhibition of hAChE, hBuChE and hMAO-B (IC50) and affinity values (Ki) at the human σ1 (hσ1) and rat σ2 (rσ2) receptors.
| Compd | hAChEa | hBuChEa | hMAO-Ba | hσ1 | rσ2 |
|---|---|---|---|---|---|
| IC50 ± SE (μM) | K i ± SE (nM) | ||||
| 7 | 5.58 ± 0.49 | 87.5 ± 11.8 | 7.20 ± 0.41 | 309 ± 45 | 1970 ± 1400 |
| 12 | 96.2 ± 7.7 | n.i.b | v.n.d.c,d | 19.6 ± 3.2 | 2630 ± 1200 |
| 20 | 10.6 ± 2.2 | 25.0 ± 4.7 | v.n.d.c,e | 27.2 ± 6.8 | 750 ± 250 |
Data from ref. 26.
n.i. (no inhibition) refers to IC50 >100 μM.
v.n.d. (values not determined). IC50 values have not been determined.
Residual activity of MAO-B @ an inhibitor concentration of 10 μM was 58%.
Residual activity of MAO-B @ an inhibitor concentration of 10 μM was 54%.
The morpholine subset (ligands 9–12) deserved particular attention, in view of the structure of S1RA (Fig. 1) and the general pharmacological properties attributed to this moiety.27 Again, chromenone 12 with the longest linker (n = 4) was the most potent σ1 receptor ligand out of this set (Ki = 19.6 nM; sel. (σ2/σ1) = 130), but showed poor cholinesterase and MAO inhibition (Table 2). Chromenone 10 was previously reported not to affect the cholinesterases, but to inhibit both MAO enzymes with IC50 values of less than 2 μM.26 This activity profile was further characterized by the observed affinity for the human σ1 receptor (Ki = 245 nM).
The most potent σ1 receptor ligand in the piperazine subset (15; Ki = 82.5 nM; sel. (σ2/σ1) = 50) was weaker than the analogous morpholine derivative 12, and in the same range than the pyrrolidine counterpart 4.
The diethylamine-containing ligands 16–18 exhibited weaker σ1 receptor affinity, e.g. when compared to the corresponding piperazine analogs, indicating the disadvantage of the acyclic amine portion.
Out of the two azepane ligands, 20 with the longer linker performed better and gave a Ki value of 27.2 nM (sel. (σ2/σ1) = 28). This compound combined the desired σ1 receptor affinity with a dual inhibitory capacity against both cholinesterases (Table 2).
In conclusion, the σ1/2 receptor radioligand binding assay of chromenones has allowed us to identify three highly potent σ1 receptor ligands, 11, 12 and 20, with Ki values lower than 30 nM. They were selective for the human σ1 receptor over the rat σ2 receptor and exhibited selectivity ratios higher than 25. Importantly, our best ligand, chromenone 12 was equipotent to the well-known morpholine-containing ligand S1RA, an advanced agent for possible therapeutic treatment of neuropathic pain.21
It is well known that σ1 receptor antagonists are highly cytotoxic, whereas σ1 receptor agonists preserve cell survival.28 This feature was employed to address the functional agonist/antagonist profile of selected chromenones. Hence, the cytotoxic activity of ligands 7, 12 and 20 was determined by the MTT assay with the SH-SY5Y neuroblastoma cell line as described.29 The results were compared with those obtained for reference compounds NE-100 and S1RA, accepted σ1 antagonists, and SA4503, a selective σ1 agonist. The outcome of the cell viability assay after 48 h compound treatment (Fig. S1, ESI†) indicated that 7 behaved as a σ1 receptor antagonist, while 12 and 20 acted as σ1 receptor agonists.
Finally, molecular modelling was carried out to shed light on the binding mode of the highly active compounds 12 (Fig. 2 and 3) and 20 (see ESI†) with the σ1 receptor protein. A validated three-dimensional model of the σ1 receptor based on the crystal structure of the human σ1 receptor in complex with the antagonist PD144418 was used for this analysis.30 The docking process was executed using AutoDock Vina software.31 The binding pocket primarily consists of hydrophobic residues of Val84, Trp89, Met93, Leu95, Leu105, Leu182, Phe107, Ile124, Trp164, and Tyr103, and the two acidic residues of Glu172 and Asp126. Receptor flexibility was introduced with respect to the rearrangement of these side chains allowing a ligand to properly adapt to the target.
Fig. 2. Modeled complex of the σ1 receptor with compound 12 (yellow), showing the key interactions.

Fig. 3. 2D schematic view of the interactions between chromenone 12 and the σ1 receptor. Intermolecular interactions are indicated with colors: green, van der Waals; orange, salt bridge and attractive charge interactions; pink, π–alkyl contacts.

Compound 12 fully occupies the active site and established a number of interactions with the σ1 receptor, as shown in Fig. 2 and 3. The morpholine moiety is perfectly encased in the hydrophobic cavity formed by residues, Ser117, Ile124, Phe133, His154, Val162, Trp164 and Tyr103. The linker alkyl chain resides in the hydrophobic region lined by residues Val84, Trp89 Phe107, Tyr120 and Phe184. The protonated nitrogen is engaged in a key bidentate salt bridge with Glu172 and in an attractive charge interaction with Asp126. On the other hand, a network of π–alkyl interactions stabilizes the position of the chromenone moiety in proximity to the residues Met93, Leu95, Leu105, Leu182 and Ala185.
The observed binding of compound 12 in the active site of the σ1 receptor fulfilled Glennon's pharmacophore model,32 preserving the central protonated group, i.e. the basic morpholine nitrogen, located between the two hydrophobic sites, i.e. the chromenone moiety and the carbon chain of the morpholine ring, at appropriate distances of 8.6 and 2.5 Å, respectively.
In order to evaluate the drug likeness and the potential ability to cross the BBB, chromenones 12 and 20 were scored in silico for their physiochemical and pharmacokinetic parameters (ADME). The results are summarized in Table S2 (ESI†). All calculated descriptors and properties are within the expected thresholds. Chromenones were found to have no Lipinski rule violations,33 thus proving their drug-likeness properties (see ESI†).
To sum up, in our search for MSMs as possible therapeutics against AD, we have successfully re-discovered and addressed the potential of chromenones as ligands of the σ1 and σ2 receptors. Noteworthy, we have identified 6-(4-(piperidin-1-yl)butoxy)-4H-chromen-4-one (7), a previously identified MSM, showing dual-target anti-hAChE and anti-hMAO-B activities,26 as a good σ1/σ2 receptor modulator. We identified 6-((5-morpholinopentyl)oxy)-4H-chromen-4-one (12) and 6-(3-(azepan-1-yl)propoxy)-4H-chromen-4-one (20) as potent σ1 receptor modulators, almost equipotent to S1RA. This promising result may pave the way for future developments on this field in our laboratories.
This work was supported by pre-doctoral FPU grants to D. D. I. from the University of Alcalá and the Spanish Ministry of Science, Innovation and Universities. JMC thanks Eli Lilly OIDD (Open Innovation Drug Discovery) program, and Universidad Camilo José Cela (Grant UCJC 2020-33) for support. W. D. C. thanks Tina Spalholz (HZDR) for supporting the radioligand binding experiments.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Electronic supplementary information (ESI) available: Supplementary Figures S1–S3 and Tables S1–S3; enzyme inhibition data (hAChE, hBuChE, hMAO-B, hMAO-A), assay protocols for the determination of hσ1 and rσ2 affinity values, cytotoxicity data, docking of 20 at the σ1 receptor, virtual ADME of 12 and 20. See DOI: 10.1039/d1md00105a
References
- Arena E. Dichiara M. Floresta G. Parenti C. Marrazzo A. Pittala V. Amata E. Prezzavento O. Future Med. Chem. 2018;10:231–256. doi: 10.4155/fmc-2017-0164. [DOI] [PubMed] [Google Scholar]
- Abate C. Niso M. Berardi F. Future Med. Chem. 2018;10:1997–2018. doi: 10.4155/fmc-2018-0072. [DOI] [PubMed] [Google Scholar]
- Brune S. Pricl S. Wünsch B. J. Med. Chem. 2013;56:9809–9819. doi: 10.1021/jm400660u. [DOI] [PubMed] [Google Scholar]
- Amata E. Dichiara M. Gentile D. Marrazzo A. Turnaturi R. Arena W. La Mantia A. Tomasello B. R. Acquaviva R. Di Giacomo C. Rescifina A. Prezzavento O. ACS Med. Chem. Lett. 2020;11:889–894. doi: 10.1021/acsmedchemlett.9b00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H. Zhang Y. Huang Y. Neurosci. Lett. 2019;691:3–10. doi: 10.1016/j.neulet.2018.07.033. [DOI] [PubMed] [Google Scholar]
- Nguyen L. Lucke-Wold B. P. Mookerjee S. A. Cavendish J. Z. Robson M. J. Scandinaro A. L. Matsumoto R. R. J. Pharmacol. Sci. 2015;127:17–29. doi: 10.1016/j.jphs.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Maurice T. Goguadze N. Adv. Exp. Med. Biol. 2017;964:213–233. doi: 10.1007/978-3-319-50174-1_15. [DOI] [PubMed] [Google Scholar]
- Meunier J. Ieni J. Maurice T. Br. J. Pharmacol. 2006;149:998–1012. doi: 10.1038/sj.bjp.0706927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almansa C. Vela J. M. Future Med. Chem. 2014;6:1179–1199. doi: 10.4155/fmc.14.54. [DOI] [PubMed] [Google Scholar]
- Entrena J. M. Cobos E. J. Nieto F. R. Cendán C. M. Gris G. Del Pozo E. Zamanillo D. Baeyens J. M. Pain. 2009;143:252–261. doi: 10.1016/j.pain.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Entrena J. M. Sánchez-Fernández C. Nieto F. R. González-Cano R. Yeste S. Cobos E. J. Baeyens J. M. Sci. Rep. 2016;6:37835. doi: 10.1038/srep37835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezzavento O. Campisi A. Parenti C. Ronsisvalle S. Aricò G. Arena E. Pistolozzi M. Scoto G. M. Bertucci C. Vanella A. Ronsisvalle G. J. Med. Chem. 2010;53:5881–5885. doi: 10.1021/jm100116p. [DOI] [PubMed] [Google Scholar]
- Marrazzo A. Parenti C. Scavo V. Ronsisvalle S. Scoto G. M. Ronsisvalle G. Life Sci. 2006;78:2449–2453. doi: 10.1016/j.lfs.2005.10.005. [DOI] [PubMed] [Google Scholar]
- McCracken K. A. Bowen W. D. Matsumoto R. R. Eur. J. Pharmacol. 1999;365:35–38. doi: 10.1016/s0014-2999(98)00876-0. [DOI] [PubMed] [Google Scholar]
- Mésangeau C. Narayanan S. Green A. M. Shaikh J. Kaushal N. Viard E. Xu Y. T. Fishback J. A. Poupaert J. H. Matsumoto R. R. McCurdy C. R. J. Med. Chem. 2008;51:1482–1486. doi: 10.1021/jm701357m. [DOI] [PubMed] [Google Scholar]
- Lan Y. Chen Y. Cao X. Zhang J. Wang J. Xu X. Qiu Y. Zhang T. Liu X. Liu B. F. Zhang G. J. Med. Chem. 2014;57:10404–10423. doi: 10.1021/jm501207r. [DOI] [PubMed] [Google Scholar]
- Utech T. Köhler J. Wünsch B. Eur. J. Med. Chem. 2011;46:2157–2169. doi: 10.1016/j.ejmech.2011.02.070. [DOI] [PubMed] [Google Scholar]
- Sun H. Shi M. Zhang W. Zheng Y. M. Xu Y. Z. Shi J. J. Liu T. Gunosewoyo H. Pang P. Gao Z. B. Yang F. Tang J. Yu L. F. J. Med. Chem. 2016;59:6329–6343. doi: 10.1021/acs.jmedchem.6b00571. [DOI] [PubMed] [Google Scholar]
- Díaz J. L. Christmann U. Fernández A. Luengo M. Bordas M. Enrech R. Carro M. Pascual R. Burgueño J. Merlos M. Benet-Buchholz J. Cerón-Bertràn J. Ramírez J. Reinoso R. F. J. Med. Chem. 2013;56:3656–3665. doi: 10.1021/jm400181k. [DOI] [PubMed] [Google Scholar]
- Díaz J. L. Cuberes R. Berrocal J. Contijoch M. Christmann U. Fernández A. Port A. Holenz J. Buschmann H. Laggner C. Serafini M. T. Burgueño J. Zamanillo D. Merlos M. Vela J. M. Almansa C. J. Med. Chem. 2012;55:8211–8224. doi: 10.1021/jm3007323. [DOI] [PubMed] [Google Scholar]
- Romero L. Zamanillo D. Nadal X. Sánchez-Arroyos R. Rivera-Arconada I. Dordal A. Montero A. Muro A. Bura A. Segalés C. Laloya M. Hernández E. Portillo-Salido E. Escriche E. Codony X. Encina G. Burgueño J. Merlos M. Baeyens J. B. Giraldo J. López-García J. A. Maldonado R. Plata-Salamán C. R. Vela J. M. Br. J. Pharmacol. 2012;166:2289–2306. doi: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vela J. M. Merlos M. Almansa C. Expert Opin. Invest. Drugs. 2015;24:883–896. doi: 10.1517/13543784.2015.1048334. [DOI] [PubMed] [Google Scholar]
- Gaspar A. Matos M. J. Garrido J. Uriarte E. Borges F. Chem. Rev. 2014;114:4960–4992. doi: 10.1021/cr400265z. [DOI] [PubMed] [Google Scholar]
- Erickson R. H. Natalie Jr. K. J. Bock W. Lu Z. Farzin F. Sherrill R. G. Meloni D. J. Patch R. J. Rzesotarski W. J. Clifton J. Pontecorvo M. J. Bailey M. A. Naper K. Karbon W. J. Med. Chem. 1992;35:1526–1535. doi: 10.1021/jm00087a005. [DOI] [PubMed] [Google Scholar]
- Oset-Gasque M. J. Marco-Contelles J. ACS Chem. Neurosci. 2018;9:401–403. doi: 10.1021/acschemneuro.8b00069. [DOI] [PubMed] [Google Scholar]
- Lemke C. Christmann J. Yin J. Alonso J. M. Serrano E. Chioua M. Ismaili L. Martìnez-Grau M. A. Beadle C. D. Vetman T. Dato F. M. Bartz U. Elsinghorst P. W. Pietsch M. Müller C. E. Iriepa I. Wille T. Marco-Contelles J. Gütschow M. ACS Omega. 2019;4:22161–22168. doi: 10.1021/acsomega.9b03409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourounakis A. P. Xanthopoulos D. Tzara A. Med. Res. Rev. 2019;40:709–752. doi: 10.1002/med.21634. [DOI] [PubMed] [Google Scholar]
- Zampieri D. Laurini E. Vio L. Fermeglia M. Pricl S. Wünsch B. Schepmann D. Mamolo M. G. Eur. J. Med. Chem. 2015;90:797–808. doi: 10.1016/j.ejmech.2014.12.018. [DOI] [PubMed] [Google Scholar]
- Estrada M. Pèrez C. Soriano E. Laurini E. Romano M. Pricl S. Morales-Garcìa J. A. Pèrez-Castillo A. Rodrìguez-Franco M. I. Future Med. Chem. 2016;8:1191–1207. doi: 10.4155/fmc-2016-0036. [DOI] [PubMed] [Google Scholar]
- Bautista-Aguilera O. M. Budni J. Mina F. Medeiros E. B. Deuther-Conrad B. Entrena J. M. Moraleda I. Iriepa I. Lopez-Munoz F. Marco-Contelles J. J. Med. Chem. 2018;61:6937–6943. doi: 10.1021/acs.jmedchem.8b00848. [DOI] [PubMed] [Google Scholar]
- Trott O. Olson A. J. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glennon R. A. Mini-Rev. Med. Chem. 2005;5:927–940. doi: 10.2174/138955705774329519. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lombardo F. Dominy B. W. Feeney P. J. Adv. Drug Delivery Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.