

Abstract
Purpose of the review:
Hypertension has been demonstrated to be a chief contributor to morbidity and mortality throughout the world. Though the etiology of hypertension is multifactorial, emerging evidence, obtained in experimental studies, as well as observational studies in humans, points to the role of inflammation and immunity. Many aspects of immune function have now been implicated in hypertension and end-organ injury; this review will focus upon the recently-described role of Th17 cells in this pathophysiological response.
Recent findings:
Studies in animal models and human genetic studies point to a role in the adaptive immune system as playing a contributory role in hypertension and renal tissue damage. Th17 cells, which produce the cytokine IL17, are strongly pro-inflammatory cells which may contribute to tissue damage if expressed in chronic disease conditions. The activity of these cells may be enhanced by physiological factors associated with hypertension such as dietary salt or Ang II. This activity may culminate in the increased sodium retaining activity and exacerbation of inflammation and renal fibrosis via multiple cellular mechanisms.
Summary:
Th17 cells are a distinct component of the adaptive immune system that may strongly enhance pathways leading to increased sodium reabsorption, elevated vascular tone and end-organ damage. Moreover, this pathway may lend itself toward specific targeting for treatment of kidney disease and hypertension.
Keywords: Angiotensin II, dietary salt, lymphocytes, fibrosis
Introduction
Many different lines of evidence point to a role of inflammation and immunity in human hypertension and renal end-organ damage. Lymphocytes (1), other mononuclear cells (2), and the deposition of immunoglobulins and complement proteins (3) have been demonstrated in the renal interstitium adjacent to damaged renal tubules and glomeruli in hypertensive humans. More recently it was shown that hypertensive individuals exhibit glomerulosclerosis and renal fibrosis which accompanies infiltration of macrophages and T-lymphocytes in the renal interstitium (4). These correlative observations are supported by functional evidence demonstrating that immunotherapeutic modulation alters blood pressure in patients with HIV (5), psoriasis (6) or rheumatoid arthritis (6). In addition, genetic markers in the regions of several genes important in immune signalling have been linked with hypertension or kidney disease in GWAS and other genetic association studies (7, 8, 9, 10, 11). These observations demonstrate the impact of inflammation and immunity in human hypertension. Mechanistically-based experimental studies, largely performed in rodents (12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24), have illustrated the importance of immune mechanisms in hypertension and renal disease (14, 20, 25, 26, 27, 28*).
T Cells in Hypertension and End-Organ Damage
Though multiple immune mechanisms have been elucidated in experimental models of hypertension (12, 13, 14, 15, 26, 27, 28*, 29*, 30, 31), several studies point specifically to the importance of adaptive immune mechanisms in the development of experimental hypertension in mice (20, 21, 22, 23, 24). Of particular note were landmark studies performed by Harrison’s group in which they observed that the degree of hypertension developed during chronic infusion of Angiotensin II was blunted in mice lacking both T- and B-lymphocytes (Rag 1−/−) (22). Following the adoptive transfer of T cells, but not B cells, hypertension caused by angiotensin II was restored in the Rag 1−/− mice (22). These studies were recently confirmed in the Dahl salt-sensitive rat, an experimental model of salt-sensitive hypertension and renal end-organ damage. Dahl SS rats deficient in T cells (SSCD247−/−) were shown to have no difference in arterial pressure or renal damage when fed low salt, but the high salt-induced hypertension and renal damage was attenuated in the SSCD247−/− which lack T cells following high-salt feeding (32). The subsequent replacement of T cells (via splenocyte transfer) in the SSCD247−/− rats lacking endogenous T cells led to a restoration of the salt-sensitive disease phenotype as both hypertension and renal damage returned to levels observed in the wild type Dahl SS rats (33**).
Though these and other studies illustrate the important role of T lymphocytes in the development of hypertension and end-organ damage (12, 13, 14, 15, 26, 27, 28*, 29*, 30, 31), there is not complete agreement in regard to these observations. Recent studies have been unable to demonstrate an attenuation of angiotension II-induced hypertension in mice lacking T and B cells (34**), while other studies point to an important role for B-cells, since activation of B cells and increased IgG production accompany an increased in angiotension II, and B cell deficiency blunted the hypertensive response to angiotension II in mice (35). In addition, multiple other immune cell types have been demonstrated to influence hypertensive disease phenotypes (12, 13, 14, 15, 26, 27, 28*, 29*, 30, 31).
Despite these observations, the preponderance of evidence points to an important role for T cells as a causative factor in hypertension and subsequent renal damage (12, 13, 14, 15, 26, 27, 28*, 29*, 30, 31). The activation of the T-cell response is initiated by engagement of antigen presenting cells with the T-cell receptor (TCR) and involvement of co-stimulatory factors such as CD80/86 or B7-CD28. Underlying injury in the kidney may serve as an initial trigger for the expression of neo-antigens and the activation of T cells in the setting of renal injury or hypertension. This point was recently illustrated in a study that aimed to distinguish effects of perfusion pressure from other circulating factors on renal inflammation in the setting of Ang II induced hypertension. In response to Ang II infusion, the resulting elevation in renal perfusion pressure was associated with peritubular capillary damage and an increase leukocyte infiltration, including T- and B-lymphocytes. Interestingly, the application of a servo-control system to maintain renal perfusion pressure to the contralateral kidney at control (pre-infusion) levels substantially reduced infiltration of lymphocytes vs the kidney exposed to elevated pressure (36**). Similar observations were previously made in the Dahl SS rat (37). These observations are consistent with the hypothesis that pressure-induced damage may initiate renal lymphocyte activation in the kidney in the setting of hypertension. However, there remains debate about the identity of specific antigens which contribute to activation of adaptive immune responses. For example, HSP70 has been identified as a common antigen in salt-sensitive hypertension, however, other potential neo-antigens (e.g, ketal adducts) have been described in models of Ang II or DOCA-salt induced hypertension (29*, 38, 39, 40)(41**) Once activated, T-cells can produce cytokines, free radicals, and other factors capable of influencing the development of hypertension and related end-organ damage (12, 13, 14, 15, 26, 27, 28*, 29*, 30, 31). Among the factors produced by different T cell subtypes are IL-17 and related cytokines which are produced by Th17 cells.
T-helper 17 cells (Th17 cells)
Overview of differentiation and function.
An important component of the adaptive immune system is the differentiation of naïve T cells into various effector T-helper cells (reviewed in (42*)). The original paradigm outlined the differentiation of naïve CD4+ T-cell cells (i.e., Th0 cells) into either T-helper 1 (Th1) or T-helper 2 (Th2) cells, classified based on the profile of cytokines each type secreted to help fulfill their function in immunity (43). Th1 cells differentiate under the control of STAT1 and T-bet. These cells secrete IFNγ, IL2, IL12 and TNFα and function primarily to promote macrophage responses in fighting intracellular pathogens such as virus or bacteria (44*). Th2 cells differentiate under the control of STAT6 and GATA3 and secrete IL4, IL10, IL13 as well as other cytokines. Th2 cells role function classically to promote host-defense against invading parasites (44*, 45).
Th17 cells are characterized by the signature cytokine IL17A, as well as IL17F, IL22, IL23 and TNFα (46, 47, 48). Similar to Th1 or Th2 cells, Th17 cell cells are thought to derive from a naïve Th0 phenotype. The factors that lead to Th17 activation have been extensively reviewed (49, 50) . Importantly, RORγt (RAR related orphan receptor gamma T) is considered the key lineage specific transcription factor regulating Th17 cell differentiation and the expression of Th17 specific cytokines such as IL17A. The process is dependent on the activity of the pro-inflammatory cytokine IL-6 and the activation of STAT3 (46, 47, 48).
Th17 cells are strongly pro-inflammatory and prominently induce the infiltration of neutrophils for defense against pathogens such as extracellular bacteria, which are not typically targeted by Th1 or Th2 cells (44*, 48*). Mouse models of IL17 deficiency show increased susceptibility to infections from a large range of different bacterial and fungal pathogens, including in the setting of urinary tract infection (51, 52). In humans, impaired Th17 differentiation has been identified in patients with primary immunodeficiencies (PID) due to mutations of IL17A or IL17F (53).
Despite the important role of Th17 cells in mediating adaptive immune responses, dysregulated activation of Th17 cells can promote tissue damage in auto-immune disorders due to their strong pro-inflammatory activity. Hyperactivation of Th17 cells may mediate inflammation in conditions such as psoriasis, inflammatory bowel disease, rheumatoid arthritis and autoimmune encephalitis (54). Th17 cells have been described as a potential contributor to inflammatory kidney diseases such as glomerular nephritis (reviewed in (55)). Systemic lupus erythematosus (SLE), is an autoimmune disorder based on the production of auto-antibodies which can deposit in the kidney and can lead to hypertension and kidney damage (56). SLE patients are reported to manifest an increase in circulating conventional Th17 cells (CD4+/IL17+) (57) and studies from animal models of SLE such as the MRL/lpr mouse, the Roquinsan/san model or pristine administration suggest that inhibition of IL17 or mutation of the IL17 gene attenuates the severity of inflammation and injury (58, 59**, 60**).
Modulation of Th17 activity
As described above, Th17 differentiation is driven primarily by the RORγT transcription factor under influence of TGFβ and IL6 and the activation of STAT3. The differentiation pathway may be dependent on the activity of the store operated calcium channel, Orai1, which was previously shown to promote nuclear accumulation of NFAT-5 and RORγT (61). Recent studies from our lab demonstrated that Orai-1 is essential for expression of IL17A in CD4+ cells, since IL17A was detected only in CD4+ cells which expressed the channel and inhibition of this channel attenuated induction of Th17 cells following renal ischemia/reperfusion injury (62**).
Th17 activity appears to be strongly modulated by dietary sodium. Following kidney ischemia reperfusion injury, subsequent exposure of rats to elevated dietary sodium (from 0.4% to 4%) results in hypertension, progression of chronic kidney disease and is associated with a dramatic expansion of Th17 cells. However, the effect of dietary salt on Th17 activity is not limited to models of kidney injury, but rather has been observed in other models of inflammation including psoriasis or autoimmune encephalomyelitis (63, 64, 65, 66). Elegant studies by Klitterfeld et al., demonstrated that elevation of extracellular sodium (to 170 mM) hastened the differentiation of Th0 to Th17 cells in vitro (67) and this response appears to be dependent on signaling through SGK1 (67, 68**). Elevated extracellular sodium, in combination with Ang II, increased IL17 production of lymphocytes isolated from post-ischemic rat kidneys or PBMCs of patients diagnosed with acute kidney injury (62**, 69). The response appeared to be specific for sodium since equimolar amounts of mannitol or choline chloride did not induce a response (62**).
Whether the biological effect of extracellular sodium on IL17 activity in vitro is directly related to the stimulatory effect of dietary sodium on Th17 activation is not clear. In recent years, it has been shown that the skin can sequester large amounts of sodium in response to high sodium intake, being complexed to extracellular glycosaminoglycans. The local extracellular sodium concentration may be higher than the extracellular fluid, and it has been suggested that leukocytes (i.e., lymphocytes or macrophages) perfusing through these areas may be exposed to higher local sodium concentrations vs extracellular fluid (70, 71). Interestingly, it was recently reported that patients with salt-losing tubulopathies have lower skin sodium content relative to non-salt losing control patients. These patients manifest reduced levels of Th17 cells and are more susceptible to a variety of infections relative to controls (72**).
We propose that other factors may potentially contribute to Th17 activation in response to high-salt intake. For example, chronic salt loading has been shown to increase renal adenosine levels, a response that was proposed to promote adaptive natriuresis (73). Adenosine, which can also be liberated from injured cells, affects lymphocytes either directly or secondarily via the activity of dendritic cells, an effect that is likely mediated by the A2A receptor subtype (74, 75). Adenosine has been suggested to increase the prevalence of Th17 cells in autoimmune disease models (74, 75, 76**). In addition, endothelin A expression is enhanced in kidney under high salt diet conditions (77, 78) and may modulate the activity of a variety of inflammatory cells including lymphocytes. Endothelin antagonists have been shown to block the induction of Th17 cells in psoriasis (41**), while they were also shown to abrogate the expression of IL17 in CD4 cells isolated from lymph nodes of mice with multiple sclerosis (79). However few studies have directly addressed the effects of endothelin A antagonists on lymphocyte activity in the kidney. In one study, Boesen et al., demonstrated that the ET-A antagonist ABT-627 significantly reduced the infiltration of lymphocytes into injured kidney and showed a strong trend at reducing CD4+/RorγT+ Th17 cells (80).
Angiotensin II may also participate in the modulation of Th17 activity. Several studies have shown that Ang II infusion can increase the expression of circulating Th17 cells and decrease T-regulatory cells and that this response is mediated in part by SGK-1 (81). Ang II synergistically increased the IL17 response to elevated sodium in isolated lymphocytes from post-ischemic rat kidneys, and the number of Th17 cells in post ischemic rats treated with high salt diet was abrogated by treatment with the AT1 receptor antagonist, losartan.
Mice overexpressing TGFβ display enhanced Th17 activation in a model of auto-immune encephalopathy (50). A recent study indicated that high glucose induced activation of latent TGFβ in T cells from mice with autoimmune disease, resulting in an increase in the expression of IL17A (82**). Finally, vitamin D status may also influence Th17 responses. Th17 cells are exacerbated in states of vitamin D deficiency, and this effect may be due to direct inhibitory effect of 1,25 hydroxy-vitamin D3 on CD4 cells (83**, 84).
Role in hypertension and tissue injury
Whether Th17 cells play a role in human hypertension remains unclear. With notable exceptions, there is a relative dearth of clinical studies highlighting Th17 cells or IL17 activity in the setting of hypertension. However, some recent studies have reported elevated levels of circulating IL17 in patients with pre-hypertension (85) or in diabetic patients with hypertension vs non-hypertensive diabetic patients (86). Ji et al., evaluated T-helper cells in hypertensive vs non-hypertensive patients and demonstrated elevated percentage of circulating Th17 cells and IL-17 and also reported that IL17 independently associated with the presence of non-dipper hypertension (87). Finally, increased circulating levels of IL17 have been demonstrated in patients with pre-eclampsia (88).
More direct support of Th17 cells in the development of hypertension derive from studies in animal models. Most notably, studies have demonstrated that Ang II infusion in mice results in an increase Th17 cells. In response to chronic infusion on Ang II, IL17−/− mice showed a reduced level of blood pressure vs. wild type control mice (86). In Dahl S rats infused with Ang II, inhibition of IL17 with a decoy IL17 receptor attenuated the development of hypertension (89). In addition to Ang II dependent models, elevated Th17 cells (and/or reduced T-regulatory cells) have been demonstrated in mineralocorticoid dependent hypertension, hypertension in response to cyclosporine A, or in a model of pre-eclampsia. In each of these examples, the importance IL17 in the hypertension and renal injury response was suggested by an attenuation in the response by using IL17−/− mice or by inhibition with IL17 receptor blockade (90, 91, 92).
Several potential mechanisms for contributions to IL17 in the setting of renal injury and the development of hypertension have been suggested by data from animal models and in vitro studies. Elegant studies performed in mice in response to Ang II infusion have shown impaired natriuretic response to an acute saline load. This response was associated with an increased expression of tubular sodium transporters NHE3 and NCC. These responses were abrogated in IL17−/− mice and were dependent on the activity of SGK1 (93, 94). IL17 has also been shown to have vascular effects favoring constriction. IL17 has been shown to negatively regulate NOS activity and has recently been shown to influence vascular remodeling of small arteries (63, 95). To the best of our knowledge, no studies have sought to evaluate the effects of IL17 on renal vascular reactivity.
In addition to its effects on blood pressure, multiple studies from a variety of renal injury models suggest IL17 contributes to end organ damage and fibrosis (55, 96, 97, 98) (99). The activation of pathways leading to inflammation and fibrosis involve the interaction of multiple cells types in the renal interstitium. IL17 is a well-known chemoattractant for neutrophils and contributes to parenchymal cell damage. IL17 stimulates endothelial cells and tubular epithelial cells into a pro-inflammatory state and promotes the release of additional cytokines (100, 101, 102). In gene array studies of human aortic smooth muscle cells, over 30 genes, including those encoding inflammatory cytokines and chemokines, are stimulated by incubation with IL17 (86) and IL17 has been shown to induce inward remodeling of blood vessels independent of blood pressure control (103).
Finally, IL17 is likely to have significant effects on scar formation via effects on macrophages. IL17 administration increased inflammation, aggravated fibrogenic scar formation and delayed wound healing, an effect that was dependent on part on macrophage activity (104). In models of renal injury by UUO or ischemia reperfusion, IL17 blockade reduces accumulation of macrophages and fibrosis (105). However, no firm consensus has developed to determine how IL17 influences macrophage phenotypes to elicit these effect, although one study suggests that IL17 mediates effects on macrophages by increasing their responsiveness to OxLDL (106).
Conclusion
Taken together, data suggest an important role for Th17 cells in the generation of hypertension and end-organ damage in the kidney. Building on prior important data on the role of the adaptive immune system in hypertension, and summarized in Figure 1, factors that lead to Th17 cell differentiation, such as Ang II or high salt diet may enhance the presence of this T-helper population. The liberation of its key cytokine IL17 may enhance sodium reabsorption or increased vascular tone and contribute to a positive sodium balance and exacerbation of hypertension. In combination with other inflammatory and interstitial cells, IL17 can also promote the development of fibrosis. Conceivably, the potential ability to target the IL17 pathway has been demonstrated for other indications such as psoriasis (107, 108) and thus this approach may enhance precision of potential treatment for conditions of hypertension or progressive chronic fibrosis.
Figure 1. Differentiation and activation factors of TH17 cells, and the effects of differential IL-17A expression.
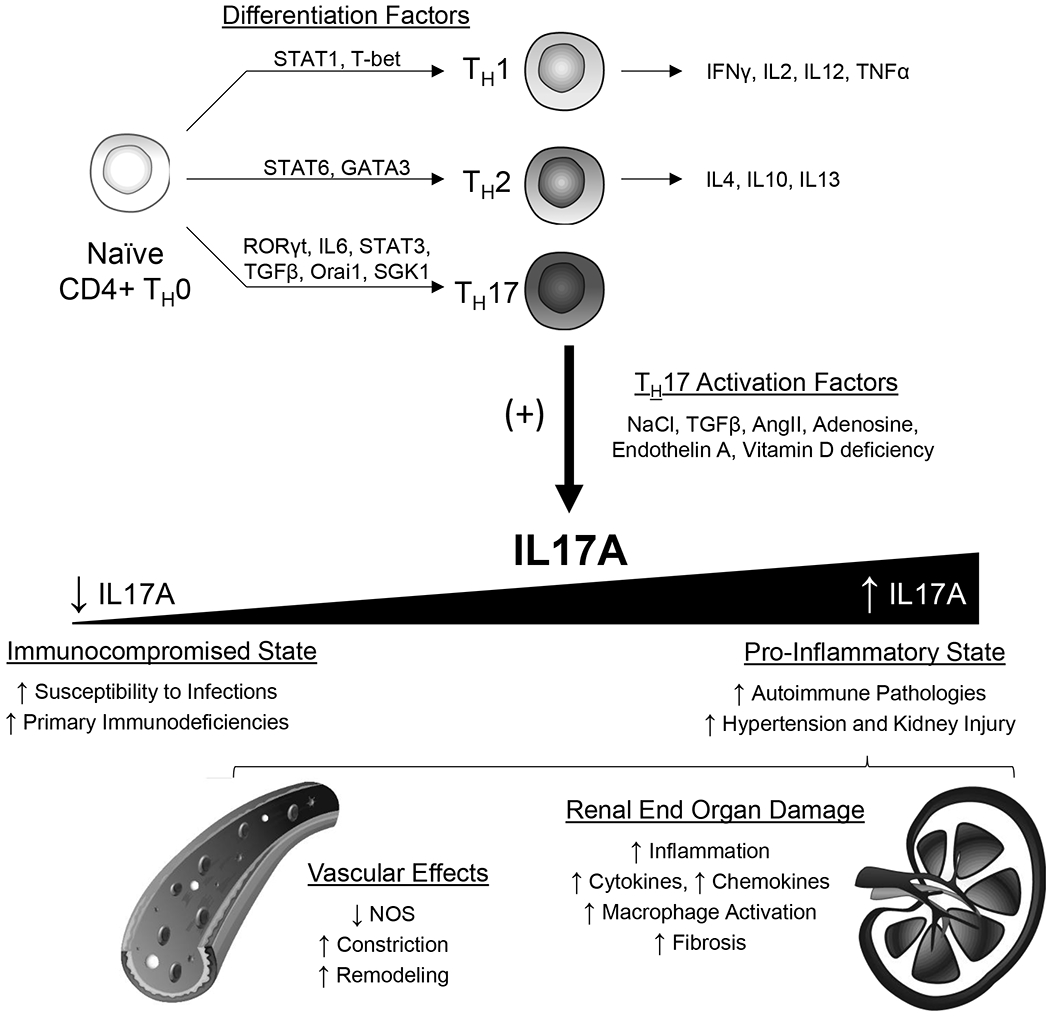
TH17 cells, under the control of RORγt, IL-6, STAT3, TGFβ, Orai1, and SGK1, are characterized by the production of the signature cytokine IL-17A. An imbalanced level of IL-17A expression dictates an immunocompromised versus pro-inflammatory state. Extensive evidence associates this pro-inflammatory state with a wide range of IL17A-induced effects, including vascular constriction and renal fibrosis, ultimately leading to the progression of hypertension and renal end organ damage.
Key Points:
There is increasing evidence from both animal models and human genetic studies indicating an important contributory role of the adaptive immune system in the development of hypertension.
IL17 producing lymphocytes, referred to as Th17 cells, are expressed in inflammatory conditions and may also be elevated in hypertension under the influence factors such as dietary salt or Angiontensin II.
The activity of IL17 may influence renal sodium handling and lead to positive sodium balance contributing to hypertension, while also promoting inflammation and fibrosis to promote hypertensive renal damage.
Financial Support and sponsorship:
Work from the authors described in this paper was supported by NIH grant DK-063114 (DB), AHA-19CDA34660184 (JA), and NIH HL116264, HL137748, and the Georgia Research Alliance (DM).
Footnotes
Conflict of Interest: DPB is an author on a patent application “Methods to treat renal disorders using calcium channel inhibitors,” US provisional application serial number 62/741,302, filed October 4, 2018. The remaining authors have no conflicts of interest.
References
*Articles of special interest
**Articles of outstanding interest
- 1.Sommers SC, Relman AS, Smithwick RH. Histologic studies of kidney biopsy specimens from patients with hypertension. Am J Pathol. 1958;34(4):685–715. [PMC free article] [PubMed] [Google Scholar]
- 2.Olsen F Inflammatory cellular reaction in hypertensive vascular disease in man. Acta Pathologica et Microbiologica Scandinavica Section A, Pathology. 1972;80(2):253–6. [PubMed] [Google Scholar]
- 3.Paronetto F Immunocytochemical observations on the vascular necrosis and renal glomerular lesions of malignant nephrosclerosis. Am J Pathol. 1965;46(6):901–15. [PMC free article] [PubMed] [Google Scholar]
- 4.Hughson MD, Gobe GC, Hoy WE, Manning RD Jr., Douglas-Denton R, Bertram JF . Associations of glomerular number and birth weight with clinicopathological features of African Americans and whites. Am J Kidney Dis. 2008;52(1):18–28. [DOI] [PubMed] [Google Scholar]
- 5.Seaberg EC, Muñoz A, Lu M, Detels R, Margolick JB, Riddler SA, et al. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS. 2005;19(9):953–60. [DOI] [PubMed] [Google Scholar]
- 6.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate Mofetil Treatment Improves Hypertension in Patients with Psoriasis and Rheumatoid Arthritis. J. Am Soc Nephrol. 2006;17(12 suppl 3):S218–S25. [DOI] [PubMed] [Google Scholar]
- 7.Ehret GB, O’Connor AA, Weder A, Cooper RS, Chakravarti A. Follow-up of a major linkage peak on chromosome 1 reveals suggestive QTLs associated with essential hypertension: GenNet study. Eur J Hum Genet 2009;17(12):1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fox ER, Young JH, Li Y, Dreisbach AW, Keating BJ, Musani SK, et al. Association of genetic variation with systolic and diastolic blood pressure among African Americans: the Candidate Gene Association Resource study. Hum Mol. Genet. 2011;20(11):2273–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shinzawa M, Yamamoto R, Nagasawa Y, Shoji T, Obi Y, Namba T, et al. Gene polymorphisms contributing to hypertension in immunoglobulin A nephropathy. Clin Exp Nephrol. 2012;16(2):250–8. [DOI] [PubMed] [Google Scholar]
- 11.Poesen R, Ramezani A, Claes K, Augustijns P, Kuypers D, Barrows IR, et al. Associations of Soluble CD14 and Endotoxin with Mortality, Cardiovascular Disease, and Progression of Kidney Disease among Patients with CKD. Clin J Am Soc Nephrol. 2015;10(9):1525–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodríguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells, and salt-sensitive hypertension: all for one and one for all. Am J Physiol-Renal Physiol. 2004;286(4):F606–F16. [DOI] [PubMed] [Google Scholar]
- 13.Johnson RJ, Rodriguez-Iturbe B, Nakagawa T, Kang D-H, Feig DI, Herrera-Acosta J. Subtle Renal Injury Is Likely a Common Mechanism for Salt-Sensitive Essential Hypertension. Hypertension. 2005;45(3):326–30. [DOI] [PubMed] [Google Scholar]
- 14.Mattson DL. Immune mechanisms of salt-sensitive hypertension and renal end-organ damage. Nat Rev Nephrol. 2019;15(5):290–300. [DOI] [PubMed] [Google Scholar]
- 15.Johnson RJ, Herrera-Acosta J, Schreiner GF, Rodriguez-Iturbe B. Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. N Engl J Med. 2002;346(12):913–23. [DOI] [PubMed] [Google Scholar]
- 16.Bravo Y, Quiroz Y, Ferrebuz A, Vaziri ND, Rodríguez-Iturbe B. Mycophenolate mofetil administration reduces renal inflammation, oxidative stress, and arterial pressure in rats with lead-induced hypertension. Am J Physiol Renal Physiol. 2007;293(2):F616–23. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Iturbe B, Johnson RJ. Role of inflammatory cells in the kidney in the induction and maintenance of hypertension. Nephrol Dial Transplant 2006;21(2):260–3. [DOI] [PubMed] [Google Scholar]
- 18.Rodríguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincón J, Chávez M, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int. 2001;59(6):2222–32. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, et al. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol-Renal Physiol. 2002;282(2):F191–201. [DOI] [PubMed] [Google Scholar]
- 20.Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madhur MS, Harrison DG. Synapses, Signals, CDs, and Cytokines. Circulation research. 2012;111(9):1113–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10):2449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Itani HA, McMaster WG, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM, et al. Activation of Human T Cells in Hypertension. Hypertension. 2016;68(1):123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, et al. CD70 Exacerbates Blood Pressure Elevation and Renal Damage in Response to Repeated Hypertensive Stimuli. Circ Res. 2016;118(8):1233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. 2017;215(1):21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schiffrin EL. T lymphocytes: a role in hypertension? Curr Opin Nephrol Hypertens. 2010;19(2):181–6 10.1097/MNH.0b013e3283360a2e. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Iturbe B, Pons H, Johnson RJ. Role of the Immune System in Hypertension. Physiol Rev. 2017;97(3):1127–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caillon A, Paradis P, Schiffrin EL. Role of immune cells in hypertension. Br J Pharmacol. 2019;176(12):1818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]; * a brief review summarizing multiple immune mechanisms implicated in the pathophysiology of hypertension
- 29.Mattson DL, Dasinger JH, Abais-Battad JM. Amplification of Salt-Sensitive Hypertension and Kidney Damage by Immune Mechanisms. Am J Hypertens. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]; *a review of immune mechanisms in the development of salt-sensitive hypertension and renal end-organ damage.
- 30.Rodríguez-Iturbe B, Pons H, Quiroz Y, Lanaspa MA, Johnson RJ. Autoimmunity in the pathogenesis of hypertension. Nat Rev Nephrol. 2014;10(1):56–62. [DOI] [PubMed] [Google Scholar]
- 31.Stewart T, Jung FF, Manning J, Vehaskari VM. Kidney immune cell infiltration and oxidative stress contribute to prenatally programmed hypertension. Kidney Int. 2005;68(5):2180–8. [DOI] [PubMed] [Google Scholar]
- 32.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout P. CD247 modulates blood pressure by altering T-lymphocyte infiltration in the kidney. Hypertension (Dallas, Tex : 1979). 2014;63(3):559–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fehrenbach DJ, Dasinger JH, Lund H, Zemaj J, Mattson DL. Splenocyte transfer exacerbates salt-sensitive hypertension in rats. Exp Physiol. 2020;105(5):864–75. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** an original article demonstrating that genetic deletion of T cells attenuates and replacement of CD4+ T cells restores salt-sensitive hypertension and end-organ damage in a rodent model of disease.
- 34.Seniuk A, Thiele JL, Stubbe A, Oser P, Rosendahl A, Bode M, et al. B6.Rag1 Knockout Mice Generated at the Jackson Laboratory in 2009 Show a Robust Wild-Type Hypertensive Phenotype in Response to Ang II (Angiotensin II). Hypertension. 2020;75(4):1110–6. [DOI] [PubMed] [Google Scholar]; ** an original article indicating that the resistance to AngII hypertension in mice lacking T- and B-cells may be modified by unidentified nongenetic modifiers in addition to the absence of functional T cells
- 35.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, et al. Obligatory Role for B Cells in the Development of Angiotensin II-Dependent Hypertension. Hypertension. 2015;66(5):1023–33. [DOI] [PubMed] [Google Scholar]
- 36.Shimada S, Abais-Battad JM, Alsheikh AJ, Yang C, Stumpf M, Kurth T, et al. Renal Perfusion Pressure Determines Infiltration of Leukocytes in the Kidney of Rats With Angiotensin II-Induced Hypertension. Hypertension. 2020;76(3):849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original article demonstrating that the infiltration of immune cells, including T cells, into the kidney in hypertension is dependent upon an initial elevation of blood pressure.
- 37.Evans LC, Petrova G, Kurth T, Yang C, Bukowy JD, Mattson DL, et al. Increased Perfusion Pressure Drives Renal T-Cell Infiltration in the Dahl Salt-Sensitive Rat. Hypertension. 2017;70(3):543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez-Iturbe B, Vaziri N, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal inflammation of immune cells, and salt-sensitivt hypertension: all for one and one for all. Am J Physiol-Renal Physiol. 2006;286(F606-F616). [DOI] [PubMed] [Google Scholar]
- 39.Abais-Battad JM, Rudemiller NP, Mattson DL. Hypertension and immunity: mechanisms of T cell activation and pathways of hypertension. Curr Opin Nephrol Hypertens. 2015;24(5):470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rudemiller NP, Crowley SD. Interactions Between the Immune and the Renin Angiotensin Systems in Hypertension. Hypertension. 2016;68(2):289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakahara T, Kido-Nakahara M, Ulzii D, Miake S, Fujishima K, Sakai S, et al. Topical application of endothelin receptor A antagonist attenuates imiquimod-induced psoriasiform skin inflammation. Sci Rep. 2020;10(1):9510. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original article indicating that an antagonist of endothelin-1 decreased Il-17A expression in human keratinocytes
- 42.Saravia J, Chapman NM, Chi H. Helper T cell differentiation. Cell Mol Immunol. 2019;16(7):634–43. [DOI] [PMC free article] [PubMed] [Google Scholar]; *a review article summarizing concepts and mechanisms of CD4+ T helper cell differentiation
- 43.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136(7):2348–57. [PubMed] [Google Scholar]
- 44.Ruterbusch M, Pruner KB, Shehata L, Pepper M. In Vivo CD4+ T Cell Differentiation and Function: Revisiting the Th1/Th2 Paradigm. Ann Rev Immunol. 2020;38(1):705–25. [DOI] [PubMed] [Google Scholar]; *a review article focused on CD4+ T cell effector function
- 45.Yu S-L, Kuan W-P, Wong C-K, Li EK, Tam L-S. Immunopathological Roles of Cytokines, Chemokines, Signaling Molecules, and Pattern-Recognition Receptors in Systemic Lupus Erythematosus. Clin Dev Immunol. 2012;2012:715190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–32. [DOI] [PubMed] [Google Scholar]
- 47.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGeachy MJ, Cua DJ, Gaffen SL. The IL-17 Family of Cytokines in Health and Disease. Immunity. 2019;50(4):892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]; *a review of Il-17 cytokine function
- 49.Bhaumik S, Basu R. Cellular and Molecular Dynamics of Th17 Differentiation and its Developmental Plasticity in the Intestinal Immune Response. Front Immunol. 2017;8:254-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang S The role of transforming growth factor β in T helper 17 differentiation. Immunology. 2018;155(1):24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sivick KE, Schaller MA, Smith SN, Mobley HLT. The Innate Immune Response to Uropathogenic Escherichia coli Involves IL-17A in a Murine Model of Urinary Tract Infection. J Immunol. 2010;184(4):2065–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sundac L, Dando SJ, Sullivan MJ, Derrington P, Gerrard J, Ulett GC. Protein-based profiling of the immune response to uropathogenic Escherichia coli in adult patients immediately following hospital admission for acute cystitis. Pathog Dis. 2016;74(6). [DOI] [PubMed] [Google Scholar]
- 53.Cypowyj S, Picard C, Maródi L, Casanova JL, Puel A. Immunity to infection in IL-17-deficient mice and humans. Eur J Immunol. 2012;42(9):2246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Monin L, Gaffen SL. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb Perspect Biol. 2018;10(4):a028522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kitching AR, Holdsworth SR. The Emergence of Th17 Cells as Effectors of Renal Injury. J Am Soc Nephrol. 2011;22(2):235–8. [DOI] [PubMed] [Google Scholar]
- 56.Taylor EB, Ryan MJ. Understanding mechanisms of hypertension in systemic lupus erythematosus. Ther Adv Cardiovasc Dis. 2016;11(1):20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shah K, Lee W-W, Lee S-H, Kim SH, Kang SW, Craft J, et al. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(2):R53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Summers SA, Odobasic D, Khouri MB, Steinmetz OM, Yang Y, Holdsworth SR, et al. Endogenous interleukin (IL)-17A promotes pristane-induced systemic autoimmunity and lupus nephritis induced by pristane. Clin Exp Immunol. 2014;176(3):341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang J, Yang X, Wang L, Li M. B cells control lupus autoimmunity by inhibiting Th17 and promoting Th22 cells. Cell Death Dis. 2020;11(3):164. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original article demonstrating that activated B cells can inhibit Th17 and promote differentiation of Th22 cells
- 60.Lee S-y, Lee SH, Seo H-B, Ryu J-G, Jung K, Choi JW, et al. Inhibition of IL-17 ameliorates systemic lupus erythematosus in Roquinsan/san mice through regulating the balance of TFH cells, GC B cells, Treg and Breg. Sci Rep. 2019;9(1):5227. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original report demonstrating the proinflammatory effects of Th17 cells as mediators of nephritis in a mouse model of systemic lupus erythematosus
- 61.Kim K-D, Srikanth S, Tan Y-V, Yee M-K, Jew M, Damoiseaux R, et al. Calcium Signaling via Orai1 Is Essential for Induction of the Nuclear Orphan Receptor Pathway To Drive Th17 Differentiation. J Immunol. 2014;192(1):110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mehrotra P, Sturek M, Neyra JA, Basile DP. Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J Clin Invest. 2019;129(11):4951–61. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original article report that activation of a store operated calcium channel, Orai1, activates Th17 and increases the severity of renal ischemia/perfusion injury in rodents; further work demonstrated elevation of Orai1 and Th17 cells in patients with acute kidney injury
- 63.Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, et al. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. 2018;21(2):240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo T, Ji W-J, Yuan F, Guo Z-Z, Li Y-X, Dong Y, et al. Th17/Treg Imbalance Induced by Dietary Salt Variation Indicates Inflammation of Target Organs in Humans. Sci Rep. 2016;6:26767-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dar HY, Singh A, Shukla P, Anupam R, Mondal RK, Mishra PK, et al. High dietary salt intake correlates with modulated Th17-Treg cell balance resulting in enhanced bone loss and impaired bone-microarchitecture in male mice. Sci Rep. 2018;8(1):2503-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haase S, Wilck N, Kleinewietfeld M, Müller DN, Linker RA. Sodium chloride triggers Th17 mediated autoimmunity. J Neuroimmunol. 2019;329:9–13. [DOI] [PubMed] [Google Scholar]
- 67.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium Chloride Drives Autoimmune Disease by the Induction of Pathogenic Th17 Cells. Nature. 2013;496(7446):518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang YH, Istomine R, Alvarez F, Al-Aubodah T-A, Shi XQ, Takano T, et al. Salt Sensing by Serum/Glucocorticoid-Regulated Kinase 1 Promotes Th17-like Inflammatory Adaptation of Foxp3/Regulatory T Cells. Cell Rep. 2020;30(5):1515–29.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original report demonstrating that high salt intake in mice drives Treg cells to a Th17-like phenotype in a process dependent upon SGK1
- 69.Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. 2015;88(4):776–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schatz V, Neubert P, Schröder A, Binger K, Gebhard M, Müller DN, et al. Elementary immunology: Na(+) as a regulator of immunity. Pediatr Nephrol. 2017;32(2):201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wenzel U, Bode M, Kurts C, Ehmke H. Salt, inflammation, IL-17 and hypertension: Salt and inflammation. Br J Pharmacol. 2018;176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Evans RDR, Antonelou M, Sathiananthamoorthy S, Rega M, Henderson S, Ceron-Gutierrez L, et al. Inherited salt-losing tubulopathies are associated with immunodeficiency due to impaired IL-17 responses. Nat Comm. 2020;11(1):4368. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original report demonstrating that patients with salt-losing tubulopathies have an increased ratio of Th2:Th17 cells
- 73.Zou A-P, Wu F, Li P-L, Cowley AW. Effect of Chronic Salt Loading on Adenosine Metabolism and Receptor Expression in Renal Cortex and Medulla in Rats. Hypertension. 1999;33(1):511–6. [DOI] [PubMed] [Google Scholar]
- 74.Li X, Liang D, Shao H, Born WK, Kaplan HJ, Sun D. Adenosine receptor activation in the Th17 autoimmune responses of experimental autoimmune uveitis. Cell Immunol. 2019;339:24–8. [DOI] [PubMed] [Google Scholar]
- 75.Wilson JM, Kurtz CC, Black SG, Ross WG, Alam MS, Linden J, et al. The A2B adenosine receptor promotes Th17 differentiation via stimulation of dendritic cell IL-6. J Immunol. 2011;186(12):6746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun D, Ko M, Shao H, Kaplan HJ. Adenosine tips the pathogenic Th1 and Th17 responses in experimental autoimmune uveitis (EAU). bioRxiv. 2020:2020.07.06.189183. [Google Scholar]; **an original report demonstrating that adenosine inhibits Th1 and enhances Th17 cell responses
- 77.Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Compr Physiol. 2011;1(2):883–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.De Miguel C, Speed JS, Kasztan M, Gohar EY, Pollock DM. Endothelin-1 and the kidney: new perspectives and recent findings. Curr Opin Nephrol Hypertens. 2016;25(1):35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tanaka K, Yoshioka K, Tatsumi K, Kimura S, Kasuya Y. Endothelin regulates function of IL-17-producing T cell subset. Life Sci. 2014;118(2):244–7. [DOI] [PubMed] [Google Scholar]
- 80.Boesen EI. ETA receptor activation contributes to T cell accumulation in the kidney following ischemia-reperfusion injury. Physiol Rep. 2018;6(17):e13865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Du Y-N, Tang X-F, Xu L, Chen W-D, Gao P-J, Han W-Q. SGK1-FoxO1 Signaling Pathway Mediates Th17/Treg Imbalance and Target Organ Inflammation in Angiotensin II-Induced Hypertension. Front Physiol. 2018;9(1581). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang D, Jin W, Wu R, Li J, Park S-A, Tu E, et al. High Glucose Intake Exacerbates Autoimmunity through Reactive-Oxygen-Species-Mediated TGF-β Cytokine Activation. Immunity. 2019;51(4):671–81.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original manuscript illustrating that high glucose intake in mice induces Th17 cells
- 83.Dankers W, Davelaar N, van Hamburg JP, van de Peppel J, Colin EM, Lubberts E. Human Memory Th17 Cell Populations Change Into Anti-inflammatory Cells With Regulatory Capacity Upon Exposure to Active Vitamin D. Front Immunol. 2019;10(1504). [DOI] [PMC free article] [PubMed] [Google Scholar]; **an original article demonstrating that illustrating that the vitamin D metabolite 1,25(OH)2D3 induced pro-inflammatory Th17 cells to switch to an anti-inflammatory phenotype in vitro
- 84.Chang SH, Chung Y, Dong C. Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. J Biol Chem. 2010;285(50):38751–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yao W, Sun Y, Wang X, Niu K. Elevated Serum Level of Interleukin 17 in a Population With Prehypertension. J Clini Hypertens. 2015;17(10):770–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ji Q, Cheng G, Ma N, Huang Y, Lin Y, Zhou Q, et al. Circulating Th1, Th2, and Th17 Levels in Hypertensive Patients. Dis Markers. 2017;2017:7146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hosseini A, Dolati S, Hashemi V, Abdollahpour-Alitappeh M, Yousefi M. Regulatory T and T helper 17 cells: Their roles in preeclampsia. J Cell Physiol. 2018;233(9):6561–73. [DOI] [PubMed] [Google Scholar]
- 89.Wade B, Petrova G, Mattson DL. Role of immune factors in angiotensin II-induced hypertension and renal damage in Dahl salt-sensitive rats. Am J Physiol-Regul Integr Comp Physiol. 2017;314(3):R323–R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Amador CA, Barrientos V, Peña J, Herrada AA, González M, Valdés S, et al. Spironolactone Decreases DOCA Salt Induced Organ Damage by Blocking the Activation of T Helper 17 and the Downregulation of Regulatory T Lymphocytes. Hypertension. 2014;63(4):797–803. [DOI] [PubMed] [Google Scholar]
- 91.Chiasson VL, Pakanati AR, Hernandez M, Young KJ, Bounds KR, Mitchell BM. Regulatory T-Cell Augmentation or Interleukin-17 Inhibition Prevents Calcineurin Inhibitor Induced Hypertension in Mice. Hypertension. 2017;70(1):183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cornelius DC, Hogg JP, Scott J, Wallace K, Herse F, Moseley J, et al. Administration of Interleukin-17 Soluble Receptor C Suppresses TH17 Cells, Oxidative Stress, and Hypertension in Response to Placental Ischemia During Pregnancy. Hypertension. 2013;62(6):1068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, et al. Interleukin-17A Regulates Renal Sodium Transporters and Renal Injury in Angiotensin II-Induced Hypertension. Hypertension. 2016;68(1):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal Transporter Activation During Angiotensin-II Hypertension is Blunted in Interferon-γ−/− and Interleukin-17A−/− Mice. Hypertension. 2015;65(3):569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2012;97(4):696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Orejudo M, Rodrigues-Diez RR, Rodrigues-Diez R, Garcia-Redondo A, Santos-Sánchez L, Rández-Garbayo J, et al. Interleukin 17A Participates in Renal Inflammation Associated to Experimental and Human Hypertension. Front Pharmacol. 2019;10(1015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lavoz C, Matus YS, Orejudo M, Carpio JD, Droguett A, Egido J, et al. Interleukin-17A blockade reduces albuminuria and kidney injury in an accelerated model of diabetic nephropathy. Kidney Int. 2019;95(6):1418–32. [DOI] [PubMed] [Google Scholar]
- 98.Mehrotra P, Collett JA, McKinney S, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion:compensatory role of Natural Killer cells in athymic rats. Am J Physiol - Renal Physiol. 2017;312 (3):F385–F97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Basile D, Ullah M, Collett J, Mehrotra P. T helper 17 cells in the pathophysiology of acute and chronic kidney disease. Kidney Res Clin Pract. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wojkowska DW, Szpakowski P, Glabinski A. Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells. Int J Mol Sci. 2017;18(5):1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chung BH, Kim KW, Kim B-M, Doh KC, Cho M-L, Yang CW. Increase of Th17 Cell Phenotype in Kidney Transplant Recipients with Chronic Allograft Dysfunction. PLOS ONE. 2016;10(12):e0145258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Weng C-H, Li Y-J, Wu H-H, Liu S-H, Hsu H-H, Chen Y-C, et al. Interleukin-17A induces renal fibrosis through the ERK and Smad signaling pathways. Biomed Pharmacother. 2020;123:109741. [DOI] [PubMed] [Google Scholar]
- 103.Orejudo M, García-Redondo Ana B, Rodrigues-Diez Raúl R, Rodrigues-Díez R, Santos-Sanchez L, Tejera-Muñoz A, et al. Interleukin-17A induces vascular remodeling of small arteries and blood pressure elevation. Clin Sci. 2020;134(5):513–27. [DOI] [PubMed] [Google Scholar]
- 104.Zhang J, Qiao Q, Liu M, He T, Shi J, Bai X, et al. IL-17 Promotes Scar Formation by Inducing Macrophage Infiltration. Am J Pathol. 2018;188(7):1693–702. [DOI] [PubMed] [Google Scholar]
- 105.Ge S, Hertel B, Susnik N, Rong S, Dittrich AM, Schmitt R, et al. Interleukin 17 Receptor A Modulates Monocyte Subsets and Macrophage Generation In Vivo. PLOS ONE. 2014;9(1):e85461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.de la Paz Sánchez-Martínez M, Blanco-Favela F, Mora-Ruiz MD, Chávez-Rueda AK, Bernabe-García M, Chávez-Sánchez L. IL-17-differentiated macrophages secrete pro-inflammatory cytokines in response to oxidized low-density lipoprotein. Lipids Health Dis. 2017;16(1):196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lavoz C, Rayego-Mateos S, Orejudo M, Opazo-Ríos L, Marchant V, Marquez-Exposito L, et al. Could IL-17A Be a Novel Therapeutic Target in Diabetic Nephropathy? J Clin Med. 2020;9(1):272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chiricozzi A, Krueger JG. IL-17 targeted therapies for psoriasis. Expert Opin Investig Drugs. 2013;22(8):993–1005. [DOI] [PubMed] [Google Scholar]