

Supplemental Digital Content is available in the text.
Keywords: arteries, endothelium, homeostasis, ischemia, transcription factor, vasodilation
Abstract
Rationale:
Understanding the mechanisms that regulate arterial flow recovery is important to design treatment options for peripheral artery disease patients ineligible for invasive revascularization. Transcriptional orchestrators of this recovery process represent an appealing target for treatment design. We previously identified Prdm (positive regulatory domain-containing protein) 16 as an arterial-specific endothelial transcription factor but its in vivo role in arteries remains completely unknown.
Objective:
To unravel the role of Prdm16 in arteries under physiological and pathological conditions, more specifically during peripheral artery disease.
Methods and Results:
Within the vasculature, Prdm16 expression was strictly confined to arterial endothelial and smooth muscle cells. Heterozygous loss of Prdm16 caused a modest reduction of the inner arterial diameter and smooth muscle cell coating without compromising vasomotor function. Upon femoral artery ligation, Prdm16+/− mice featured significantly impaired flow recovery to ischemic limbs. This impairment was recapitulated in mice with a Prdm16 deletion specifically in endothelial cells (EC-Prdm16−/−) but not smooth muscle cells. Structural collateral remodeling was normal in both Prdm16+/− and EC-Prdm16−/− mice, but significant endothelial dysfunction postligation was present in EC-Prdm16−/− mice as evidenced by impaired endothelial-dependent relaxation. Upon ligation, endothelial Prdm16 deficiency altered the expression of genes encoding endothelial cell function regulators, many related to nitric oxide bioavailability and Ca2+ homeostasis. Accordingly, Prdm16 overexpression in cultured endothelial cells affected both total cellular Ca2+ levels and store-operated Ca2+ entry.
Conclusions:
We showed that Prdm16 is indispensable for arterial flow recovery under pathological challenge not because it affects structural remodeling but due to its role in maintaining endothelial function. It, therefore, represents an appealing target for designing novel therapeutic strategies for no-option patients with peripheral artery disease.
Editorial, see p 78
Meet the First Author, see p 4
The blood vascular system has a dual structure composed of an arterial and venous arm.1 The former provides oxygenated blood to tissues, and the latter recirculates deoxygenated blood to heart and lungs for reoxygenation. These opposite functions are mirrored by distinct molecular and morphological signatures in endothelial cells (ECs) lining the inside of arteries and veins. The existence of arterial-specific diseases (eg, atherosclerosis) is a likely consequence of these distinct features.2 Understanding the mechanisms determining the arterial EC signature is equally important for development of treatments tailored towards improving growth of arteries and preserving their specific functions during ischemic diseases caused by insufficient arterial blood supply.
Limb ischemia, known as peripheral artery disease (PAD), affects 200 million people worldwide, and this number will further increase in the aging population.3 Current revascularization strategies are mainly invasive procedures for which many PAD patients are ineligible due to the presence of risk factors or multiple-vessel occlusion.4 Although noninvasive alternatives, for example, administration of general angiogenic growth factors like VEGF (vascular endothelial growth factor) and FGF (fibroblast growth factor) have been tested for no-option patients, these have shown limited success,5 potentially due to lack of specificity towards an arterial response. Hence, the advent of more tailored molecular therapies is eagerly awaited. Designing such therapies requires a thorough mechanistic understanding of the arterial flow recovery process upon arterial obstruction.
To cope with such an obstruction, the vascular system has a collateral reserve network in which collateral arteries connect 2 nurturing arteries such that flow can be redirected. Sudden exposure to flow imposes shear stress on collateral ECs which triggers structural and functional changes, that is, luminal expansion and acquisition of a thicker smooth muscle cell (SMC) coat.6 This remodeling process is known as adaptive arteriogenesis and converges on transcription factors. Msx (muscle segment homeobox) 1 is part of a mechanosensing pathway that fuels collateral arterial growth. It induces a proinflammatory state in collateral ECs, enabling them to recruit monocytes to initiate collateral expansion.7 In addition to shear stress-driven collateral growth proximal to the obstruction, hypoxia triggers capillary growth more distally. This angiogenic response is also mediated through transcription factors (eg, TFEB [trancription factor-EB]).8
Inspired by the finding that Msx1 triggered structural collateral growth during PAD,7 we sought to determine whether other transcription factors from our previously established arterial EC signature were also involved.9 Prdm (positive regulatory domain-containing protein) 16 emerged as a likely candidate as it was the most differentially expressed transcription factor with an unknown role in arteries in vivo.9 Prdm16 is part of a family of 17 members that are involved in a spectrum of biological processes including cell fate decision making.10 Although initially identified as an oncogene in myelodysplastic syndrome and acute myeloid leukemia,11 Prdm16 is currently mostly known for its roles in brown/beige adipose tissue fate decision,10,12 craniofacial development,13 hematopoietic/neuronal stem cell maintenance, and homeostasis14,15 and in the context of certain cardiomyopathies.16 Accordingly, Prdm16 expression has been documented in brown and beige adipose tissue, the skeletal, hematopoietic, and nervous systems and the heart.10,11,13–15 Constitutive homozygous deficiency of Prdm16 in mice causes respiratory failure and perinatal death.13 Recently, Prdm16 expression was reported in retinal arterioles,17 yet, a detailed analysis of its expression and role in the adult arterial system, more specifically during adaptive arteriogenesis, remains unexplored.
Methods
Extended methods and a Major Resources Table (Table I in the Data Supplement) are provided in the Data Supplement.
Data Availability
The authors declare that the majority of supporting data are presented within this article and in the Data Supplement. Data not directly presented are available from the corresponding author upon reasonable request. Single-cell RNA sequencing data have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress; accession number E-MTAB-9703).
Mouse Strains and Cell Type–Specific Prdm16 Deletion
Animal experiments were approved by the KU Leuven Ethics Committee on Animal Use. Three mouse lines were used: ubiquitous constitutive Prdm16 knockout mice (Prdm16Gt(OST67423)Lex; or Prdm16+/−)13; EC-specific Prdm16 knockout mice (Cdh5-CreERT2;Prdm16fl/fl; or EC-Prdm16−/−) generated by intercrossing the Cdh5-CreERT2 driver line18 with Prdm16fl/fl mice19; mice with Prdm16 deficiency in SMCs (Sm22α-Cre;Prdm16fl/fl; or SMC-Prdm16−/−) generated by crossing the Sm22α-Cre driver line20 with Prdm16fl/fl mice. EC-specific Prdm16 deletion was induced by tamoxifen (4 intragastric injections on postnatal day 1 to 4 for long-term deletion or, for short-term deletion, 5 daily injections in week 10, starting 14 days before hindlimb ischemia (HLI) induction). Tamoxifen-induced deletion efficacy was ≈80% and Prdm16 loss in collaterals was restricted to ECs or SMCs in EC-Prdm16−/− and SMC-Prdm16−/− mice, respectively (Figure I in the Data Supplement).
Prdm16 Expression Analysis
Prdm16 expression was examined by X-gal 5-bromo-4-chloro-3-indolyl-D-galactopyranoside (X-gal) staining (taking advantage of the presence of a gene-trap cassette in the Prdm16 locus encoding β-galactosidase in Prdm16+/− mice)13 or by an in-house optimized immunofluorescence staining.
Structural, Biomechanical, and Blood Pressure Measurements
Inner diameter and SMC cross-sectional area of descending aorta (DA) and femoral artery (FA) were measured in cross-sections of adult Prdm16+/+ and Prdm16+/− mice. Biomechanical measurements were obtained by mounting DA segments in a Rodent Oscillatory Tension Set-Up to Study Arterial Compliance.21 Blood pressure was determined via noninvasive tail-cuff measurements on a CODA system (Kent Scientific).
HLI Induction and Follow-Up
HLI studies were performed in 8- to 12-week old Prdm16+/−, EC-Prdm16−/−, SMC-Prdm16−/− mice and their wild-type (WT) littermates by FA ligation.7 Reperfusion recovery was monitored by laser doppler imaging (Lisca, PIMII), and necrosis was assessed by an in-house developed necrosis score. At day 3 or day 21 postligation, adductor or gastrocnemius muscles were dissected out and processed for histology, RNA, or protein extraction. Gene expression was quantified by quantitative real-time polymerase chain reaction as described online using primer sets listed in Table II in the Data Supplement. Protein quantification was performed by Western blotting, as described online. Fibrosis/fibro-adipose tissue22 in day 21 postligation gastrocnemius muscles was investigated on Sirius red-stained cross-sections. Collateral remodeling in the adductor region was evaluated by studying inflammatory cell recruitment around growing collaterals at day 3 postligation on Cd45- or Cd68-αSMA (smooth muscle actin) double-stained cross-sections and by determining luminal and medial expansion at day 21 postligation on Cd31-αSMA double-stained cross-sections. To determine capillary density in the gastrocnemius muscle, an identical Cd31/αSMA staining was used. Morphometry was performed by a blinded investigator.
Nano–Computed Tomography
Prdm16+/− and Prdm16+/+ littermates were subjected to FA ligation and perfused 7 days later with barium sulfate/gelatin. After overnight solidification, limbs were dissected out and scanned with a Phoenix Nanotom M, and scans were optimized during three-dimensional reconstruction (Datos|x, Waygate Technologies). Images were analyzed as described online.
Organ Bath and Myograph Measurements
DA and FA segments of Prdm16+/− or long-term EC-Prdm16−/− mice and their corresponding WT littermates were dissected out and mounted in an organ bath or on a myograph (DMT, 610M), respectively.23,24 Active contractions and relaxations were monitored during cumulative dose exposure to phenylephrine (contraction), acetylcholine (EC-dependent relaxation), or diethylamine NONOate (EC-independent relaxation). To study vasomotor function upon HLI, right FA segments downstream of the ligation site of Prdm16+/− or long-term EC-Prdm16−/− mice, and their corresponding WT littermates were isolated and mounted on a myograph day 3 postligation. FA segments of the unligated left side served as control. For EC-Prdm16−/− and WT littermates, DA vasomotor function was recorded from the same mice.
Single-Cell RNA Sequencing
On day 3 postligation, FA segments downstream of the ligation and corresponding segments on the unligated side from EC-Prdm16−/− or EC-Prdm16+/+ littermates (n=4 each) were dissected out and collected on ice. FA segments were pooled per sample type (ligated and unligated, for both EC-Prdm16−/− and EC-Prdm16+/+), converted into single-cell suspensions, and barcoded libraries were prepared using kits from 10X Genomics and sequenced on an Illumina Nextseq and NovaSeq6000 as described online. Raw data processing, quality control (Figure II in the Data Supplement), comparative analysis of Cd45+ (Ptprc+) clusters, and differential expression/functional annotation analyses of the arterial EC cluster was performed as described online using Cell Ranger, Harmony, and MAST software packages/algorithms.25–27
Lentiviral Transduction and Ca2+ Measurements
A lentivirus overexpressing full-length mouse Prdm16 (clone ID 6409778; Thermo Scientific Molecular Biology) was used.9 Human umbilical vein ECs (generated after obtaining informed consent from donors)9 were transduced and after 6 days, harvested and seeded for Ca2+ measurements. As control, cells were transduced with a Cherry-expressing lentivirus. Human umbilical vein ECs were loaded with Fura-2AM (Fura-2 acetoxymethyl) and total cellular and endoplasmic reticulum (ER) Ca2+ content and store-operated Ca2+ entry were measured 24 hours later as described online.
Statistics
Continuous data are presented as mean±SEM. Detailed information on statistics is provided in the Data Supplement (Table III in the Data Supplement).
Results
Prdm16 Universally Marks Adult Arteries
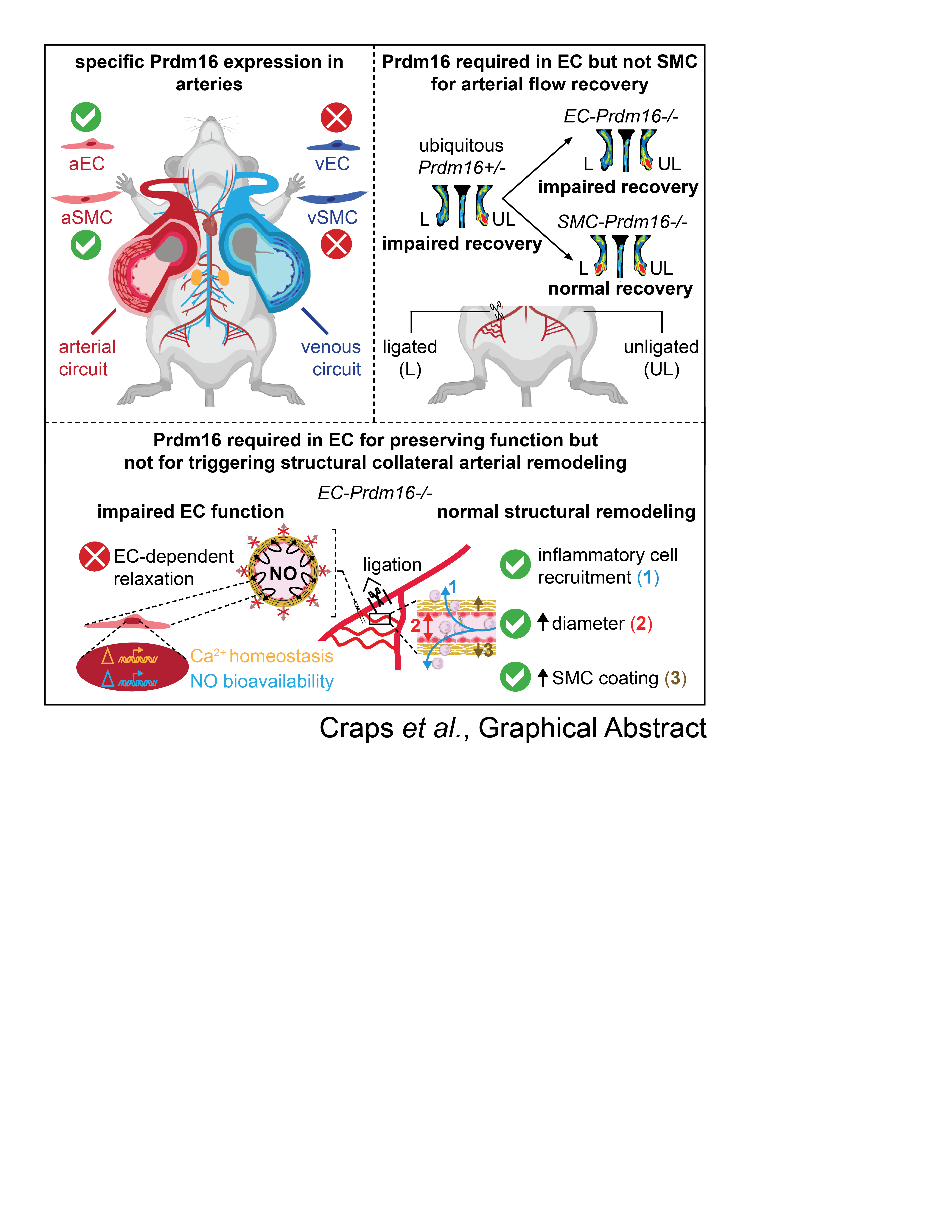
From comparative profiling of human umbilical cord ECs, PRDM16 emerged as an arterial EC-enriched transcription factor.9 This expression pattern was conserved in adult mice, as Prdm16 was only detectable in arteries and not veins, with strong expression in both ECs and SMCs. This arterial-exclusive expression was present in the intima and media of multiple adult blood vessel types with strong expression in the carotid artery, thoracic aorta, and FA, the former 2 representing elastic conduit arteries, the third one being a smaller caliber muscular artery (Figure 1A through 1F). Similar observations were made in other arteriovenous pairs (Figure IIIA through IIID in the Data Supplement). Whole-mount staining of the retina revealed that arterial expression was retained down to the arteriolar level (Figure 1G), in accordance with a previous study.17 Next to physiological conditions, Prdm16 expression was also prominent in ECs and SMCs of collateral arteries following HLI induction (Figure 1H). Compared with the unligated side, Prdm16 expression in collateral ECs and SMCs was relatively stable during the first 3 days postligation, then subsequently declined followed by a slow recovery from day 7 onwards (Figure IIIE and IIIF in the Data Supplement). Thus, Prdm16 universally marked ECs and SMCs of arteries and not veins, independent of anatomic location, vessel caliber, artery type, or health status.
Figure 1.
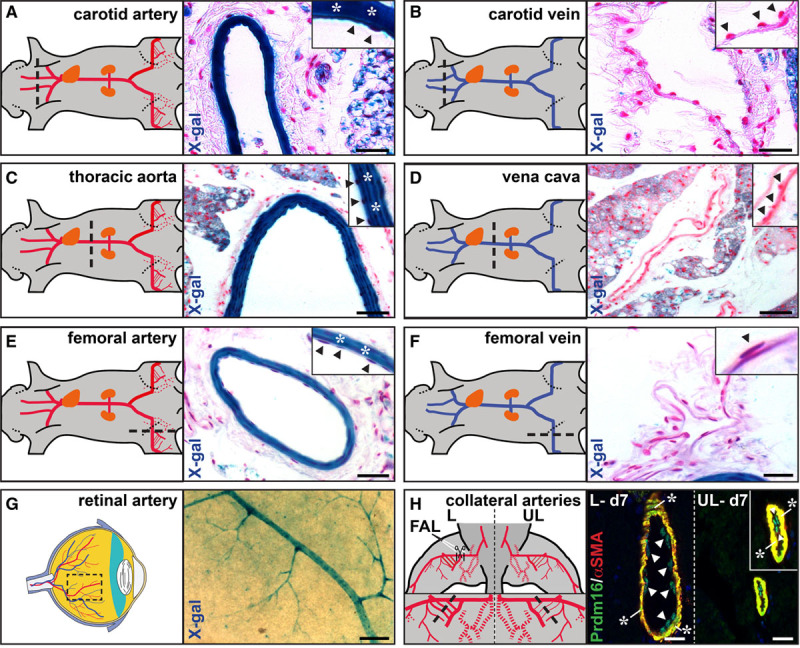
Prdm (positive regulatory domain-containing protein) 16 is a universal arterial marker in health and disease. A–F, X-gal (5-bromo-4-chloro-3-indolyl–D-galactopyranoside) staining on cross-sections of adult Prdm16+/− arteriovenous pairs. Arteries are shown in (A, C, and E), venous counterparts in (B, D, and F). G, Whole-mount X-gal staining on a flat-mounted retina of a Prdm16+/− adult mouse. H, Immunofluorescence staining for Prdm16 (green) and αSMA (smooth muscle actin; red) on adductor sections of the unligated (UL) and ligated (L) side 7 d after femoral artery ligation (FAL). Arrowheads and asterisks indicate endothelial and smooth muscle cells, respectively, in A–F, and H. Black dashed lines in A–F, and H and dashed box in G show location of the cross-section or whole-mount staining. Scale bars: 50 μm in A–F, 25 μm in G, and 10 μm in H.
Ubiquitous Heterozygous Prdm16 Deficiency Does Not Affect Arterial Vasomotor Function
Next, we questioned whether the arterial-restricted expression pattern of Prdm16 reflected a functional role in adult arteries under physiological conditions. Although homozygous Prdm16 deficiency causes respiratory failure and death around birth,13 heterozygous Prdm16 mice were viable and fertile. DA and FA dimensions were slightly smaller in Prdm16+/− versus Prdm16+/+ mice (Figure 2A through 2C). SMC cross-sectional area was significantly decreased in the FA and showed a trend towards decrease in the DA in Prdm16+/− mice (Figure 2D through 2F). Despite reduced arterial dimensions, baseline arterial vasomotor function was normal, as evidenced by identical blood pressure (not shown) and similar maximal contraction and EC-dependent or EC-independent relaxation in the DA and FA of Prdm16+/− and Prdm16+/+ littermates (Figure 2G through 2O). Further examinations showed normal biomechanical properties of Prdm16+/− DA segments at physiological stretch frequency and pressure (Figure IV in the Data Supplement). Thus, Prdm16 heterozygous deficiency did not affect baseline arterial vasomotor function or biomechanics.
Figure 2.
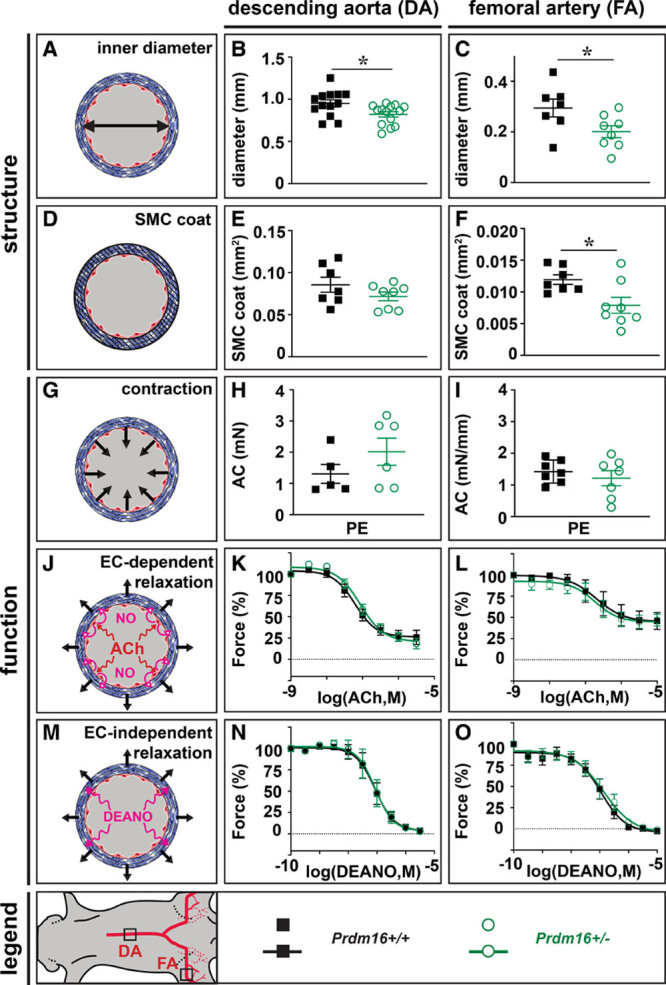
Ubiquitous heterozygous Prdm (positive regulatory domain-containing protein) 16 deficiency slightly affects arterial geometry but not vasomotor function. A–C, Schematic representation (A) and quantification of descending aorta (DA; B) and femoral artery (FA; C) diameter of Prdm16+/+ (n=13 for DA; n=7 for FA) and Prdm16+/− (n=14 for DA; n=8 for FA) mice. D–F, Schematic representation (D) and quantification of DA (E) and FA (F) smooth muscle cell (SMC) cross-sectional area of Prdm16+/+ (n=7 for DA and FA) and Prdm16+/− (n=8 for DA and FA) mice. G–I, Schematic representation (G) and quantification of the phenylephrine (PE)-induced absolute contraction (AC) in DA (H) and FA (I) of Prdm16+/+ (n=5 for DA; n=7 for FA) and Prdm16+/− (n=6 for DA; n=7 for FA) mice (concentration PE: 3×10-6 mol/L for DA and FA). J–L, Schematic representation (J) and quantification of relative acetylcholine (ACh)-mediated relaxation of DA (K) and FA (L) from Prdm16+/+ (n=6 for DA; n=7 for FA) and Prdm16+/− (n=5 for DA; n=7 for FA) mice. M–O, Schematic representation (M) and quantification of relative diethylamine NONOate (DEANO)–mediated relaxation of DA (N) and FA (O) from Prdm16+/+ (n=6 for DA; n=7 for FA) and Prdm16+/− (n=6 for DA; n=7 for FA) mice. Data represent mean±SEM. *P<0.05: vs corresponding control (Table III in the Data Supplement). EC indicates endothelial cell, and M in (K,L,N,O), molar.
Ubiquitous or EC-Specific Prdm16 Deficiency Impairs Arterial Flow Recovery Upon HLI
To test whether Prdm16 had a role in arteries during PAD, FAs of Prdm16+/− and Prdm16+/+ littermates were ligated to trigger adaptive arteriogenesis. Prdm16+/+ mice had spontaneous arterial flow recovery to >80% at day 21, whereas recovery was impaired from day 7 onwards in Prdm16+/− mice (Figure 3A and 3B; Figure VA and VB in the Data Supplement). Accordingly, Prdm16+/− mice had more tissue necrosis, fibrosis, and fibro-adipose tissue in their gastrocnemius muscles than Prdm16+/+ littermates (Figure 3C and 3D; Figure VC through VE in the Data Supplement). Because Prdm16 was expressed in both EC and SMC compartments, we investigated which cell type was mainly responsible for the defective flow recovery by inducing HLI in mice with Prdm16 deficiency in ECs or SMCs using Cre-mediated excision driven by the tamoxifen-inducible Cdh5 or the constitutive Sm22α promoter, respectively. Long-term (ie, from birth onwards) Prdm16 deficiency in ECs caused a significant impairment in arterial flow recovery, compared with their corresponding WT littermates, which was accompanied by worsened tissue necrosis, more fibrosis, and fibro-adipose tissue in the gastrocnemius muscle (Figure 3E through 3H; Figure VF through VJ in the Data Supplement). When shortening the duration of Prdm16 deficiency in ECs by starting tamoxifen administration only 14 days before HLI induction, the impairment in flow recovery was shifted towards a later time-point (Figure 3I and 3J; Figure VK and VL in the Data Supplement) and follow-up until 6 weeks after HLI induction revealed that this impairment did not resolve later on (not shown). This less severe recovery deficit also correlated with lower scores for necrosis, fibrosis, and fibro-adipose tissue (Figure 3K and 3L; Figure VM through VO in the Data Supplement). In contrast, constitutive Prdm16 loss in SMCs did not impair arterial flow recovery (Figure 3M and 3N; Figure VP and VQ in the Data Supplement). Accordingly, there was no alteration in necrosis, fibrosis, or fibro-adipose tissue in SMC-Prdm16−/− versus SMC-Prdm16+/+ gastrocnemius muscles (Figure 3O and 3P; Figure VR through VT in the Data Supplement). Thus, Prdm16 deficiency in ECs but not SMCs had a detrimental time-dependent effect on arterial flow recovery.
Figure 3.
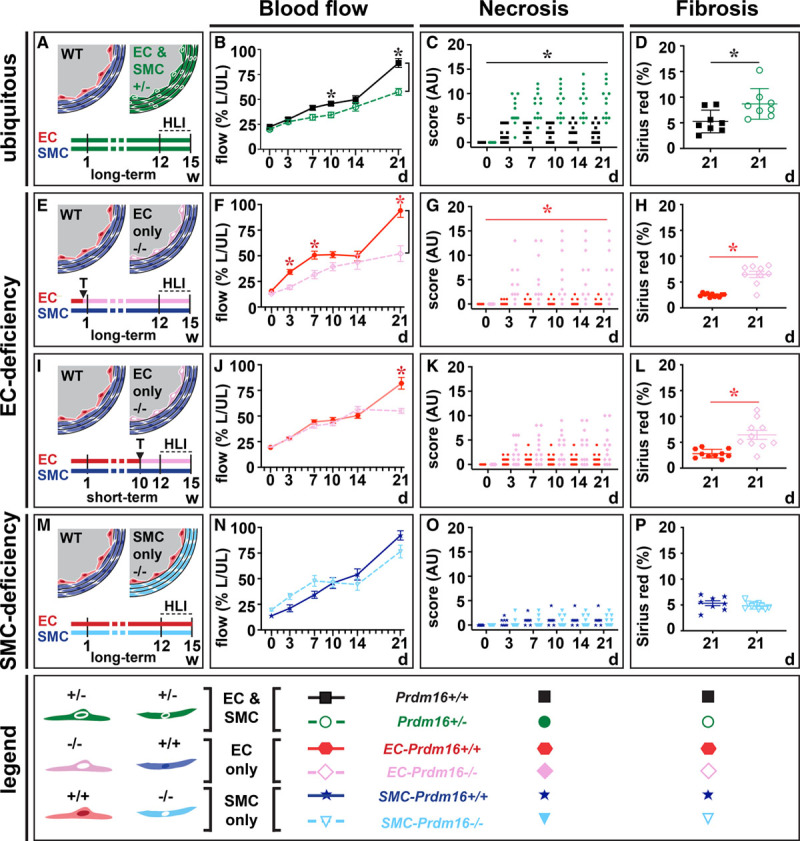
Ubiquitous or endothelial cell (EC)–specific Prdm (positive regulatory domain-containing protein) 16 deficiency impair flow recovery and boost necrosis and fibrosis upon hindlimb ischemia (HLI). A–D, Schematic representation/time-line (A; w [weeks]; wild-type [WT]) and quantification of flow recovery (B; expressed as % of ligated [L] over unligated [UL]), d 21 necrosis (C; expressed in arbitrary units [AU]) and d 21 fibrosis scores (D) in Prdm16+/+ (n=8–12) or Prdm16+/− (n=8–12) mice after HLI induction. E–H, Schematic representation/time-line (E; T: tamoxifen) and quantification of flow recovery (F; expressed as % of L over UL), d 21 necrosis (G; expressed in AU), and d 21 fibrosis scores (H) in long-term EC-Prdm16+/+ (n=11–12) or EC-Prdm16−/− (n=9–10) mice after HLI induction. I–L, Schematic representation/time-line (I) and quantification of flow recovery (J; expressed as % of L over UL), d 21 necrosis (K; expressed in AU) and d 21 fibrosis scores (L) in short-term EC-Prdm16+/+ (n=10–16) or EC-Prdm16−/− (n=10–13) mice after HLI induction. M–P, Schematic representation/time-line (M) and quantification of flow recovery (N; expressed as % of L over UL), d 21 necrosis (O; expressed in AU), and d 21 fibrosis scores (P) in smooth muscle cell (SMC)-Prdm16+/+ (n=8) or SMC-Prdm16−/− (n=10) mice after HLI induction. Data in B, D, F, H, J, L, N, and P represent mean±SEM. *P<0.05: vs corresponding control (Table III in the Data Supplement).
Ubiquitous or EC-Specific Prdm16 Deficiency Does Not Impair Vessel Remodeling Upon HLI
Structural vascular remodeling during adaptive arteriogenesis is initiated by inflammatory cell recruitment towards the endothelium of collaterals which primes them for luminal and medial expansion.7,28 Recruitment of Cd45+ cells around growing collaterals at the ligated side at day 3 postligation in the adductor region was unaltered in Prdm16+/− and long-term EC-Prdm16−/− mice compared with their corresponding WT littermates (Figure 4A through 4C; Figure VIA through VID in the Data Supplement). Accordingly, the increase in inner diameter and SMC cross-sectional area of the remodeled collaterals was not affected by ubiquitous or EC-restricted Prdm16 deficiency (Figure 4D through 4I; Figure VIE through VIH in the Data Supplement). Analysis of the day 3 Cd45+ cell infiltrate around EC-Prdm16−/− arteries at single-cell resolution revealed 4 Cd45+ (Ptprc+) cell types among the 14 annotated clusters, that is, Cd68+ monocyte/macrophages, Ly6g+ monocyte/neutrophils, Cd3+ T, and Cd79a+ B lymphocytes (Figure VIIA through VIID in the Data Supplement). There were no major genotypic differences in extent or composition of the lymphoid/myeloid cell groups, and the comparable Cd68+ monocyte/macrophage recruitment around EC-Prdm16−/− and EC-Prdm16+/+ collaterals was confirmed by immunofluorescence staining (Figure VIIE through VIIJ in the Data Supplement). The lack of a remodeling deficit in Prdm16+/− adductors was confirmed by 3-dimensional reconstruction of the vessel network by nano-computed tomography (CT), as evident from a comparable expansion of the total vascular volume or growth/remodeling of the collaterals (Figure VIII in the Data Supplement).
Figure 4.
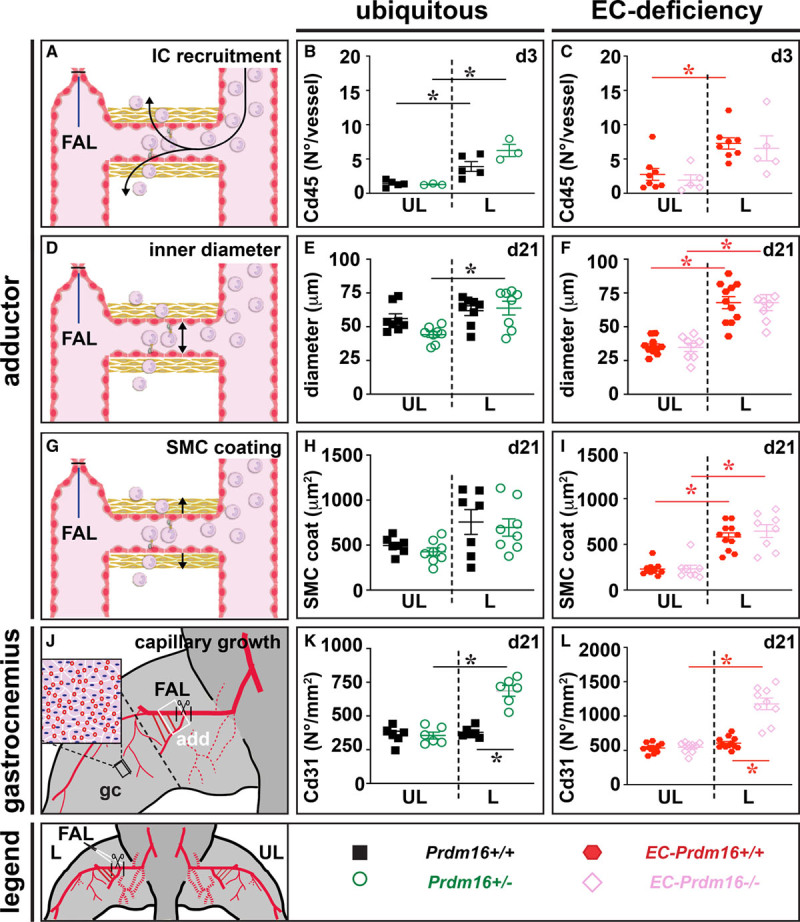
Ubiquitous or endothelial cell (EC)–specific Prdm (positive regulatory domain-containing protein) 16 deficiency do not affect structural vascular remodeling upon hindlimb ischemia. A–C, Schematic representation (A) and quantification of Cd45+ inflammatory cell (IC) recruitment around expanding collaterals in the adductor region of the ligated (L) and unligated (UL) side at 3 d after femoral artery ligation (FAL) in Prdm16+/+ (n=5) or Prdm16+/− (n=3) mice (B) and long-term EC-Prdm16+/+ (n=8) or EC-Prdm16−/− (n=5) mice (C). D–F, Schematic representation (D) and quantification of collateral inner diameter at d 21 after FAL in Prdm16+/+ (n=8) or Prdm16+/− (n=8) mice (E) and long-term EC-Prdm16+/+ (n=11) or EC-Prdm16−/− (n=8) mice (F). G–I, Schematic representation (G) and quantification of smooth muscle cell (SMC)–coating (measured as cross-sectional medial area) of remodeled collateral arteries at d 21 after FAL in Prdm16+/+ (n=7) or Prdm16+/− (n=8) mice (H) and long-term EC-Prdm16+/+ (n=11) or EC-Prdm16−/− (n=8) mice (I). J–L, Schematic representation (J) and quantification of capillary density in gastrocnemius (gc; black box) muscle at d 21 after FAL in Prdm16+/+ (n=6) or Prdm16+/− (n=6) mice (K) and long-term EC-Prdm16+/+ (n=12) or EC-Prdm16−/− (n=9) mice (L). Data represent mean±SEM. *P<0.05: vs indicated condition (Table III in the Data Supplement). Adductor region (add) studied in A–I is indicated by a white box in J.
In addition to adaptive arteriogenesis in the adductor region, flow distribution to the downstream ischemic muscle can be improved by increasing capillary density in the gastrocnemius region.8 Unlike in their Prdm16+/+ littermates, Cd31+ capillary density in the gastrocnemius muscle of the ligated limb was significantly increased in Prdm16+/− and long-term EC-Prdm16−/− mice, likely representing a compensatory (but insufficient) response to aggravated ischemia driven by increased Vegfa levels (Figure 4J through 4L; Figure VII through VIL in the Data Supplement; not shown). Altogether, the deficit in flow recovery in Prdm16+/− and long-term EC-Prdm16−/− mice could not be explained by structural vascular remodeling impairment.
EC-Specific Prdm16 Deficiency Causes Attenuated EC-Dependent Relaxation Upon HLI
To study vasomotor function upon HLI induction, FA segments downstream of the ligation site were isolated from Prdm16+/−, long-term EC-Prdm16−/− and their corresponding WT littermates at day 3 postligation and mounted on a myograph. Phenylephrine-mediated contractions tended to be lower in FA segments of the ligated versus the unligated side but Prdm16 deficiency did not affect contraction (Figure 5A through 5C). Likewise, EC-dependent relaxation in response to acetylcholine was altered in the FA segment of the ligated side, but this response was not impaired in Prdm16+/− compared with their Prdm16+/+ littermates (Figure 5D and 5E). In contrast, HLI triggered a significantly lowered response to acetylcholine in long-term EC-Prdm16−/− compared with EC-Prdm16+/+ FA segments on the ligated side (Figure 5D and 5F). Vice versa, EC-independent relaxation upon exposure to NO-donor diethylamine NONOate was not significantly affected by HLI or by Prdm16 deficiency (Figure 5G through 5I). Vasomotor function of the FA at the unligated side (Figure 5C, 5F, and 5I) and the DA upstream of the ligation (Figure IX in the Data Supplement) was very similar between long-term EC-Prdm16−/− mice and their EC-Prdm16+/+ littermates. Altogether, endothelial Prdm16 deficiency resulted in early-onset EC dysfunction only when challenged by HLI.
Figure 5.
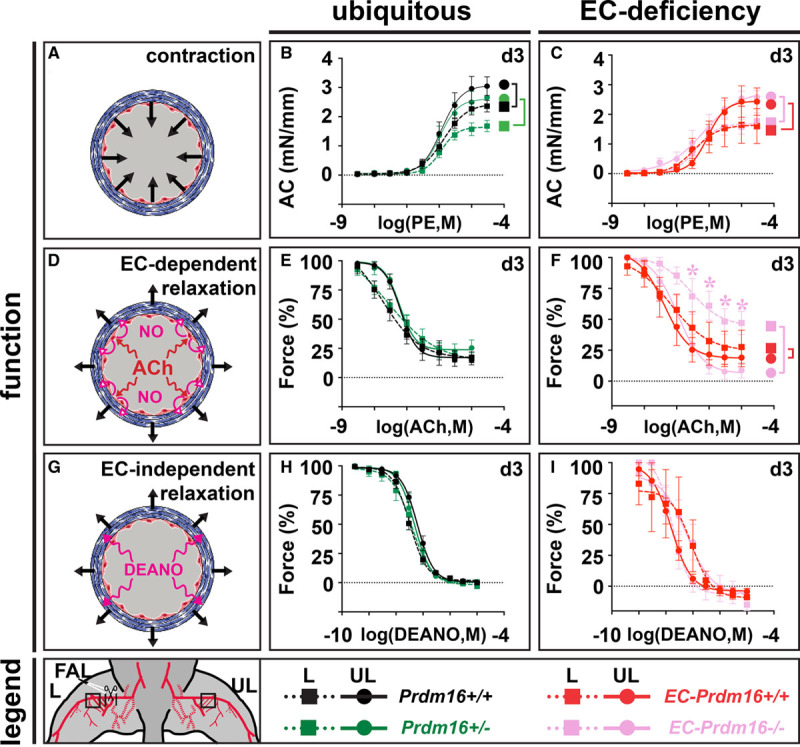
Endothelial cell (EC)–specific Prdm (positive regulatory domain-containing protein) 16 deficiency causes impaired EC-dependent relaxation upon hindlimb ischemia. A–C, Schematic representation (A) and quantification of phenylephrine (PE)-mediated absolute contraction (AC) in femoral artery (FA) segments of the ligated (L) and unligated (UL) side 3 d after FA ligation (FAL) in Prdm16+/+ (n=7) or Prdm16+/− (n=5–6) mice (B) and long-term EC-Prdm16+/+ (n=3–5) or EC-Prdm16−/− (n=4–5) mice (C). D–F, Schematic representation (D) and quantification of relative EC-dependent relaxation upon administration of acetylcholine (ACh) in FA segments at d 3 after FAL in Prdm16+/+ (n=7) or Prdm16+/− (n=5–6) mice (E) and long-term EC-Prdm16+/+ (n=3–5) or EC-Prdm16−/− (n=4–5) mice (F). G–I, Schematic representation (G) and quantification of relative EC-independent relaxation upon administration of diethylamine NONOate (DEANO) in FA segments at d 3 after FAL in Prdm16+/+ (n=7) or Prdm16+/− (n=5–6) mice (H) and long-term EC-Prdm16+/+ (n=3–5) or EC-Prdm16−/− (n=4–5) mice (I). Data represent mean±SEM. *P<0.05: vs indicated condition (significance is only indicated in case of a differential genotypic response to FAL; Table III in the Data Supplement). M in (B, C, E, F, H, I) indicates molar.
Prdm16 in ECs Regulates Their Function Through Ca2+ and NO Homeostasis
To explore mechanisms of this early-onset EC dysfunction in an unbiased way, we performed in situ differential expression analysis in EC-Prdm16−/− and EC-Prdm16+/+ littermates at day 3 postligation by single-cell RNA sequencing of FA segments from the ligated and unligated side. Cluster analysis revealed 14 cell populations, including Sox17+ arterial ECs (Figure 6A and 6B; Figure VIIA through VIIC in the Data Supplement). To identify genes with altered expression specifically in arterial ECs by induction of HLI in a Prdm16-dependent or Prdm16-independent way, we performed a 2-step analysis (Figure 6C). First, we shortlisted (based on statistical significance, degree of differential expression, and percentage of cells featuring expression changes) the genes altered by FA ligation by comparing the ligated and unligated side for EC-Prdm16+/+ and EC-Prdm16−/− separately. Next, we overlapped these shortlists to distinguish genes commonly regulated (type A genes) from those that were uniquely changed (type B genes) among genotypes. This revealed 218 type A and 398 type B genes, of which 217 were uniquely altered in EC-Prdm16+/+ (type B1 genes) and 181 were exclusively changed in EC-Prdm16−/− mice (type B2 genes). We considered both type B1 and B2 genes to be potentially involved in the observed EC dysfunction (Figure 6C; Tables IV and V in the Data Supplement). We next performed functional annotation analysis on type B1 and B2 genes to identify and prioritize unique genotype-dependent processes known to be involved in EC function. Among prioritized functions related to type B1 genes, we identified gap junctions and endocrine and other factor-related Ca2+ reabsorption, whereas Ca2+ binding and multiple functional terms related to fat(ty acid) handling were found associated with type B2 genes (Figure 6D: Tables VI and VII in the Data Supplement).
Figure 6.

In situ analyses upon hindlimb ischemia induction in endothelial cell (EC)–specific Prdm (positive regulatory domain-containing protein) 16–deficient mice and their wild-type littermates reveal differential expression changes related to NO bioavailability and Ca2+ homeostasis. A, Schematic representation of the study design for the in situ differential expression analysis based on single-cell RNA sequencing 3 d after femoral artery ligation (FAL). The regions of interest (ROI) from which single-cell suspensions from the ligated (L) or unligated (UL) side were generated for EC-Prdm16+/+ (red) and EC-Prdm16−/− (dark pink) mice are indicated by a black box. The ROI material from 4 animals was pooled to obtain sufficient cells. B, Uniform Manifold Approximation and Projection (UMAP) plot highlighting cell clusters annotated as arterial endothelial cell (aEC) in red. C, Schematic representation of the 2-step model used for identification of differentially expressed genes (DEGs) in aECs that were commonly (type A; red) or genotype-dependently (type B; light pink) altered when comparing the ligated (L) vs the unligated (UL) side. D, Diagrams showing results of the combined functional annotation analysis by Database for Annotation, Visualization and Integrated Discovery and Gene Set Enrichment Analysis for Kyoto Encyclopedia of Genes and Genomes (KEGG; left) and for Gene Ontology terms molecular_functions (GO-MF; right), expressed as % of genes of the total number of DEGs. Functional categories prioritized because of their known association with EC (dys)function are named. E, Heat maps (upper left) showing the 30 genes represented by the prioritized functional terms in D. Color scale code is shown on top. Table (lower left) showing the documented involvement of the genes in regulation of eNOS (endothelial NO synthase)/NO production, redox state, and Ca2+ homeostasis. Cartoon (right) reveals location of genes encoding for structural components of caveolae or clathrin-coated pits. Arrows indicate caveolae recycling. LDL indicates low-density lipoprotein.
Accordingly, an in-depth literature analysis on the 30 type B genes—the majority of which were downregulated upon ligation—associated with the prioritized functions revealed that as many as 50% of them related to Ca2+ handling (Figure 6E; Table VIII in the Data Supplement). Next, because the vascular relaxation deficit in EC-Prdm16−/− mice was no longer apparent after NO-donor supplementation, we hypothesized that changes in prioritized genes might reflect altered regulation of NO bioavailability. NO decrease can be caused by reduced levels/activity of eNOS (endothelial NO synthase) or by Nox (NADPH-oxidase)-induced eNOS uncoupling, the latter resulting in production of radical oxygen species rather than NO.29 In support of this hypothesis, 33% of prioritized genes have a documented involvement in eNOS regulation/NO production, and 17% are known to be involved in redox status regulation (Figure 6E; Table VIII in the Data Supplement). Finally, ≈43% of the prioritized genes have been associated with the process of clathrin- or caveolae-related endocytosis, the latter known to play an important role in EC function, including regulation of eNOS/NO production, redox status as well as Ca2+ homeostasis (Figure 6E; Table VIII in the Data Supplement).29 Altogether, our in situ differential expression analysis identified functions and associated genes converging on several pathways that previously have been associated with regulation of EC functionality, most notably, Ca2+ and NO.
To verify a role for endothelial Prdm16 in Ca2+ and NO homeostasis upon HLI, we performed additional experiments. Given the technical complexity of in vivo Ca2+ measurements in the FA, and because ECs in culture rapidly lose Prdm16 expression,9 we used a complementary approach based on intracellular Ca2+ measurements upon Prdm16 overexpression in human umbilical vein ECs. Prdm16 overexpression increased intracellular Ca2+ storage by ≈18% (Figure 7A) as measured by exposing cells to the ionophore ionomycin upon chelation of extracellular Ca2+. Repeating these experiments using thapsigargin, which causes depletion of the ER Ca2+ store, instead of ionomycin, showed that this increase in stored Ca2+ was not at the level of the ER (Figure 7B). Finally, store-operated Ca2+ entry, measured by readdition of extracellular Ca2+ after ER depletion with thapsigargin, was increased by ≈21% upon Prdm16 overexpression (Figure 7C). To further support the involvement of Prdm16 in NO bioavailability upon HLI, we investigated the levels of eNOS and Nox in situ. Although induction of eNOS protein expression in the adductor region was not affected by endothelial Prdm16 deficiency (eNOS protein, expressed as ratio of ligated versus unligated side of EC-Prdm16+/+ versus EC-Prdm16−/− mice: 1.4±0.5 versus 1.1±0.4; n=3–4, P=8.57×10−1 by Mann-Whitney test; Figure X in the Data Supplement), induction of Nox2 and Nox4 expression was increased in EC-Prdm16−/− mice suggesting more eNOS uncoupling (Figure 7D and 7E). Altogether, these findings suggest that Prdm16 controls EC function by affecting Ca2+ signaling and NO availability.
Figure 7.
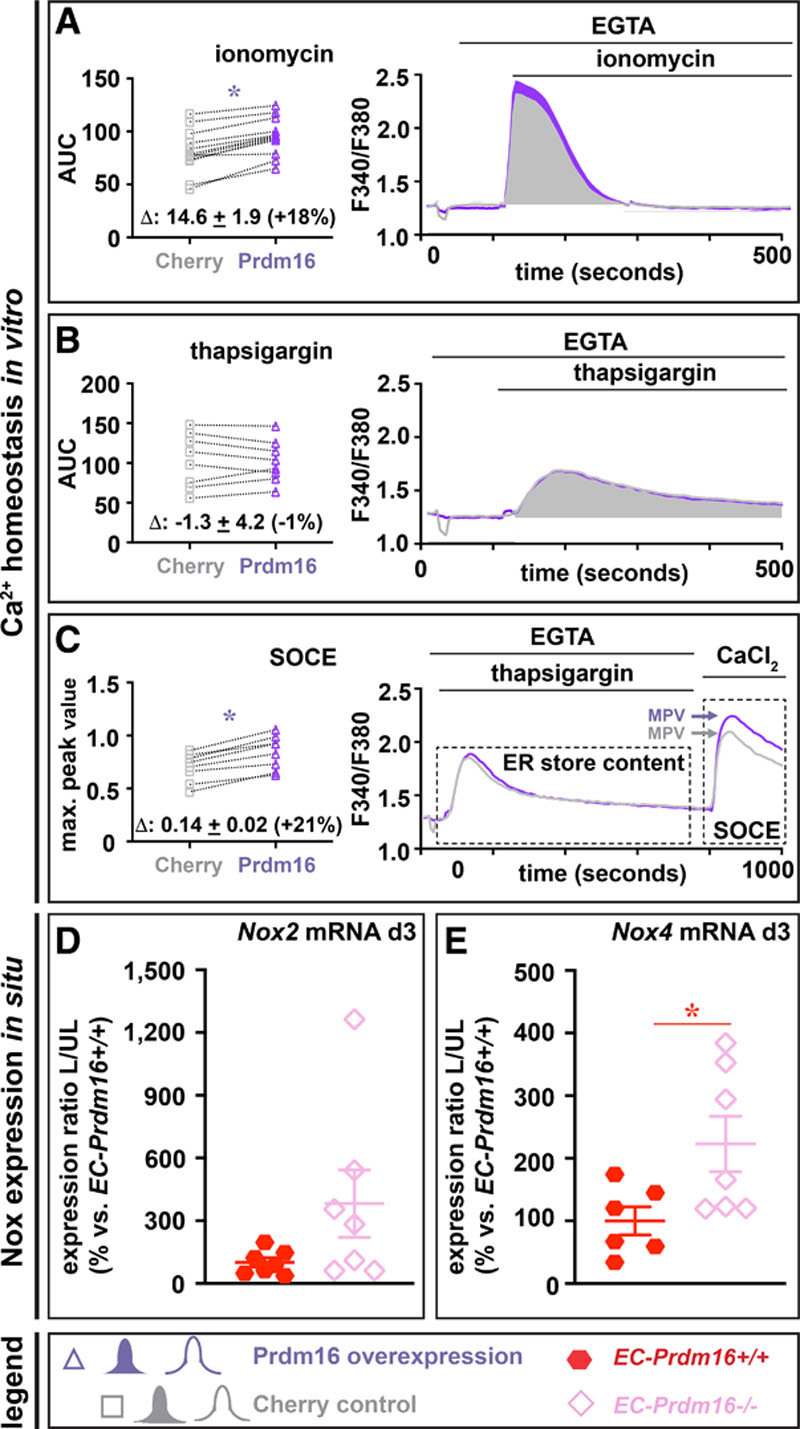
Prdm (positive regulatory domain-containing protein) 16 regulates Ca2+ homeostasis and Nox (NADPH-oxidase) gene expression. A–C, In vitro Ca2+ handling experiments following treatment of Prdm16- or Cherry-overexpressing human umbilical vein endothelial cells (ECs) with ionomycin in Ca2+-free medium (A; n=12; quantified as area under the curve [AUC]), thapsigargin in Ca2+-free medium (B; n=8; quantified as AUC) and exposure to high extracellular Ca2+ following endoplasmic reticulum (ER) Ca2+ depletion using thapsigargin treatment as a measure for store-operated calcium entry (C; n=8; quantified as maximum peak value (MPV) of the response to CaCl2). Mean differences between Cherry and Prdm16 overexpression conditions and corresponding % change are indicated above the X-axis in diagrams on the left. D and E, Diagram showing expression of Nox2 (D) and Nox4 (E; expressed as ratio of ligated (L) vs unligated (UL) side and as % of EC-Prdm16+/+) in the adductor muscle of long-term EC-Prdm16+/+ (red; n=6–7) or EC-Prdm16−/− (pink; n=7) mice at 3 d postligation. Data represent mean±SEM. *P<0.05 vs indicated condition (Table III in the Data Supplement). EGTA indicates ethylene glycol tetraacetic acid; and SOC, store-operated Ca2+ entry.
Discussion
Currently, there are no effective options to restore arterial flow in PAD patients ineligible for invasive revascularization.4,5 The lack of specificity towards the arterial part of the vasculature may be one reason for the inefficacy of VEGF- or FGF-based angiogenic growth factor monotherapy.30 Identifying transcriptional regulators orchestrating arterial flow recovery may lead to development of more tailored strategies. Here, we report that loss of the arterial-restricted transcription factor Prdm16 significantly impaired flow recovery in a mouse PAD model. Intriguingly, Prdm16 deficiency did not compromise the arterial remodeling response that classically occurs in this model, but its absence in ECs significantly attenuated EC-dependent relaxation upon HLI induction. Our unbiased screen for molecular targets downstream of Prdm16 unveiled a potential role in regulating NO bioavailability and Ca2+ homeostasis.
Apart from its expression in mouse retinal arterioles,17 this transcription factor has not been reported in adult blood vessels nor was its role known in this compartment. Here, we performed a systematic analysis of Prdm16 expression in adult blood vessels and found it to be universally restricted to arteri(ol)es, independent of artery type, location, caliber, or health status. Due to this arterial-specific expression, we hypothesized a crucial role for Prdm16 during adaptive arteriogenesis. Arterial flow recovery was severely impaired, even when only one Prdm16-allele was missing. Unexpectedly, this impairment caused by Prdm16 deficiency was not due to an attenuated arterial remodeling response, unlike we previously reported for Msx1.7 The increased angiogenic response in the gastrocnemius muscle of Prdm16+/− mice likely represented an insufficient compensatory attempt to improve blood distribution in this area. In contrast to Msx1,7 Prdm16 expression was not triggered by shear stress or hypoxia in vitro (not shown) nor in vivo, which is in line with our current observation that Prdm16 is not directly involved in driving the shear stress-induced collateral remodeling or the hypoxia-induced angiogenic response in the adductor and gastrocnemius region, respectively. Rather, Prdm16 has an early role in arterial flow recovery by preservation of endothelial function. This not only demonstrates that the mechanism by which arterial transcription factors affect arterial flow recovery can be fundamentally different but it also suggests that proper collateral remodeling and the angiogenic response alone are not sufficient for full arterial flow restoration. Our findings are in line with a previous study in mice with a heterozygous deficiency for Notch ligand Delta-like ligand 4 (Dll4) from which the authors concluded that ischemic recovery is not simply determined by the extent of the collateral network but rather by normal vascular function.31 Although it remains to be determined whether adaptive arteriogenesis also encompasses changes in arterial specification, this process is likely more important during development than in adult, as previously reported in the same study.31
The flow recovery deficit upon Prdm16 deficiency resulted in fibrosis, fibro-adipose tissue accumulation, and toe loss. Although these could be manifestations indirectly caused by lack of oxygen supply, they may also result from a direct effect of Prdm16 deficiency. Quantitative real-time polymerase chain reaction analysis of gastrocnemius tissue at day 3 postligation revealed that endothelial Prdm16 deficiency tended to alter expression of genes encoding fibrosis regulators (not shown), some of which (ie, tissue inhibitor of metalloproteinases (Timp)1 and Timp4) were altered in the same way in our single-cell RNA sequencing dataset (Table V in the Data Supplement), suggesting those genes may be directly targeted by Prdm16. However, fibro-adipose tissue accumulation in Prdm16+/− gastrocnemius muscles was likely not resulting from a direct effect of Prdm16 on adipogenic differentiation of mesenchymal precursors since lack of Prdm16 in the latter is known to decrease adipogenesis.12 The fact that we found a similar increase in fibro-adipose tissue upon loss of Prdm16 in nonmesenchymal cells, that is, ECs, further argues against this direct effect scenario. Finally, the increased occurrence of toe loss upon endothelial Prdm16 deficiency may potentially be related to neuropathy, as the latter has been associated with EC dysfunction through breakdown of the peripheral blood-nerve barrier.32
In search of the cell type(s) responsible for the flow recovery deficit, we used cell type–specific knockout mice. Although EC-specific loss of Prdm16 recapitulated—and even somewhat aggravated—the reperfusion deficit observed in Prdm16+/− mice, Prdm16 loss in SMCs did not. This suggests that Prdm16 expression in ECs had a dominant role in guaranteeing normal arterial flow recovery after FA ligation. Although this precludes an important role for SMC-Prdm16 in the current PAD model, our findings do not exclude its potential importance in other (arterial) disease models. Like in Prdm16+/− mice, mice completely lacking Prdm16 in their ECs had a normal collateral growth response. However, EC-Prdm16−/− mice showed significantly attenuated EC-dependent vasorelaxation of the FA segment connecting downstream of the ligation with the collateral network but not in the upstream DA or the contralateral unligated FA segments. This indicates that the dysfunction was specifically unveiled in the region affected by the pathological challenge and that proper EC function is a critical determinant of normal arterial flow recovery, in line with the previous findings in Dll4+/− mice.31 To our knowledge, this is the first report of vasomotor function recordings in a PAD model. Our recordings did not only reveal a defective response in EC-Prdm16−/− mice compared with their WT littermates but also showed that HLI per se caused the FA to become less capable of responding.
Although Prdm16+/− and EC-Prdm16−/− mice both had impaired flow recovery, EC-dependent relaxation was only affected in the latter. This could be explained in different ways. First, there may be gene dose-dependency, such that abnormal vasomotor responses are only present upon homozygous lack of Prdm16 in ECs. Second, as mentioned above, Prdm16 deficiency in non-ECs may also contribute to the flow recovery deficit in Prdm16+/− mice. Third, Prdm16 deficiency in ECs may encompass other features of EC dysfunction not revealed by the used set of vasomotor tests. When looking in more detail at our vasomotor recordings, we noticed that contraction-relaxation curves of the ligated FA segments of 80% of Prdm16+/− (Figure XI in the Data Supplement) and 75% of EC-Prdm16−/− mice featured spikes, suggestive for abnormal vascular function. A broader interference with EC homeostasis is also supported by our unbiased screen for molecular mechanisms that could account for a Prdm16-related role in preserving EC function.
We performed this unbiased screen by an in situ differential expression analysis at single-cell resolution in arterial ECs. The Prdm16-dependent functions and associated genes emerging from this analysis converged on a broad role for Prdm16 in processes previously associated with many aspects of EC (dys)function, including regulation of eNOS production/activation, redox and Ca2+ homeostasis, gap junctions, and fat(ty)acid handling.29,33–35 Indeed, endothelial Prdm16-deficiency affected the in situ expression of several genes previously associated with eNOS activation, including Cavin2 and EH domain-containing (Ehd)2.36,37 Furthermore, a role for Prdm16 in promoting NO bioavailability during HLI was supported by the fact that endothelial loss of Prdm16 upregulated Nox genes, known to cause eNOS uncoupling and oxidative stress29 and downregulated several genes that protect against oxidative stress (eg, fibulin [Fbln]5 and retinol-binding protein [Rbp]7).38,39 A role for Prdm16 in Ca2+ homeostasis also emerged from our in situ expression analysis, and we validated this by in vitro functional studies. In the PAD model used in the current study, the sudden increased perfusion in collaterals and the connecting downstream FA alters extracellular Ca2+ concentrations to which ECs are exposed and the shear stress sensed by ECs triggers an intracellular Ca2+ response through release from internal stores.40 Prdm16 overexpression did not seem to affect the Ca2+ content of the major intracellular store, that is, the ER, suggesting that other compartments, for example, mitochondria (representing the second-largest Ca2+ store), may be involved.40,41 Intriguingly, endothelial Prdm16 regulated several genes encoding structural components of endocytotic organelles, that is, caveolae and clathrin-coated pits (Figure 6E; Table VIII in the Data Supplement) and clathrin-related functional terms emerged from our annotation analysis (Table VII in the Data Supplement). It is known that caveolae play an important role in EC function, including regulation of eNOS/NO production and oxidative stress through recruitment of Nox as well as in Ca2+ homeostasis.29 Although clathrin-mediated endocytosis has not been associated with eNOS/NO production, several structural genes localized in clathrin-coated pits have been associated with Ca2+ handling (Figure 6E; Table VIII in the Data Supplement).
In conclusion, our study demonstrates an unprecedented role for Prdm16 in sustaining arterial flow recovery in a mouse PAD model, mainly by preserving endothelial function, rather than affecting structural remodeling. Our findings may have important implications for the design of future therapeutic avenues for no-option PAD patients. Inducing collateral growth or angiogenesis per se may be insufficient for recovery from an ischemic insult, but approaches aimed at collateral arterial network expansion should be complemented with those aiming at preserving arterial functionality. Our data indicate that the arterial-restricted transcription factor Prdm16 is an appealing target to design such complementary approaches.
Acknowledgments
We thank P. Vandervoort, B. Hoekman, E. Caluwé, M. Lox, T. Van Brussel, and R. Schepers for technical support and G. Bultynck for helpful discussions. The nano–computed tomography (CT) images have been generated on the X-ray CT facility of the Department of Development and Regeneration of KU Leuven, financed by the Hercules Foundation (project AKUL/13/47). The computational resources and services related to single-cell RNA sequencing were provided by the VSC (Vlaams Supercomputer Centrum), funded by the Fonds voor Wetenschappelijk Onderzoek (FWO), and the Flemish Government–Department Economie, Wetenschap en Innovatie (EWI).
Sources of Funding
This work was supported by a European Commission Grant (FP7-StG-IMAGINED203291), research grants from Fonds voor Wetenschappelijk Onderzoek (FWO; G0B0913N, G0A2214N, G099521N to A. Luttun; and G091018N to E.A.V. Jones), a Program Financing grant (PF/10/014) to A. Luttun; KU Leuven Geconcerteerde Onderzoeksacties (GOA11/012), Interuniversity Attraction Poles (IUAP/P7/07) and KU Leuven Research Coordination (C12/16/023; C14/19/095) grants to A. Luttun, A. Zwijsen, and E.A.V. Jones; ERA-Net ([LYMIT-DYS] funding to E.A.V. Jones; FWO Strategic Basic Research predoctoral fellowships to J. Van Wauwe (1S25817N) and D. De Munck (1S05216N); FWO predoctoral fellowships to W. Dheedene (1157318 N) and N. Criem (1127815 N); a Bijzonder Onderzoekfonds voor Geconcentreerde Onderzoeksacties grant to S. De Moudt (33931), an Agentschap voor Innovatie door Wetenschap en Technologie (IWT; SB/0881071) predoctoral fellowship to M. Beerens and an FWO postdoctoral fellowship to T. Vervliet (12H2120N).
Disclosures
None.
Supplemental Materials
Expanded Methods
Data Supplement Figures I–XI
Data Supplement Tables I–VIII
References 6,7,9,13,18–27
Supplementary Material
{kind=link}
Footnotes
Nonstandard Abbreviations and Acronyms
- DA
- descending aorta
- DEG
- differentially expressed gene
- Dll4
- delta-like ligand 4
- EC
- endothelial cell
- Ehd2
- EH domain-containing 2
- eNOS
- endothelial NO synthase
- ER
- endoplasmic reticulum
- FA
- femoral artery
- Fbln5
- fibulin 5
- FGF
- fibroblast growth factor
- HLI
- hindlimb ischemia
- Msx
- muscle segment homeobox
- Nox
- NADPH-oxidase
- PAD
- peripheral artery disease
- Prdm
- positive regulatory domain-containing protein
- SMA
- smooth muscle actin
- SMC
- smooth muscle cell
- Timp
- tissue inhibitor of metalloproteinases
- VEGF
- vascular endothelial growth factor
- WT
- wild-type
M. Beerens and A. Luttun contributed equally.
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.318501.
For Sources of Funding and Disclosures, see page 76.
Novelty and Significance
What Is Known?
Endothelial cells (ECs) in arteries have a distinct expression pattern including transcription factors.
During peripheral artery disease (PAD), the impaired arterial blood supply triggers a rescue response that is orchestrated by transcription factors.
Prdm (positive regulatory domain-containing protein) 16 is a transcription factor mostly known for its role in the skeletal, adipose, hematopoietic, and nervous systems.
What New Information Does This Article Contribute?
Prdm16 is a general marker of ECs and smooth muscle cells in the arterial but not venous arm of the adult vascular system.
Prdm16 in arterial ECs but not smooth muscle cells (SMCs) is indispensable for normal arterial blood flow recovery during PAD.
Prdm16 supports normal arterial blood flow recovery not by mediating the structural collateral arterial remodeling response but rather by preserving arterial endothelial function.
Arterial ECs have a distinct profile that makes them vulnerable to artery-restricted diseases like PAD. Upon PAD, limb blood flow is impaired which causes damage sometimes necessitating amputation. The impaired blood flow triggers a rescue mechanism driven by transcription factors during which flow is redirected through collateral arteries that thereby feature structural changes. Identifying these transcription factors and revealing their mode of action is crucial for designing novel strategies to improve arterial blood flow and avoid amputation in PAD patients. Here, we found that Prdm16 is a transcription factor highly expressed in arterial ECs and SMCs. Through its expression in arterial ECs, Prdm16 rescued arterial blood flow upon PAD. Surprisingly, the mechanism of action was not related to governing the structural collateral remodeling process but rather to preserving endothelial function. Unlike most research in the PAD field, the present study establishes that supporting structural collateral remodeling is not the only important action to guarantee normal arterial blood flow recovery as the latter equally relies on preserving EC functionality. We identified Prdm16 as a key promoter of arterial EC function during PAD. Hence, Prdm16 represents an interesting target to design novel PAD treatment strategies that ideally should combine structural and functional rescue.
References
- 1.Dejana E, Hirschi KK, Simons M. The molecular basis of endothelial cell plasticity. Nat Commun. 2017;8:14361. doi: 10.1038/ncomms14361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Potente M, Mäkinen T. Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol. 2017;18:477–494. doi: 10.1038/nrm.2017.36 [DOI] [PubMed] [Google Scholar]
- 3.Fowkes FG, Rudan D, Rudan I, Aboyans V, Denenberg JO, McDermott MM, Norman PE, Sampson UK, Williams LJ, Mensah GA, et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: a systematic review and analysis. Lancet. 2013;382:1329–1340. doi: 10.1016/S0140-6736(13)61249-0 [DOI] [PubMed] [Google Scholar]
- 4.Gupta R, Tongers J, Losordo DW. Human studies of angiogenic gene therapy. Circ Res. 2009;105:724–736. doi: 10.1161/CIRCRESAHA.109.200386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol. 2013;10:387–396. doi: 10.1038/nrcardio.2013.70 [DOI] [PubMed] [Google Scholar]
- 6.Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, Schaper W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34:775–787. doi: 10.1006/jmcc.2002.2013 [DOI] [PubMed] [Google Scholar]
- 7.Vandersmissen I, Craps S, Depypere M, Coppiello G, van Gastel N, Maes F, Carmeliet G, Schrooten J, Jones EA, Umans L, et al. Endothelial Msx1 transduces hemodynamic changes into an arteriogenic remodeling response. J Cell Biol. 2015;210:1239–1256. doi: 10.1083/jcb.201502003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan Y, Lu H, Liang W, Garcia-Barrio MT, Guo Y, Zhang J, Zhu T, Hao Y, Zhang J, Chen YE. Endothelial TFEB (Transcription Factor EB) positively regulates postischemic angiogenesis. Circ Res. 2018;122:945–957. doi: 10.1161/CIRCRESAHA.118.312672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aranguren XL, Agirre X, Beerens M, Coppiello G, Uriz M, Vandersmissen I, Benkheil M, Panadero J, Aguado N, Pascual-Montano A, et al. Unraveling a novel transcription factor code determining the human arterial-specific endothelial cell signature. Blood. 2013;122:3982–3992. doi: 10.1182/blood-2013-02-483255 [DOI] [PubMed] [Google Scholar]
- 10.Chi J, Cohen P. The multifaceted roles of PRDM16: adipose biology and beyond. Trends Endocrinol Metab. 2016;27:11–23. doi: 10.1016/j.tem.2015.11.005 [DOI] [PubMed] [Google Scholar]
- 11.Mochizuki N, Shimizu S, Nagasawa T, Tanaka H, Taniwaki M, Yokota J, Morishita K. A novel gene, MEL1, mapped to 1p36.3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t(1;3)(p36;q21)-positive leukemia cells. Blood. 2000;96:3209–3214. [PubMed] [Google Scholar]
- 12.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. 2010;19:774–789. doi: 10.1093/hmg/ddp543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguilo F, Avagyan S, Labar A, Sevilla A, Lee DF, Kumar P, Lemischka IR, Zhou BY, Snoeck HW. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117:5057–5066. doi: 10.1182/blood-2010-08-300145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12:999–1006. doi: 10.1038/ncb2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arndt AK, Schafer S, Drenckhahn JD, Sabeh MK, Plovie ER, Caliebe A, Klopocki E, Musso G, Werdich AA, Kalwa H, et al. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet. 2013;93:67–77. doi: 10.1016/j.ajhg.2013.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groman-Lupa S, Adewumi J, Park KU, Brzezinski JA., IV. The transcription factor Prdm16 marks a single retinal Ganglion cell subtype in the mouse retina. Invest Ophthalmol Vis Sci. 2017;58:5421–5433. doi: 10.1167/iovs.17-22442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002 [DOI] [PubMed] [Google Scholar]
- 19.Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell. 2014;156:304–316. doi: 10.1016/j.cell.2013.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A. 2002;99:7142–7147. doi: 10.1073/pnas.102650499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leloup AJ, Van Hove CE, Kurdi A, De Moudt S, Martinet W, De Meyer GR, Schrijvers DM, De Keulenaer GW, Fransen P. A novel set-up for the ex vivo analysis of mechanical properties of mouse aortic segments stretched at physiological pressure and frequency. J Physiol. 2016;594:6105–6115. doi: 10.1113/JP272623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meisner JK, Annex BH, Price RJ. Despite normal arteriogenic and angiogenic responses, hind limb perfusion recovery and necrotic and fibroadipose tissue clearance are impaired in matrix metalloproteinase 9-deficient mice. J Vasc Surg. 2015;61:1583–94.e1. doi: 10.1016/j.jvs.2014.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Moudt S, Leloup A, Van Hove C, De Meyer G, Fransen P. Isometric stretch alters vascular reactivity of mouse aortic segments. Front Physiol. 2017;8:157. doi: 10.3389/fphys.2017.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leloup AJ, Van Hove CE, Heykers A, Schrijvers DM, De Meyer GR, Fransen P. Elastic and muscular arteries differ in structure, basal NO production and voltage-gated Ca(2+)-channels. Front Physiol. 2015;6:375. doi: 10.3389/fphys.2015.00375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278. doi: 10.1186/s13059-015-0844-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, 3rd, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive integration of single-cell data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scholz D, Cai WJ, Schaper W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis. 2001;4:247–257. doi: 10.1023/a:1016094004084 [DOI] [PubMed] [Google Scholar]
- 29.Billaud M, Lohman AW, Johnstone SR, Biwer LA, Mutchler S, Isakson BE. Regulation of cellular communication by signaling microdomains in the blood vessel wall. Pharmacol Rev. 2014;66:513–569. doi: 10.1124/pr.112.007351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conway EM, Carmeliet P. The diversity of endothelial cells: a challenge for therapeutic angiogenesis. Genome Biol. 2004;5:207. doi: 10.1186/gb-2004-5-2-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cristofaro B, Shi Y, Faria M, Suchting S, Leroyer AS, Trindade A, Duarte A, Zovein AC, Iruela-Arispe ML, Nih LR, et al. Dll4-Notch signaling determines the formation of native arterial collateral networks and arterial function in mouse ischemia models. Development. 2013;140:1720–1729. doi: 10.1242/dev.092304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roustit M, Loader J, Deusenbery C, Baltzis D, Veves A. Endothelial dysfunction as a link between cardiovascular risk factors and peripheral neuropathy in diabetes. J Clin Endocrinol Metab. 2016;101:3401–3408. doi: 10.1210/jc.2016-2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chabowski DS, Cohen KE, Abu-Hatoum O, Gutterman DD, Freed JK. Crossing signals: bioactive lipids in the microvasculature. Am J Physiol Heart Circ Physiol. 2020;318:H1185–H1197. doi: 10.1152/ajpheart.00706.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson WF. Levitan I, Dopico AM, eds. Endothelial cell Ion channel expression and function in arterioles and resistance arteries. In: Vascular Ion Channels in Physiology and Disease. 2016. Springer International Publishing; 3–36. [Google Scholar]
- 35.Kameritsch P, Pogoda K, Ritter A, Münzing S, Pohl U. Gap junctional communication controls the overall endothelial calcium response to vasoactive agonists. Cardiovasc Res. 2012;93:508–515. doi: 10.1093/cvr/cvr345 [DOI] [PubMed] [Google Scholar]
- 36.Boopathy GTK, Kulkarni M, Ho SY, Boey A, Chua EWM, Barathi VA, Carney TJ, Wang X, Hong W. Cavin-2 regulates the activity and stability of endothelial nitric-oxide synthase (eNOS) in angiogenesis. J Biol Chem. 2017;292:17760–17776. doi: 10.1074/jbc.M117.794743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthaeus C, Lian X, Kunz S, Lehmann M, Zhong C, Bernert C, Lahmann I, Müller DN, Gollasch M, Daumke O. eNOS-NO-induced small blood vessel relaxation requires EHD2-dependent caveolae stabilization. PLoS One. 2019;14:e0223620. doi: 10.1371/journal.pone.0223620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu C, Keen HL, Lu KT, Liu X, Wu J, Davis DR, Ibeawuchi SC, Vogel S, Quelle FW, Sigmund CD. Retinol-binding protein 7 is an endothelium-specific PPARγ cofactor mediating an antioxidant response through adiponectin. JCI Insight. 2017;2:e91738. doi: 10.1172/jci.insight.91738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen AD, Itoh S, Jeney V, Yanagisawa H, Fujimoto M, Ushio-Fukai M, Fukai T. Fibulin-5 is a novel binding protein for extracellular superoxide dismutase. Circ Res. 2004;95:1067–1074. doi: 10.1161/01.RES.0000149568.85071.FB [DOI] [PubMed] [Google Scholar]
- 40.Scheitlin CG, Julian JA, Shanmughapriya S, Madesh M, Tsoukias NM, Alevriadou BR. Endothelial mitochondria regulate the intracellular Ca2+ response to fluid shear stress. Am J Physiol Cell Physiol. 2016;310:C479–C490. doi: 10.1152/ajpcell.00171.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013;112:1171–1188. doi: 10.1161/CIRCRESAHA.111.300233 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the majority of supporting data are presented within this article and in the Data Supplement. Data not directly presented are available from the corresponding author upon reasonable request. Single-cell RNA sequencing data have been deposited in the ArrayExpress database at EMBL-EBI (www.ebi.ac.uk/arrayexpress; accession number E-MTAB-9703).