

Summary
p53-binding protein 1 (53BP1) regulates both the DNA damage response and p53 signaling. Although 53BP1’s function is well established in DNA double-strand break repair, how its role in p53 signaling is modulated remains poorly understood. Here, we identify the scaffolding protein AHNAK as a G1 phase-enriched interactor of 53BP1. We demonstrate that AHNAK binds to the 53BP1 oligomerization domain and controls its multimerization potential. Loss of AHNAK results in hyper-accumulation of 53BP1 on chromatin and enhanced phase separation, culminating in an elevated p53 response, compromising cell survival in cancer cells but leading to senescence in non-transformed cells. Cancer transcriptome analyses indicate that AHNAK-53BP1 cooperation contributes to the suppression of p53 target gene networks in tumors and that loss of AHNAK sensitizes cells to combinatorial cancer treatments. These findings highlight AHNAK as a rheostat of 53BP1 function, which surveys cell proliferation by preventing an excessive p53 response.
Keywords: 53BP1, AHNAK, p53, p21, DNA repair, phase-separation, cell-cycle progression, cancer, senescence, apoptosis
Graphical abstract
Highlights
-
•
AHNAK is a G1-enriched interactor of 53BP1
-
•
AHNAK controls 53BP1-mediated G1-S phase transition upon DNA damage
-
•
AHNAK restrains 53BP1 oligomerization and phase separation
-
•
AHNAK balances between apoptosis and senescence in cancer and non-transformed cells
Ghodke et al. identify the large scaffolding protein AHNAK as a G1-enriched interactor of 53BP1 that ensures optimal partitioning of 53BP1 into phase-separated condensates and limits excessive interaction with p53, which would otherwise lead to apoptosis in cancer cells and senescence in non-transformed cells.
Introduction
The tumor suppressor protein p53 plays a pivotal role in triggering multiple signaling pathways in response to a wide range of cellular stresses. Mechanistically, upon sensing stress, p53 rewires pan-genomic transcriptional programs, which includes the induction of cell-cycle arrest (e.g., CDKN1A/p21, 14-3-3σ), pro-apoptotic (e.g., BAX, TP53I3, PUMA,), and senescence (e.g., FAS, PDID) genes. In response to DNA damage, p53 activation is triggered by the ATM/ATR kinases, the apical responders to DNA double-stranded breaks (DSBs), and replication stress, respectively. ATM/ATR-mediated phosphorylation of p53 and of its inhibitor MDM2 initiate a converging mechanism, leading to the stabilization of p53 (Meek, 2004). The primary target of activated p53 is a multifunctional protein, p21, which attenuates cyclin/CDK activity and instigates G1/S cell-cycle arrest. p21 is also known to activate genes involved in senescence (Meek, 2004; Mirzayans et al., 2012). In a p53-deficient background, cells have an impaired G1/S checkpoint, resulting in the propagation of structural aneuploidies and early onset of carcinogenesis (Soto et al., 2017). Other studies (Abbas and Dutta, 2009) have shown that the strength of the p53 response dictates the balance between cell division and cell-cycle arrest.
p53-binding protein 1 (53BP1) was identified as an interactor of p53 (Iwabuchi et al., 1994) and is positioned at the crossroads of DSB repair and p53 signaling. It harbors key structural elements, including 28 N-terminal Ser/Thr-Gln (S/T-Q) sites, a central minimal focus-forming region (MFFR) composed of an oligomerization domain (OD), a Gly- and Arg-rich (GAR) motif, a tandem Tudor domain, a ubiquitylation-dependent recruitment (UDR) motif, and a C-terminal BRCT domain. Whereas both the OD and BRCT domains are critical for p53 activation, the BRCT domain is dispensable for DNA repair (Mirman and de Lange, 2020). 53BP1 is stabilized on chromatin by UDR motif-mediated binding to H2AUb15, and via binding of its tandem Tudor domain to mono- and dimethylated H4K20 (Panier and Boulton, 2014). Domains flanking the Tudor domain (i.e., OD and the LC8 domain) drive multimerization of 53BP1 and promote DNA damage-dependent 53BP1 recruitment to the chromatin (Becker et al., 2018, Sundaravinayagam et al., 2019; Ward et al., 2006; Zgheib et al., 2009).
Previous studies have implicated critical roles of the 53BP1-p53 pathway in cell proliferation. For example, 53BP1 and p53 deficiency predispose mice to T cell lymphoma and genomic instability, suggesting a synergy between 53BP1 and p53 function (Morales et al., 2006; Ward et al., 2005). 53BP1 shields spontaneously arising DNA lesions from endogenous stress during DNA replication (Harrigan et al., 2011; Lukas et al., 2011). In the subsequent G1 phase, this enriched 53BP1 on damaged chromatin in foci known as 53BP1 nuclear bodies (NBs) triggers a p53 response, thereby controlling G1 duration and cell cycle fate by activating p21 (Arora et al., 2017; Barr et al., 2017; Lezaja and Altmeyer, 2018). In addition, 53BP1-p53 cooperation is extended beyond DNA repair in global cellular stress generated after the loss of histone demethylase LSD1, which is shown to negatively regulate 53BP1-dependent p53 function (Huang et al., 2007). Although 53BP1 is critical in regulating p53-dependent trans-activating functions (Cuella-Martin et al., 2016; Kilic et al., 2019), how this response is triggered in a controlled fashion in G1 phase of the cell cycle remains elusive.
Results
Identification of AHNAK as a G1-enriched interactor of 53BP1
To identify cell cycle stage-specific regulators of 53BP1, we performed label-free quantitative mass spectrometry of 53BP1 under unperturbed conditions. We established U2OS cell lines stably expressing the MFFR of 53BP1 (amino acids 1220–1711) fused to mCherry and carrying a BioTag (henceforth denoted as mCherry-53BP1MFFR-BioTag) and co-expressing the biotin ligase BirA and as a negative control, a U2OS cell line co-expressing mCherry-BioTag and BirA (Figures S1A–S1C). We then used in vivo biotinylation tagging followed by streptavidin immunoprecipitation (Figures 1A, 1B, and S1D) in cells arrested either in G0/G1 or released in S/G2, as described previously (Javanmoghadam-Kamrani and Keyomarsi, 2008). In addition to the known interactors of 53BP1, such as TIRR, RUVBL2, DYNLL1, and DYNLL2, which were enriched throughout the cell cycle, mass spectrometry analysis led to the identification of AHNAK, which was reproducibly enriched in G1 phase (Figure 1A), but not in S-G2 phase (Figure 1B). The interaction of MFFR with AHNAK was further validated by streptavidin immunoprecipitation (Figure 1C). Moreover, the interaction was confirmed with the endogenous proteins (Figure 1D). AHNAK harbors three structurally distinct regions: the N-terminal 500 amino acids, a large central region with 4,388 amino acids composed of 36 repeated units, and the C-terminal region of 1,003 amino acids (Figure 1E). Multiple studies have demonstrated that the central repeated units (CRUs) perform the majority of AHNAK functions. (Jin et al., 2020; Lee et al., 2004, 2008, 2014; Lim et al., 2013). To obtain further insights into AHNAK-53BP1 interaction, we transiently overexpressed strep-tagged versions of the N-terminal or the C-terminal domains or four central repeating units of human AHNAK (henceforth denoted as N-AHNAK, C-AHNAK, and AHNAK-4CRU, respectively) in U2OS cells and found that endogenous 53BP1 interacts with the AHNAK-4CRUs but not with the N- or C-terminal parts of the protein (Figure 1F). This result was further confirmed using the GFP-tagged AHNAK-4CRU. (Figure S1E). Consistent with the mass spectrometry analysis, AHNAK displayed a robust interaction with 53BP1 primarily in the G1 phase, while the interaction in S/G2 phase was feeble (Figure 1G). In concordance with these results, synchronization of U2OS cells revealed elevated expression of AHNAK in G1, while its expression is substantially reduced in S/G2 (Figure S1F). Interestingly, treatment with benzonase did not affect the AHNAK-53BP1 interaction, suggesting that it is a putative protein-protein interaction, and it is not mediated by DNA or chromatin (Figures 1H and 1I).
Figure 1.

Identification of AHNAK as a G1-enriched interactor of 53BP1
(A and B) Volcano plots depicting cell cycle-specific TP53BP1-MFFR (53BP1MFFR) interactor proteins identified using mass spectrometry. Each circle represents an identified 53BP1 interactor protein. The x axis (log2 fold change) represents the fold upregulation over the BioTag control. The y axis (−log10 [p value]) represents significance. Red circles represent proteins that are enriched over the control, and gray circles represent proteins that are not enriched. Synchronization is depicted as fluorescence-activated cell sorting (FACS) profiles. (A) Proteins identified in G1 phase. (B) Proteins identified in the S-G2 phase.
(C) Western blot (WB) using anti-mCherry and anti-AHNAK antibodies after immunoprecipitation (IP) using streptavidin beads.
(D) Western blot (WB) using anti-mCherry and anti-AHNAK antibodies after immunoprecipitation (IP) with IgG or Anti-AHNAK beads as indicated.
(E) Schematic illustration of the domain architecture of AHNAK, N-terminal fragment (N-AHNAK), central repetitive units (CRU), and C-terminal fragment (C-AHNAK).
(F) U2OS cells expressing strep-tagged N, 4CRU, or C-terminal AHNAK domains. Co-precipitated 53BP1 and p53 were detected using immunoblotting as indicated.
(G) WB analysis using anti-mCherry and anti-AHNAK antibodies after IP in extracts of U2OS-mCherry-53BP1MFFR-BioTag cells arrested in G1 or released in S/G2.
(H) WB analysis with the indicated antibodies after IP of chromatin fractions from U2OS-mCherry-53BP1MFFR-BioTag cells in the presence or not of benzonase.
(I) WB analysis with the indicated antibodies after IP of chromatin fractions of NCS-treated U2OS-mCherry-53BP1MFFR-BioTag cells with or without benzonase.
(J) U2OS cells transiently transfected with GFP or AHNAK-4CRU-GFP were subjected to immunoprecipitation using GFP trap beads in the presence and absence of NCS (100 ng), and bound complexes were analyzed using immunoblot using indicated antibodies.
(K) Effect of ATMi (KU55933) on AHNAK and 53BP1 interaction. U2OS-mCherry-53BP1MFFR-BioTag cells were treated or not with NCS (100 ng) and KU55933 (10 μM, 1 h) as indicated, and chromatin fractions were subjected to streptavidin pull-down and bound complexes analyzed using immunoblotting with indicated antibodies.
See also Figure S1.
As 53BP1’s function and chromatin binding are regulated by DNA damage, we next studied whether DNA damage alters the AHNAK-53BP1 interaction. Remarkably, neocarzinostatin (NCS) treatment led to an increased association of AHNAK with chromatin, and this was more pronounced in G1 cells compared with S/G2 cells (Figure S1G), which can be ascribed to its elevated levels in G1 phase (Figure S1G). In line with the higher association of AHNAK with chromatin upon DNA damage, NCS treatment led to the increased association of GFP-AHNAK-4CRU with endogenous 53BP1 (Figure 1J). Notably, the AHNAK-53BP1 interaction depends on the catalytic activity of the ATM, a central regulator of DNA damage response (Figure 1K). Collectively, these results establish that AHNAK expression is elevated in the G1 phase of the cell cycle, and its large scaffolding CRU repeats mainly mediate its direct interaction with 53BP1. Moreover, DNA damage and activated ATM positively influence their interaction.
AHNAK is a component of the USP28-53BP1-p53-p21 circuit
The previously described interaction of AHNAK with p53 and its role in regulating p53 function (Gu et al., 2019), and the findings that 53BP1 promotes genome-wide p53-dependent gene transactivation events (Cuella-Martin et al., 2016; Iwabuchi et al., 1998), led us to postulate that AHNAK might function in cooperation with 53BP1 in regulating the p53 pathway. To test this, we used CRISPR-Cas9 to generate AHNAK knockout (KO) lines in U2OS and MCF7 cells by specifically deleting exon 3 (Figures S2A and S2B). To assess the impact of AHNAK on p53 activity, we examined the levels of cyclin-dependent kinase inhibitor p21, a surrogate proxy for p53-dependent activation. Strikingly, regardless of exogenous DNA damage, the absence of AHNAK leads to p21 induction (Figure 2A). This effect was not due to elevated endogenous DNA damage caused by the depletion of AHNAK (Figures S2C and S2D). Interestingly, depletion of 53BP1 in AHNAK−/− cells largely reverted the p21 protein levels associated with loss of AHNAK (Figure 2A), suggesting that the increased p21 level in AHANK −/−cells is 53BP1 dependent. Additionally, the results obtained in AHNAK−/− U2OS cells were confirmed in MCF7 AHNAK−/− cell lines (Figure S2E) and depletion of AHNAK, 53BP1, or the combination of AHNAK and 53BP1 by small interfering RNA (siRNA) in U2OS (Figure S2F) and MCF7 (Figure S2G) cells. Additionally, quantification of p21 nuclear intensity confirmed the increase in AHNAK−/− U2OS (Figure 2B) and MCF7 (Figure S2H) cells being triggered by 53BP1. Furthermore, pulse labeling with EdU (Figure S2I) and cell cycle-resolved high-content microscopy by quantitative image-based cytometry (QIBC) confirmed that p21 levels are elevated throughout the cell cycle, with a more pronounced increase in the G1 phase (Figures 2C and 2D).
Figure 2.

AHNAK restrains 53BP1-mediated p53 activation
(A) WB of WT and two independent AHNAK−/− U2OS cell lines transfected with control (Scr, scrambled siRNA) or 53BP1 siRNA. WB of cell lysates from untreated and NCS-treated (100 ng/mL) cells using the indicated antibodies. Vinculin was used as a loading control. pATM antibody was used as a control for DNA damage induction.
(B) Quantification of mean nuclear p21 fluorescence intensity in single WT or AHNAK−/− U2OS cells transfected with control or the indicated siRNAs. The solid line denotes median. A.U., arbitrary units (n = 895).
(C) Cell cycle-resolved p21 nuclear intensity using QIBC in WT and AHNAK−/− U2OS cells. A.U., arbitrary units (n ≥ 2,500 cells per condition).
(D) Quantification of fold enrichment of p21 signal across cell cycle phases from (C).
(E–H) p53 target genes expression analysis at WT and AHNAK−/− U2OS cell lines following transfection with scramble or 53BP1 siRNA. Cells exposed or not to NCS. (E) CDKN1A (p21), (F) BBC3 (PUMA), (G) BAX, and (H) TP53I3. Fold change was calculated after normalization to β-ACTIN transcript levels.
(I) Immunoblot analysis of BJ fibroblast following transfection with control or indicated siRNA. Cell lysates prepared from untreated and NCS-treated cells were analyzed using immunoblot using the specified antibodies. Vinculin was used as a loading control.
(J) p21 fluorescence intensity in BJ fibroblasts transfected with control or AHNAK siRNA. The solid line denotes median. A.U., arbitrary units (n ≥ 513, unpaired t test).
(K) Immunoblot analysis of WT and two independent AHNAK−/− U2OS cell lines following transfection with control or indicated siRNA. Cell lysates prepared from untreated and NCS-treated (100 ng, 4 h recovery) cells were then analyzed using the specified antibodies. GAPDH was used as a loading control.
(L) WB analysis using anti-GFP antibody of lysates from of U2OS cells transfected with GFP or AHNAK-4CRU-GFP.
(M) WB analysis of the lysates in (L) with the indicated antibodies. Vinculin was used as a loading control.
See also Figures S2 and S3.
To study the kinetics of p21 activation in wild-type (WT) and AHNAK−/− cells, we quantified p21 levels upon increasing doses of the radiomimetic drug NCS (Figure S2J). p21 expression displayed a linear increase until 1,000 ng/mL in both WT and AHNAK−/− cells, thereafter reaching saturation. Interestingly, WT cells could not attain p21 expression level similar to that of AHNAK−/− cells at any given NCS concentration, thus pointing toward the role of AHNAK in surveying p53-p21 response in the absence and presence of DNA damage (Figure S2J).
Moreover, the absence of AHNAK led to significant induction of a wide range of p53 target genes, such as cell-cycle (CDKN1A, TP53I3) and pro-apoptotic (BAX, PUMA/BBC3) genes that were largely dependent on 53BP1 in both U2OS (Figures 2E–2H) and MCF7 (Figures S2K–S2N) cells.
We next performed clonogenic survival assays in the presence of Nutlin-3, a disruptor of p53-Mdm2 interaction, which leads to p53 stabilization (Vassilev et al., 2004). Nutlin-treated AHNAK−/− cells displayed a survival defect compared with DMSO-treated cells (Figures S3A and S3B). In concordance with previous findings (Cuella-Martin et al., 2016), depletion of 53BP1 displayed resistance to Nutlin-3 treatment compared with both AHNAK−/− and WT cells. Notably, depletion of 53BP1 in AHNAK−/− cells led to Nutlin-3-induced resistance comparable with that of si53BP1 alone (Figures S3A and S3B), further supporting the notion that AHNAK modulates 53BP1-mediated p53 activation. Depletion of AHNAK in non-transformed BJ (Figures 2I and 2J) and MCF10A (Figures S3C and S3D) fibroblasts resulted in a robust 53BP1-dependent increase in p21 levels and p53 stabilization, suggesting that the role of AHNAK is not unique to cancer cells.
We next examined the effect of another p53 regulator, USP28, a deubiquitinating enzyme known to interact with 53BP1 (Zhang et al., 2006) and to cooperate with 53BP1 in regulating p53 transactivation (Cuella-Martin et al., 2016). As expected, USP28- and 53BP1-depleted cells were defective in mounting a p21 response (Figure 2K). Interestingly, increased p21 expression in cells lacking AHNAK was suppressed following depletion of USP28 and 53BP1 alone or in combination (Figure 2K). Similar results were obtained in MCF7 cells (Figure S3E), suggesting an epistatic relationship between 53BP1 and USP28 in alleviating p53 responses in AHNAK−/− cells.
To further demonstrate the specificity of the AHNAK in p53 stabilization, we ectopically expressed AHNAK-4CRU-GFP or GFP alone (Figure 2L). We observed reduced p53 and p21 levels in cells expressing AHNAK-4CRU-GFP in the presence and absence of DNA damage (Figure 2M). In agreement, WT and AHNAK−/− cells expressing AHNAK-4CRU-GFP showed reduced p21 intensity compared with GFP alone (Figure S3F), indicating that the observed effects were specific to loss of AHNAK and the CRU domain. MCF7 cells transiently overexpressing AHNAK-4CRU-GFP exhibited resistance to Nutlin-3 treatment compared with cells expressing GFP alone (Figures S3G and S3H), and clonogenic survival assays in p53-deficient MCF7 cell line following Nutlin-3 treatment (Figures S3I and S3J) confirmed that sensitivity displayed in the absence of AHNAK is mediated through p53. Taken together, these results establish AHNAK as a component of the USP28-53BP1-p53-p21 signaling pathway both in non-transformed and cancer cells.
AHNAK triggers 53BP1-mediated G1-S checkpoint function
p21 controls the transition from G1 phase to S phase of the cell cycle (Martín-Caballero et al., 2001). To assess whether the AHNAK-imposed regulation of 53BP1 is vital for G1-S checkpoint activation, we performed G1 checkpoint recovery assay in MCF7 and BJ cells as previously described (Cuella-Martin et al., 2016) (Figures 3A and 3D). In agreement with previous findings (Bigot et al., 2019; Cescutti et al., 2010; Cuella-Martin et al., 2016), following exogenous DNA damage by ionizing radiation (IR), both MCF7 53BP1−/− cells and BJ cells depleted of 53BP1 by siRNA reached S/G2 phase faster compared with control cells (Figures 3B, 3C, 3E, and 3F). In contrast, both AHNAK−/− MCF7 cells and AHNAK-depleted BJ cells displayed robust block in G1 phase (Figures 3B, 3C, 3E, and 3F), indicating the pivotal role of AHNAK in the regulation of damaged induced G1 checkpoint both in cancer and non-transformed cells. Intriguingly, 53BP1 had a stronger impact on G1 checkpoint in cells lacking AHNAK, as its depletion resulted in removal of G1 checkpoint block (Figures 3B, 3C, 3E, and 3F). Collectively, these results revealed that following DNA damage, both transformed and non-transformed cell lines are dependent on AHNAK to control the 53BP1-mediated delay in G1-S phase transition.
Figure 3.

AHNAK regulates 53BP1-mediated G1-S transition upon DSB induction in transformed and non-transformed cells
(A) Schematic representation of the G1 checkpoint assay. Briefly, 24 h post-transfection with the indicated siRNA, MCF7 cells were serum arrested in G0 for 24 h and subsequently released in serum-containing medium containing nocodazole. At 4 h, cells were treated with IR (4 Gy) and collected at 0, 4, 16, and 22 h time points before examined by flow cytometry. Solid green bars denote experimental time points.
(B) Cell-cycle distributions were analyzed using flow cytometry in WT, 53BP1 −/−, and two independent AHNAK−/− MCF7 cell lines, transfected with control or 53BP1 siRNA.
(C) Quantification of the relative distribution of cells in S-G2 phase at the experimental time points, relative to 0 h.
(D) Schematic representation of the G1 checkpoint assay for BJ fibroblast.
(E) Cell-cycle distributions were analyzed using flow cytometry in BJ cell lines, transfected with control or indicated siRNA.
(F) Quantification of the relative distribution of cells in S-G2 phase at the experimental time points, relative to 0 h.
AHNAK limits the accumulation of 53BP1 on chromatin
To investigate how AHNAK regulates 53BP1 functions, we assessed 53BP1-NBs’ fluorescence intensity on the chromatin. Strikingly, regardless of exogenous DNA damage, the absence of AHNAK led to a significant increase in chromatin-associated 53BP1-NB intensity in U2OS (Figures 4A and 4B), MCF7 (Figures S4A and S4B), BJ (Figure S4C), and MCF10A (Figure S4D) cells. The enrichment of 53BP1 on chromatin in AHNAK−/− U2OS cells was validated by cellular fractionation (Figure 4C). Single-cell QIBC analysis revealed that 53BP1 enrichment in NBs is 2-fold higher in G1 than in S and G2 phases (Figure S4E). Moreover, DNA damage leads to a time-dependent increase in the intensity of discernible nuclear 53BP1 foci (Figures S4F and S4G) in AHNAK−/− cells compared with the WT cells. In line with the above observations, AHNAK-4CRU-GFP overexpression led to a significant decrease in 53BP1 accumulation under unperturbed conditions, with the effect being more pronounced following DNA damage (Figures S4H and S4I). We next asked whether ATM or ATR activity is involved in 53BP1 chromatin enrichment. Interestingly, in both WT and AHNAK−/− cells, 53BP1 chromatin enrichment was dependent on the ATM kinase activity but not ATR (Figures S4J and S4K). As previous studies had found a correlation between high p21 levels and 53BP1 (Barr et al., 2017; Moser et al., 2018), we assessed whether elevated 53BP1 has a direct impact on the expression level of p21 in cells lacking AHNAK. Strikingly, higher intensity of 53BP1 on the chromatin associates with elevated p21 levels (Figures 4D and 4E). RNF8/RNF168-dependent H2A ubiquitination is important for the binding of 53BP1 to chromatin. To further assess the functional attributes of chromatin-bound 53BP1 in p53 activation, we manipulated 53BP1’s chromatin binding by depleting TRIP12, an E3 ubiquitin ligase, which maintains an optimal nuclear pool of RNF168 and leads to increased H2A ubiquitination and therefore 53BP1 chromatin retention (Gudjonsson et al., 2012) without causing extensive DNA damage (Figure S4L). As expected, depletion of TRIP12 led to increased enrichment of 53BP1 on the chromatin. Interestingly, loss of TRIP12 was sufficient to increase the expression of p21 in the absence of DNA damage (Figures 4F and 4G). Similar results were obtained when p53 levels were analyzed following pre-extraction (Figures S4M and S4N). Collectively, these results suggest that enhanced p53 responses observed are due to chromatin-enriched 53BP1 protein.
Figure 4.

AHNAK limits 53BP1 accumulation on chromatin
(A) Representative confocal images following immunostaining of 53BP1 (fluorescein green) in WT and two independent AHNAK−/− U2OS cell lines. Dashed line delineates the edge of the nucleus.
(B) Quantification of integrated fluorescence intensity (see STAR Methods) of 53BP1-NBs in individual cells of the indicated cell lines. The solid line denotes median. A.U., arbitrary units (n ≥ 1,376).
(C) Western blot analysis with the specified antibodies of chromatin fractions from WT and AHNAK−/− U2OS cells. LaminA and H4 were used as a loading control for chromatin fractions.
(D) Representative confocal images following co-immunostaining of p21 (red) and 53BP1 (fluorescein green) in WT and AHNAK−/− U2OS cells. Dashed line delineates the edge of the nucleus.
(E) Quantification of mean nuclear p21 fluorescence intensity in individual cells categorized into low and high chromatin-enriched integrated fluorescence intensity of 53BP1-NBs in two independent AHNAK−/− U2OS cell lines. The solid line denotes median. A.U., arbitrary units (n ≥ 514).
(F) Representative confocal images of U2OS cells co-immunostained with p21 (red) and 53BP1 (fluorescein green) antibodies following siRNA-mediated depletion of TRIP12. Dashed line delineates the edge of the nucleus.
(G) Quantification of mean nuclear p21 fluorescence intensity in single U2OS cells after the indicated siRNA-mediated depletion. Solid line denotes median. A.U., arbitrary units (n = 707).
(H) WB analysis using the indicated antibodies of chromatin fractions of U2OS cells stably expressing the indicated 53BP1 truncations and transfected with scramble or AHNAK siRNA. LaminA was used as a loading control for chromatin fractions.
(I) WB using anti-mCherry and anti-AHNAK antibodies after IP from U2OS cells stably expressing 53BP1 MFFR.
See also Figure S4.
53BP1 binds nucleosomes by interacting with H4K20me2 and H2AUb15. Additionally, the OD-driven multimeric assembly of 53BP1 manifests in its chromatin enrichment (Fradet-Turcotte et al., 2013). Therefore, to discriminate whether the AHNAK-imposed regulation of 53BP1 is by interfering with its interaction with histone marks (e.g., H4K40me2/H2AUb15) or restraining its oligomerization potential, we performed chromatin fractionation in U2OS cells stably expressing truncations of the 53BP1. Interestingly, AHANK depletion marginally affected the binding of the Tudor domain (1,271–1,711 aa) alone to chromatin but substantially increased the binding of every 53BP1 fragment that includes the OD (Figure 4H). Along the same lines, immunoprecipitation analysis revealed that 53BP1-OD is required for its stable interaction with AHNAK (Figure 4I).
These results suggest that AHNAK plays a critical role in attenuating OD-driven 53BP1 assembly on the chromatin.
AHNAK-regulated 53BP1 phase separation culminates in optimal p53 response
Recent studies demonstrated that 53BP1 nuclear compartments show key features of liquid-liquid phase separation (LLPS) (Kilic et al., 2019, Pessina et al., 2019). Motivated by our observation that 53BP1 NBs in AHNAK−/− cells were significantly larger than that of WT cells (Figures S5A and S5B), we studied the functional significance of AHNAK in 53BP1 phase separation properties. We first used a CRY2-53BP1 fusion to examine the impact of AHNAK on light-induced 53BP1 condensation (Figure 5A), as done previously (Kilic et al., 2019). Under unperturbed conditions, we observed enhanced 53BP1 optoDroplet formation along with increased nucleation of p53 molecules in AHNAK−/− cells compared with WT cells (Figures 5B and 5C). As expected, the potential of 53BP1 to form optoDroplets was dependent strictly on the 53BP1 OD domain both in WT and AHNAK−/− cells (Figures 5D and 5E). Along the same lines, tethering of 53BP1 at lacO array through lacI resulted in the accrual of p53 in G1 cells that was augmented after siRNA-mediated depletion of AHNAK (Figures 5F and 5G). Similarly, we observed an increased accumulation of p53 in the chromatin fraction in AHNAK−/− cells (Figure S5C). To determine the impact of 53BP1 condensation on p53 signaling, we exposed WT and AHNAK−/− cells to 1,6-hexanediol (1,6-HD), an aliphatic alcohol often used to disrupt LLPS. As shown in Figure S5D, following immunoblotting, we observed a robust decrease in p21 levels after 1,6-HD treatment, implicating LLPS in p53 stabilization.
Figure 5.

AHNAK-regulated 53BP1 phase separation culminates in optimal p53 response
(A) Schematic illustration of blue light-induced optoDroplet formation by 53BP1.
(B) Representative image of Cry2-mCherry-53BP1-MFFR-BRCT-W1495A optoDroplet formation in WT and AHNAK−/− U2OS cells. After optoDroplet induction cells, were fixed and stained for p53 (fluorescein green) and DAPI (blue).
(C) Quantification of sum 53BP1 optoDroplet fluorescence intensity in individual cells of the specified genotype. Solid line denotes mean. A.U., arbitrary units (WT, n = 222; ΔAHNAK-1, n = 1,083; and ΔAHNAK-2, n = 412).
(D) Representative image of Cry2-mCherry optoDroplet formation in WT and AHNAK−/− U2OS cells transfected with CRY2 harboring or not the OD domain. Nuclei are depicted in blue (DAPI).
(E) Quantification of sum 53BP1 optoDroplet fluorescence intensity in individual cells of the specified genotype from (D). Solid line denotes median. A.U., arbitrary units (n ≥ 2,650).
(F) Schematic illustration of the LacO-LacI tethering system.
(G) Co-localization of control GFP-LacI or GFP-53BP1-LacI (fluorescein green) with p53 (red) on the LacO array in U2OS19 cells after depletion with indicated siRNAs (n = 2 independent experiments). The white numbers represent the percentage of cells that exert colocalization of the indicated protein with the lacO array.
(H) Fluorescence recovery after photobleaching (FRAP) analysis on the 53BP1-NBs. Graphical representation for normalized fluorescence intensity curves versus time in seconds for WT-U2OS-GFP-53BP1 and two independent U2OS-GFP-53BP1-AHNAK−/− cell lines. Error bars represent the standard error of the mean (n = 18).
(I) WB analysis using the indicated antibodies after IP of nuclear extracts using anti-HA beads from WT and AHNAK−/− U2OS-GFP-53BP1 cells transfected with FLAG-HA-53BP1.
See also Figure S5.
To further validate the role of AHNAK in modulating the ability of 53BP1 to condensate on the chromatin, we compared the mobility of GFP-53BP1 within NBs in AHNAK−/− and WT cells stably expressing GFP-53BP1 protein. Interestingly, AHNAK−/− cells displayed decreased 53BP1 mobility within NBs compared with WT cells (Figure 5G), thus suggesting enhanced 53BP1 condensation and denser 53BP1 chromatin compartments in the absence of AHNAK.
Moreover, the potential of 53BP1 to self-associate was increased in (Figures 5A–5G) in AHNAK−/− cells compared with control cells (Figure 5I). This was concomitant with increased interaction of p53 with 53BP1 in AHNAK−/− cells (Figure 5I). In line with these data, we observed an increase association of GFP-53BP1-LacI with HA-53BP1 at the LacO array following AHNAK depletion (Figures S5E and S5F).
Knowing that AHNAK and p53 interact (Gu et al., 2019), we sought to determine whether this interaction depends on 53BP1. As expected, we found that AHNAK-4CRU interacts with p53. Notably, this occurs independently of 53BP1 (Figure S5G). Surprisingly, overexpression of the MFFR domain of 53BP1, which interacts with p53 as it contains the OD domain, did not alter the AHNAK-p53 interaction in both WT and 53BP1−/− cells (Figure S5H), suggesting that although AHNAK modulates the 53BP1-p53 interaction, this effect is not reciprocal. Collectively, these results point to a role of AHNAK in restraining 53BP1 oligomerization-driven condensation, which, if lost, results in enhanced interaction with p53, thus culminating in perpetuated p53 activation.
AHNAK dictates the choice between apoptosis and senescence in transformed and non-transformed cells
p53 activation can result in apoptosis or senescence (Horn and Vousden, 2007; Levine et al., 2006). We have shown that AHNAK depletion leads to reduced entrance to S/G2 and arrest in G1 (Figure 3). To further investigate the outcome of this arrest, we assessed Nutlin-3-induced apoptosis in transformed U2OS and non-transformed BJ fibroblasts. Nutlin-3 treatment induced apoptosis to a substantial population of U2OS cells (more than 15%) but had a minor effect in BJ cells (Figures 6A–6C). Interestingly, AHNAK depletion increased cell death in both undamaged and Nutlin-3-treated cells, and co-depletion of 53BP1 rescued the AHNAK-dependent increase in cell death in both the presence and absence of DNA damage (Figures 6A–6C), thus suggesting that AHNAK triggers apoptosis mainly in cancer cells.
Figure 6.

AHNAK dictates the choice between apoptosis and senescence in transformed and non-transformed cells
(A) BJ and U2OS cells transfected with the indicated siRNAs and treated with either DMSO or Nutlin-3 (20 μM). Apoptosis was assayed using FACS with antibody to annexin V-Alexa 488.
(B) Percentage of annexin-V-positive cells in BJ.
(C) Percentage of annexin-V-positive cells in U2OS.
(D) Representative bright-field images of SA-β-gal staining of DMSO or Nutlin-3-treated BJ fibroblast transfected with indicated siRNAs.
(E) Graphical representation of percentage of β-gal-positive cells from (D).
The fact that non-transformed BJ cells display resistance to apoptosis following treatment with the same doses of Nutlin-3 has prompted us to investigate whether AHNAK regulates senescence in non-transformed cells by using β-galactosidase staining. As shown in Figures 6D and 6E, depletion of AHNAK in BJ cells increased the percentage of cells positive for β-gal in undamaged cells and cells treated with Nutlin-3. Collectively, these results suggest that AHNAK constrains the p53-p21 axis to control cell death in cancer cells and senescence in non-transformed cells.
High expression of AHNAK is associated with suppression of p53 target genes in multiple cancer types
Given the impact of AHNAK on p53-mediated gene regulation in our cancer cell line models, we investigated whether this mechanism is reflected in primary tumor data. Using 28 gene expression datasets from 26 different cancer types, we interrogated the relationship between AHNAK expression and p53 target network (Figures 7A and 7B). We found that 25 of the 28 datasets showed significant enrichment of TP53 target genes among the transcripts that anticorrelated with AHNAK mRNA levels, suggesting that higher expression of AHNAK is associated with suppression of the p53 network in multiple cancer types (Figures 7A and 7B). Although AHNAK had been described as tumor suppressor (Chen et al., 2017; Lee et al., 2014; Zhao et al., 2017), the low frequency of single-nucleotide variants in AHNAK argues against point mutations in AHNAK as strong cancer drivers (Gonzalez-Perez et al., 2013). On the other hand, we found that AHNAK is overexpressed at mRNA levels in patient subgroups of multiple cancer types. Notably, AHNAK was overexpressed in 3.9% of stomach adenocarcinoma (The Cancer Genome Atlas [TCGA]; Z score = 2–6.5), and of all cancer types analyzed, stomach cancer exhibited the most substantial anticorrelation between expression of AHNAK and p53 target genes (Figures 7A and 7B). Furthermore, in stomach cancer, AHNAK and 53BP1 showed mild co-expression (Spearman’s R = 0.29) and, importantly, a joint anticorrelation with p53 network (Spearman’s R = 0.61; Figure 7C). Interestingly, the joint anticorrelation between AHNAK-53BP1 and p53 target genes is more apparent in tumors with intact p53 (Figure 7C). Moreover, the fact that AHNAK expression is increased when comparing WT TP53 (n = 198) versus mutated (n = 209; p = 0.001, Wilcoxon rank-sum test) and particularly between WT TP53 without copy number alteration (n = 153) and TP53 mutated and deletion of at least one allele (n = 118); p = 0.0001, Wilcoxon rank-sum test) (Figure 7D) further strengthens the notion that AHNAK-dependent attenuation of the p53 network may be required for cancer cell survival, specifically in tumors with otherwise intact p53 function. Our findings that AHNAK expression in a large number of 53BP1-expressing cancers and irrespective of cancer type anticorrelates with p53 target gene expression point to a general mechanism of AHNAK-mediated attenuation of the p53 response via 53BP1 modulation.
Figure 7.

AHNAK attenuates cellular global p53 response, and its loss makes cancer cells more sensitive to cancer treatment
(A and B) Gene set enrichment analysis for co-expression of AHNAK with genes of the ontology “transcriptional regulation by TP53” in 28 different cancer transcriptome datasets and differentiation by TP53 mutation in stomach cancer.
(C) Comparison of correlation coefficients (Spearman’s R) between co-expressed genes with AHNAK versus co-expression with TP53BP1, differentiated by TP53 mutation status in stomach cancer. Density lines indicate the density of genes in the two-dimensional (2D) space, where “other genes” (green) are all genes not assigned to the ontology of “transcriptional regulation by p53” (orange).
(D) Differentiation of AHNAK expression in stomach cancer by TP53 status (i.e., mutation and/or deletion). The p value is derived from Wilcoxon testing.
(E) WT and two independent AHNAK−/− U2OS cell lines were treated with fixed concentration of Nutlin and increasing concentration of etoposide.
(F) BJ fibroblast transfected with AHNAK siRNA or control and treated with fixed concentration of Nutlin-3 and increasing concentration of etoposide.
(G) Coefficient of drug interaction (CDI) analysis (see STAR Methods) of Nutlin and etoposide as in (E) and (F).
Having established the connection between AHNAK expression and p53 signaling in cancers, we sought to investigate the response of AHNAK-depleted cells to combinatorial anticancer therapies. To this end, we treated transformed U2OS and non-transformed BJ cells with Nutlin-3 in combination with increasing concentrations of etoposide. As shown in Figure 7E, U2OS cells are more sensitive to the combination of Nutlin-3 and etoposide, and depletion of AHNAK increases substantially their sensitivity. In marked contrast, BJ cells displayed marginal sensitivity to the combination treatment in the presence and absence of AHNAK (Figure 7F). Furthermore, to determine the nature of the interaction between the known anticancer drug etoposide and Nutlin-3, we calculated the coefficient of drug interaction (CDI), as described previously (Hao et al., 2008). Interestingly, AHNAK-depleted U2OS cells displays higher synergistic interaction (i.e., lower CDI) over increasing etoposide concentration compared with non-transformed BJ fibroblasts (Figure 7G).
These results together suggest that AHNAK expression is relevant for cancer cell survival, in particular when p53 function has not been lost. Additionally, interfering with AHNAK’s function to restrain the 53BP1-p53 response potentiates the effect of combined etoposide and Nutlin-3 treatment in transformed cells compared with non-transformed cells.
Discussion
AHNAK was previously identified as a structural scaffold protein and has been implicated in a range of cellular processes, including cell architecture, intracellular trafficking, and membrane repair (Amagai, 2004; Lim et al., 2013). In this study, we identify a previously concealed role of AHNAK in curbing spontaneous activation of p53 response by directly restraining 53BP1, a known mediator of p53 activity. Such an intrinsic cell response enforced by AHNAK fine-tunes the levels of “G1-S checkpoint” under unstressed conditions in cancer and non-transformed cells, thereby conceding an exponential pace of cell proliferation.
Previously, AHNAK mRNA was shown to be significantly enriched in G0 and G1 phase of the cell cycle (Shtivelman and Bishop, 1993). In line with this report, our findings demonstrate enhanced protein levels of AHNAK and chromatin binding in G1, which substantially reduce in S/G2. Moreover, AHNAK displays robust interaction with 53BP1 in G1 phase on chromatin, where it controls 53BP1 self-assembly and 53BP1 condensation-associated p53 activation. Interestingly, DNA damage increases AHNAK chromatin association and its interaction with 53BP1 in an ATM-dependent manner, suggesting that the DNA damage response deploys a counterbalance of p53-p21 response through AHANK to prevent permanent cell-cycle arrest or cell death.
Our results are consistent with previous finding showing that AHNAK interacts with p53 and inhibits p53-mediated target gene expression (Gu et al., 2019). Our study goes beyond by mechanistically underpinning the p53 activation in AHNAK−/− cells by 53BP1, a master regulator of p53 activation in response to intrinsic cell stress, such as during replication (Arora et al., 2017; Barr et al., 2017; Lezaja and Altmeyer, 2018). Notably, although the activation of p21 upon AHNAK depletion is apparent in every experimental system used (KO cells versus siRNA-depleted cells), p53 stabilization was more apparent upon acute AHNAK depletion by siRNA (Figures 2I, S2F, and S3C). A possible explanation for the difference between transient depletion by siRNA and AHNAK-KO clones could be the dynamics of rapid siRNA-mediated depletion and activation of p53 signaling cascade and the adaptation experienced by our cell lines after knocking out AHNAK.
AHNAK depletion in mouse embryonic fibroblasts (MEFs) increases cell growth (Lee et al., 2014), yet we observed elevated G1 arrest and increased apoptosis after AHNAK depletion in human cancer cells. This discrepancy may stem from species-specific differences in 53BP1-p53 regulation and function; for example, the sequence identity between mouse and human 53BP1 is about 80%, with marked differences particularly at the C-terminal p53-interacting BRCT domain. Additionally, our results suggest that non-cancer cells and cancer cells respond differently to deregulation of AHNAK, indicating a cancer cell-specific role of AHNAK in regulating p53 response. The data in the current literature implicating AHNAK status to carcinogenesis are ambiguous. On one hand, AHNAK has been implicated as a tumor suppressor in breast cancer (Chen et al., 2017; Lee et al., 2014), glioma (Zhao et al., 2017), and non-stem cell lung cancers (Gu et al., 2019). On the other hand, AHNAK is associated with poor outcome of pancreatic adenocarcinoma (Zhang et al., 2019). Here we observe that although AHNAK is encoded by a very long gene, single-nucleotide mutations are surprisingly not primarily responsible for tumorigenesis. However, in a substantial fraction of cancers, AHNAK is found to be overexpressed. Strikingly, our analysis of RNA sequencing (RNA-seq)-derived cancer transcriptomes has revealed that in the large majority of cancers with detectable 53BP1 expression, p53 target genes are significantly enriched among the transcripts that are anticorrelated with elevated AHNAK mRNA levels, consistent with the function of AHNAK in alleviating of 53BP1-p53 responses by modulation of 53BP1 in human cancers.
How does AHNAK influence 53BP1-dependent p53 regulation? We found that the absence of AHNAK enforced elevated levels of 53BP1 oligomerization and condensation. Interestingly, AHNAK restrains 53BP1 accrual on chromatin through its interaction with the OD. AHNAK is a protein of exceptional size (more than 5,000 residues; ∼627 kDa), and because of its elongated and rigid structure, it can facilitate interaction with multiple proteins (Dempsey et al., 2012; Shtivelman et al., 1992; Stiff et al., 2004). Therefore, it is tempting to speculate that AHNAK might act as a scaffold for 53BP1 and may favor structural changes in 53BP1 that limit the ability of 53BP1 to dynamically undergo LLPS, thus retaining their optimal assembly. Another obvious possibility is that AHNAK competes with 53BP1 for binding to the 53BP1 OD domain. Similarly, one cannot exclude the possibility that AHNAK competes with p53 for binding to the 53BP1 OD domain and the increased 53BP1-p53 interaction in the absence of AHNAK is due to the increased physical interaction with OD domain and to increased potential of 53BP1 to undergo LLPS.
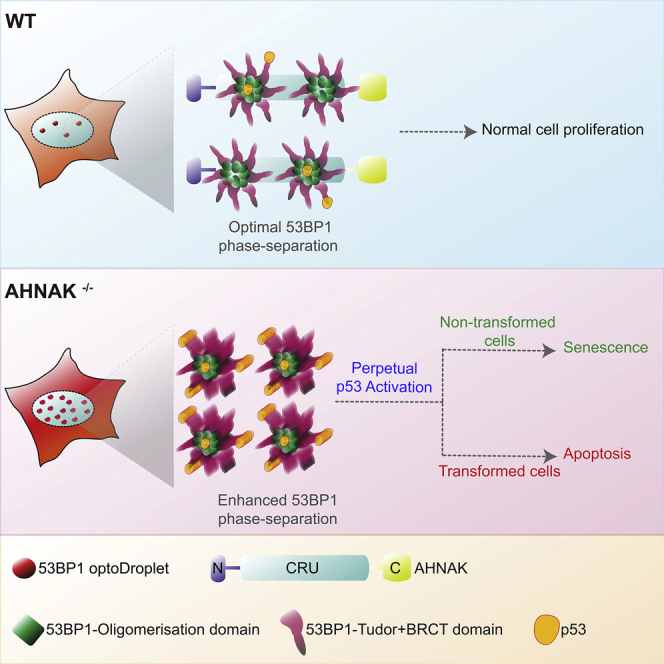
Upon DNA damage, AHNAK is further recruited to chromatin, where its interaction with 53BP1 is increased in an ATM-dependent manner. In the absence of AHNAK, uncontrolled 53BP1 multimerization leads to massive 53BP1 condensates and excessive interaction with p53, simulating conditions of high DNA damage, leading to cell-cycle arrest (Figure S6). Interestingly, although AHNAK depletion leads non-transformed cells to terminal cell-cycle arrest through senescence, it confers decreased cancer cell viability and sensitivity to drug treatments (Figure S6). Therefore, further addressing the dynamic interplay between AHNAK and the 53BP1-p53 axis and how it can be manipulated in cells might offer a novel therapeutic approach in anticancer therapies.
Limitations of study
Our results demonstrate AHNAK interacts with both 53BP1 and p53 to restrain their interaction and to prevent excessive 53BP1 oligomerization on chromatin. Our study does not explore whether this assembly occurs merely at DNA damage sites or also at p53 bound regulatory elements in the genome, or at reservoir places in the nucleus. Interestingly, the supra-accumulation of 53BP1 in NBs in G1 has already been implicated in attracting and locally activating p53 (Feng and Jasin, 2018), thus suggesting that the enlarged nuclear area occupied by 53BP1 in NBs or in DNA repair foci increase the chances for p53 activation in the vicinity rather than physically interacting with the DNA breaks. Another possibility is that 3D genome folding allows the interaction of p53 regulatory elements with the 53BP1 NBs. Further studies are necessary to shed light on these issues.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-AHNAK, 1:1000 (WB) | Santacruz | Cat# sc-390743 |
| Rabit anti-53BP1, 1:1000 (IF), 1:5000 (WB) | Novus Biologicals | Cat# 100-304, RRID:AB_350221 |
| Streptavidin HRP, 1:5000 (WB) | Invitrogen | Cat# S911 |
| Mouse anti-Vinculin, 1:5000 (WB) | Sigma | Cat# V9131, RRID:AB_477629 |
| Mouse anti-GAPDH, 1:10000 (WB) | Millipore | Cat# MAB374, RRID:AB_2107445 |
| Goat anti-GFP, 1:3000 (WB) | Abcam | Cat# 6673-100, RRID:AB_305643 |
| Mouse anti-p21, 1:3000 (WB), 1:1000 (IF) | GeneTex | Cat# GTX62953 |
| Mouse anti-p53, 1:200 (IF), 1:1000 (WB) | Santacruz | Cat# sc-126, RRID:AB_628082 |
| Rabit anti-USP28, 1:5000 (WB) | Abcam | Cat# ab126604, RRID:AB_11127442 |
| Rabit anti-pATM S1981, 1:5000 (WB) | Abcam | Cat# ab81292, RRID:AB_1640207 |
| Rabit anti-LaminA, 1:5000 (WB) | Abcam | Cat# ab26300, RRID:AB_775965 |
| Rabit anti-H4, 1:20000 (WB) | Abcam | Cat# ab7311, RRID:AB_305837 |
| Rabit anti-mCherry, 1:5000 (WB) | Abcam | Cat# ab167453, RRID:AB_2571870 |
| Rabit anti-p21, (1:500) (IF) | Santa Cruz | Cat# sc-756, RRID:AB_2229243 |
| Mouse anti-p53, (1:500) (IF) | Invitrogen | Cat# MA5-12571, RRID:AB_10986581 |
| Rabit anti-HA, (1:500) (IF) | Cell Signaling Technology | Cat# mAb-3724, RRID:AB_1549585 |
| FITC AnnexinV, (1:500) (FACS) | Ozyme | Cat# 640945, |
| Rabit anti-phospho-Histone H3 (Ser10) | Millipore | Cat# 06-570 RRID:AB_310177 |
| Mouse anti-Strep-tag II | IBA Lifesciences | Cat# 2-1507-001, RRID:AB_513133 |
| Goat anti-mouse Alexa Fluor 488 IgG, (1:1000) (IF) | Thermo Fischer Scientific | Cat# A11029, RRID:AB_138404 |
| Goat anti-rabbit Alexa Fluor 568 IgG, (1:1000) (IF) | Thermo Fischer Scientific | Cat# A11036, RRID:AB_10563566 |
| Goat anti-rabbit Alexa Fluor 488 IgG, (1:1000) (IF) | Thermo Fischer Scientific | Cat# A11034, RRID:AB_2576217 |
| Goat anti-mouse Alexa Fluor 568 IgG, (1:1000) (IF) | Thermo Fischer Scientific | Cat# A11031, RRID:AB_144696 |
| HRP horse anti-mouse IgG antibody, (1:10000) (WB) | Thermo Fischer Scientific | Cat# PI-2000, RRID:AB_2336177 |
| HRP goat anti-rabbit IgG antibody, (1:10000) (WB) | Thermo Fischer Scientific | Cat# PI-1000, RRID:AB_2336198 |
| HRP Mouse anti-goat IgG antibody, (1:10000) (WB) | Santacruz | Cat# sc-2354, RRID:AB_628490 |
| Chemicals, peptides, and recombinant proteins | ||
| Neocarzinostatin (NCS) | Sigma | Cat# N9162-100 |
| KU 55933 (ATM inhibitor) | Selleckchem | Cat# 1092 |
| AZD6738 (ATR inhibitor) | Selleckchem | Cat# S7693 |
| cOmplete, Mini Protease Inhibitor Cocktail | Roche | Cat# 11836153001 |
| PhosSTOP | Roche | Cat# 4906837001 |
| Mevinolin/ Lovastatin | Sigma | Cat# M2147 |
| Mevalonolactone | Sigma | Cat# M4667 |
| Benzonase | Millipore | Cat# 70746 |
| λ-Phosphatase | Santacruz | Cat# sc-200312A |
| Crystal Violet | Sigma | C0775 |
| Experimental models: Cell lines | ||
| U2OS | ATCC | Cat# HTB-96 |
| U2OS -ΔAHNAK-1 | This paper | N/A |
| U2OS -ΔAHNAK-2 | This paper | N/A |
| MCF7 | Ross Chappman lab, University of Oxford, UK. | N/A |
| MCF7-ΔAHNAK-1 | This paper | N/A |
| MCF7-ΔAHNAK-2 | This paper | N/A |
| BJEH = BJEH (hTERT) | William C. HAHN, Department of Medical Oncology, Dana-Farber Cancer Institute, BOSTON, USA | N/A |
| MCF10A | ATCC | Cat# CRL-10317 |
| hTERT IMR-90 immortalized | Dr Vincent DION, University of Lausanne, Center for Integrative Genomics, LAUSANNE, SWITZERLAND | N/A |
| MCF7-Δ53BP1 | Ross Chappman lab, University of Oxford, UK. | N/A |
| MCF7-Δp53 | Ross Chappman lab, University of Oxford, UK. | N/A |
| U2OS-GFP-53BP1-WT | Jiri Lukas lab, University of Copenhagen, Denmark | N/A |
| U2OS-GFP-53BP1-ΔAHNAK-1 | This paper | N/A |
| U2OS-GFP-53BP1-ΔAHNAK-2 | This paper | N/A |
| Oligonucleotides | ||
| scramble | Dharmacon | Cat#D-001810-01-10 |
| scramble | siTOOL biotech | Cat# negative control siPOOL |
| AHNAK | siTOOL biotech | Cat#79026 - AHNAK (human) |
| TRIP12 | Dharmacon | Cat# L-007182-00-0005 |
| 53BP1 | Dharmacon | Cat# L-003548-00-0005 |
| USP28 | Dharmacon | Cat# L-006076-00-0005 |
| Primers for cloning, see Table S1 | This paper | N/A |
| Sequence of CRISPR-Cas9 guide-RNAs (gRNAs) and primer pairs used to generate AHNAK knockout, see Table S2 | This paper | N/A |
| Sequences of primer pairs used for RT-qPCR in this study, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| AHNAK-4CRU-GFP | This paper | N/A |
| pAAV-CBA- AHNAK -N-Strep | Yong Kim lab, Rockefeller University: New York, USA. | N/A |
| pAAV-CBA- AHNAK −4CRU-Strep | Yong Kim lab, Rockefeller University: New York, USA. | N/A |
| pAAV-CBA- AHNAK -C-Strep | Yong Kim lab, Rockefeller University: New York, USA. | N/A |
| 53BP1-Tudor (1271-1771 aa) | This paper | N/A |
| 53BP1-MFFR (1220-1771 aa) | This paper | N/A |
| 53BP1-MFFR-BRCT (1220-1972 aa) | This paper | N/A |
| Software and algorithms | ||
| ScanR Analysis software, V 2.7.1. QIBC data analysis | Olympus Corporation | https://www.olympus-ims.com/en/microscope/software/ |
| Spotfire, data visualization software, V7.8.0.1.20/V10.5.0.72 | TIBCO Software Inc. | https://account.cloud.tibco.com/signup/spotfire |
| Fiji/ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| CellProfiler 4.1.3, Cell profiler image analysis Software | Carpenter Lab at the Broad Institute of Harvard and MIT | https://cellprofiler.org/releases |
| MaxQuant, Quantitative analysis of mass-spectrometric data | Max planck institute of biochemistry | https://www.maxquant.org/ |
| Perseus, Visualization of mass-spectrometric data | Max planck institute of biochemistry | https://www.maxquant.org/ |
| GraphPad Prism 9.00 for Windows, data visualization and statistics | GraphPad Software, LLC | https://www.graphpad.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Evi Soutoglou (E.Soutoglou@sussex.ac.uk).
Materials availability
The plasmids and cell lines generated in this study are available from the Lead Contact upon request.
Data and code availability
Original code and data for complete RNaseq analysis from primary tumors is available at https://github.com/arpoe/GhodkeI_MolCell_2020. Original data have been deposited to Mendeley Data: https://dx.doi.org/10.17632/rrjnz8rwc8.1
Method details
Plasmid construction
The DNA sequences encoding 53BP1 (MRC PPU Reagents and Services, DU49658) and repetitive elements of human AHNAK (amino acids 1068–1579) pAAV-CBA-Ahnak-R-Strep (Jin et al., 2019) were used as a template for amplification by PCR and cloned by megawhop cloning (Miyazaki, 2011). (See Table-S1 for primers). All plasmids (STAR Methods) were assembled by golden gate cloning (Engler et al., 2009).
Cell lines and culture conditions
U2OS and cell lines derived from U2OS were grown in DMEM (1g/l glucose) medium supplemented with 10% fetal calf serum and 50 μg/ml Gentamycin. MCF7 and cell lines derived from MCF7 were grown in high Glucose DMEM (4.5 g/L glucose) medium supplemented with 10% fetal calf serum, 2 mM L-glutamine, 1% penicillin and streptomycin. All cells were cultured in a humidified incubator at 37°C with 5% CO2. For quantitative label-free mass spectrometry experiment cell were cultured in the presence of 150 μM of Biotin.
Cell line generation
Following approach was used to generate U2OS cells stably expressing the protein of interest. Plasmid DNA was transfected with Lipofectamine 2000 (Invitrogen, 11668019) as per the manufacturer’s instructions. After 18 h, cells were sorted in BD FACSAria Fusion as a single cell in each well of 96 well microtiter plates. After 2 days, sorted cells were selected with medium containing 1.5 μg/ml puromycin (Invivogen, ant-pr-1). After incubation of approximately 2 weeks to obtain colonies, cells were expanded and screened for the expression by microscopy and confirmed with western blot for the desired size of protein of interest.
CRISPR/Cas9 editing
AHNAK−/− MCF7, U2OS and U2OS-GFP-53BP1 cells were obtained by high-fidelity Cas9. Briefly, cells were co-transfected with plasmids expressing two gRNAs targeting the AHNAK locus in exon 3 (See Table-S2 for gRNA sequence), and high-fidelity Cas9 was fused to EGFP (For U2OS and MCF7 cells) or mCherry (For U2OS-GFP-53BP1 cells), and following 2 days after transfection EGFP+ or mCherry+ cells were sorted by fluorescence-activated cell sorting (FACS) in 96 well plate, grown for three weeks and confirmed by western blot analysis.
Cell treatment
siRNA transfection
siRNAs were transfected with Lipofectamine RNAiMAX (Invitrogen, 13778150) at a final concentration of 10 nM for a minimum period of 48 h as per the manufacturer’s instructions. For AHNAK, pool of siRNA was purchased from siTOOLS Biotech, and used at a final concentration of 2 nM. (KEY RESOURCES TABLE) western blotting or RT-qPCR were performed to analyze the knockdown efficiency. All western blot analysis and imaging experiments were performed 48 h post knockdown and 16 h post plasmid DNA transfection unless otherwise indicated.
Cell irradiation
Cells were irradiated using the CELLRAD - Precision X-Ray at the following settings: 130 kV, 5 mA, 13 Gy/min. Soft X-rays were filtered out with a 0.5-mm aluminum filter (CELLRAD - Precision X-Ray).
DNA damage by NCS
Neocarzinostatin (NCS; N9162-100 UG; Sigma) was added (100 ng/ml), 15 min later cells were washed three times with PBS and released into fresh medium and harvested at the indicated time points.
Cell cycle analysis
Following harvesting, cells were fixed in ice-cold 70% ethanol overnight at −20°C. After centrifugation cells were treated with RNaseA (100 μg/ml) at 42°C for 90 min, and DNA was stained with propidium iodide (50 μg/ml). Flow cytometry was performed by FACS Calibur (Becton Dickinson), Aggregates were gated out, and the data were analyzed with Flowjo software (TreeStar).
Cell cycle synchronization
U2OS cells were synchronized in G1/S and G2 phase of the cell cycle with Mevinolin/ Lovastatin (Sigma, M2147-25MG) and Mevalonolactone (Sigma, M4667-1G), as previously described 21. Briefly, U2OS cells were plated at 80% confluency, then cells were treated with 40 μM of lovastatin for 40 h, subsequently, after washing with 1X PBS, cells were resuspended in 4 mM Mevalonolactone (M4667-1G) and collected after 6 h for G1 and 22 h for S-G2 enrichment. Cell synchronizations were confirmed by flow cytometry as described above.
Quantitative label-free mass spectrometry
Sample were reduced, alkylated and digested with trypsin at 37°C overnight. Extracted peptides were analyzed using an Ultimate 3000 nano-RSLC (Thermo Scientific, San Jose California) coupled in line with an Orbitrap ELITE (Thermo Scientific, San Jose California). Each sample was analyzed in triplicate. Briefly, peptides were separated on a C18 nano-column with a linear gradient of acetonitrile and analyzed with Top 20 CID method. MS spectra were identified and quantified by MaxQuant 1.6.5.0 (Cox and Mann, 2008) using Uniprot human protein database (20169 reviewed sequences). During analysis, trypsin was set as an enzyme, oxidation (M) and N-term acetylation were set as variable modifications, and carbamidomethylation (C) as a fixed modification. Proteins were identified with a minimum of two peptides and were filtered with False Discovery Rate < 1%. Lastly, MaxQuant results were exported to Perseus 1.6.6.0 for data cleaning and statistics before graphical representation (Volcano plots). Venn diagram was used to discriminate protein population according to the phase of the cell cycle.
Real-time qPCR
RNA extraction (QIAGEN, 74106) and cDNA (ThermoFisher Scientific, 12574026) were prepared as per the manufacturer’s instructions. qPCR was performed in triplicate using SyberGreen (QIAGEN, 204143) and a LightCycler 480 (Roche, 05015278001) as previously described (Pankotai et al., 2012). Relative quantification of transcript quantities were calculated from standard after normalizing it to β-ACTIN (U2OS) and GAPDH (MCF7) mRNA (see Table-S3 for primers).
Immunoprecipitation
After harvesting, cells were washed in PBS and lysed in ice-cold RIPA buffer containing 50 mM Tris (pH 7.5), 150 mM sodium chloride, 0.5% NP-40, 0.1% sodium deoxycholate, 5% Glycerol, 1 mM EDTA, supplemented with complete protease (Roche, 11697498001) and Phosphatase Inhibitor Cocktail (Roche, 4906837001) and benzonase (750 U/ml) (Millipore, E1014-25KU). After 30 minutes of incubation on ice, lysates were clarified by centrifugation at 13,000 rpm for 20 min at 4°C. Bradford Protein Assay (Bio-Rad) was used to quantify protein concentration, and ∼5mg of clarified lysates were incubated with appropriate beads for 12-16 h at 4°C. The Ig–antigen complexes were washed once with ice-cold RIPA lysis buffer and twice with TBS buffer (50mM Tris-HCl, pH 7.4, with 150 mM NaCl). Subsequently, the bound proteins were eluted in 4x NuPAGE LDS sample buffer (Invitrogen) supplemented with 1 mM DTT at 95°C for 30 min before SDS-PAGE.
To analyze the interaction between AHNAK-4CRU-GFP and 53BP1, U2OS cells were transiently transfected with either GFP or AHNAK-4CRU-GFP for 18 h and cell lysates were prepared as described above. About 5 mg of clarified lysates were incubated with 25 μL GFP-Trap magnetic beads (Chromotek, gtd-20) for 18 h on a rotating wheel at 4°C. Subsequently, the protocol is followed according to the manufacturer’s instructions.
For immunoprecipitation of endogenous AHNAK, Anti-AHNAK (Santacruz, sc-390743) was immobilized on Dynabeads Protein G (Invitrogen, 10004D) as per the manufacturer’s instructions.
For immunoprecipitation from soluble and chromatin fractions, the cell pellets were resuspended in (10 mM HEPES, pH 7, 200 mM NaCl, 1 mM EDTA, and 0.5% NP-40) supplemented by protease and phosphatase inhibitors and incubated for 20 min on ice and then centrifuged at 16,000 g for 10 min. The soluble fraction was collected. Pellets were washed twice (10 mM HEPES, pH 7, 200 mM NaCl, 1 mM EDTA with protease and phosphatase inhibitors), centrifuged at 16,000 g for 5 min, resuspended in chromatin-lysis buffer (10 mM HEPES, pH 7, 200 mM NaCl, 1 mM EDTA, 1% NP-40, and protease-inhibitor cocktail and 750 U/ml benzonase), incubated for 30 min on ice, sonicated at low amplitude, and centrifuged at 16,000 g for 10 min. Soluble and chromatin fractions were applied on respective magnetic beads.
Western blotting
The cells were lysed and protein was quantified as per above description. The cell lysate was resolved on precasted SDS-PAGE gels (NuPAGE 3%–8% Tris-acetate for large proteins and NuPAGE 4%–12% Bis-Tris for smaller proteins, Invitrogen). Proteins were transferred onto nitrocellulose membranes, and the membranes were blocked with 5% BSA (Biorad) solution in PBS for 1h at room temperature. The membranes were incubated with antibodies diluted in 5% BSA for overnight at 4°C. Horseradish peroxidase-conjugated secondary antibodies were incubated at room temperature for 1 h, and Novex ECL chemiluminescence (Invitrogen, WP20005) was used for immunodetection.
Immunofluorescence and Confocal microscopy of fixed and live cells
Cells were cultured on coverslips and fixed in 4% paraformaldehyde/1X PBS for 15 min at room temperature. After the fixation step, cells were permeabilized in 0.5% Triton/1X in PBS for 10 min. For EDU incorporation Click-iT EdU Alexa Fluor 488 Imaging Kit (Invitrogen, C10338) was used as per the manufacturer’s instructions. Subsequently, the cells were blocked in 5% BSA/1X PBS-0.1% Tween for 60 min and incubated with primary antibody for 1 h (see Table-5 for antibodies) and secondary antibody for 1 h. Cells were counterstained with DAPI (1 mg/ml) and mounted on slides. Cells were observed on a confocal laser scanning microscope (TCS SP5; Leica).
Chromatin fractionation
After indicated treatments, cells were washed three times with ice-cold PBS and harvested by scraping. The cytosolic protein fraction was removed by incubation in hypotonic buffer (10 mM HEPES-KOH, pH 7, 50 mM NaCl, 0.3 M sucrose, and 0.5% Triton X-100, supplemented with protease and phosphatase inhibitor cocktail, Roche) for 10 min on ice, and centrifuged at 1500 g for 5 min. The remaining pellet was resuspended in nuclear buffer (10 mM HEPES-KOH, pH 7, 200 mM NaCl, 1 mM EDTA, 0.5% NP-40, and supplemented with protease and phosphatase inhibitor cocktail, Roche), incubated on ice for 10 min and centrifuged at 13000 rpm for 2 min. The final pellet containing chromatin fraction was resuspended in lysis buffer (10 mM HEPES-KOH, pH 7, 500 mM NaCl, 1 mM EDTA, 1% NP-40, and supplemented with protease and phosphatase inhibitor cocktail, Roche, and Benzonase, Millipore), sonicated at low amplitude for three times, and incubated for 15 min on ice and centrifuged for 1 min at 13000 rpm. Total protein was quantified through Bradford’s method, and a total of 10 μg protein from chromatin fraction was used for western blots.
Nutlin-3 survival assay
Cells were seeded in triplicate at a number of 5 × 103 cells/well in 6-well plates, were DMSO treated or treated with 4 μM Nutlin-3 (Cayman Chemicals, 10004372) for 16 h. After 10 to 14 days, the emerging colonies were fixed with 4% formaldehyde and stained with crystal violet (0.5% [w/v] in 20% methanol). The cell culture plates were scanned to obtain digital images. The relative growth was calculated by dissolving bound crystal violet in 10% (v/v) acetic acid, and absorbance of 1:50 dilutions was measured at 595 nm.
Interaction analysis between Nutlin-3 and etoposide
Coefficient drug interaction (CDI) was used to analyze interaction between Nutlin-3 and Etoposide as descried previously (Zhou et al., 2012)
Where AB is the percentage cell viability ratio of Nutlin-3 and Etoposide together, A is the percentage cell viability ratio of Nutlin-3 alone, and B is the percentage cell viability ratio of Etoposide alone. CDI values < 1 indicates synergistic drug effect, for CDI = 1 indicate additive drug effect and for CDI values > 1 indicates antagonistic drug effect.
Annexin-V apoptosis assay
For quantitation of Nutlin-3 induced apoptosis in BJ and U2OS cells, following 24 hour transfection of siRNA, the cells were left treated with DMSO or Nutlin-3 (20 μM) for 3 days. 20 Gy of IR treatment was used as a positive control for both the cell lines. Subsequently, according cells were resuspended in annexin-V binding buffer and stained with FITC-annexin-V and PI, according to the manufacturer’s instructions (Ozyme life sciences). LSR Fortessa (Becton Dickinson, USA) flow cytometer was used to analyze stained cells. Annexin-V positive cells was used to calculate the extent of apoptosis.
SA-β galactosidase assay for senescence
To induce senescence by Nutlin-3, BJ cells were seeded at low density, and then transfected with indicated siRNA. Subsequently, cells were left treated with DMSO or Nutlin-3 (20 μM) for indicated days, and SA-β galactosidase assay is performed as described previously (Simboeck et al., 2013). Briefly, following treatment, cells were washed with 2 mL PBS in 6-well plate and were fixed with 0.5% glutaraldehyde in PBS for 15 min at room temperature. Subsequently, cells were washed twice with 1 mM MgCl2 in PBS pH 6.0. Cells were incubated in 2 mL of freshly prepared 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside X-gal staining solution (1 mg/ml X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside), 0.2 M K4Fe (CN)6-3H2O, and 0.2 M K3Fe(CN)6 in PBS/MgCl2 pH 6) at 37°C, sealed, and protected from light for 4 h and brightfield pictures were taken at various magnifications. Approximately 100–300 cells were counted to quantify SA-β galactosidase-positive cells.
Fluorescence recovery after photobleaching
The FRAP experiment was carried out on a Nikon Ti-E inverted microscope (with Perfect Focus System, PFS) equipped with a Yokogawa CSU-X1 spinning disk scan head, as CMOS Prime 95B camera (Photometrics), the iLas2 FRAP unit and a 100 X 1.4 NA oil immersion objective lens, configured by Gataca Systems (Massy,France) and run using Metamorph 7.10.2 (Molecular Devices). During all FRAP measurements, cells were maintained at 37°C in 5% CO2 using the Tokai Hit INUBG2E-TIZ stage top incubator. Cell fluorescence was excited using a 491 nm laser line, 200 ms exposure time, 300 ms time interval. Recovery of fluorescence was monitored with the same frequency and exposure time until 2 minutes. Analysis of the FRAP kinetics was performed using custom macros developed with the Fiji image processing package. Three ROI was used to quantify over time the background signal, the loss of fluorescence due to photobleaching outside from the bleach area (cell in the same field of view) and the average intensity in the bleach ROI. FRAP curves were generated after background subtraction, photobleaching correction, and normalization to the pre-bleach values. Averages ± standard error mean were plotted. FRAP efficiency, and time kinetics were extracted by fitting the curves with a 2-exponential diffusion model to extract y(t) = Y1(1−exp(−t/τ1)) +Y2(1−exp(−t/τ2)).
G1/S checkpoint analysis
Following 24 h siRNA transfection, the cells were synchronized in G0 by incubation for 24 h in serum starved medium. Then, the cells were released in normal medium with 0.25 μg/ml nocodazole (Sigma, M1404-10MG). To activate the G1/S checkpoint, 4 h post-release, the cells were inflicted with DNA damage by ionizing radiations (4 Gy), and the experiment was followed until 22 h by collecting the cells at every indicated time point. The DNA was stained with propidium iodide (50 μg/ml). Flow cytometry was performed by FACS Calibur (Becton Dickinson), Aggregates were gated out, and the cell cycle distribution was analyzed with built-in cell cycle plugin in Flowjo software (TreeStar).
OptoDroplet experiments
8’000 U-2 OS cells per well were seeded into 96-well plates (Greiner μclear). On the next day, cells were transfected with Cry2-mCherry-53BP1 FFR W1495A to induce 53BP1 optoDroplets as performed previously (Kilic et al., 2019). 100 ng of the expression plasmid, 0.3 μl of TransIT (Mirus Bio) and 9 μl of OptiMEM (ThermoFisher) per well were incubated for 15 min at room temperature, before 92 μl of Fluorobrite DMEM (ThermoFisher) with 10% FCS and Glutamax (ThermoFisher) was added. The cell culture medium on the cells was then removed and replaced with the transfection mixture. 24 h after transfection, optoDroplet formation was induced at a GE INCell Analyzer 2500HS, using a CFI Plan Apo Lambda 10x objective (NA 0.45) and 25 ms pulses of blue light (excitation BP 390/18, emission BP 432.5/47) every 15 s for a total duration of 10 min. Upon optoDroplet induction, cells were washed with PBS and immediately fixed with 4% formaldehyde in PBS for 15 min, and immunofluorescence staining was performed for p53 detection. Images were acquired on an Olympus ScanR high-content screening system using a UPLSAPO 20x air objective (NA 0.75) and analyzed by QIBC. The optoDroplet data were normalized to mCherry expression levels for appropriate comparison between samples.
Quantitative image-based cytometry (QIBC)
Automated multichannel wide-field microscopy for high-content imaging and quantitative image-based cytometry (QIBC) was performed using the Olympus ScanR System as described previously (Michelena et al., 2019). Images were analyzed with the Olympus ScanR Image Analysis software version 3.0.1, a dynamic background correction was applied, and nuclei segmentation was performed using an integrated intensity-based object detection module based on the DAPI signal. Downstream analyses focused on properly detected nuclei containing a 2C-4C DNA content as measured by total and mean DAPI intensities. OptoDroplets were detected using an integrated intensity-based spot detection module. Nuclear fluorescence intensities were quantified and are depicted as arbitrary units. Color-coded scatterplots of asynchronous cell populations were generated with TIBCO Spotfire data visualization software version 7.0.1. Within one experiment, similar cell numbers were compared for the different conditions. For visualizing discrete data in scatterplots, mild jittering (random displacement of data points along discrete data axes) was applied in order to demerge overlapping data points.
Co-expression analysis of cancer transcriptomes
Gene expression data from 28 datasets were obtained as co-expression matrices, z-scores, and metadata for TP53 mutation/ copy number status were obtained through www.cbioportal.org on 05/23/2020. Data were selected for their presence of RNA-Seq data and a cohort size of at least n = 100. Array-based gene expression data were limited to the Metabric dataset, while all others are derived from RNA-Seq and comprised of the Cancer Cell Line Enzyclopedia, the Neuroblastoma and Willms Tumor datasets from TARGET and TCGA Pancancer Atlas data from the indicated cancer types. For Stomach cancer, the data were also obtained for wild-type TP53 (n = 198) and mutated (n = 209) separately. The entire analysis was performed in R. Gene ontology was derived from reactome.org with the R package “reactomePA.” Gene set enrichment was performed using “fgsea” and visualized with custom “ggplot2” based visualization. Heatmaps were generated using “pheatmap.” Statistical assessment is based on the Wilcoxon test and Spearman correlation. Original code and data for the full analysis will be available at https://github.com/arpoe/AHNAK.
LacO array experiments
The cell line U2OS19 containing 256 lac operator and 9s6 tetracycline response element copies was grown on glass coverslip as previously described (Lemaître et al., 2012). Following, 24 h siRNA transfection, 53BP1-GFP-LacI and LacI-GFP was transfected using Jet Pei (Polyplus transfection). Twenty-four hours after transfection, cells were processed for immunofluorescence and visualized on a confocal laser scanning microscope (TCS SP5; Leica).
Image analysis
We have expanded Cell profiler to construct our own image analysis pipelines. Due to even nucleoplasmic distribution of p21, we measured mean p21 intensity per nucleus and due to variable sizes of 53BP1 foci/Nuclear Bodies, we measured integrated intensity per nucleus. Briefly, following segmentation, DAPI image was used to create mask and isolated the fluorescence intensity from p21. In addition, by ‘identify speckle pipelines’, we have measured the 53BP1 number and its integrated intensity per nucleus. For the analysis involving the correlation of 53BP1 and p21 integrated intensity, we manually categorised low and high integrated intensity of 53BP1 per nucleus, and fluorescence intensity of p21 was plotted in GraphPad prism 9.0
Quantification and statistical analysis
Prism 9.00 software (GraphPad Software Inc.) was typically used for statistical analysis of datasets. For all statistical analysis, unless mentioned otherwise, were evaluated by ordinary one-way ANOVA. All proteomic related statistical analyses were performed using Perseus.
Acknowledgments
We thank R. Chapman for MCF7 WT, P53−/−, 53BP1−/− cells and FLAG-HA-53BP1 plasmid, Jiri Lukas for U2OS GFP-53BP1 cells, and Yong Kim for AHNAK plasmids. We acknowledge the Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC) proteomics and Imaging Center for their support. We are also grateful to Karen Meaburn for critical reading of the manuscript and the Soutoglou lab for comments. I.G. was supported by Fondation pour la Recherche Medical (FRM). The E.S. lab was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (ERC-2015-COG-682939) and by ANR-10-LABX-0030-INRT, managed by Agence Nationale de la Recherche under the program Investissements d’Avenir (ANR-10-IDEX-0002-02). M.A. acknowledges research funding from the Swiss National Science Foundation (PP00P3_179057) and the ERC under the European Union’s Horizon 2020 research and innovation program (ERC-2016-STG 714326). A.R.P. was supported by St. Anna-Kinderkrebsforschung and the Mildred Scheel Early Career Center Dresden P2, funded by German Cancer Aid.
Author contributions
Conceptualization, E.S. and I.G.; Methodology, I.G., M.R., and S.K.; Investigation, I.G., M.R., A.F., and A.R.P.; Formal Analysis, A.R.P.; Resources, B.R.-S.-M.; Writing – Original Draft, I.G. and E.S.; Writing – Review and Editing, A.R.P., B.R.-S.-M., M.A., and M.R.; Supervision, E.S. and M.A.; Funding Acquisition, E.S.
Declaration of interests
The authors declare no competing interests.
Published: May 6, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2021.04.010.
Contributor Information
Indrajeet Ghodke, Email: ghodkei@igbmc.fr.
Evi Soutoglou, Email: e.soutoglou@sussex.ac.uk.
Supplemental information
References
- Abbas T., Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagai M. A mystery of AHNAK/desmoyokin still goes on. J. Invest. Dermatol. 2004;123 doi: 10.1111/j.0022-202X.2004.23432.x. xiv–xv. [DOI] [PubMed] [Google Scholar]
- Arora M., Moser J., Phadke H., Basha A.A., Spencer S.L. Endogenous replication stress in mother cells leads to quiescence of daughter cells. Cell Rep. 2017;19:1351–1364. doi: 10.1016/j.celrep.2017.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr A.R., Cooper S., Heldt F.S., Butera F., Stoy H., Mansfeld J., Novák B., Bakal C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017;8:14728. doi: 10.1038/ncomms14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J.R., Cuella-Martin R., Barazas M., Liu R., Oliveira C., Oliver A.W., Bilham K., Holt A.B., Blackford A.N., Heierhorst J. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun. 2018;9:5406. doi: 10.1038/s41467-018-07855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigot N., Day M., Baldock R.A., Watts F.Z., Oliver A.W., Pearl L.H. Phosphorylation-mediated interactions with TOPBP1 couple 53BP1 and 9-1-1 to control the G1 DNA damage checkpoint. eLife. 2019;8:e44353. doi: 10.7554/eLife.44353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cescutti R., Negrini S., Kohzaki M., Halazonetis T.D. TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J. 2010;29:3723–3732. doi: 10.1038/emboj.2010.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Wang J., Dai D., Zhou Q., Guo X., Tian Z., Huang X., Yang L., Tang H., Xie X. AHNAK suppresses tumour proliferation and invasion by targeting multiple pathways in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2017;36:65. doi: 10.1186/s13046-017-0522-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cuella-Martin R., Oliveira C., Lockstone H.E., Snellenberg S., Grolmusova N., Chapman J.R. 53BP1 integrates DNA repair and p53-dependent cell fate decisions via distinct mechanisms. Mol. Cell. 2016;64:51–64. doi: 10.1016/j.molcel.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey B.R., Rezvanpour A., Lee T.W., Barber K.R., Junop M.S., Shaw G.S. Structure of an asymmetric ternary protein complex provides insight for membrane interaction. Structure. 2012;20:1737–1745. doi: 10.1016/j.str.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Engler C., Gruetzner R., Kandzia R., Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W., Jasin M. 53BP1 nuclear body-marked replication stress in a human mammary cell model of BRCA2 deficiency. bioRxiv. 2018:462119. [Google Scholar]
- Fradet-Turcotte A., Canny M.D., Escribano-Díaz C., Orthwein A., Leung C.C., Huang H., Landry M.C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F., Durocher D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A., Perez-Llamas C., Deu-Pons J., Tamborero D., Schroeder M.P., Jene-Sanz A., Santos A., Lopez-Bigas N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods. 2013;10:1081–1082. doi: 10.1038/nmeth.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J., Mao W., Ren W., Xu F., Zhu Q., Lu C., Lin Z., Zhang Z., Chu Y., Liu R., Ge D. Ubiquitin-protein ligase E3C maintains non-small-cell lung cancer stemness by targeting AHNAK-p53 complex. Cancer Lett. 2019;443:125–134. doi: 10.1016/j.canlet.2018.11.029. [DOI] [PubMed] [Google Scholar]
- Gudjonsson T., Altmeyer M., Savic V., Toledo L., Dinant C., Grøfte M., Bartkova J., Poulsen M., Oka Y., Bekker-Jensen S. TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell. 2012;150:697–709. doi: 10.1016/j.cell.2012.06.039. [DOI] [PubMed] [Google Scholar]
- Hao J.Q., Li Q., Xu S.P., Shen Y.X., Sun G.Y. Effect of lumiracoxib on proliferation and apoptosis of human nonsmall cell lung cancer cells in vitro. Chin. Med. J. (Engl.) 2008;121:602–607. [PubMed] [Google Scholar]
- Harrigan J.A., Belotserkovskaya R., Coates J., Dimitrova D.S., Polo S.E., Bradshaw C.R., Fraser P., Jackson S.P. Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn H.F., Vousden K.H. Coping with stress: multiple ways to activate p53. Oncogene. 2007;26:1306–1316. doi: 10.1038/sj.onc.1210263. [DOI] [PubMed] [Google Scholar]
- Huang J., Sengupta R., Espejo A.B., Lee M.G., Dorsey J.A., Richter M., Opravil S., Shiekhattar R., Bedford M.T., Jenuwein T., Berger S.L. p53 is regulated by the lysine demethylase LSD1. Nature. 2007;449:105–108. doi: 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K., Bartel P.L., Li B., Marraccino R., Fields S. Two cellular proteins that bind to wild-type but not mutant p53. Proc. Natl. Acad. Sci. U S A. 1994;91:6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwabuchi K., Li B., Massa H.F., Trask B.J., Date T., Fields S. Stimulation of p53-mediated transcriptional activation by the p53-binding proteins, 53BP1 and 53BP2. J. Biol. Chem. 1998;273:26061–26068. doi: 10.1074/jbc.273.40.26061. [DOI] [PubMed] [Google Scholar]
- Javanmoghadam-Kamrani S., Keyomarsi K. Synchronization of the cell cycle using lovastatin. Cell Cycle. 2008;7:2434–2440. doi: 10.4161/cc.6364. [DOI] [PubMed] [Google Scholar]
- Jin J., Bhatti D.L., Lee K.W., Medrihan L., Cheng J., Wei J., Zhong P., Yan Z., Kooiker C., Song C. Ahnak scaffolds p11/Anxa2 complex and L-type voltage-gated calcium channel and modulates depressive behavior. Mol. Psychiatry. 2019;25:1035–1049. doi: 10.1038/s41380-019-0371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J., Bhatti D.L., Lee K.W., Medrihan L., Cheng J., Wei J., Zhong P., Yan Z., Kooiker C., Song C. Ahnak scaffolds p11/Anxa2 complex and L-type voltage-gated calcium channel and modulates depressive behavior. Mol. Psychiatry. 2020;25:1035–1049. doi: 10.1038/s41380-019-0371-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019;38:e101379. doi: 10.15252/embj.2018101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I.H., You J.O., Ha K.S., Bae D.S., Suh P.G., Rhee S.G., Bae Y.S. AHNAK-mediated activation of phospholipase C-gamma1 through protein kinase C. J. Biol. Chem. 2004;279:26645–26653. doi: 10.1074/jbc.M311525200. [DOI] [PubMed] [Google Scholar]
- Lee I.H., Lim H.J., Yoon S., Seong J.K., Bae D.S., Rhee S.G., Bae Y.S. Ahnak protein activates protein kinase C (PKC) through dissociation of the PKC-protein phosphatase 2A complex. J. Biol. Chem. 2008;283:6312–6320. doi: 10.1074/jbc.M706878200. [DOI] [PubMed] [Google Scholar]
- Lee I.H., Sohn M., Lim H.J., Yoon S., Oh H., Shin S., Shin J.H., Oh S.H., Kim J., Lee D.K. Ahnak functions as a tumor suppressor via modulation of TGFβ/Smad signaling pathway. Oncogene. 2014;33:4675–4684. doi: 10.1038/onc.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaître C., Fischer B., Kalousi A., Hoffbeck A.S., Guirouilh-Barbat J., Shahar O.D., Genet D., Goldberg M., Betrand P., Lopez B. The nucleoporin 153, a novel factor in double-strand break repair and DNA damage response. Oncogene. 2012;31:4803–4809. doi: 10.1038/onc.2011.638. [DOI] [PubMed] [Google Scholar]
- Levine A.J., Hu W., Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- Lezaja A., Altmeyer M. Inherited DNA lesions determine G1 duration in the next cell cycle. Cell Cycle. 2018;17:24–32. doi: 10.1080/15384101.2017.1383578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H.J., Kang D.H., Lim J.M., Kang D.M., Seong J.K., Kang S.W., Bae Y.S. Function of Ahnak protein in aortic smooth muscle cell migration through Rac activation. Cardiovasc. Res. 2013;97:302–310. doi: 10.1093/cvr/cvs311. [DOI] [PubMed] [Google Scholar]
- Lukas C., Savic V., Bekker-Jensen S., Doil C., Neumann B., Pedersen R.S., Grøfte M., Chan K.L., Hickson I.D., Bartek J., Lukas J. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- Martín-Caballero J., Flores J.M., García-Palencia P., Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–6238. [PubMed] [Google Scholar]
- Meek D.W. The p53 response to DNA damage. DNA Repair (Amst.) 2004;3:1049–1056. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- Michelena J., Gatti M., Teloni F., Imhof R., Altmeyer M. Basal CHK1 activity safeguards its stability to maintain intrinsic S-phase checkpoint functions. J. Cell Biol. 2019;218:2865–2875. doi: 10.1083/jcb.201902085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirman Z., de Lange T. 53BP1: a DSB escort. Genes Dev. 2020;34:7–23. doi: 10.1101/gad.333237.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayans R., Andrais B., Scott A., Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J. Biomed. Biotechnol. 2012;2012:170325. doi: 10.1155/2012/170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol. 2011;498:399–406. doi: 10.1016/B978-0-12-385120-8.00017-6. [DOI] [PubMed] [Google Scholar]
- Morales J.C., Franco S., Murphy M.M., Bassing C.H., Mills K.D., Adams M.M., Walsh N.C., Manis J.P., Rassidakis G.Z., Alt F.W., Carpenter P.B. 53BP1 and p53 synergize to suppress genomic instability and lymphomagenesis. Proc. Natl. Acad. Sci. U S A. 2006;103:3310–3315. doi: 10.1073/pnas.0511259103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser J., Miller I., Carter D., Spencer S.L. Control of the restriction point by Rb and p21. Proc. Natl. Acad. Sci. U S A. 2018;115:E8219–E8227. doi: 10.1073/pnas.1722446115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Pankotai T., Bonhomme C., Chen D., Soutoglou E. DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 2012;19:276–282. doi: 10.1038/nsmb.2224. [DOI] [PubMed] [Google Scholar]
- Pessina F., Giavazzi F., Yin Y., Gioia U., Vitelli V., Galbiati A., Barozzi S., Garre M., Oldani A., Flaus A. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat Cell Biol. 2019;21:1286–1299. doi: 10.1038/s41556-019-0392-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E., Bishop J.M. The human gene AHNAK encodes a large phosphoprotein located primarily in the nucleus. J. Cell Biol. 1993;120:625–630. doi: 10.1083/jcb.120.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtivelman E., Cohen F.E., Bishop J.M. A human gene (AHNAK) encoding an unusually large protein with a 1.2-microns polyionic rod structure. Proc. Natl. Acad. Sci. U S A. 1992;89:5472–5476. doi: 10.1073/pnas.89.12.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simboeck E., Gutierrez A., Cozzuto L., Beringer M., Caizzi L., Keyes W.M., Di Croce L. DPY30 regulates pathways in cellular senescence through ID protein expression. EMBO J. 2013;32:2217–2230. doi: 10.1038/emboj.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto M., Raaijmakers J.A., Bakker B., Spierings D.C.J., Lansdorp P.M., Foijer F., Medema R.H. p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep. 2017;19:2423–2431. doi: 10.1016/j.celrep.2017.05.055. [DOI] [PubMed] [Google Scholar]
- Stiff T., Shtivelman E., Jeggo P., Kysela B. AHNAK interacts with the DNA ligase IV-XRCC4 complex and stimulates DNA ligase IV-mediated double-stranded ligation. DNA Repair (Amst.) 2004;3:245–256. doi: 10.1016/j.dnarep.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Sundaravinayagam D., Rahjouei A., Andreani M., Tupiņa D., Balasubramanian S., Saha T., Delgado-Benito V., Coralluzzo V., Daumke O., Di Virgilio M. 53BP1 supports immunoglobulin class switch recombination independently of its DNA double-strand break end protection function. Cell Rep. 2019;28:1389–1399.e6. doi: 10.1016/j.celrep.2019.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L.T., Vu B.T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Ward I.M., Difilippantonio S., Minn K., Mueller M.D., Molina J.R., Yu X., Frisk C.S., Ried T., Nussenzweig A., Chen J. 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Mol. Cell. Biol. 2005;25:10079–10086. doi: 10.1128/MCB.25.22.10079-10086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward I., Kim J.E., Minn K., Chini C.C., Mer G., Chen J. The tandem BRCT domain of 53BP1 is not required for its repair function. J. Biol. Chem. 2006;281:38472–38477. doi: 10.1074/jbc.M607577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgheib O., Pataky K., Brugger J., Halazonetis T.D. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell. Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D., Zaugg K., Mak T.W., Elledge S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell. 2006;126:529–542. doi: 10.1016/j.cell.2006.06.039. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Liu X., Huang R., Liu X., Liang Z., Liu T. Upregulation of nucleoprotein AHNAK is associated with poor outcome of pancreatic ductal adenocarcinoma prognosis via mediating epithelial-mesenchymal transition. J. Cancer. 2019;10:3860–3870. doi: 10.7150/jca.31291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z., Xiao S., Yuan X., Yuan J., Zhang C., Li H., Su J., Wang X., Liu Q. AHNAK as a prognosis factor suppresses the tumor progression in glioma. J. Cancer. 2017;8:2924–2932. doi: 10.7150/jca.20277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Zhang Y., Li Y., Hao X., Liu X., Wang Y. Azithromycin synergistically enhances anti-proliferative activity of vincristine in cervical and gastric cancer cells. Cancers (Basel) 2012;4:1318–1332. doi: 10.3390/cancers4041318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original code and data for complete RNaseq analysis from primary tumors is available at https://github.com/arpoe/GhodkeI_MolCell_2020. Original data have been deposited to Mendeley Data: https://dx.doi.org/10.17632/rrjnz8rwc8.1