

Abstract
Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder caused by a trinucleotide expansion in exon 1 of the huntingtin (HTT) gene. Cell death in HD occurs primarily in striatal medium spiny neurons (MSNs), but the involvement of specific MSN subtypes and of other striatal cell types remains poorly understood. To gain insight into cell type-specific disease processes, we studied the nuclear transcriptomes of 4524 cells from the striatum of a genetically precise knock-in mouse model of the HD mutation, HttQ175/+, and from wild-type controls. We used 14- to 15-month-old male mice, a time point at which multiple behavioral, neuroanatomical, and neurophysiological changes are present but at which there is no known cell death. Thousands of differentially expressed genes (DEGs) were distributed across most striatal cell types, including transcriptional changes in glial populations that are not apparent from RNA-seq of bulk tissue. Reconstruction of cell type-specific transcriptional networks revealed a striking pattern of bidirectional dysregulation for many cell type-specific genes. Typically, these genes were repressed in their primary cell type, yet de-repressed in other striatal cell types. Integration with existing epigenomic and transcriptomic data suggest that partial loss-of-function of the polycomb repressive complex 2 (PRC2) may underlie many of these transcriptional changes, leading to deficits in the maintenance of cell identity across virtually all cell types in the adult striatum.
SIGNIFICANCE STATEMENT Huntington's disease (HD) is a dominantly inherited neurodegenerative disorder characterized by specific loss of medium spiny neurons (MSNs) in the striatum, accompanied by more subtle changes in many other cell types. It is thought that changes in transcriptional regulation are an important underlying mechanism, but cell type-specific gene expression changes are not well understood, particularly at time points relevant to the onset of disease-related symptoms. Single-nucleus (sn)RNA-seq in a genetically precise mouse model enabled us to identify novel patterns of transcriptional dysregulation because of HD mutations, including bidirectional dysregulation of many cell type identity genes that may be driven by partial loss-of-function of the polycomb repressive complex (PRC). Identifying these regulators of transcriptional dysregulation in HD can be leveraged to design novel disease-modifying therapeutics.
Keywords: gene regulation, Huntington's disease, polycomb repressive complex 2, single-nucleus RNA-seq, striatum
Introduction
Huntington's disease (HD) is a fatal neurodegenerative disorder caused by dominant inheritance of trinucleotide repeat expansion mutations in the huntingtin (HTT) gene (MacDonald et al., 1993). Clinical symptoms include deficits in motor control and cognition, as well as psychiatric symptoms. Although the causal mutation has been known for >25 years, there are no existing treatments that dramatically alter disease progression. In the absence of treatment, symptoms progressively worsen, leading inevitably to death 10–15 years after the symptomatic age at onset.
An enduring mystery in HD biology is why HD mutations lead to selective neurodegeneration in specific subtypes of neurons, while other nearby cells remain largely spared, despite the fact that the HTT gene is robustly expressed in most or all cell types. HD progression is linked to the selective cell death of medium spiny neurons (MSNs) in the striatum (Vonsattel et al., 1985). Among MSNs, Drd2-expressing MSNs that project to the lateral segment of the globus pallidus (termed D2 MSNs) are thought to be more vulnerable than Drd1-expressing MSNs that project to the entopenduncular nucleus and the substantia nigra pars reticulata (termed D1 MSNs; Reiner et al., 1988). Striatal interneurons are less vulnerable than MSNs but may undergo disease-related changes that remain little studied (Zucker et al., 2005; Deng and Reiner, 2016). Neurodegeneration in HD is accompanied by neuroinflammatory processes. Microglial activation occurs before the clinical manifestation of the disease (Tai et al., 2007). Reactive astrogliosis occurs relatively late in the natural progression of HD pathophysiology but may still contribute to neurotoxicity (Liddelow et al., 2017; Diaz-Castro et al., 2019). Changes in forebrain white matter and striatal oligodendrocytes begin early in disease progression, including deficits in myelinated axon fibers and increased oligodendrocyte cell number (Myers et al., 1991; Bourbon-Teles et al., 2019). However, the regulation and impact of these changes in glial cell types remain poorly understood.
Transcriptional changes are among the earliest phenotypes in cells and tissues expressing mHTT and are highly reproducible in human HD (Hodges et al., 2006; Langfelder et al., 2016; Ament et al., 2017). Studies in bulk striatal tissue have shown that MSN-specific genes and components of synapses are downregulated, while upregulated genes include signatures of neuroinflammation (Seredenina and Luthi-Carter, 2012; Liddelow et al., 2017). Notably, there is evidence that some transcriptional changes are directly related to functions of HTT in the nucleus, including interactions of both wild-type and mutant HTT with transcriptional regulatory proteins (Zuccato et al., 2003; Seong et al., 2010; Ament et al., 2018). However, previous transcriptomic studies using bulk tissue failed to illuminate the cell type-specificity of many disease processes.
Single-nucleus RNA-seq (snRNA-seq) has emerged as a scalable technology enabling an unprecedented view of cell types and cell states in the mammalian brain. Several published studies have applied this approach to neurodegenerative diseases, but the regulation of cell type-specific molecular changes remains poorly characterized (Grubman et al., 2019; Mathys et al., 2019; Al-Dalahmah et al., 2020; Habib et al., 2020; Lee et al., 2020; Wertz et al., 2020). Here, we analyzed the nuclear transcriptomes of 4524 striatal cells from a genetically precise knock-in mouse model of a juvenile-onset HD mutation, HttQ175/+. The 14- to 15-month-old mice in our study are considerably older than those studied in previously published transcriptomic datasets from knock-in mouse models of the HD mutation, sampling a time point at which striatal atrophy is detectable, resembling early changes in human HD without yet any neuronal cell death (Deng et al., 2021). Our analyses of these data reveal numerous insights into cell type-specific disease processes.
Materials and Methods
Experimental design and statistical analysis
We generated snRNA-seq from 14- to 15-month-old male HttQ175/+ and Htt+/+ mice (n = 4 per genotype). Following rigorous quality control, statistical analyses were performed using the integrated, smoothed, and normalized unique molecular identifier (UMI) counts from 3014 cells in n = 4 HttQ175/+ mice and 1314 cells in n = 3 Htt+/+ mice. Cells were assigned to molecular cell types using clustering algorithms in the Seurat v3.0 R package (Butler et al., 2018). Proportions of cells assigned to each cell type in HttQ175/+ versus Htt+/+ mice were compared using generalized linear models with a binomial link. Differentially expressed genes (DEGs) in each cell type were identified using Wilcoxon signed-rank tests implemented in the Seurat R package. Gene co-expression networks were reconstructed by applying k-means clustering to an adjacency matrix derived from pairwise Pearson correlations among all pairs of genes. A gene regulatory network was derived using the GENIE3 R package (Huynh-Thu et al., 2010; Aibar et al., 2017), which implements a random forest regression approach. Three types of gene set enrichment analyses were performed to evaluate DEGs and network models. For comparisons between two categorical gene lists, we used hypergeometric tests. For comparisons between a ranked gene list and a categorical gene list, we used the geneSetTest() function in the limma R package (Smyth, 2005). For comparisons between two ranked gene lists, we used rank-rank hypergeometric overlap (RRHO), implemented in the RRHO R package (Plaisier et al., 2010). Details are provided below.
Mice
HttQ175/+ mice and wild-type littermate controls on the C57BL6/J background (B6J.129S1-Htttm1.1Mfc/190ChdiJ; JAX stock #029928) were bred and aged in the colony of the Carroll lab at Western Washington University. The B6J.zQ175DN KI used in this study also lack a floxed neo cassette 1.3 kbp upstream of exon 1 that is not known to have an effect on the phenotypic characteristics of the mice. Three 14-month-old and one 15-month-old male mice of each genotype were used to generate the primary dataset described here. Mice were deeply anesthetized using a phenobarbital-based euthanasia solution (Fatal Plus, Henry Schein Medical) and striatal tissue dissected on ice. Tissue was immediately flash frozen in liquid nitrogen and stored at −80°C. Experiments were performed following National Institutes of Health animal care guidelines and approved by Western Washington University's Institutional Animal Care and Use Committee under protocol 16-011.
Isolation of nuclei from frozen brain tissue
Nuclei were isolated from flash-frozen striatal tissue as described in previous protocols (Krishnaswami et al., 2016; Lake et al., 2017; Matson et al., 2018), with slight modifications. A detergent-mechanical cell lysis method was used, involving three major steps: lysis, homogenization, and density barrier centrifugation. In a laminar hood, a ∼50-mg piece of frozen brain tissue was placed into a prefrozen BioPulverizer (BioSpec) and smashed to a thin frozen layer, then immediately transferred to lysis buffer (250 mm sucrose, 25 mm KCl, 5 mm MgCl2, 1uM DTT, 1× RNase inhibitor, 0.1% Triton X-100, and 10 mm tricine buffer, pH 8.0). Tissue was disaggregated, flushing up and down, first using a 1 ml pipette tip, then with a 30-G needle in a 3-ml syringe. Homogenized tissue was diluted to 10 ml with lysis buffer and filtered through a 70-μm filter. Homogenate was spun at 1000 × g for 8 min at 4°C. Pellet was then re-suspended and 500 µl of pellet suspension was diluted with 500 µl 50% iodixanol solution (50% iodixanol, 250 mm sucrose, 150 mm KCl, 30 mm MgCl2, 1× RNase inhibitor, and 60 mm tricine buffer, pH 8.0), filtered for second time with a 70-μm mesh and placed on top of 500-µl layer of 29% iodixanol solution (29% iodixanol, 250 mm sucrose, 150 mm KCl, 30 mm MgCl2, 1× RNase inhibitor, and 60 mm tricine buffer, pH 8.0). Density barrier was centrifuged at 13,500 × g for 20 min at 4°C. The pellet was collected and washed with 10 ml of PBS, 2% BSA, 1× RNase inhibitor, centrifuged at 1000 × g for 8 min at 4°C, and finally suspended in 1 ml of PBS, 2% BSA, 1× RNase inhibitor and filtered using a 40-μm Flowmi cell strainer. Single nuclei suspensions were counted and evaluated for integrity using Propidium Iodide in a MoxiGo cytometer using 650-nm filter. Nuclei count was adjusted to 5000 nuclei/ml.
Library preparation and sequencing
A total of 13,000 nuclei were loaded into each well of a chromium microfluidics controller (10× Genomics) using PBS + 2% BSA. Sequencing libraries were generated using the Chromium Single Cell Gene Expression 3′ kit with version 3 chemistries. Samples were sequenced across two lanes of an Illumina HiSeq4000 Sequencer to obtain 75 base pair paired-end reads.
Cell quality control and data preprocessing
Raw sequencing reads were processed to counts of UMIs in each droplet with cellranger v3.0.2 (10× Genomics). Artifacts from ambient RNA were reduced with SoupX (Young and Behjati, 2018), based on a uniform contamination fraction estimate of 10%. This contamination estimate was derived using marker genes >20-fold enriched in each cell type from DropViz (Saunders et al., 2018). Also using SoupX, we derived the following sample-specific thresholds at which we observed relatively low contamination, and we removed cells outside these ranges: 1260–6300 UMIs/cell for BBY2, BCR2, BCR3, BCR4, BCR5 and BCR6, and 1260–4470 nUMIs for BCR1. One Htt+/+ sample, BBY3, was deemed to be an outlier based on very low UMI counts across most cells and was dropped from further analysis. In addition, using Seurat, we removed libraries with >5% of read counts from mitochondrial genes, as the presence of these non-nuclear transcripts indicates both incomplete fractionation of nuclei and cellular stress. To obtain the final set of cells for clustering, we manually removed any remaining doublets with high expression of mutually exclusive markers.
Normalization and batch effect correction
UMI counts from each cell were normalized to log (counts per million). A uniform set of highly-variable genes was selected by identifying the top 5000 most variable genes in each sample separately using the FindVariableFeatures() function in Seurat, then taking the intersect across samples, resulting in a final set of 937 highly-variable genes. Expression levels for these highly-variable genes were centered and scaled within each sample. Then, batch effects correction was performed across samples with MNN (Haghverdi et al., 2018), implemented with the mnnCorrect() function in the scran R package. MNN normalization is performed sequentially across samples and lacks a built-in function to optimize sample order. Therefore, 17 MNN iterations were performed using different sample ordering, and the best ordering was selected based on cell clustering results. In the final ordering, samples were in the order BBY2, BCR1, BCR2, BCR3, BCR4, BCR5, and BCR6.
Cell type clustering and labeling
MNN-adjusted counts from 5429 cells passing the initial QC were used for clustering, using the RunPCA(), FindNeighbors(), and FindClusters() functions in Seurat v3.0. The clusters were visualized using the RunTSNE() function in Seurat; npcs = 10 principal components, k = 30 shared nearest neighbors, and a Louvain clustering resolution of 0.4 were empirically determined to produce optimal clustering. Marker genes identified in each cluster using the FindAllMarkers() function were then used to assign a cell type label to each molecularly-defined cluster based on known marker genes for striatal cell types (https://dropviz.org). Three clusters were dropped from further analysis because they included cells from only one sample, were marked by high expression of mitochondrial transcripts, or expressed markers from mixed cell types, resulting in the final set of 4524 cells.
Identifying subtypes of MSNs presented a particular challenge, since the small number of markers distinguishing D1 versus D2 MSNs were dwarfed by the massive effects of HD mutations in these cells. Therefore, we performed subclustering of MSNs using 36 marker genes with >8-fold difference in expression between D1 versus D2 MSNs in the DropViz atlas (Saunders et al., 2018). Using the normalized counts from these 36 genes, we computed pairwise Spearman rank correlations among all canonical MSNs, and we performed average-distance hierarchical clustering using 1 – Spearman's ρ as a distance metric, revealing two major groups of cells with high expression of markers for D1 versus D2 MSNs, respectively. 87 MSNs were in smaller clusters that lacked high expression for either subtype and were discarded.
Similarly, clusters of reactive versus non-reactive astrocytes were derived by subclustering with 87 markers of reactive and non-reactive subtypes (Zamanian et al., 2012). In this case, subclustering was performed in Seurat v3, using the top 5 principal components and a Louvain modularity resolution of 0.5.
Smoothing of read counts
For the purpose of assessing gene expression differences between genotypes, and subsequent analyses of pseudotime trajectories and gene co-expression clustering, we smoothed read counts to reduce dropout effects and improve gene-gene correlation structure. Smoothing was performed with knn-smoothing (Wagner et al., 2018), using k = 15 neighbors and 30 PCs. Smoothed counts were normalized using the Seurat NormalizeData() function for downstream analyses.
Cell type-specific DEGs
We identified cell type-specific DEGs in HttQ175/+ versus wild-type mice by two methods. In our primary analysis, we compared cells of each genotype using Wilcoxon Rank Sum tests implemented with the Seurat FindMarkers() function, testing all genes with non-zero imputed counts in at least 10% of the cells from at least one of the two genotypes. This analysis treats the cell (not the mouse) as the primary unit of analysis. We believe this choice is justified, as both the neurodegenerative processes in MSNs and the activation of inflammatory states in glial cells are thought to occur in a relatively cell-autonomous fashion, resulting in a mosaic of cell states within each mouse that would not be captured in an analysis treating the mouse as the primary unit of analysis. To confirm that DEGs detected by our primary approach were robust, we also conducted a secondary analysis in which the Seurat FindMarkers() function was applied to non-smoothed read counts.
Trajectory analysis
We used Seurat v3.0 to perform principal components analysis on the centered and scaled expression levels of the 5000 most variable genes within each cell type. We eliminated any principal component for which the strongest loadings were dominated by mitochondrial genes, as these vectors typically correspond to technical variation among cells rather than biological signal. Using the embeddings of the top five remaining principal components, we then identified a non-branching pseudotime trajectory with the slingshot() function in the Slingshot v1.4.0 R package (Street et al., 2018). We tested for associations of slingPseudoTime and genotype using the t.test() function in R.
Gene set enrichment analysis
DEGs in each cell type and genes in each gene co-expression module were tested for enrichment in curated gene sets from four sources. First, we tested for enrichments in 2368 curated gene sets from the HD molecular Signatures Database (https://www.hdinhd.org/2018/05/22/hdsigdb/), including lists of DEGs and weighted gene co-expression network analysis (WGCNA) modules from RNA-seq experiments, as well as known marker genes for striatal cell types. Second, we tested for enrichments in 12,177 Gene Ontology terms from the org.Mm.egGO2ALLEGS object in the org.Mm.eg.db R package. Third, we tested for enrichments in transcription factor regulons from ChIP-seq experiments in the ChEA 2016 database (Lachmann et al., 2010), downloaded from the Enrichr website (https://amp.pharm.mssm.edu/Enrichr/geneSetLibrary?mode=text&libraryName=ChEA_2016). Fourth, we tested for enrichments with evolutionarily conserved sequence motifs from RcisTarget (Aibar et al., 2017). For the first three sources, enrichments among downregulated and upregulated genes were tested separately using the geneSetTest() function in the limma R package with ranks.only = TRUE and type = “t” and using genes ranked from most strongly downregulated to most strongly upregulated based on the -log10(p value) multiplied by the sign of the log2(fold change). Motif analysis was performed with RcisTarget using default parameters.
Statistical overlap of cell type-specific DEGs across cell types
Statistical overlap among ranked lists of downregulated and upregulated genes in each pair of cell types was evaluated using RRHO, implemented using the RRHO() function from the RRHO R package (Plaisier et al., 2010). The RRHO algorithm steps through two gene lists ranked by the degree of differential expression in two independent experiments, successively measuring the statistical significance of the number of overlapping genes. Each comparison between cell types used the set of genes expressed with a non-zero read count in at least 10% of the cells from both cell types, with the genes ranked from most strongly downregulated to most strongly upregulated within each cell type based on the -log10(p value) multiplied by the sign of the log2(fold change). We used a step size such that each gene list was divided into 100 equally sized bins.
Gene co-expression modules
Gene-gene correlation structure in snRNA-seq is notoriously weak because of the sparsity of the data. Therefore, for network analyses we used smoothed counts, as described above, to impute a more complete representation of gene expression in each cell. We selected 8971 genes with non-zero counts in at least 10% of the cells from any cell type. We applied three distinct clustering algorithms to the smoothed, log-normalized, centered and scaled counts to derive gene modules and compared the results.
WGCNA
WGCNA was performed using the blockwiseModules() function from the WGCNA R package (Langfelder and Horvath, 2008), with the following parameters: maxBlockSize = 10,000, corType = “pearson,” power = 8, TOMType = “none,” minModuleSize = 15, mergeCutHeight = 0.15, minKMEtoStay = 0.1. This implementation of WGCNA assigns genes to modules by hierarchical clustering of an adjacency matrix defined by Pearson correlations.
K-means
We applied the kmeans() function to smoothed read counts with k = 50, algorithm = “Lloyd,” max iterations = 10,000, and 10 starts. We removed genes with Pearson's r < 0.3 to the module centroid. We removed modules with fewer than 10 remaining genes and merged modules whose average expression was strongly correlated with r > 0.85, resulting in a final network with 5647 genes in 42 modules.
K-means “hybrid”
Sparseness is a challenge in clustering single-cell RNA sequencing data, resulting in lost information and biases in network reconstruction. One solution to this problem is to cluster genes based on metrics that are proportional to the number of neighbors that a pair of nodes share in common, since this may be more robust to dropouts. For instance, Seurat assigns cells to clusters by clustering a shared nearest neighbors graph (Butler et al., 2018). Topological overlap metrics are implemented for gene co-expression clustering in WGCNA (Langfelder and Horvath, 2008), but empirically we have found that WGCNA's topological overlap matrices yield very few modules. As an alternate approach to cluster genes based on their network neighborhood, we computed Pearson correlations among all pairs of genes, then applied k-means clustering to the resulting adjacency matrix to derive gene modules. k = 150 was manually determined to capture some of the finer structure in the network without over-splitting. Each module's eigengene, the first principal component, was computed with the moduleEigengenes() function in the WGCNA R package. We dropped modules for which the eigengene explained <10% of the variance, and we merged modules whose eigengenes were >85% correlated. For comparison, we also summarized modules by the average expression of the component genes, obtaining very similar results. These procedures resulted in 77 modules spanning 5874 genes.
Gene regulatory network
A gene regulatory network model describing predicted interactions between TFs and their potential target genes in the mouse striatum was derived using GENIE3 (Huynh-Thu et al., 2010). The GENIE3 algorithm performs network reconstruction by using a random forest regression model to select sets of TFs whose combined expression predicts the expression of each gene. We started with the smoothed counts of 8971 genes with non-zero counts in at least 10% of the cells from at least one cell type, as for gene co-expression networks above. We downloaded a curated list of all 1639 known and likely human TFs from http://humantfs.ccbr.utoronto.ca/download/v_1.01/TFs_Ensembl_v_1.01.txt. We identified 1373 mouse orthologs of these TFs using the biomaRt R package (accessed April 21, 2020). We then intersected this list of putative mouse TFs with the 8971 striatally-expressed genes from our dataset, producing a list of 589 striatally-expressed TFs. GENIE3 was run using default parameters as implemented in the GENIE3 BioConductor package v1.1. The algorithm produces a very long list of potential TF-gene interactions ranked by the randomForest importance score, and many of these potential interactions are very weak. This importance score is agnostic to whether the TF-gene interaction is positive (“activating”) or negative (“inhibitory”), but it has been suggested that inhibitory interactions are less reliable (Aibar et al., 2017). We therefore retained only the predicted interactions between pairs of genes whose expression were positively correlated (Pearson's r > 0), and we trimmed this list to the top remaining 180,000 TF-gene interactions with the strongest importance scores, corresponding to a mean in-degree of ∼20 TFs per gene.
Overlap with polycomb repressive complex 2 (PRC2)-related datasets
We analyzed PRC2 target genes derived from nine ChIP-seq datasets in four cell types to test for over-representation in gene co-expression modules. When available, we used published target gene lists. Otherwise, we downloaded aligned sequence reads, performed peak-calling with MACS v2.1 (Zhang et al., 2008), and annotated peaks to genes with transcription start sites within ±5 kb. The nine datasets are as follows: (1) ChIP-seq of EZH2 in mouse embryonic stem cells (Kloet et al., 2016), obtained from ChEA (Lachmann et al., 2010); (2) ChIP-seq of SUZ12 in mouse embryonic stem cells (Kloet et al., 2016), obtained from ChEA (Lachmann et al., 2010); (3) ChIP-seq of H3K27me3 in mouse embryonic stem cells, generated by Bing Ren's lab (UCSD) for the ENCODE consortium (ENCFF055QNY); (4) ChIP-seq of H3K27me3 in mouse MSNs (Von Schimmelmann et al., 2016), obtained from HDSigDB; (5) ChIP-seq of H3K27me3 in bulk striatal tissue from four-month-old HttQ111/+ and Htt+/+ mice (Pearl et al., 2020); (6) ChIP-seq of EZH2 in human astrocytes, generated by Bradley Bernstein's lab for the ENCODE consortium (reproducible peaks from ENCFF254DFD and ENCFF831JFC); (7) ChIP-seq of H3K27me3 in human astrocytes, generated by Bradley Bernstein's lab for the ENCODE consortium (ENCFF315BVX); (8) ChIP-seq of EZH2 in mouse corpus callosum (enriched for oligodendrocytes; Bardile et al., 2019; their Table S3); (9) ChIP-seq of EZH2 in mouse corpus callosum (enriched for oligodendrocytes; Bardile et al., 2019; their Table S3). We tested for over-representation in gene co-expression modules using Fisher's exact tests. We also analyzed published microarray gene expression profiles (Affymetrix 430_2 array) of MSNs from six-week-old, three-month-old, and six-month-old Ezh1−/−; Ezh2fl/fl; Camk2a-cre versus control mice, in which PRC2 had been conditionally silenced in adult MSNs (Von Schimmelmann et al., 2016). Normalized data were downloaded from the Gene Expression Omnibus (GSE84243). We fit a linear model using the lmFit() function in the limma R package, followed by post hoc contrasts contrasts.fit() and eBayes() to estimate the effect of genotype at each time point. Over-representation analysis of gene co-expression modules among upregulated and downregulated genes was performed with the geneSetTest() function.
Replication in independent datasets
We downloaded snRNA-seq read counts and sample-level metadata from six-month-old HttQ175/+ and HttQ20/+ and from HD cases and controls from Lee et al., 2020 (GSE152058). We selected cells with >1000 UMIs and applied uniform preprocessing to each dataset, including ambient RNA reduction with SoupX, followed by integration and clustering with the Seurat Standard Workflow (Butler et al., 2018). Cell clusters were assigned to cell types based on known markers, as described above. Markers of MSNs in the matrix versus patch compartments in mouse striatum included Ebf1 and Oprm1 (Lobo et al., 2008; Märtin et al., 2019). We downloaded read counts from translatome profiling of striatal D1 MSNs, D2 MSNs, astrocytes, and Chat+ interneurons in six-month-old mice from an HD allelic series (GSE152058; Lee et al., 2020). Read counts were converted to log2(transcripts per million) using the cpm() in the edgeR R package (Robinson et al., 2010). We downloaded normalized read counts (FPKM) from mRNA-seq of bulk striatal tissue in an HD allelic series at 2, 6, and 10 months (GSE65774; Langfelder et al., 2016). In each dataset, we calculated the activity of the 10 PRC2-regulated gene modules using the module Eigengenes() function from the WGCNA R package (Langfelder and Horvath, 2008). Effects of HD genotype on each module in each cell type were calculated in each cell type or type point separately using the lmFit() and eBayes() functions in the limma R package. For allelic series studies, we treated Q-length as a quantitative variable.
Results
snRNA-seq of 14- to 15-month-old HttQ175/+ HD knock-in mice and wild-type controls
We generated snRNA-seq from the striatum of four male 14- to 15-month-old HttQ175/+ mice and four male wild-type controls using the 10× Genomics Chromium system. HttQ175/+ is a widely used genetically precise mouse model for a mutation associated with juvenile-onset HD. A humanized HTT exon 1 fragment with 140 CAG repeats was knocked into the endogenous Htt locus. The repeats spontaneously expanded to ∼175 CAG repeats and were later stabilized at ∼190 repeats. These mice have normal lifespan, with progressive behavioral, neuroanatomical, and transcriptomic deficits (Smith et al., 2014; Alexandrov et al., 2016; Langfelder et al., 2016). At 14- to 15-month-old, striatal atrophy is detectable, resembling early changes in human HD without yet any neuronal cell death (Deng et al., 2021). The mice in our study are considerably older than those studied in previously published transcriptomic datasets from knock-in mouse models of the HD mutation, which have often focused on the earliest signs of transcriptional dysregulation in two- to six-month-old mice (Langfelder et al., 2016; Ament et al., 2017; Lee et al., 2020). However, even at 18 months, striatal pathology in HttQ175/+ is less severe than in premanifest HD (Deng et al., 2021), suggesting a need for studies of aging HttQ175/+ mice to adequately model the pathologic processes leading to the onset of overt symptoms.
Following QC and normalization, we analyzed 4524 high-quality cells, of which 3210 were derived from HttQ175/+ mice and 1314 from wild-type mice. A web portal for visualization and analysis of these snRNA-seq data are available at the Gene Expression Analysis Resource (Orvis et al., 2020; https://umgear.org/p?l=1d76bf3e&g=Penk). Louvain clustering and annotation with known marker genes (Saunders et al., 2018) revealed well-defined clusters corresponding to each of the major cell populations in the striatum, including 3003 MSNs, 288 Sst+ interneurons, 120 Pvalb+ interneurons, 73 Chat+ interneurons, 468 oligodendrocytes, 300 astrocytes, 112 endothelial cells, 82 microglia, and 78 polydendrocytes (Fig. 1A,B; Extended Data Fig. 1-1). Subclustering of MSNs using 36 genes with >8-fold differences in expression between MSN subtypes in prior scRNAseq of mouse striatum (Saunders et al., 2018) revealed 1809 D1 MSNs and 941 D2 MSNs (Fig. 1C), as well as 166 MSNs whose expression profiles mach the recently described “eccentric” subtype (Saunders et al., 2018). Most cell types were represented at similar proportions in HttQ175/+ versus wild-type mice (Fig. 1D). Notably, however, ∼30% fewer cells in HttQ175/+ mice were identified as D2 MSNs (generalized linear model: p = 1.6e-6), while a higher proportion of cells were identified as microglia (p = 9.6e-3) and Chat+ interneurons (p = 3.3e-4). Our results in D2 MSNs are consistent with immunostaining of neuronal subtypes in HttQ175/+ versus Htt+/+ mice, which revealed a progressive decrease in the number of neurons expressing the D2 MSN marker enkephalin beginning as early as six months of age, while no changes in cell counts were observed for markers of D1 MSNs or interneurons (Deng et al., 2021). However, stereological experiments in the same study failed to detect a decrease in the overall number of neurons. Therefore, a conservative interpretation of the decreased numbers of cells identified as D2 MSNs both by snRNA-seq and immunostaining is that these patterns represent a loss of cell identity marker genes rather than loss of neurons per se
Figure 1.

Identification of cell types in snRNA-seq of 4524 cells from the striatum of 14- to 15-month-old HttQ175/+ versus Htt+/+ mice. A, tSNE plot showing clustering of nuclei into major cell types. B, Dot plot of top marker genes for all cell types. C, Dot plot showing marker genes for D1 versus D2 MSN subtypes. D, Proportion of cells from HttQ175/+ versus Htt+/+ mice in each cell type. Generalized linear model: ***p < 0.001, **p < 0.01.
Cell type marker genes and DEGs in nine striatal cell types from 14- to 15-month-old HttQ175/+ versus Htt+/+ mice. Tab 1, Marker genes in each cell type. Tab 2, Gene expression changes in each cell type based on smoothed counts. Tab 3, Gene expression changes in each cell type based on non-smoothed counts. Tabs show all genes in each analysis, which included in each cell type that set of genes with non-zero counts in at least 10% of cells (Excel file). Download Figure 1-1, XLSX file (12.9MB, xlsx) .
Cell type-specific gene expression changes in HD knock-in mice
Next, we studied cell type-specific gene expression changes in HttQ175/+ versus wild-type mice. We identified 13,897 cell type-specific gene expression changes, involving 8124 distinct genes [DEGs; false discovery rate (FDR) < 0.05; Fig. 2A; Extended Data Fig. 1-1]. In our primary analysis, we detected DEGs by applying Wilcoxon rank-sum tests to smoothed read counts. A second approach, applying Wilcoxon signed-rank tests to non-smoothed read counts yielded a similar rank-ordering of DEGs but with reduced statistical power. Microglia and Chat+ interneurons were excluded from this analysis because of insufficient cell numbers.
Figure 2.

Characterization of DEGs in nine cell types from the striatum of 14- to 15-month-old HttQ175/+ versus Htt+/+ mice. A, Counts of DEGs in each cell type (red = upregulated; blue = downregulated; saturated color, FDR < 0.05; desaturated color, p < 0.01). B, Statistical overlap of upregulated and downregulated genes with published RNA-seq of bulk striatal tissue from 10-month-old HttQ175/+ versus HttG20/+ mice and from HttQ140/+ versus HttG20/+ mice. C, Scatterplots and RRHO heatmaps indicating shared and unique gene expression changes in selected pairs of cell types. D, Slingshot pseudotime trajectory analysis in MSN subtypes, astrocytes, oligodendroocytes, and SST interneurons. E, Statistical overlap of upregulated and downregulated genes among pairs of cell types.
Gene set enrichment analysis of cell type-specific DEGs. Enrichments in gene sets from Gene Ontology (Tab 1) and HDSigDB (Tab 2), and for the predicted target genes of each striatally expressed transcription factor in our GENIE3 gene regulatory network model (Tab 3; Excel file). Download Figure 2-1, XLSX file (1.6MB, xlsx) .
We found 5181, 3666, and 685 DEGs in D1, D2, and eccentric MSNs, respectively. Comparison of these cell type-specific DEGs to previously described lists of DEGs from RNA-seq of bulk striatal tissue from 10-month-old HttQ175/+ mice versus HttQ20/+ controls (Langfelder et al., 2016) indicated that both upregulated and downregulated DEGs in all MSN subtypes were strongly enriched for known DEGs from bulk tissue RNA-seq (limma geneSetTest: p < 1e-100 for DEGs in D1 and D2 MSNs; p < 1e-30 in eccentric MSNs; Fig. 2B; Extended Data Fig. 2-1). Genes with the lowest p values included Pde10a, Rgs9, Wnt8b, Trank1, Scn4b, Rap1gap, Pde1b, Ptpn5, Adcy5, Atp2b1, and Arpp21, all of which are also among the strongest and most-consistently observed DEGs from previous studies in bulk tissue. Downregulated genes in MSNs were enriched for synaptic functions (e.g., the Gene Ontology term “neuron spine,” p = 7.95e-7, p = 1.95e-8, and p = 8.1e-4 in D1, D2, and eccentric MSNs, respectively; Extended Data Fig. 2-1). Upregulated genes in MSNs were enriched for genes localized to the “nucleus” (p = 4.1e-11, p = 2.0e-10, and p = 0.014 in D1, D2, and eccentric MSNs, respectively), especially genes related to “histone modification” (p = 3.8e-4, p = 8.8e-4, and p = 3.1e-3 in D1, D2, and eccentric MSNs, respectively). Comparing the fold changes of DEGs in D1, D2, and eccentric MSNs revealed that with very few exceptions these fold changes were nearly identical in magnitude and direction [Pearson correlation comparing the log2(fold changes) of the top 15% of genes ranked by p value in D1 vs D2 MSNs, r = 0.97, p < 1e-308; D1 vs eccentric MSNs, r = 0.77, p = 3.0e-242; Fig. 2C, left]. The slope of the regression line is ∼1 in all of these comparisons among MSN subtypes. Down-sampling analyses suggested that the somewhat weaker correlation coefficient in D1 versus eccentric MSNs was primarily because of the smaller eccentric MSN sample size and is not biologically meaningful. Pseudotime trajectory analysis with Slingshot (Street et al., 2018) indicated that nearly all MSNs in these 14- to 15-month-old HttQ175/+ mice exist in a disease-specific transcriptional state that is never observed in wild-type mice (Fig. 2D). Thus, snRNA-seq revealed strong transcriptional effects of the HD mutation in all three MSN subtypes, including eccentric MSNs, yet the enhanced vulnerability of D2 versus D1 MSNs is not reflected in the magnitude of transcriptional effects, at least not in the current dataset.
We found 2351 DEGs in oligodendrocytes from HttQ175/+ versus Htt+/+mice. Neither upregulated nor downregulated DEGs in oligodendrocytes strongly overlapped known DEGs from bulk tissue RNA-seq of 10-month-old HttQ175/+ versus HttQ20/+ mice (p = 0.01 and p = 0.06, respectively). By contrast, our DEGs in oligodendrocytes strongly overlapped DEGs from RNA-seq of sorted oligodendrocytes in the striatum of BACHD mice – a transgenic mouse model of the HD mutation – compared with wild-type controls (Bardile et al., 2019; p = 6.6e-19 and p = 1.8e-13 for downregulated and upregulated DEGs, respectively). Downregulated DEGs in oligodendrocytes were strongly enriched for oligodendrocyte-specific functions such as “myelin sheath” (p = 6.75e-14), as well as more basic cellular processes such as “structural constituent of ribosome” (p = 8.64e-12) and “mitochondrion” (p = 3.16e-10). Upregulated DEGs were enriched for several categories of genes that are typically associated with neurons, including “ion channel complex” (p = 2.0e-8) and “synaptic membrane” (p = 1.2e-6). Trajectory analysis of oligodendrocytes suggested that oligodendrocytes exist on a continuum from normal to disease-associated states, i.e., in contrast to the discrete disease-associated transcriptional states of MSNs, the disease-associated transcriptional states in oligodendrocytes are also present in Htt+/+ mice, but at a lower frequency (Fig. 2D). In summary, we identified thousands of reproducible DEGs in oligodendrocytes that are obscured in bulk tissue RNA-seq.
We found 1987 DEGs in astrocytes from HttQ175/+ versus Htt+/+mice. The upregulated genes in astrocytes overlapped known upregulated DEGs from bulk tissue RNA-seq of 10-month-old HttQ175/+ versus HttQ20/+ mice (p = 2.3e-12), whereas downregulated gene sets in astrocytes did not significantly overlap these known DEGs (p > 0.05). Prior work has revealed neurotoxic reactive astrocytes in postmortem striatal brain tissue from HD patients, but their presence in mouse models of the HD mutation is controversial (Liddelow et al., 2017; Diaz-Castro et al., 2019). To identify reactive astrocytes in our dataset, we subclustered astrocytes on the basis of 87 genes previously shown to be induced in reactive astrocytes (Zamanian et al., 2012), revealing a cluster of 134 reactive astrocytes, enriched for pan-reactive markers such as Gfap (p = 3.6e-14) and Vim (p = 1.2e-9), as well as two clusters of non-reactive astrocytes that do not express these markers (Fig. 3). While reactive astrocytes were present in both genotypes, they were significantly more abundant in HttQ175/+ mice (Fisher's exact test: OR = 2.3; p = 0.001). Many reactive astrocytes expressed markers of the “A1” neurotoxic subtype (e.g., H2-D1, p = 1.2e-8), whereas very few cells expressed markers of “A2” neuroprotective reactive astrocytes. While these results support the presence of neurotoxic reactive astrocytes in the striatum of HttQ175/+ mice aged more than one year, several analyses suggest that prototypical reactive astrogliosis explains only a subset of the HD-related transcriptional changes in astrocytes. Trajectory analyses showed a strong shift in astrocyte cell states in HttQ175/+ versus Htt+/+mice (Fig. 2D), but this trajectory was only weakly correlated with reactive versus non-reactive subtypes (Fig. 3H). Instead, upregulated DEGs in astrocytes were most strongly enriched for the GO term “synapse” (p = 8.1e-32), while downregulated DEGs were most strongly enriched for GO terms related to transcriptional regulation (e.g., “negative regulation of transcription by RNA polymerase II,” p = 2.7e-9). Thus, there are profound changes in the transcriptomes of astrocytes from HttQ175/+ versus Htt+/+mice, only some of which reflect known neuroinflammatory processes.
Figure 3.

Characterization of reactive astrocytes in the striatum of HttQ175/+ versus Htt+/+ mice. Reactive astrocytes were identified by subclustering astrocytes on the expression of 87 markers of reactive populations. A–D, Expression of marker genes for all astrocytes (Slc1a3), pan-reactive astrocytes (Gfap), A1 reactive astrocytes (H2-D1), and A2 reactive astrocytes (Tm4sf1). E, Assignments of astrocyte subclusters as reactive versus non-reactive based on known markers. F, Distribution of cells from HttQ175/+ versus Htt+/+ mice across astrocyte subclusters. G, Proportion of reactive versus non-reactive astrocytes in HttQ175/+ versus Htt+/+ mice. H, Violin plots showing the relationship between the reactive state and genotype of astrocytes and Slingshot pseudotime.
At an FDR < 0.05, we detected fewer than 10 DEGs in Sst+ and Pvalb+ interneurons, endothelial cells, and polydendrocytes (Fig. 2A). Moreover, trajectory analysis indicated that the principal curve in these cell types was not correlated with genotype (p > 0.05). We note that although these cell types are relatively rare, we were able to detect hundreds of DEGs in comparably rare eccentric MSNs. Therefore, our data indicate that these cell types are less vulnerable to the transcriptional effects of the HD mutation.
Comparisons of gene expression changes across cell types detected a marked difference in cell type-specificity of downregulated versus upregulated genes. Downregulated genes were largely non-overlapping across cell types, with the only strong overlaps occurring among MSN subtypes (Fig. 2E). Many of these downregulated DEGs were “cell identity” genes that are expressed specifically in that same cell type. That is, top genes downregulated in MSNs included MSN marker genes such as Ppp1r1b, Pde10a, and Rgs9. Genes downregulated in astrocytes were enriched for astrocyte marker genes (astrocyte marker genes (p = 1.11e-212), including Hes5, Gjb6, and Ddhd1. And genes downregulated in oligodendrocytes were enriched for oligodendrocyte-specific genes (p = 3.32e-155), including Mog, Gjb1, and Cldn11.
By contrast, many upregulated DEGs were shared across cell types, with statistically significant overlap among upregulated DEGs in D1 MSNs, D2 MSNs, eccentric MSNs, astrocytes, oligodendrocytes, endothelial cells, polydendrocytes, and Sst+ interneurons (Fig. 2C). Thirty genes were upregulated (FDR < 0.05) in five distinct cell types. These included inflammation-related genes such as colony stimulating factor 2 receptor subunit α (Csf2ra), histocompatibility 2, D region locus 1 (H2-D1), and myocardial infarction associated transcript (Miat), suggesting that some shared changes are because of the broadly acting effects of pro-inflammatory molecules such as cytokines. However, broadly upregulated genes also included genes that are not typically associated with inflammation, including synaptic genes like the GABAA receptor α 1 subunit (Gabra1) and the voltage-gated sodium channel α 9 subunit (Scn9a). Thus, upregulation of certain transcripts across multiple striatal cell types is a prominent feature of gene expression changes in HttQ175/+ versus Htt+/+mice, involving both neuroinflammation-related and non-neuroinflammation-related genes.
Network analyses reveal principles of transcriptional dysregulation
We reconstructed and analyzed gene co-expression networks to gain deeper insight into the processes driving transcriptional dysregulation within and across cell types. Gene co-expression networks are widely employed in RNA-seq with bulk tissue, but the sparseness of scRNA-seq data masks gene-gene correlation structure (Crow et al., 2016). To overcome this issue, we used knn-smoothing (Wagner et al., 2018) to impute read counts across cells (k = 15, n = 30 principal components). We confirmed that this approach produced strong correlations among known markers of D1 MSNs and among known markers of D2 MSNs, without inducing spurious correlations among markers across subtypes (Fig. 4). We compared three approaches to compute gene co-expression modules using these smoothed read counts: WGCNA (Langfelder and Horvath, 2008), k-means clustering (k = 50; Pearl et al., 2019), and a novel approach in which we applied k-means clustering to a gene-gene correlation matrix to cluster genes based on shared interactions partners (henceforth “k-means hybrid”; details in Materials and Methods). After merging highly similar modules (r > 0.85), these approaches identified 26, 42, and 77 modules, respectively (Extended Data Fig. 4-1).
Figure 4.

Smoothing rescues gene-gene correlation structure among markers of D1 versus D2 MSNs. A, Pearson correlations among 36 marker genes for D1 versus D2 MSNs in normalized, non-smoothed counts from the 4524 cells in our dataset. B, Pearson correlations among the same 36 marker genes using smoothed counts. The 36 marker genes are the same genes shown in Figure 1C and are ranked in order of subtype specificity based on data from the DropViz atlas.
Gene co-expression modules. Tab 1, Modules derived by the k-means hybrid method. Tab 2, Modules derived by WGCNA. Tab 3, Modules derived by k-means (k = 50). Download Figure 4-1, XLSX file (88.3KB, xlsx) .
Five analyses support the relevance of these gene co-expression modules to gene regulation and biology and indicate that module detection is robust to the network reconstruction approach (Fig. 5; Extended Data Fig. 5-1). First, gene regulatory network reconstruction with GENIE3 (Huynh-Thu et al., 2010) using the smoothed expression profiles from the same cells and genes revealed TF-target gene regulons that significantly overlapped (p < 0.001) every gene co-expression module from each of the three network models, predicting specific TFs as key regulators of their activity (Fig. 5A). Second, most modules overlapped direct target genes of TFs inferred by ChIP-seq in the ChEA 2016 database (Fig. 5B; Lachmann et al., 2010). Third, most modules were enriched for at least one Gene Ontology functional annotation (Fig. 5C). Fourth, most modules overlapped a published gene co-expression module from bulk RNA-seq of striatal tissue in knock-in mouse models of the HD mutation (Fig. 5D; Langfelder et al., 2016). Fifth, modules were highly reproducible across the three network models, with every module significantly overlapping a cognate module in the other network models (Fig. 5E). We describe subsequent analyses using the 77 modules from the k-means hybrid model, which had the largest number of modules validated in the analyses above. Similar results were obtained using the alternate network models.
Figure 5.
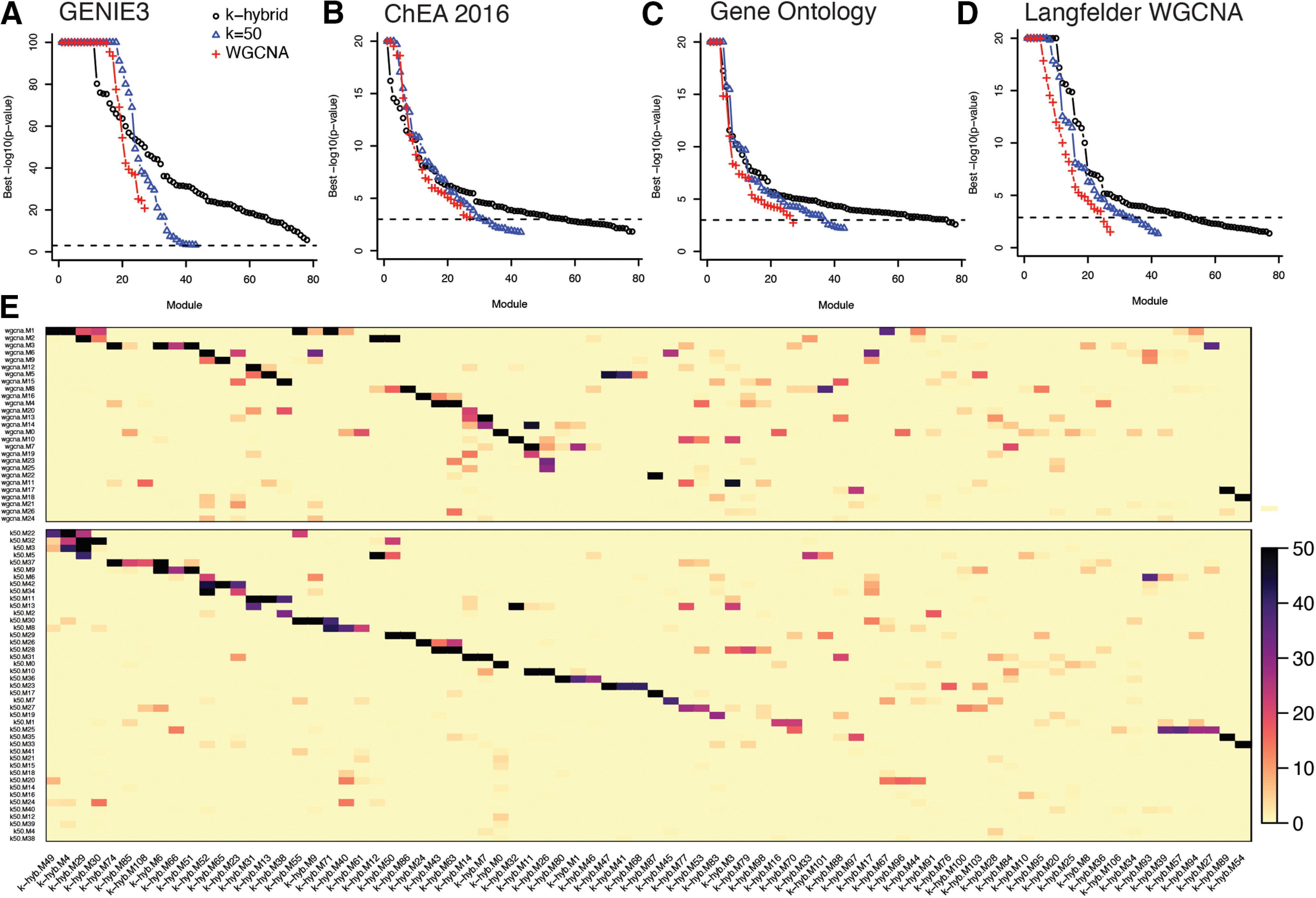
Validation and comparison of gene co-expression modules. A, Enrichment of gene co-expression modules for TF-target regulons derived with GENIE3 (Huynh-Thu et al., 2010). B, Enrichment of gene co-expression modules for TF-target regulons from ChIP-seq experiments in the ChEA 2016 database (Lachmann et al., 2010). C, Enrichment of gene co-expression modules for Gene Ontology terms. D, Enrichment of gene co-expression modules for published gene co-expression modules derived bulk RNA seq of HTT knock-in mice (Langfelder et al., 2016). The points in A–D indicate the -log10(p value) for the most significant regulon or GO term for each module, with the modules ranked from strongest to weakest enrichment. E, Overlap between gene co-expression modules derived with WGCNA, k-means (k = 50), and the k-means hybrid approach.
Gene set enrichment analysis of gene co-expression modules. Tabs 1–6, Enrichments for k-means hybrid modules. Tab 1, Enrichment for Gene Ontology terms. Tab 2, HDSigDB terms. Tab 3, WGCNA modules from Langfelder et al. (2016; subset of HDSigDB). Tab 4, Predicted target genes of TFs based on ChIP-seq experiments in ChEA. Tab 5, Predicted target genes of striatally expressed TFs in our GENIE3 gene regulatory network model. Tab 6, Evolutionarily conserved cis-motifs from RcisTarget. Tab 7, Enrichments in modules derived by WGCNA. Tab 8, Enrichments in modules derived by k-means clustering, k = 50. Download Figure 5-1, XLSX file (8.8MB, xlsx) .
Next, we characterized the activity of the 77 gene co-expression modules across cell types and genotypes. Our primary analyses summarized each module's expression pattern based on its eigengene (Fig. 6). Similar results were obtained by summarizing the modules' expression based on the average expression of their component genes (Extended Data Fig. 6-1). Many modules were expressed specifically in a single cell type (Fig. 6B). Notably, gene co-expression modules derived from snRNA-seq appeared to have greater fidelity to specific cell types than the published network derived from bulk RNA-seq (Extended Data Fig. 5-1). For instance, genes from a large neuronally-enriched bulk RNA-seq gene co-expression module, “bulk M2,” previously shown to be downregulated in HD knock-in mice, were enriched in 13 distinct snRNA-seq modules from the k-means hybrid network (modules M12, M50, M29, M101, M95, M86, M33, M46, M10, M70, M1, M76, and M36), all of which were downregulated in HttQ175/+ versus Htt+/+ mice but with varying specificity across MSN subtypes and in other striatal cells (Fig. 6B). Similarly, a downregulated non-neuronal bulk RNA-seq module, “bulk M11,” overlapped five distinct snRNA-seq modules expressed specifically in astrocytes (M11), oligodendrocytes (M13, M31), or endothelial cells (M43), and across all glial cell types (M32). Thus, network reconstruction from single-cell RNA-seq provides complementary information about cell type-specific gene regulation that is not readily apparent from standard RNA-seq.
Figure 6.
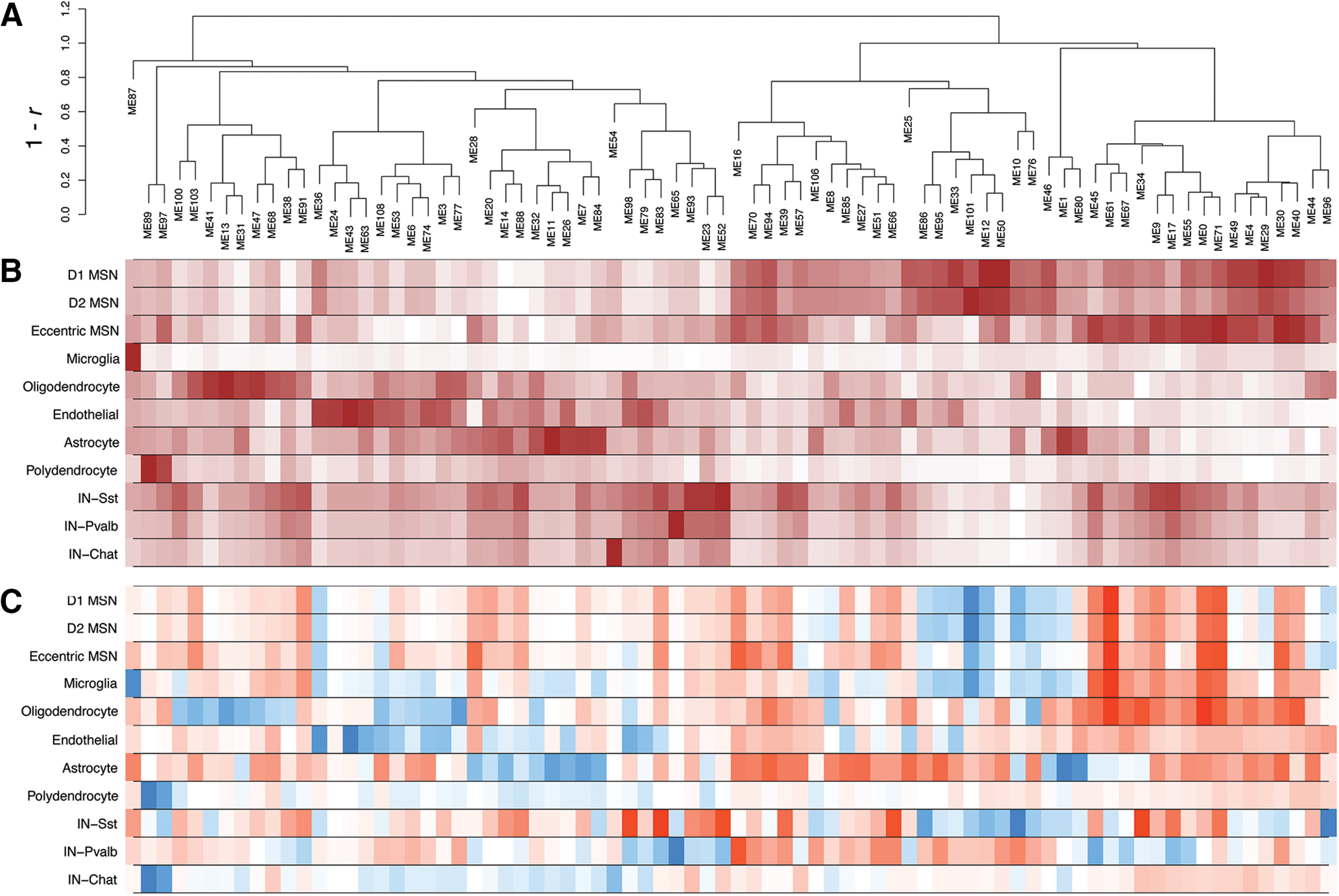
Expression patterns of gene co-expression modules in mouse striatum. A, Average-linkage hierarchical clustering of modules based on their first principal components (module eigengenes). B, Mean expression level of each module eigengene in each cell type. C, Change in module eigengene activity in cells from HttQ175/+ versus Htt+/+ mice.
Associations of gene co-expression modules with Htt genotype. Tab 1, Module differential expression statistics using the module eigengene. Tab 2, Module differential expression statistics using the average expression of the module's component genes. In each tab, “averageExpr” gives the mean value in of the module in the given cell type in standard units, where 0 represents the mean across all cell types. “Change in expression Q175 versus WT” indicates the effect of genotype. “p Q175 versus WT” indicates the raw p value for the difference in Q175 versus WT, and “bonf Q175 versus WT” indicates the Bonferroni-adjusted p value. Download Figure 6-1, XLSX file (116.5KB, xlsx) .
A total of 55 of the 77 modules were differentially expressed [adjusted p < 0.01, > 0.5 SD (z score) change in expression; Fig. 6C] in at least one cell type. These included 13 differentially expressed modules in D1 MSNs, 15 in D2 MSNs, 9 in eccentric MSNs, 39 in astrocytes, and 27 oligodendrocytes. Consistent with findings from DEGs (above), we identified numerous “cell identity” modules that were specifically expressed in one cell type and downregulated in cells of that same type from HttQ175/+ versus Htt+/+ mice (Fig. 7, columns 1–4). We also identified modules that were broadly expressed and upregulated across most or all cell types (Fig. 7, columns 5, 6). A striking and less expected finding was that many of the cell identity modules were also upregulated in incorrect cell types. For instance, the MSN identity module M12 (Fig. 7, first column) was downregulated in D1 (effect size in standard units = −0.74, p < 1e-308), D2 (effect size = −0.74, p = 7.0e-217) and eccentric MSNs (effect size = −0.34, p = 3.7e-6), but upregulated in astrocytes (effect size = 0.56, p = 3.5e-7) and oligodendrocytes (effect size = 0.27, p = 7.4e-3). The astrocyte identity module M11 (Fig. 7, second column) was downregulated in astrocytes (effect size = −1.06, p = 2.1e-11), but slightly upregulated in MSNs (effect size = 0.02, 0.03; p = 4.5e-10 and p = 3.0e-4, in D1 and D2 subtypes, respectively). The oligodendrocyte identity module M13 (Fig. 7, third column) was downregulated in oligodendrocytes (effect size = −0.96, p = 1.3e-9), but upregulated in MSNs (effect size = 0.083, 0.08; p = 1.4e-27 and p = 5.5e-18 in D1 and D2 subtypes) and astrocytes (effect size = 0.26, p = 2.0e-4). Further examples include the parvalbumin interneuron identity module M65 (Fig. 7, fourth column) and the endothelial cell identity module M43. Examining the expression of individual genes from these modules confirmed that they follow these same bi-directional patterns of transcriptional dysregulation (Fig. 7D). Notably, our gene regulatory network model predicted that many of these cell identity modules are regulated by canonical cell type-specific hub transcription factors, such as FOXP1 in M12, SOX9 in M11, MYRF in M13, and NKX2.1 in M65 (Fig. 7A,C), which are required for the development of MSNs, astrocytes, oligodendrocytes, and interneurons, respectively (Marín et al., 2000; Kang et al., 2012; Hornig et al., 2013; Precious et al., 2016).
Figure 7.
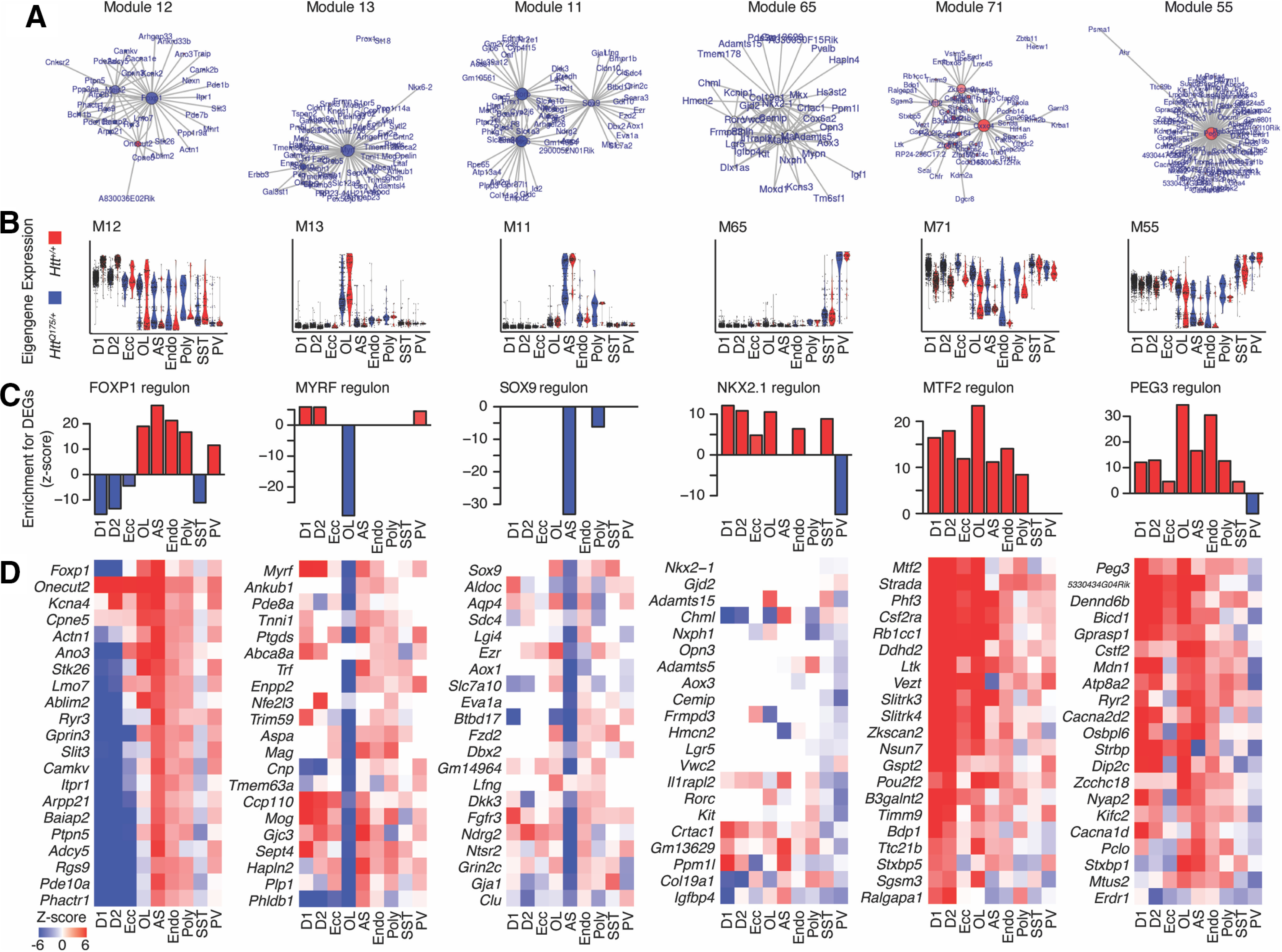
mHTT-associated cell type identity-related gene co-expression modules. A, Graphs of the top 100 gene-gene interactions within each module. Node size corresponds to eigencentrality. Node color corresponds to the change in expression between cells from HttQ175/+ versus Htt+/+ mice in the major cell type for that cluster (blue = downregulated; red = upregulated). B, Violin plots of module eigengenes. C, Over-representation of the predicted target genes from GENIE3 for the hub transcription factors in each module for upregulated and downregulated genes in each cell type. D, Z-scores for cell type-specific upregulation or downregulation of the top 20 genes in each regulon shown in panel C.
We postulated that these bidirectional changes in gene expression may reflect aberrant repression and de-repression of cell type identity genes in HttQ175/+ mice. One mechanism by which this could occur is through interactions of wild-type and mutant HTT with the polycomb repressive complex 2 (PRC2). PRC2 facilitates gene repression via trimethylation of histone H3 at lysine 27 (H3K27me3), particularly in the promoters of genes involved in the development and maintenance of cell types. The HTT protein has genotype-specific interactions with PRC2 in vitro (Seong et al., 2010) and in vivo (Pearl et al., 2020), and conditional knock-out of PRC2 in striatal MSNs causes gene expression changes that mimic the effects of HD mutations (Von Schimmelmann et al., 2016). To test whether dysregulated cell type identity modules in HttQ175/+ mice involve PRC2, we assembled nine ChIP-seq datasets profiling the genomic occupancy for components of the PRC2 complex (EZH2, SUZ12) or for H3K27me3 in four disease-relevant cell types: MSNs (Von Schimmelmann et al., 2016; Pearl et al., 2020), astrocytes (Davis et al., 2018), oligodendrocytes (Bardile et al., 2019), and embryonic stem cells (Kloet et al., 2016; Davis et al., 2018). We tested for over-representation of each of our 77 gene co-expression modules among putative PRC2 target genes, defined by the presence of a PRC2-related ChIP-seq peak ±5 kb from a gene's transcription start site. Ten modules were robustly overrepresented for these PRC2 target genes (adjusted ps < 0.01 in at least four of the ChIP-seq datasets). All of these 10 PRC2-regulated modules were expressed specifically in a single striatal cell type (Fig. 8A), including modules specific to MSNs (M12, M29, M50), all interneurons (M23, M52), Pvalb+ interneurons (M65), Chat+ interneurons (M54), endothelial cells (M43), oligodendrocytes (M13), and astrocytes (M11). All of these PRC2-regulated modules except for those specific to interneurons were significantly downregulated in that same cell type in HttQ175/+ mice (adjusted p < 0.01), while the expression of interneuron-specific modules trended downward in cells from HttQ175/+ mice (Fig. 8B). All 10 PRC2-regulated modules (including interneuron-specific modules) were significantly upregulated in at least one other cell type in which these genes are not normally expressed. As expected, the dynamics of PRC2 occupancy across cell types was negatively correlated with cell type-specific gene expression (Fig. 8C). PRC2 target genes in embryonic stem cells, in which genes for all differentiated cell types are repressed, were overrepresented in all 10 modules. PRC2 target genes in MSNs were overrepresented in interneuron-specific and glial-specific modules, but not in MSN-specific modules. PRC2 target genes in astrocytes and oligodendrocytes were primarily enriched in neuron-specific modules, but not in glial-specific modules.
Figure 8.
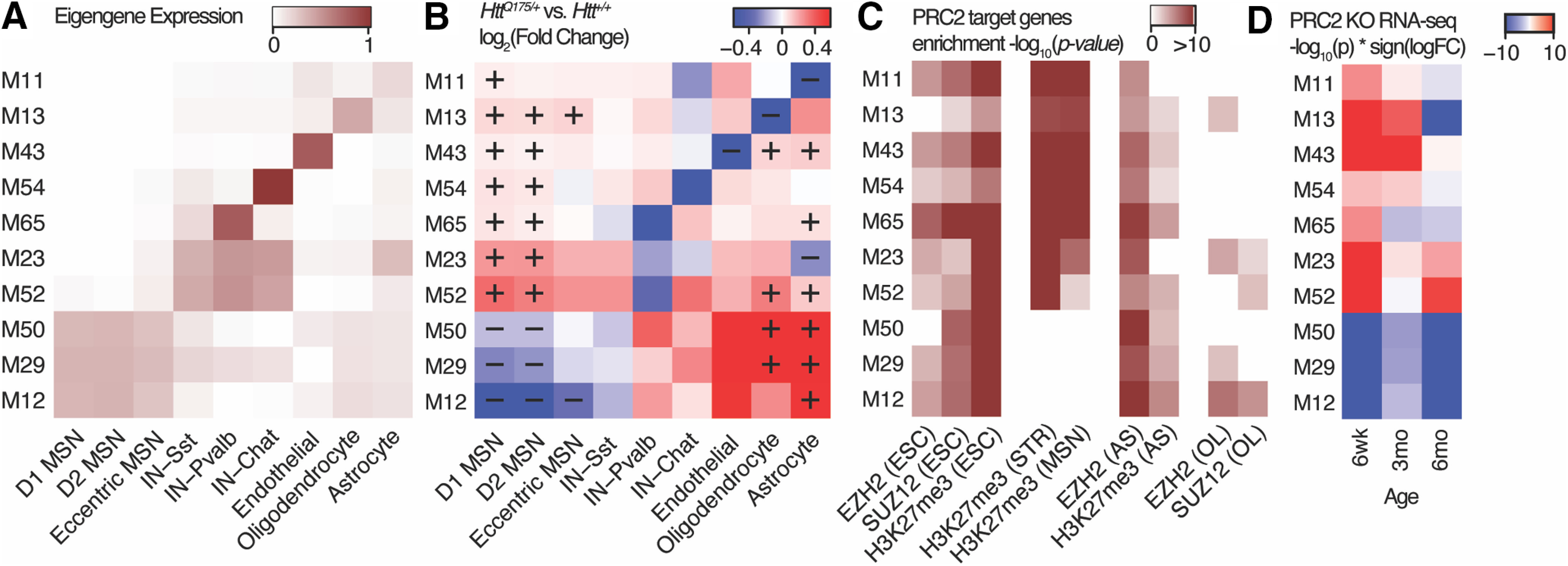
Regulation of HD-associated cell type identity modules by the PRC2. A, Cell type-specific expression of module eigengenes for 10 gene co-expression modules robustly enriched for PRC2 target genes. B, Change in expression of module eigengenes in cells from HttQ175/+ versus Htt+/+ mice; “+” and “–” indicate statistically significant upregulation or downregulation of eigengene expression in each cell types (adjusted p < 0.01). C, Over-representation of the genes in each module for PRC2 target genes defined by nine ChIP-seq experiments in embryonic stem cells (ESCs), MSNs, astrocytes (AS), and oligodendrocytes (OL). D, Over-representation of the genes in each module for upregulated and downregulated genes in MSNs from six-week-old, three-month-old, and six-month-old PRC2-silenced versus control mice.
To more directly establish the regulation of these modules by PRC2, we analyzed published microarray gene expression profiles of MSNs from six-week-old, three-month-old, and six-month-old EZH2/EZH1 double conditional knock-out mice, in which PRC2 was silenced specifically in adult MSNs (Von Schimmelmann et al., 2016). PRC2 silencing in MSNs strongly dysregulated all 10 modules in the same direction observed in MSNs from HttQ175/+ versus Htt+/+ mice, i.e., modules specific to other cell types were ectopically expressed in PRC2-silenced MSNs, while MSN-specific modules were repressed (Fig. 8D). These analyses extend prior analyses of these data, which had also noted the bidirectional overlap with genes dysregulated in HD. Our analysis indicates that a much larger proportion of the transcriptional dysregulation in HD knock-in mice may be explained by altered interactions with PRC2 than had previously been appreciated and suggests that these interactions occur in multiple striatal cell types, not just MSNs. Thus, PRC2 regulates the cell type-specific expression patterns of many gene co-expression modules that are bidirectionally dysregulated in HttQ175/+ versus Htt+/+ mice, and our data support a model in which PRC2 loss-of-function because of the HD mutation causes both the de-repression of these modules in inappropriate cell types, as well as their repression in their primary cell type.
PRC2-regulated modules are bidirectionally dysregulated in multiple cell types from six-month-old HD knock-in mice and in HD patients
Our results above suggest that bidirectional changes in the expression of PRC2-regulated cell identity modules is a prominent effect of the HD mutation in 15-month-old HttQ175/+ mice. We tested the robustness and generalizability of this finding in independent datasets. First, we asked whether the cell type-specific effects of HD mutations on PRC2-regulated modules could also be observed at earlier time points in HD knock-in mice, using snRNA-seq of six-month-old HttQ175/+ versus HttQ20/+ mice, translatome profiling of D2 MSNs, D1 MSNs, astrocytes, and Chat+ interneurons from an HD allelic series at six months (Lee et al., 2020); and RNA-seq of bulk striatum tissue from an HD allelic series at 2, 6, and 10 months (Langfelder et al., 2016). The snRNA-seq data confirmed that all 10 PRC2-regulated modules were expressed in specific neuronal or non-neuronal cell types, as in our primary dataset (Fig. 9A,B). snRNA-seq also confirmed that in D1 MSNs, D2 MSNs, eccentric MSNs, oligodendrocytes, astrocytes, endothelial cells, and PV interneurons the cognate module was downregulated (p < 8.8e-11 in each cell type; Fig. 9C; Extended Data Fig. 9-1), while modules specific to other cell types were aberrantly upregulated. The magnitude of the changes in glia and interneurons appeared smaller in 6- versus 15-month-old mice. These results indicate that bidirectional changes in the expression of PRC2-regulated cell identity modules in multiple striatal cell types are an early and progressive feature in HD knock-in mice, beginning by six months and expanding in magnitude in 15-month-old mice.
Figure 9.

Dynamics of PRC2-regulated modules in the striatum of six-month-old HttQ175/+ versus HttQ20/+ mice (snRNA-seq). A, Clustering of cells from Lee et al. (2020) on uniform manifold approximation and projection (UMAP) coordinates. B, Cell type-specific expression of module eigengenes for PRC2-regulated modules. C, Change in expression of module eigengenes in cells from HttQ175/+ versus Htt+/+ mice; “+” and “–” indicate statistically significant upregulation or downregulation of eigengene expression in each cell types (adjusted p < 0.05).
Dynamics of PRC2-regulated modules in independent cohorts. Linear models were used to test effects of HD mutations on module eigengene expression in each cell type and condition (Excel file). Download Figure 9-1, XLSX file (47KB, xlsx) .
Translatome profiling data of D1 and D2 MSNs also confirmed both downregulation of MSN cell identity modules and aberrant upregulation of modules specific to other cell types in six-month-old HttQ170/+ and HttzQ175/+ mice (Fig. 10). However, translatome profiling of astrocytes and Chat+ interneurons failed to detect equivalent changes. We speculate that the higher levels of ambient RNA inherent to translatome profiling data masked bidirectional changes in these rare cell types. Similarly, in bulk RNA-seq of 6- and 10-month-old mice, we detected downregulation of MSN-specific modules accompanied by upregulation or no change in the expression of other modules (Fig. 11). These patterns mirror the changes in MSNs, while changes in other cell types are masked. No expression changes in these modules were detected in RNA-seq of two-month-old mice. Thus, bidirectional dysregulation of PRC2-regulated cell identity modules can be detected by transcriptional profiling with multiple technologies, but its full breadth across cell types is most apparent with snRNA-seq.
Figure 10.
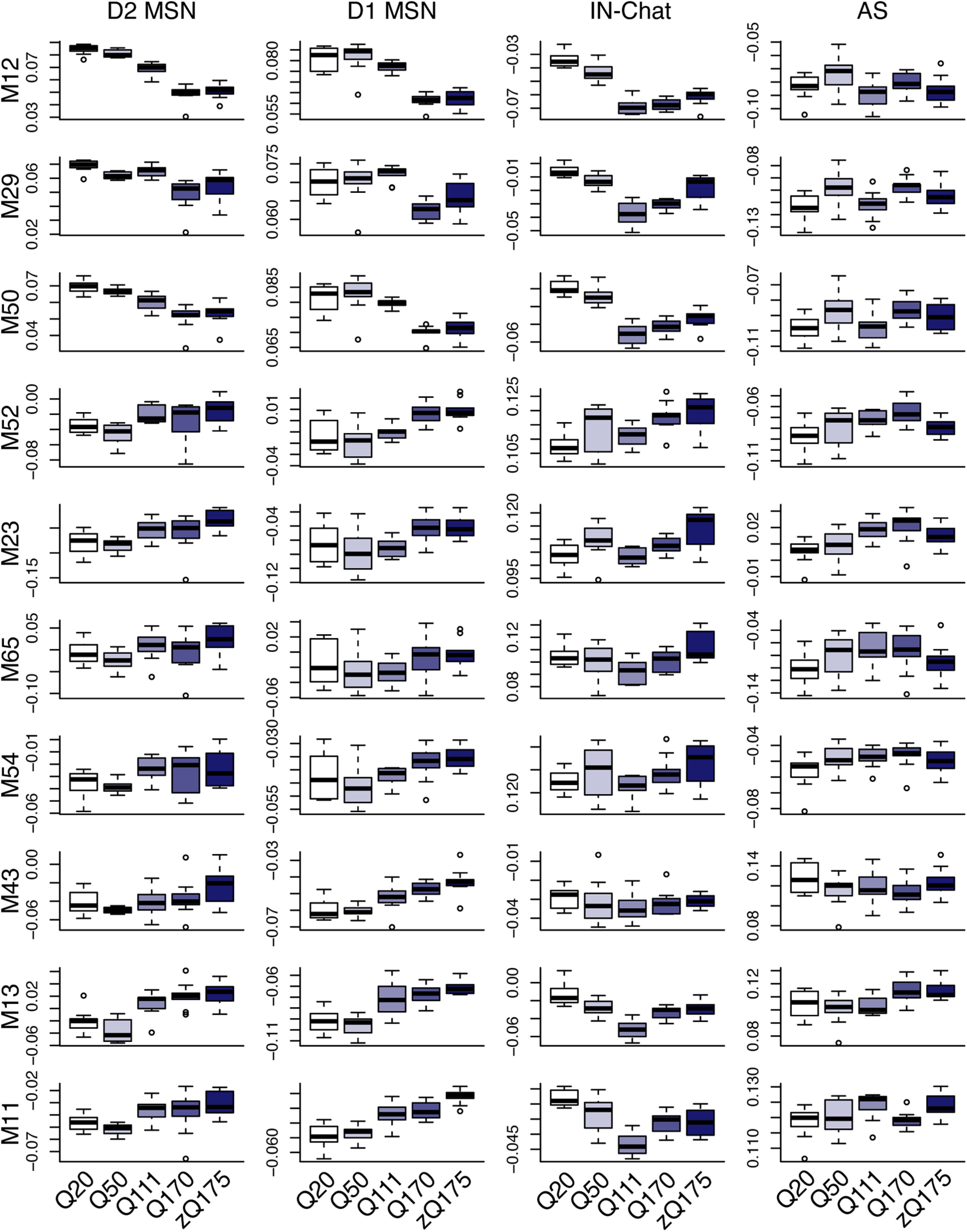
Dynamics of PRC2-regulated modules in the striatum of six-month-old HD knock-in mice (translatome profiling). Network analysis of translatome profiling data from Lee et al. (2020). Expression of eigengenes for PRC2-regulated modules in striatal D1 MSNs, D2 MSNs, Chat+ interneurons (IN Chat), and astrocytes (AS) from six-month-old HttQ20/+, HttQ50/+, HttQ111/+, HttQ170/+, and HttzQ175/+ mice.
Figure 11.

Dynamics of PRC2-regulated modules in the striatum of six-month-old HD knock-in mice (bulk tissue RNA-seq). Network analysis of mRNA-seq from (Langfelder et al., 2016). Expression of eigengenes for PRC2-regulated modules in striatal tissue from six-month-old HttQ20/+, HttQ80/+, HttQ92/+, HttQ111/+, HttzQ140/+, and HttzQ175/+ mice.
Finally, we examined snRNA-seq of dorsal and ventral striatum from (human) HD cases versus controls (Lee et al., 2020). We confirmed expression changes in PRC2-regulated modules that were similar to those in 15-month-old HttzQ175/+ mice in most respects (Fig. 12). Cognate PRC2-regulated cell identity modules were downregulated (adjusted p < 0.05) in D1 MSNs, D2 MSNs, eccentric MSNs, oligodendrocytes, astrocytes, endothelial cells, Chat+ interneurons, and PV interneurons. Moreover, in MSNs and Chat+ interneurons, we also detected aberrant upregulation (FDR < 0.05) of multiple modules specific to other cell types. Interestingly, though, in oligodendrocytes and astrocytes nearly all PRC2-regulated modules were downregulated, perhaps reflecting a more extensive loss of cellular function. The overall similarity of these patterns to the HD knock-in mice is remarkable, given that postmortem tissue from HD patients has much greater pathology than is observed in the HD knock-in mice (Vonsattel et al., 1985; Deng et al., 2021).
Figure 12.

Dynamics of PRC2-regulated modules in the striatum of human HD cases versus controls (snRNA-seq). A, Clustering of cells from Lee et al. (2020) on uniform manifold approximation and projection (UMAP) coordinates. B, Cell type-specific expression of module eigengenes for PRC2-regulated modules. C, Change in expression of module eigengenes in cells from HD cases versus controls; “+” and “–” indicate statistically significant upregulation or downregulation of eigengene expression in each cell types (adjusted p < 0.05).
Discussion
Several findings from our study merit particular attention. We observe pronounced transcriptional dysregulation in nearly all striatal cell types, which we have here defined and compared with genes identified with RNA-seq in bulk striatal tissue. Analysis of HD-associated transcriptional networks across cell types reveals a striking downregulation of cell identity genes, coupled with aberrant upregulation of these genes in incorrect cell types. Integration of these analyses with existing transcriptomic and epigenomic data suggests that altered PRC2 function may underlie these bidirectional changes, leading to dysregulation of cell identity across essentially all cell types in the adult striatum.
The early phases of disease progression in HD are dominated by hyperkinetic motor features, notably chorea. This has been attributed to the selective atrophy and early loss of D2 MSNs, revealed by the preferential loss of enkephalin-containing fibers across disease severity (Sapp et al., 1995; André et al., 2011; Deng et al., 2013). Our data reveal profound transcriptional changes across all subtypes of MSNs. However, our data do not reveal significant differences among MSN subtypes with respect to the genes that are differentially expressed nor in the magnitude of the fold changes. By contrast, Lee et al. (2020) detected certain differences in the transcriptional effects of HD mutations in D1 versus D2 MSNs at an earlier time point (six-month-old HttQ175/+ mice). Future studies should continue to characterize the trajectory and timing of changes in each MSN subtype.
The role of various types of glial cells in the progression of HD has been an important avenue of research. Our data reveal novel aspects of transcriptional dysregulation in glial cell types, which rely on the ability to examine transcriptional changes within specific cell types. Specifically, we find increased numbers of microglia, consistent with previous reports that microgliosis begins in presymptomatic HD mutation carriers (Tai et al., 2007). In addition, we detected thousands of cell type-specific DEGs in glia, especially astrocytes and oligodendrocytes, many of which are not readily apparent in RNA-seq of bulk striatal tissue. It is notable that these changes were detected in our dataset despite a relatively small sample size. Additional changes, especially in microglia, may be observed as snRNA-seq sample sizes increase. Across glial cell types, we observed a spectrum of transcriptional states, comprising of nuclei in similar transcriptional states to wild-type nuclei, nuclei in discernably different transcriptional states, as well as intermediate states between the two that may indicate a gradual shift in cell states, opening new areas of study. This is distinct from our trajectory analysis of MSNs, in which we observed nearly complete separation of wild-type and HttQ175/+ cellular states. Activation of neuroinflammatory genes is observed across several cell types, including astrocytes, oligodendrocytes, and MSNs. However, the transcriptional states of glia are distinct from classical neuroinflammatory states such as A1 and A2 reactive astrocytes, suggesting disease-specific mechanisms.
Across cell types, a common feature is the transcriptional dysregulation of cell identity genes, consistent with dysfunction of cell fate commitment. Previous studies, based on RNA-seq of bulk striatal tissue, have suggested that loss of cell fate commitment may drive MSN vulnerability in HD (Langfelder et al., 2016; Von Schimmelmann et al., 2016). Reconstruction of gene co-expression networks and gene regulatory networks from our data revealed numerous gene modules that were dysregulated in HD knock-in mice. HD-associated gene modules that are expressed primarily in MSNs overlapped strongly with disease-associated gene modules from bulk RNA-seq of HD and HD mouse models, which is to be expected since MSNs represent a large percentage of cells in the striatum. However, the improved resolution of our network models across cell types provides greater insight into this mode of pathology. Notably, we confirm that aged MSNs expressing mHTT have reduced expression of MSN-specific genes, e.g., the FOXP1 regulon in module M12. In addition, our findings reveal reduced cellular identity genes in other cell types, including oligodendrocytes (MYRF regulon in module M13) and astrocytes (SOX9 regulon in module 11). Here, we observe, for the first time, that in addition to being downregulated in their correct cell type, cell identity genes are also aberrantly upregulated in other cell types. This expands the model of cell fate commitment changes in HD, suggesting it may be a shared feature of aged cells expressing mHTT, affecting virtually all cell types in the striatum.
Given the importance of PRC2 in establishing and maintaining cell fate and the known interactions between HTT and PRC2, we compared our results to PRC2-related ChIP-seq and RNA-seq datasets. We find that 10 of our 77 gene co-expression modules are enriched with PRC2 target genes, all of which were expressed and downregulated in specific cell types. Extending these analyses to published snRNA-seq suggest that dysregulation of these modules in multiple cell types begins at early time points and accurately reflects the dysregulation in human HD. Our results suggest that PRC2 helps restrict expression of these genes to their appropriate cell type in the adult striatum. Conditional knock-out of PRC2 and the presence of mHTT each result in highly similar patterns of bidirectional dysregulation across cell types. Thus, many of the transcriptional effects of mHTT may be mediated by PRC2 loss-of-function. Consistent with this idea, we recently described decreased H3K27me3 levels at hundreds of loci in striatal tissue from HD knock-in mice (Pearl et al., 2020). Future studies should test whether restoring PRC2 function can rescue the phenotypic consequences of HD mutations.
Footnotes
This work was supported by contracts from the CHDI Foundation (S.A.A. and J.B.C.) and by the BRAIN Initiative Grant R24 MH114815.
The authors declare no competing financial interests.
References
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, Van Den Oord J, Atak ZK, Wouters J, Aerts S (2017) SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14:1083–1086. 10.1038/nmeth.4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, Adorjan I, Menon V, Goldman JE (2020) Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol Commun 8:19. 10.1186/s40478-020-0880-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov V, Brunner D, Menalled LB, Kudwa A, Watson-Johnson J, Mazzella M, Russell I, Ruiz MC, Torello J, Sabath E, Sanchez A, Gomez M, Filipov I, Cox K, Kwan M, Ghavami A, Ramboz S, Lager B, Wheeler VC, Aaronson J, Rosinski J, et al. (2016) Large-scale phenome analysis defines a behavioral signature for Huntington's disease genotype in mice. Nat Biotechnol 34:838–844. 10.1038/nbt.3587 [DOI] [PubMed] [Google Scholar]
- Ament SA, Pearl JR, Grindeland A, St. Claire J, Earls JC, Kovalenko M, Gillis T, Mysore J, Gusella JF, Lee JM, Kwak S, Howland D, Lee MY, Baxter D, Scherler K, Wang K, Geman D, Carroll JB, MacDonald ME, Carlson G, et al. (2017) High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington's disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet 26:913–922. 10.1093/hmg/ddx006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ament SA, Pearl JR, Cantle JP, Bragg RM, Skene PJ, Coffey SR, Bergey DE, Wheeler VC, MacDonald ME, Baliga NS, Rosinski J, Hood LE, Carroll JB, Price ND (2018) Transcriptional regulatory networks underlying gene expression changes in Huntington's disease. Mol Syst Biol 14:e7435. 10.15252/msb.20167435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- André VM, Fisher YE, Levine MS (2011) Altered balance of activity in the striatal direct and indirect pathways in mouse models of Huntington's disease. Front Syst Neurosci 5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardile CF, Garcia-Miralles M, Caron NS, Rayan NA, Langley SR, Harmston N, Rondelli AM, Yi Teo RT, Waltl S, Anderson LM, Bae HG, Jung S, Williams A, Prabhakar S, Petretto E, Hayden MR, Pouladi MA (2019) Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc Natl Acad Sci USA 116:9622–9627. 10.1073/pnas.1818042116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbon-Teles J, Bells S, Jones D, Coulthard E, Rosser A, Metzler-Baddeley C (2019) Myelin breakdown in human Huntington's disease: multi-modal evidence from diffusion MRI and quantitative magnetization transfer. Neuroscience 403:79–92. 10.1016/j.neuroscience.2017.05.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R (2018) Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36:411–420. 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow M, Paul A, Ballouz S, Huang ZJ, Gillis J (2016) Exploiting single-cell expression to characterize co-expression replicability. Genome Biol 17:101. 10.1186/s13059-016-0964-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, Onate KC, Graham K, Miyasato SR, Dreszer TR, Strattan JS, Jolanki O, Tanaka FY, Cherry JM (2018) The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res 46:D794–D801. 10.1093/nar/gkx1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YP, Reiner A (2016) Cholinergic interneurons in the Q140 knockin mouse model of Huntington's disease: reductions in dendritic branching and thalamostriatal input. J Comp Neurol 524:3518–3529. 10.1002/cne.24013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YP, Wong T, Bricker-Anthony C, Deng B, Reiner A (2013) Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington's disease mice. Neurobiol Dis 60:89–107. 10.1016/j.nbd.2013.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Wang H, Joni M, Sekhri R, Reiner A (2021) Progression of basal ganglia pathology in heterozygous Q175 knock-in Huntington's disease mice. J Comp Neurol 529:1327–1371. 10.1002/cne.25023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Castro B, Gangwani MR, Yu X, Coppola G, Khakh BS (2019) Astrocyte molecular signatures in Huntington's disease. Sci Transl Med 11:eaaw8546. [DOI] [PubMed] [Google Scholar]
- Grubman A, Chew G, Ouyang JF, Sun G, Choo XY, McLean C, Simmons RK, Buckberry S, Vargas-Landin DB, Poppe D, Pflueger J, Lister R, Rackham OJLL, Petretto E, Polo JM (2019) A single-cell atlas of entorhinal cortex from individuals with Alzheimer's disease reveals cell-type-specific gene expression regulation. Nat Neurosci 22:2087–2097. 10.1038/s41593-019-0539-4 [DOI] [PubMed] [Google Scholar]
- Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, Green G, Dionne D, Nguyen L, Marshall JL, Chen F, Zhang F, Kaplan T, Regev A, Schwartz M (2020) Disease-associated astrocytes in Alzheimer's disease and aging. Nat Neurosci 23:701–706. 10.1038/s41593-020-0624-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Lun ATL, Morgan MD, Marioni JC (2018) Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 36:421–427. 10.1038/nbt.4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RLM, et al. (2006) Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 15:965–977. 10.1093/hmg/ddl013 [DOI] [PubMed] [Google Scholar]
- Hornig J, Fröb F, Vogl MR, Hermans-Borgmeyer I, Tamm ER, Wegner M (2013) The transcription factors Sox10 and Myrf define an essential regulatory network module in differentiating oligodendrocytes. PLoS Genet 9:e1003907. 10.1371/journal.pgen.1003907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P (2010) Inferring regulatory networks from expression data using tree-based methods. PLoS One 5:e12776. 10.1371/journal.pone.0012776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang P, Lee HK, Glasgow SM, Finley M, Donti T, Gaber ZB, Graham BH, Foster AE, Novitch BG, Gronostajski RM, Deneen B (2012) Sox9 and NFIA coordinate a transcriptional regulatory cascade during the initiation of gliogenesis. Neuron 74:79–94. 10.1016/j.neuron.2012.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloet SL, Makowski MM, Baymaz HI, Van Voorthuijsen L, Karemaker ID, Santanach A, Jansen PWTC, Di Croce L, Vermeulen M (2016) The dynamic interactome and genomic targets of Polycomb complexes during stem-cell differentiation. Nat Struct Mol Biol 23:682–690. 10.1038/nsmb.3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, Linker SB, Pham S, Erwin JA, Miller JA, Hodge R, McCarthy JK, Kelder M, McCorrison J, Aevermann BD, Fuertes FD, Scheuermann RH, Lee J, Lein ES, Schork N, et al. (2016) Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc 11:499–524. 10.1038/nprot.2016.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A (2010) ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26:2438–2444. 10.1093/bioinformatics/btq466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Codeluppi S, Yung YC, Gao D, Chun J, Kharchenko PV, Linnarsson S, Zhang K (2017) A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci Rep 7:6031. 10.1038/s41598-017-04426-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, Deverasetty S, Tebbe A, Schaab C, Lavery DJ, Howland D, Kwak S, Botas J, Aaronson JS, Rosinski J, Coppola G, et al. (2016) Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci 19:623–633. 10.1038/nn.4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, Garcia FJ, Therrien M, Novis HS, Gao F, Wilkinson H, Vogt T, Kellis M, LaVoie MJ, Heiman M (2020) Cell type-specific transcriptomics reveals that mutant Huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron 107:891–818. 10.1016/j.neuron.2020.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Yeh C, Yang XW (2008) Pivotal role of early B-cell factor 1 in development of striatonigral medium spiny neurons in the matrix compartment. J Neurosci Res 86:2134–2146. 10.1002/jnr.21666 [DOI] [PubMed] [Google Scholar]
- MacDonald M, Ambrose C, Duyao M (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72:971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- Marín O, Anderson SA, Rubenstein JLR (2000) Origin and molecular specification of striatal interneurons. J Neurosci 20:6063–6076. 10.1523/JNEUROSCI.20-16-06063.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Märtin A, Calvigioni D, Tzortzi O, Fuzik J, Wärnberg E, Meletis K (2019) A spatiomolecular map of the striatum. Cell Rep 29:4320–4333.e5. [DOI] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai LHH (2019) Single-cell transcriptomic analysis of Alzheimer's disease. Nature 570:332–337. 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson K, Sathyamurthy A, Johnson KR, Kelly MC, Kelley MW, Levine AJ (2018) Isolation of adult spinal cord nuclei for massively parallel single-nucleus RNA sequencing. J Vis Exp. Advance online publication. Retrieved Oct 12, 2018. doi: 10.3791/58413. 10.3791/58413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers R, Vonsattel J, Paskevich PA, Kiely DK, Stevens BA, Cupples LA, Richardson EP, Bird ED (1991) Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus. J Neuropathol Exp Neurol 50:729–742. 10.1097/00005072-199111000-00005 [DOI] [PubMed] [Google Scholar]
- Orvis J, Gottfried B, Kancherla J, Adkins RS, Song Y, Dror AA, Olley D, Rose K, Chrysostomou E, Kelly MC, Milon B, Matern MS, Azaiez H, Herb B, Colantuoni C, Carter RL, Ament SA, Kelley MW, White O, Corrada Bravo H, et al. (2020) gEAR: gene expression analysis resource portal for community-driven, multi-omic data exploration. bioRxiv 2020.08.28.272039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl JR, Colantuoni C, Bergey DE, Funk CC, Shannon P, Basu B, Casella AM, Oshone RT, Hood L, Price ND, Ament SA (2019) Genome-scale transcriptional regulatory network models of psychiatric and neurodegenerative disorders. Cell Syst 8:122–135.e7. [DOI] [PubMed] [Google Scholar]
- Pearl JR, Shetty AC, Cantle JP, Bergey DE, Bragg RM, Coffey SR, Kordasiewicz HB, Hood LE, Price ND, Ament SA, Carroll JB (2020) Altered Huntingtin-chromatin interactions predict transcriptional and epigenetic changes in Huntington's disease. bioRxiv 2020.06.04.132571. [Google Scholar]
- Plaisier SB, Taschereau R, Wong JA, Graeber TG (2010) Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res 38:e169. 10.1093/nar/gkq636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Precious SV, Kelly CM, Reddington AE, Vinh NN, Stickland RC, Pekarik V, Scherf C, Jeyasingham R, Glasbey J, Holeiter M, Jones L, Taylor MV, Rosser AE (2016) FoxP1 marks medium spiny neurons from precursors to maturity and is required for their differentiation. Exp Neurol 282:9–18. 10.1016/j.expneurol.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB (1988) Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci USA 85:5733–5737. 10.1073/pnas.85.15.5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapp E, Ge P, Aizawa H, Bird E, Penney J, Young AB, Vonsattel JP, Difiglia M (1995) Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grade Huntington's disease using high resolution image analysis. Neuroscience 64:397–404. 10.1016/0306-4522(94)00427-7 [DOI] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, Goeva A, Nemesh J, Kamitaki N, Brumbaugh S, Kulp D, McCarroll SA (2018) Molecular diversity and specializations among the cells of the adult mouse brain. Cell 174:1015–1030.16. 10.1016/j.cell.2018.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee J-M, Wheeler VC, Walz T, Kingston RE, Gusella JF, Conlon RA, MacDonald ME (2010) Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet 19:573–583. 10.1093/hmg/ddp524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seredenina T, Luthi-Carter R (2012) What have we learned from gene expression profiles in Huntington's disease? Neurobiol Dis 45:83–98. 10.1016/j.nbd.2011.07.001 [DOI] [PubMed] [Google Scholar]
- Smith G, Rocha E, McLean J, Hayes MA, Izen SC, Isacson O, Hallett PJ (2014) Progressive axonal transport and synaptic protein changes correlate with behavioral and neuropathological abnormalities in the heterozygous Q175 KI mouse model. Hum Mol Genet 23:4510–4527. 10.1093/hmg/ddu166 [DOI] [PubMed] [Google Scholar]
- Smyth GKG (2005) Limma: linear models for microarray data. In: Bioinformatics and computational biology solutions using R and bioconductor, pp 397–420. New York: Springer-Verlag. [Google Scholar]
- Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S (2018) Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 19:477. 10.1186/s12864-018-4772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai Y, Pavese N, Gerhard A, Tabrizi S, Barker RA, Brooks DJ, Piccini P (2007) Microglial activation in presymptomatic Huntington's disease gene carriers. Brain 130:1759–1766. 10.1093/brain/awm044 [DOI] [PubMed] [Google Scholar]
- von Schimmelmann M, Feinberg PA, Sullivan JM, Ku SM, Badimon A, Duff MK, Wang Z, Lachmann A, Dewell S, Ma'ayan A, Han MH, Tarakhovsky A, Schaefer A (2016) Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat Neurosci 19:1321–1330. 10.1038/nn.4360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44:559–577. [DOI] [PubMed] [Google Scholar]
- Wagner F, Yan Y, Yanai I (2018) K-nearest neighbor smoothing for high-throughput single-cell RNA-seq data. bioRxiv 217737. [Google Scholar]
- Wertz MH, Pineda SS, Lee H, Kulicke R, Kellis M, Heiman M (2020) Interleukin-6 deficiency exacerbates Huntington's disease model phenotypes. Mol Neurodegener 15:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Behjati S (2018) SoupX removes ambient RNA contamination from droplet based single cell RNA sequencing data. bioRxiv 303727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, Barres BA (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410. 10.1523/JNEUROSCI.6221-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35:76–83. 10.1038/ng1219 [DOI] [PubMed] [Google Scholar]
- Zucker B, Luthi-Carter R, Kama A, Dunah A, Stern E, Fox JH, Standaert DG, Young AB, Augood SJ (2005) Transcriptional dysregulation in striatal projection-and interneurons in a mouse model of Huntington's disease: neuronal selectivity and potential neuroprotective role. Hum Mol Genet 14:179–189. 10.1093/hmg/ddi014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell type marker genes and DEGs in nine striatal cell types from 14- to 15-month-old HttQ175/+ versus Htt+/+ mice. Tab 1, Marker genes in each cell type. Tab 2, Gene expression changes in each cell type based on smoothed counts. Tab 3, Gene expression changes in each cell type based on non-smoothed counts. Tabs show all genes in each analysis, which included in each cell type that set of genes with non-zero counts in at least 10% of cells (Excel file). Download Figure 1-1, XLSX file (12.9MB, xlsx) .
Gene set enrichment analysis of cell type-specific DEGs. Enrichments in gene sets from Gene Ontology (Tab 1) and HDSigDB (Tab 2), and for the predicted target genes of each striatally expressed transcription factor in our GENIE3 gene regulatory network model (Tab 3; Excel file). Download Figure 2-1, XLSX file (1.6MB, xlsx) .
Gene co-expression modules. Tab 1, Modules derived by the k-means hybrid method. Tab 2, Modules derived by WGCNA. Tab 3, Modules derived by k-means (k = 50). Download Figure 4-1, XLSX file (88.3KB, xlsx) .
Gene set enrichment analysis of gene co-expression modules. Tabs 1–6, Enrichments for k-means hybrid modules. Tab 1, Enrichment for Gene Ontology terms. Tab 2, HDSigDB terms. Tab 3, WGCNA modules from Langfelder et al. (2016; subset of HDSigDB). Tab 4, Predicted target genes of TFs based on ChIP-seq experiments in ChEA. Tab 5, Predicted target genes of striatally expressed TFs in our GENIE3 gene regulatory network model. Tab 6, Evolutionarily conserved cis-motifs from RcisTarget. Tab 7, Enrichments in modules derived by WGCNA. Tab 8, Enrichments in modules derived by k-means clustering, k = 50. Download Figure 5-1, XLSX file (8.8MB, xlsx) .
Associations of gene co-expression modules with Htt genotype. Tab 1, Module differential expression statistics using the module eigengene. Tab 2, Module differential expression statistics using the average expression of the module's component genes. In each tab, “averageExpr” gives the mean value in of the module in the given cell type in standard units, where 0 represents the mean across all cell types. “Change in expression Q175 versus WT” indicates the effect of genotype. “p Q175 versus WT” indicates the raw p value for the difference in Q175 versus WT, and “bonf Q175 versus WT” indicates the Bonferroni-adjusted p value. Download Figure 6-1, XLSX file (116.5KB, xlsx) .
Dynamics of PRC2-regulated modules in independent cohorts. Linear models were used to test effects of HD mutations on module eigengene expression in each cell type and condition (Excel file). Download Figure 9-1, XLSX file (47KB, xlsx) .