

Inflammation induces macrophage histone lactylation uncoupled from arginine metabolism or M2 polarization.
Abstract
Lactate is an end point of Warburg-type metabolism found in inflammatory macrophages. Recently, lactate was shown to modify histones of lipopolysaccharide (LPS)–activated macrophages in a time-dependent way and promote the expression of genes linked to tissue repair, including arginase-1 (Arg1). We tested the interrelationships between histone lactylation (Kla) and tissue reparative gene expression and found that Kla was uncoupled from changes in gene expression linked to resolving M2 macrophage activation but correlated with Arg1 expression. LPS-induced Arg1 was instead dependent on autocrine-paracrine interleukin-6 (IL6) production, the IL6 receptor, and Stat3 signal transduction. We found that Kla increases as macrophages prepare to die under inflammatory stress, and Kla was absent in macrophages that cannot generate reactive nitrogen or have defects in diverse macrophage death pathways. Thus, Kla is a consequence rather than a cause of macrophage activation but occurs coincidently with an IL6- and Arg1-dependent metabolic rewiring under inflammatory duress.
INTRODUCTION
Inflammatory activation of macrophages by bacterial lipopolysaccharide (LPS) triggers increased glycolysis and the coincident production of lactate (1). The shift to glycolysis occurs partly at the expense of mitochondrial oxidative phosphorylation (OX-PHOS) and is a hallmark of activated myeloid cells and required for inflammatory cytokine production and microbial control and killing (2). Recently, lactate was shown to modify histones at lysine residues (histone lactylation or “Kla” hereafter), and increased Kla was associated with a time-dependent change in macrophage phenotype, where increased expression of genes involved in tissue repair and immune regulation such as arginase-1 (Arg1) occurred (3–5). Other recent findings have pointed to enrichment of Arg1 and programmed cell death 1-L1 (PD-L1)–expressing macrophages in inflammatory regions within hypoxic and lactate-rich tumor microenvironments (6, 7). In this context, lactate from Warburg-type metabolism in tumor cells increased macrophage Arg1 expression and has been further linked to both protumor and tissue repair myeloid phenotypes in different systems (8–11). Precisely how lactate and Kla contribute to macrophage activation is not entirely clear, but establishing the relationships between glycolysis, lactate, arginine metabolism, and macrophage phenotypic plasticity may provide insights into why macrophages are commonly linked with cancer progression. Accordingly, we evaluated the mechanistic linkage between glycolysis, lactate, and macrophage activation.
RESULTS AND DISCUSSION
Lactate and M2 gene expression are uncoupled
We began by reproducing key findings (3) that linked lactate, sourced from LPS-activated macrophages, and increased Arg1 expression. In primary bone marrow–derived macrophages (BMDMs), both LPS and LPS + interferon-γ (IFNγ) increased intracellular lactate concentrations relative to controls (Fig. 1A). By contrast, the addition of lactate alone or treatment with interleukin-4 (IL4) + IL13 and IL4 + IL13, a stimulus that favors OX-PHOS (1), had no substantial effect on cellular lactate amounts. Consistent with previous findings, Arg1 expression was strongly induced by LPS but independent of signal transducer and activator of transcription 6 (STAT6), which controls Arg1 expression from the IL4 and IL13 pathways (Fig. 1B and fig. S1A) (12–14). Prolonged stimulation with IL4/IL13 also increased Arg1 protein expression over time but to a lower relative degree than LPS (fig. S1B). Thus, a time-dependent increase of Arg1 expression in LPS-stimulated cells correlated with increased intracellular lactate, confirming a key experimental finding of Zhang et al. (3). A central supposition of the lactate “timer” model is that multiple mRNAs encoding wound healing and tissue repair functions also increase with time as the inflammatory stimulus wanes (3). These mRNAs are associated with IL4- and IL13-stimulated or “M2-like” macrophages (15, 16). However, our examination of the deposited RNA sequencing (RNA-seq) data from Zhang et al. and our own quantitative evaluation of cardinal M2 gene expression (15, 16) failed to reveal any clear association between inflammatory stimulation, lactate, and time (Fig. 1C and fig. S1C). Hallmark M2 mRNAs like Mrc1 or Fn1 were not regulated or even decreased by LPS/IFNγ stimulation. However, Arg1 expression was clearly induced by LPS inflammation or IL4 + IL13 signals (Fig. 1C). Collectively, our experimental and bioinformatic evaluation of the lactate timer phenomenon revealed a link to Arg1 that was independent of changes in polarization state to an M2-like phenotype. These findings prompted us to define the basis of the specific connections between lactate and Arg1 expression during inflammatory glycolysis.
Fig. 1. Inflammation-associated lactate and M2-like gene expression are biochemically uncoupled.
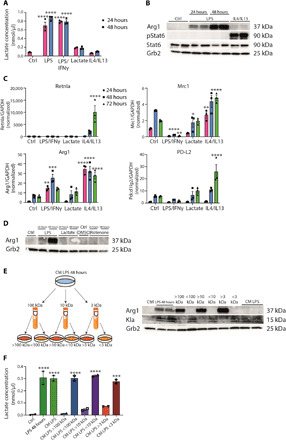
(A) Intracellular lactate concentration was determined in wild-type BMDMs left untreated (Ctrl), and LPS/IFNγ-, LPS-, lactate-, or IL4/IL13-treated for 24 or 48 hours. Data shown are representative of three independent experiments. (B) BMDMs, which were treated with solvent (Ctrl), LPS, or IL4/IL13, were used to isolate whole-cell lysates and analyzed by Western blotting for the indicated proteins. (C) RNA from wild-type BMDMs left untreated (Ctrl), stimulated with LPS/IFNγ, lactate, or IL4/IL13 was used for quantitative reverse transcription polymerase chain reaction. Data shown are the mean fold increase of the 24-hour Ctrl group. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (D) Wild-type BMDMs were stimulated with LPS, lactate, or rotenone. DMSO, dimethyl sulfoxide. (E) Wild-type BMDMs were stimulated with LPS for 48 hours (LPS 48 hours). Supernatant was used unfractionated (CM LPS) or fractionated for 100-, 10-, or 3-kDa protein size. Unfractionated and fractionated supernatant was transferred to naïve BMDMs for further 24 hours. (F) Intracellular lactate concentration was determined in the described groups. Statistically significant differences were determined by one-way or two-way (A and C) analysis of variance (ANOVA) with Tukey correction; n = 2 biological replicates. All values are means ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Superscripts indicate statistical significance compared to the control group. If not indicated otherwise, n = 3 biological replicates.
Arg1 expression is controlled by IL6
Given that exogenous lactate had no effect on Arg1 mRNA (Fig. 1C), we manipulated macrophage glycolysis reasoning that ectopically increasing intracellular as opposed to exogenous lactate would induce Arg1. We treated macrophages with rotenone, which uncouples complex 1 from the electron transport chain and forces adenosine 5′-triphosphate generation via glycolysis, resulting in lactate production (3). While LPS induced Arg1 expression by time as noted above, neither rotenone nor exogenous lactate increased Arg1 protein expression (Fig. 1D). Therefore, the extent of LPS-induced Arg1 expression was independent of accumulated extracellular lactate or enforced lactate production. On the basis of these experiments, we reasoned that LPS may induce a different factor(s) that controlled Arg1 expression with the same kinetics as lactate but was independent of lactate itself. To test this, we used size-dependent filtration approaches to separate conditioned media (CM) from LPS-stimulated macrophages into different molecular weight fractions, noting that lactate has an Mr of 90.08 and would pass through a 3-kDa cutoff filter. We stimulated macrophages with LPS (48 hours) and collected the CM, which was sequentially filtered to collect distinct molecular weight fractions, followed by the transfer of each fraction to recipient “naïve” macrophages. We then quantified Arg1 or Kla in the recipient cells. The results of these experiments revealed that an LPS-regulated factor >3 kDa controlled Arg1 expression that we suspected was a protein (Fig. 1E). By contrast, Kla and lactate amounts were associated with all fractions <3 kDa (Fig. 1, E and F), conceivably controlled by the transfer of heterogeneous molecular weight components of LPS and its proinflammatory contaminants. Regardless of the factor(s) controlling Kla, our findings suggested that an LPS-regulated soluble factor(s) larger than lactate induced Arg1.
Inflammatory factors, such as LPS, induce a myriad of soluble mediators via Toll-like (TLR) and NOD-like receptor (NLR) signaling, and any one or combinations of these factors could control Arg1 expression. Because several cytokines induced by LPS activate Stat3 signaling and this pathway was previously linked to Arg1 expression in different inflammatory contexts, we tested whether decreasing Stat3 activity via Janus kinase 2 (JAK2) inhibition in the presence of LPS regulated Arg1 (12). Tyrphostin AG490 is a JAK2 inhibitor and has suppressive activity toward JAK2/STAT3 signaling (17, 18). JAK2 inhibition blocked LPS-induced Arg1 but not Kla, which increased with time of LPS treatment and was uncoupled from Arg1 expression, as anticipated from the filtration experiments (Fig. 2A). We therefore suspected that the soluble protein(s) that regulated Arg1 was a Stat3-activating cytokine. IL6 is a key Stat3-activating inflammatory cytokine induced in the first hours of inflammation and linked to hypoxia (19–22). Conceivably, IL6 could accrue in the media and control Arg1 expression via autocrine-paracrine signaling. To test this hypothesis, we prepared BMDMs from control, Il6−/−, or Myd88−/− mice (because MyD88 is “upstream” of IL6 gene expression but downstream of TLR4 signaling), stimulated them with LPS for 48 hours, and transferred the CM to unstimulated control, Il6−/−, or Myd88−/− BMDMs. The CM retain LPS (fig. S2A), which therefore is also transferred to the recipient cells. Arg1 and phosphorylated Stat3 were quantified in the recipient BMDMs by immunoblotting (Fig. 2B). The results of these reciprocal CM transfer experiments indicated that Myd88-dependent IL6 production is the main factor responsible for Arg1 expression in glycolytic macrophages. Nevertheless, the transfer of CM also showed that additional factor(s), which are Myd88 dependent, are involved in the LPS-stimulated regulation of Arg1 expression. The key experimental condition in these experiments is the transfer of CM from Il6−/− BMDMs to control BMDMs, which showed a complete absence of Arg1 expression and highlights the importance of the IL6 pathway in the CM. CM fractions >100 kDa were also capable of inducing Arg1 expression, which is greater than the normal size of IL6 (~25 kDa) (Fig. 1E). However, IL6 can form a heterodimer with soluble IL6Rα chain to “trans” signal to gp130, the signal transduction chain of the IL6R (23, 24). Therefore, we repeated the key CM transfer experiments using BMDMs from the Il6ra-deficient mice and also observed the expected dependency on the IL6 pathway (Fig. 2C). In this setting, the informative experimental condition was the transfer of control CM (containing IL6) to Il6ra−/− BMDMs (25). RNA analysis of BMDMs stimulated with the supernatant showed that there was no difference in the induction of the tumor necrosis factor mRNA in either Il6R−/− BMDMs versus control BMDMs (fig. S2B), which was equivalent because of the transfer of LPS. These genetic-based results emphasize the essential nature of IL6 pathway to induce Arg1 expression.
Fig. 2. Arg1 expression is controlled by IL6.
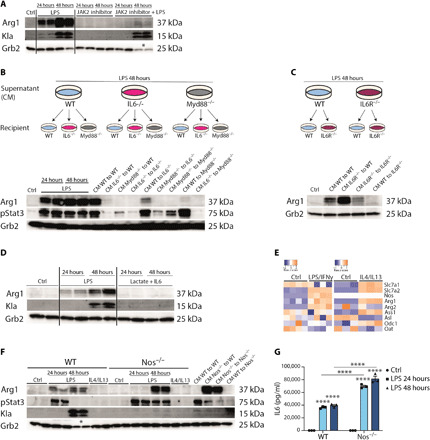
(A) Wild-type (WT) BMDMs were stimulated with LPS and treated with the JAK2 inhibitor AG 490 or both. Data shown are representative of three experiments. (B) Wild-type, Il6−/−, and Myd88−/− BMDMs were stimulated with LPS for 24 or 48 hours. CM of LPS, 48-hour–stimulated BMDMs of all genotypes, were transferred to naïve wild-type and Il6−/− and Myd88−/− BMDMs for further 24 hours. (C) Wild-type and Il6r−/− BMDMs were stimulated with LPS for 48 hours. CM were transferred to naïve wild-type and Il6r−/− BMDMs. (D) Whole-cell lysates of wild-type BMDMs, which were left untreated (Ctrl) or treated with LPS or lactate + IL6, were analyzed by Western blotting. (E) Heatmaps showing transcriptomic data from profile GSE115354 and RNA-seq analysis of Ctrl and IL4/IL13-stimulated BMDMs. (F) Wild-type and Nos2−/− BMDMs were left untreated or stimulated with LPS or IL4/IL13. CM of both genotypes were transferred to untreated wild-type and Nos2−/− BMDMs. (G) Concentration of IL6 in the supernatant of wild-type and Nos2−/− BMDMs stimulated with LPS was measured by enzyme-linked immunosorbent assay (ELISA). n = 3 biological replicates. Statistically significant differences were determined by a two-way ANOVA with Tukey correction; * displays that increased cell death was observed and therefore loading control (Grb2) was decreased. All values are means ± SEM; ****P < 0.0001. If not indicated otherwise, superscripts show statistical significance compared to the control group.
Given that IL6 and IL6R signaling was necessary for Arg1 expression, we asked whether this pathway was also sufficient. We incubated wild-type BMDMs with IL6 in the presence of lactate for 24 or 48 hours and found that, compared to LPS-treated controls, no Arg1 or Kla was detected (Fig. 2D). These data provide unequivocal evidence that IL6 is necessary but not sufficient to induce Arg1. Additional signals downstream of the TLR pathways are needed for IL6 to regulate Arg1, which is consistent with the residual requirement for MyD88 observed in the reciprocal CM transfers (Fig. 2B). Taken collectively, our experiments argue that the lactate timer is coincident with inflammatory Arg1 expression but not the cause; instead, IL6 controls Arg1 expression via the IL6Rα and Stat3 signaling with an additional contribution from other MyD88-dependent pathways.
NO drives Kla
Arginine metabolism is central to diverse metabolic fates of inflammatory macrophages, including tempering antimicrobial nitric oxide (NO) production, suppressing T cell proliferation by local arginine depletion, acting as a source of polyamines, and supplying intermediary metabolism (26, 27). To gain information about the arginine metabolic pathways elicited by inflammation, we used the BMDM LPS/IFNγ stimulation RNA-seq data from Zhang et al. and compared it to our own BMDM datasets from IL4 + IL13 stimulation of BMDMs (3). Either stimulation caused qualitative and quantitative changes in different nodes of arginine metabolic gene expression (Fig. 2E). For example, LPS induced inducible NO synthase (iNOS) (encoded by Nos2) expression as expected, while both diverse stimulations induced Arg1 and expression of cation transporters such as SLC7A2, which import arginine and are essential for NO production and Arg1 activity (28, 29). In activated macrophages, NO production poisons the respiratory chain and causes dependency on glycolysis (2, 30, 31). By contrast, macrophages that cannot make NO maintain OX-PHOS and mitochondrial respiratory integrity that functions concurrently with glycolysis. Accordingly, lactate production (and therefore Kla) should differ between wild-type and NO-deficient macrophages. Therefore, we next asked whether the main enzymes that consume arginine, Arg1, and iNOS could be used to separate the effects of glycolysis-induced lactate and the IL6-mediated production of Arg1. We isolated BMDMs from Nos2−/− mice and stimulated them with LPS over time. LPS-treated Nos2−/− BMDMs or BMDMs treated with the iNOS inhibitor L-Nil expressed higher amounts of Arg1 compared to controls (Fig. 2F and fig. S2C). However, Kla expression was only detected in LPS-stimulated wild-type BMDMs. Furthermore, Nos2−/− BMDMs produced twofold more IL6 than controls (Fig. 2G), and the transfer of CM from these cells stimulated an equivalent increase in Arg1 expression. These experiments confirm that Kla and Arg1 expression are uncoupled and that the biochemical utilization of arginine via NO dictates Kla expression independently from Arg1.
IFNs and histone lactylation
In our hands, Kla increases were coincident with inflammation-induced macrophage death. By contrast, Zhang et al. (3) did not note this association even at 48 hours after LPS stimulation, prompting us to investigate the differences in macrophage cell death between the experimental systems. Therefore, we used an annexin V/propidium iodide (PI) staining to detect viable and apototic cells. We sorted for viable and late apototic/necroptotic cells in BMDMs treated with LPS for 48 hours (Fig. 3A and fig. S3A). Late apototic/necroptotic cells from LPS-stimulated cells showed an increased Kla expression in comparison to the viable cells of the same sample, highlighting the correlation of cell death and Kla. In addition, we noticed variation in the timing of LPS-induced cell death, which was correlated with increased Kla expression. Because of the primary cell system that we used, a variability between the cell death and timing from mouse to mouse was detected, which has an influence on the Kla expression. Therefore, to measure inflammation-induced BMDM death using a different methodology, we used a lactate dehydrogenase (LDH) release assay across a 72-hour window. In all experiments where cellular death was detected, LDH release is presented as fold change to time point zero such that different genotypes and conditions could be compared. In addition, we included the presentation as percentage cytotoxicity (fig. S3C). As a result of the experimental design, where BMDMs have to be replated to use the same cell numbers in all genotypes, we detected some baseline LDH at time point zero. As a partly death-resistant control, we used BMDMs lacking iNOS to confirm that these cells had increased viability even when stimulated for 72 hours with LPS (Fig. 3B) (32). LPS-stimulated macrophages were resistant to Ras-selective lethal (RSL3)–mediated ferroptosis (Fig. 3C and fig. S3B), as anticipated from recent findings (33) and thus ruling out this redox-dependent pathway as a mediator of inflammatory macrophage death. While these experiments indicated modes of inflammation-induced macrophage death, they did not provide insight into the possible differences between our experimental system and that of Zhang et al. (3). Examining the methodology of the BMDM production by Zhang et al. more closely revealed that they used CM from L cells [fibroblasts, L929-cell conditioned media (LCCM)] as the source of colony-stimulating factor 1 (CSF1) required to promote macrophage development and survival, while we use highly purified mammalian-expressed recombinant human CSF1. We therefore generated LCCM and compared it to CSF1 following LPS stimulation. CSF1 was associated with increased Arg1 expression and higher IL6 relative to LCCM, in agreement with previous findings (Fig. 3, D and E) (34). In addition, BMDMs grown in LCCM had increased viability over time of LPS stimulation relative to CSF1-generated cells (Fig. 3F), possibly explaining the fact that we observed Kla expression in the same time window as BMDMs begin to die. In this regard, previous findings demonstrated that BMDMs differentiated by LCCM appear primed and show down-regulation of oxidative stress response, which indicates a less naïve population of macrophages (35).
Fig. 3. Type 1 IFNs and cell death suppression are inversely associated with histone lactylation.
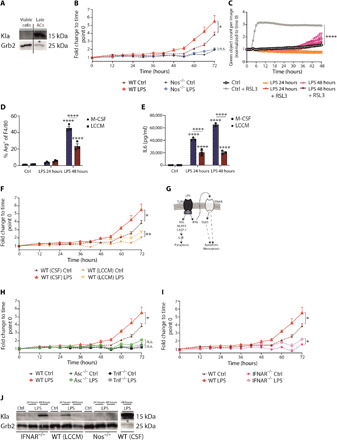
(A) Wild-type BMDMs were treated with LPS for 48 hours, and live/dead staining was used. Cells were sorted for viable cells and late apototic cells (ACs). (B) Wild-type and Nos2−/− BMDMs were left untreated (Ctrl) or were stimulated with LPS, and cell death was assayed by LDH release. (C) Wild-type BMDMs were left untreated or were stimulated with LPS, and where indicated, the ferroptosis inducer RSL3 was added. Cell death was detected via CellTox green cytotoxicity assay. (D) Wild-type BMDMs differentiated with macrophage colony-stimulating factor (M-CSF) or LCCM were stimulated with LPS, and the percentages of Arg1+ of F4/80+ macrophages were determined. (E) The concentration of IL6 in the supernatant was measured by ELISA. (F) Wild-type BMDMs were differentiated with M-CSF or LCCM and stimulated with LPS. LDH release was evaluated over time. (G) Overview of LPS-induced cell death pathways. Wild-type, Asc−/−, and Trif−/− BMDMs (H) or Ifnar−/− BMDMs (I) were stimulated with LPS, and LDH release was evaluated. (J) Ifnar−/−, Nos2−/−, and wild-type BMDMs, differentiated with LCCM or CSF, were left untreated or were stimulated with LPS. * displays that increased cell death was observed. All values are means ± SEM; *P < 0.05; **P < 0.01; ****P < 0.0001. Statistically significant differences were determined by a one-way (A, E, G, and H) or two-way (B to D) ANOVA with Bonferroni correction; n = 3 biological replicates. n.s., not significant.
PANoptosis refers to the combinatorial effects of distinct pathways that drive macrophage death in inflammation and involves coordinated regulation of apoptosis, necroptosis, and pyroptosis as occurs in the cytokine storm during severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections (36). To test links between Kla and different modes of death in PANoptosis (Fig. 3G), we generated BMDMs lacking Trif, a key adapter of TLR signaling linked to death induction, as well as BMDMs deficient of Asc, which controls caspase-1–mediated pyroptosis and a substantial fraction of NLR signaling, and given the key role of type 1 IFNs in NO-induced macrophage death, we used also CSF1-differentiated Ifnar−/− BMDMs (lacking the type 1 IFN receptor) (32). We stimulated each macrophage population with LPS over 72 hours and observed that they were each resistant to death (Fig. 3, H and I). We next tested the hypothesis that there would be a correlation between Kla expression and IFN-regulated cell survival. We therefore generated wild-type BMDMs in CSF1 or LCCM, Ifnar−/− BMDMs, and Nos2−/− BMDMs. Following LPS stimulation, Kla was readily detected in wild-type CSF1 BMDMs (Fig. 3J, right). However, Ifnar−/− and wild-type LCCM BMDMs showed a reduced induction of Kla relative to the wild-type CSF1 BMDMs, showing circumstances where Kla expression was correlated with minimal increase in cell death (Fig. 3I), and Nos2−/− BMDMs had a complete absence of Kla as noted before (Fig. 2F). We isolated human monocyte-derived macrophages (MDMs) from healthy donors and showed that human macrophages, which express NO only at a very low level or not at all, fail to express Kla (fig. S3D) (37). Thus, Kla inversely correlated with the observed reduced cell death in macrophages bearing mutations in central PANoptosis pathways.
Arg1 and macrophage bioenergetics
Macrophages residing in tumor and hypoxic lactate-rich inflammatory microenvironments express high amounts of Arg1 (8–10). Recently, these Arg1+ macrophages were shown to coexpress PD-L1 and Trem2 and termed regulatory macrophages (Mreg), consistent with their potential to suppress T cell proliferation via local arginine depletion and the PD-1 pathway (6, 7). A key question therefore concerned the relationships between Arg1, glycolysis, and lactate of the Mreg phenotype. To probe these relationships, we first modified our LPS stimulation conditions to engender a phenotype consistent with Mreg. We treated BMDMs with LPS for 24 hours and then extensively washed them to reduce the LPS content. These cells were then maintained in low CSF1 for further 24 hours and termed LPS* BMDMs (fig. S4A). LPS* BMDMs expressed Arg1, Trem2, PD-L1, and PD-L2, consistent with the Mreg phenotype (Fig. 4A and fig. S4D). Notably, the expression of Arg1 and PD-L1 was increased relative to BMDMs treated with IL4 + IL13 for 48 hours, which are generally considered “regulatory” macrophages in type 2 immune responses. LPS* BMDMs were expanded in size (fig. S4B), consistent with the Trem2+ macrophages in the tumor microenvironment and completely resistant to ferroptosis (fig. S4, E to G), possibly suggesting that the Mreg phenotype may have an increased survival advantage under redox stress, and were highly phagocytic in both single and double efferocytosis assays (fig. S5, A and B). We next compared LPS* to macrophages treated under the same conditions with BMDMs treated with lactate. Unlike LPS*, lactate did not induce Arg1 as expected and inhibited PD-L1 expression relative to unstimulated baseline BMDM controls (Fig. 4B). Lactate was only able to increase PD-L2 expression to a minor degree. Together, these data support a model where lactate is insufficient to cause an Mreg phenotype.
Fig. 4. Arg1 controls the bioenergetic state of inflammation-induced regulatory macrophages.
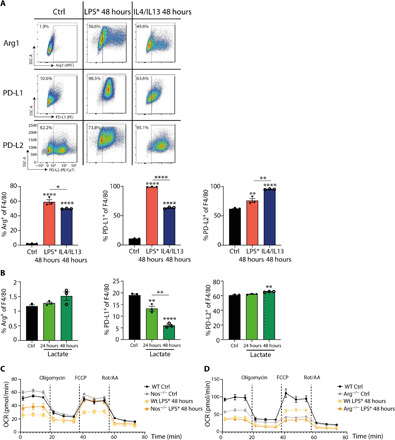
(A) Representative blots of wild-type BMDMs stimulated with LPS or IL4/IL13 for the indicated time and percentages of Arg1+, PD-L1+ and PD-L2+ or F4/80+ macrophages were determined by flow cytometry analysis. Data shown are representative of three independent experiments. SSC-A, side scatter area; APC, allophycocyanin; PE, phycoerythrin. (B) Wild-type BMDMs were stimulated with solvent (Ctrl) or lactate for the indicated time, and the percentages of Arg1+, PD-L1+, and PD-L2+ or F4/80+ macrophages were determined by flow cytometry analysis. Data represent two independent experiments. (C) Wild-type and Nos2−/− BMDMs were left untreated or were treated with LPS*. The cells were sequentially treated with oligomycin, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), and rotenone/antimycin A (Rot/AA), and the oxygen consumption rate (OCR) was determined. (D) Wild-type or Arg1-deficient BMDMs were left untreated or were treated with LPS*, and mitochondrial respiration was measured. All values are means ± SEM; *P < 0.05; **P < 0.01; and ****P < 0.0001. Statistically significant differences were determined by one-way ANOVA with Tukey correction. If not indicated otherwise, superscripts show statistical significance compared to the control group. n = 3 biological replicates.
To determine whether the Mreg phenotype is also NO dependent in pyruvate-elicited respiration (30), we stimulated wild-type and Nos2−/− BMDMs with LPS* and analyzed their mitochondrial metabolism and observed that the Mreg phenotype is dependent on the presence of NO in LPS-induced reduction of mitochondrial respiration (Fig. 4C). Unlike the absence of iNOS, the lack of Arg1 resulted in a reduction of mitochondrial respiration in LPS*-treated BMDMs (Fig. 4D). Thus, arginine metabolism through the iNOS and Arg1 pathways is required for the overall bioenergetic state of macrophages during prolonged inflammatory stimulation. Therefore, Mreg, which express high Arg1, may be protected from inflammatory duress in the hypoxic and lactate-rich tumor microenvironments, potentially accounting for their association with protumor functions.
In summary, Kla and lactate in general are uncoupled from changes in macrophage activation state and gene expression associated with tissue repair and, especially, Arg1. Instead, autocrine-paracrine cytokine signaling via IL6 is responsible for the effects observed on arginine metabolism and Arg1 expression. By contrast, arginine metabolism through NO in collaboration with other death pathways is primarily responsible for macrophage death and accrual of Kla. Furthermore, the iNOS and Arg1 pathway is necessary for an intact bioenergetic state of proinflammatory macrophages, which is especially in the context of hypoxic tumor-associated macrophages with an increased Arg1 expression of relevance. Additional in vivo genetic-based studies are needed to investigate correlations between Arg1+ macrophages and tumor development and outcomes.
MATERIALS AND METHODS
Experimental design
Mice
C57BL/6 (wild type), Stat6−/− (purchased from the Jackson Laboratory, no. 002828; Bar Harbor, ME), Arg1 conditional knockout mice (14) and actin–green fluorescent protein mice were mated and maintained under specific pathogen–free conditions at the animal facility of the Max Planck Institute of Biochemistry. Il6−/−, Myd88−/−, and Nos2−/− mice were bred within the Animal Resource Center at St. Jude Children’s Research Hospital, and bone marrow was frozen and transferred to the Max Planck Institute of Biochemistry. Il6ra−/− mice were bred at the Institute of Biochemistry and Molecular Biology II, Medical Faculty, Heinrich Heine University, Düsseldorf, Germany. Bones from Asc−/− and Trif−/− were a gift from V. Hornung (Gene Center and Department of Biochemistry, Ludwig-Maximilians-Universität München, Munich, Germany). Ifnar−/− mice were bred at the Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna, Vienna, Austria. Use of mice for breeding and organ isolation was approved by the Government of Upper Bavaria and North Rhine-Westphalia, the policies of the St. Jude Children’s Research Hospital Institution Animal Care and Use Committee, and the guidelines at the animal facility of the Institute of Animal Breeding and Genetics, University of Veterinary Medicine Vienna.
Macrophage culture
BMDMs were grown in Dulbecco’s modified Eagle’s medium (DMEM) (41966-029; Thermo Fisher Scientific) composed of 10% fetal bovine serum (S0115; Biochrome), penicillin-streptomycin, and CSF1 as described (13). All cell culture was done at 37°C with 5% CO2. LPS stimulation (5 ng/ml) was performed with LPS from Escherichia coli O111:B4 (L4391; Sigma-Aldrich) for the indicated time. Where indicated, the BMDMs were stimulated with IFNγ (2 ng/ml) (315-05; Peprotech), IL4 (10 ng/ml) (produced in insect cells), IL13 (10 ng/ml) (210-13; Peprotech), 25 mM lactate (83601; Carl Roth), and IL6 (2 ng/ml) or 500 nM rotenone (R8875; Sigma-Aldrich). As JAK2 inhibitor, 20 μM tyrphostin AG490 (T3434; Sigma-Aldrich) was used and added 30 min before LPS treatment.
Human MDMs
All experiments with blood from human volunteer donors were approved by the blood donation service from Boehringer Ingelheim Pharma GmbH and Co. KG following ethical standards and local regulation. Human MDMs were isolated from healthy donors and differentiated for 7 days in the presence of macrophage CSF (M-CSF).
Column filtration experiments
The supernatant of LPS-stimulated BMDMs (48 hours) was fractionated with Amicon-Ultra 100-, 10-, and 3-kDa columns (Merck). If the volume was too low (like in >100 and >10 kDa fractions), unconditioned media was added. The fractionated CM was left for further 24 hours on before unstimulated BMDMs.
Ferroptosis experiments
The protocol was adopted from (33). A total of 0.02 × 106 BMDMs were plated on a 24-well plate. To induce ferroptotic cell death, 500 nM RSL3 (S8155; Selleckchem), which was added for the last 5 hours of LPS stimulation time, and 2 μM ML162 (SML2561; Sigma-Aldrich) and 20 μM erastin (21910-3406; Tebu), which were both added for the last 18 hours of stimulation, were used. Where indicated, 400 nM ferrostatin-1 (SML0583; Sigma-Aldrich), for the last 5 hours, was used to protect for ferroptotic cell death. Cell death was detected via CellTox Green Cytotoxicity Assay (G8731; Promega) and monitored using the IncuCyte Live Cell Analysis System (Sartorius, USA). The green object count was detected with an IncuCyte software. With the help of positive and negative controls, we defined an analysis mask to detect green objects. The analysis parameters were identical for all groups and time points.
RNA sequencing
RNA samples were subjected to RNA-seq and quantified using Qubit 2.0 according to the manufacturer’s instructions. Libraries were prepared and sequenced on an Illumina NovaSeq 6000 platform. Raw mapped reads were processed in R to determine differentially expressed genes and generate normalized read counts. For comparison of untreated versus LPS-/IFNγ-treated BMDMs, available RNA-seq data from GSE115354 were used.
Seahorse bioenergetic measurements
Real-time oxygen consumption rate and extracellular acidification rate measurements were made with a Seahorse XF HS Mini Analyzer (Agilent). A total of 0.04 × 106 BMDMs were plated into each well of Seahorse cell culture plates and preincubated at 37°C for a minimum of 45 min in the absence of CO2 in Seahorse XF DMEM medium (103575-100; Agilent) supplemented with glutamine, glucose, and pyruvate. After analysis, normalization was made by protein quantification.
RNA isolation and analysis
The BMDMs were lysed in TRIzol (15596018; Invitrogen) for subsequent RNA isolation according to the manufacturer’s instructions. Complementary DNA was synthesized using SuperScript IV reverse transcriptase (18090050; Invitrogen), random hexamers (N8080127; Invitrogen), and oligo(dT) primers (N8080128; Invitrogen) and analyzed by quantitative reverse transcription polymerase chain reaction. Arg1, Retnla, and Mrc1 primer sets were purchased from Qiagen. Primer sequences specific for Pdcd1lg2 (written 5′ to 3′) are CCT CTG AGT CGG ATG GAA CC (forward) and GGC CAG GAA GAT CAA AGC GA (reverse). All values were normalized to glyceraldehyde-3-phosphate dehydrogenase (4352932E; Applied Biosystems).
Immunoblotting
Lysates were prepared from BMDMs on ice in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors (78444; Thermo Fisher Scientific). Proteins were separated on tris-HCl gradient gels (5678084; Bio-Rad) and transferred to nitrocellulose. Membranes were blocked in 5% bovine serum albumin (A2153; Sigma-Aldrich) or 5% nonfat milk (T145.2; Carl Roth) and probed with primary antibodies overnight at 4°C. Anti-pStat6 (56554), Stat6 (5397), pStat3 (9131), and Stat3 (4904) antibodies were obtained from Cell Signaling Technology. Anti-Arg1 (GTX109242) antibody was purchased from Genetex and anti-Kla (PTM-1401) antibody from PTM Biolabs. As a loading control, anti-Grb2 (610112) antibody was used from BD Biosciences. Membranes were washed and probed with secondary antibodies at a 1:10,000 dilution and developed using chemiluminescence reagents.
Flow cytometry
BMDMs were suspended in fluorescence-activated cell sorting (FACS) buffer and incubated with Fc block (anti-mouse CD16/32; BioLegend). Cell surface markers were stained with anti-F4/80 (123108 or 123124; BioLegend), anti–PD-L1 (124308; BioLegend), anti–PD-L2 (107214; BioLegend), anti-Trem2 (FAB17291G; R&D) and, for live/dead staining, with LDFA (LIVE/DEAD Fixable Aqua Dead Cell Stain Kit; L34957; Thermo Fisher Scientific). Afterward, the cells were washed, permeabilized with the Foxp3/Transcription Factor Staining Buffer Set (00-5523-00; Thermo Fisher Scientific), blocked with 2% normal mouse serum (Invitrogen), and stained for intracellular Arg1 (17-3697-82; Thermo Fisher Scientific). The cells were washed twice and then resuspended for analysis on a BD LSRFortessa FACS flow cytometer. Gating strategy is shown in fig. S4C. For live/dead staining, APC Annexin V Apoptosis Detection Kit with PI (640932; BioLegend) was used, and the cells were sorted on a BD FACSAria. Data analysis was carried out using FlowJo software.
IL6 enzyme-linked immunosorbent assay
Analysis of IL6 levels in cell supernatant was performed with an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s protocol (M6000B; R&D Systems).
Intracellular lactate concentration
Intracellular l-lactate concentrations were determined by using a colorimetric lactate assay according to the manufacturer’s protocol (ab65331; Abcam).
LDH assay
Intracellular LDH concentrations were determined by using a CyQuant LDH cytotoxicity assay according to the manufacturer’s protocol (C20301; Thermo Fisher Scientific).
Statistical analysis
The significance of differences in the experimental data were determined using GraphPad Prism software. All data involving statistics are presented as means ± SEM. The number of replicates and the statistical test used are described in the figure legends.
Acknowledgments
The Asc−/− and Trif−/− mice were provided by V. Hornung. We thank A. Sahu for experimental assistance and insight with lactate biology. Funding: This work was supported by the Deutsche Forschungsgemeinschaft FOR 2599 (Type 2 Tissue Immunity), the Max Planck Gesellschaft, and an Alexander von Humboldt Fellowship (S.D.). Author contributions: S.D. performed most of the experiments; L.L., L.Z., and D.S. performed experiments. K.B. and J.S. bred and supplied the Il6R –/– mice. B.S. bred and supplied the Ifnar–/– mice. K.C.E.K. contributed to data interpretation. S.D. and P.J.M. conceived the project and wrote the manuscript. Competing interests: D.S. and K.C.E.K. are employees of Boehringer Ingelheim Pharma GmbH & Co KG. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. RNA-seq data from GSE115354 and GSE169348 were used. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg3505/DC1
REFERENCES AND NOTES
- 1.O’Neill L. A., Pearce E. J., Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Everts B., Amiel E., van der Windt G. J., Freitas T. C., Chott R., Yarasheski K. E., Pearce E. L., Pearce E. J., Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang D., Tang Z., Huang H., Zhou G., Cui C., Weng Y., Liu W., Kim S., Lee S., Perez-Neut M., Ding J., Czyz D., Hu R., Ye Z., He M., Zheng Y. G., Shuman H. A., Dai L., Ren B., Roeder R. G., Becker L., Zhao Y., Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diskin C., Ryan T. A. J., O’Neill L. A. J., Modification of proteins by metabolites in immunity. Immunity 54, 19–31 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Irizarry-Caro R. A., McDaniel M. M., Overcast G. R., Jain V. G., Troutman T. D., Pasare C., TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc. Natl. Acad. Sci. U.S.A. 117, 30628–30638 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katzenelenbogen Y., Sheban F., Yalin A., Yofe I., Svetlichnyy D., Jaitin D. A., Bornstein C., Moshe A., Keren-Shaul H., Cohen M., Wang S.-Y., Li B., David E., Salame T.-M., Weiner A., Amit I., Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell 182, 872–885.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Molgora M., Esaulova E., Vermi W., Hou J., Chen Y., Luo J., Brioschi S., Bugatti M., Omodei A. S., Ricci B., Fronick C., Panda S. K., Takeuchi Y., Gubin M. M., Faccio R., Cella M., Gilfillan S., Unanue E. R., Artyomov M. N., Schreiber R. D., Colonna M., TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell 182, 886–900.e17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmona-Fontaine C., Deforet M., Akkari L., Thompson C. B., Joyce J. A., Xavier J. B., Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. U.S.A. 114, 2934–2939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colegio O. R., Chu N.-Q., Szabo A. L., Chu T., Rhebergen A. M., Jairam V., Cyrus N., Brokowski C. E., Eisenbarth S. C., Phillips G. M., Cline G. W., Phillips A. J., Medzhitov R., Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arlauckas S. P., Garren S. B., Garris C. S., Kohler R. H., Oh J., Pittet M. J., Weissleder R., Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 8, 5842–5854 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J., Muri J., Fitzgerald G., Gorski T., Gianni-Barrera R., Masschelein E., D’Hulst G., Gilardoni P., Turiel G., Fan Z., Wang T. T., Planque M., Carmeliet P., Pellerin L., Wolfrum C., Fendt S.-M., Banfi A., Stockmann C., Soro-Arnáiz I., Kopf M., De Bock K., Endothelial lactate controls muscle regeneration from ischemia by inducing M2-like macrophage polarization. Cell Metab. 31, 1136–1153.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qualls J. E., Neale G., Smith A. M., Koo M. S., DeFreitas A. A., Zhang H., Kaplan G., Watowich S. S., Murray P. J., Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci. Signal. 3, ra62 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutschman R., Lang R., Hesse M., Ihle J. N., Wynn T. A., Murray P. J., Cutting edge: Stat6-dependent substrate depletion regulates nitric oxide production. J. Immunol. 166, 2173–2177 (2001). [DOI] [PubMed] [Google Scholar]
- 14.El Kasmi K. C., Qualls J. E., Pesce J. T., Smith A. M., Thompson R. W., Henao-Tamayo M., Basaraba R. J., Konig T., Schleicher U., Koo M.-S., Kaplan G., Fitzgerald K. A., Tuomanen E. I., Orme I. M., Kanneganti T. D., Bogdan C., Wynn T. A., Murray P. J., Toll-like receptor–induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 9, 1399–1406 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray P. J., Macrophage Polarization. Annu. Rev. Physiol. 79, 541–566 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Murray P. J., Allen J. E., Biswas S. K., Fisher E. A., Gilroy D. W., Goerdt S., Gordon S., Hamilton J. A., Ivashkiv L. B., Lawrence T., Locati M., Mantovani A., Martinez F. O., Mege J.-L., Mosser D. M., Natoli G., Saeij J. P., Schultze J. L., Shirey K. A., Sica A., Suttles J., Udalova I., van Ginderachter J. A., Vogel S. N., Wynn T. A., Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 41, 14–20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang C., Yang G., Jiang T., Huang K., Cao J., Qiu Z., Effects of IL-6 and AG490 on regulation of Stat3 signaling pathway and invasion of human pancreatic cancer cells in vitro. J. Exp. Clin. Cancer Res. 29, 51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J.-S., Lee J., Lim M.-A., Kim E.-K., Kim S.-M., Ryu J.-G., Lee J. H., Kwok S.-K., Park K.-S., Kim H.-Y., Park S.-H., Cho M.-L., JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T Cells and Th17 cells. J. Immunol. 192, 4417–4424 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Tong A.-J., Liu X., Thomas B. J., Lissner M. M., Baker M. R., Senagolage M. D., Allred A. L., Barish G. D., Smale S. T., A stringent systems approach uncovers gene-specific mechanisms regulating inflammation. Cell 165, 165–179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qing H., Desrouleaux R., Israni-Winger K., Mineur Y. S., Fogelman N., Zhang C., Rashed S., Palm N. W., Sinha R., Picciotto M. R., Perry R. J., Wang A., Origin and function of stress-induced IL-6 in murine models. Cell 182, 372–387.e14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hargreaves D. C., Horng T., Medzhitov R., Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138, 129–145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El Kasmi K. C., Pugliese S. C., Riddle S. R., Poth J. M., Anderson A. L., Frid M. G., Li M., Pullamsetti S. S., Savai R., Nagel M. A., Fini M. A., Graham B. B., Tuder R. M., Friedman J. E., Eltzschig H. K., Sokol R. J., Stenmark K. R., Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J. Immunol. 193, 597–609 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baran P., Hansen S., Waetzig G. H., Akbarzadeh M., Lamertz L., Huber H. J., Ahmadian M. R., Moll J. M., Scheller J., The balance of interleukin (IL)-6, IL-6.soluble IL-6 receptor (sIL-6R), and IL-6.sIL-6R.sgp130 complexes allows simultaneous classic and trans-signaling. J. Biol. Chem. 293, 6762–6775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jostock T., Müllberg J., Özbek S., Atreya R., Blinn G., Voltz N., Fischer M., Neurath M. F., Rose-John S., Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 268, 160–167 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Schumacher N., Meyer D., Mauermann A., von der Heyde J., Wolf J., Schwarz J., Knittler K., Murphy G., Michalek M., Garbers C., Bartsch J. W., Guo S., Schacher B., Eickholz P., Chalaris A., Rose-John S., Rabe B., Shedding of endogenous interleukin-6 receptor (IL-6R) is governed by a disintegrin and metalloproteinase (ADAM) proteases while a full-length IL-6R isoform localizes to circulating microvesicles. J. Biol. Chem. 290, 26059–26071 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murray P. J., Amino acid auxotrophy as a system of immunological control nodes. Nat. Immunol. 17, 132–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qualls J. E., Murray P. J., Immunometabolism within the tuberculosis granuloma: Amino acids, hypoxia, and cellular respiration. Semin. Immunopathol. 38, 139–152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson R. W., Pesce J. T., Ramalingam T., Wilson M. S., White S., Cheever A. W., Ricklefs S. M., Porcella S. F., Li L., Ellies L. G., Wynn T. A., Cationic amino acid transporter-2 regulates immunity by modulating arginase activity. PLOS Pathog. 4, e1000023 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicholson B., Manner C. K., Kleeman J., MacLeod C. L., Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J. Biol. Chem. 276, 15881–15885 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Palmieri E. M., Gonzalez-Cotto M., Baseler W. A., Davies L. C., Ghesquière B., Maio N., Rice C. M., Rouault T. A., Cassel T., Higashi R. M., Lane A. N., Fan T. W.-M., Wink D. A., McVicar D. W., Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 11, 698 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McNeill E., Stylianou E., Crabtree M. J., Harrington-Kandt R., Kolb A.-L., Diotallevi M., Hale A. B., Bettencourt P., Tanner R., O’Shea M. K., Matsumiya M., Lockstone H., Müller J., Fletcher H. A., Greaves D. R., McShane H., Channon K. M., Regulation of mycobacterial infection by macrophage Gch1 and tetrahydrobiopterin. Nat. Commun. 9, 5409 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zwaferink H., Stockinger S., Reipert S., Decker T., Stimulation of inducible nitric oxide synthase expression by beta interferon increases necrotic death of macrophages upon Listeria monocytogenes infection. Infect. Immun. 76, 1649–1656 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapralov A. A., Yang Q., Dar H. H., Tyurina Y. Y., Anthonymuthu T. S., Kim R., St. Croix C. M., Mikulska-Ruminska K., Liu B., Shrivastava I. H., Tyurin V. A., Ting H.-C., Wu Y. L., Gao Y., Shurin G. V., Artyukhova M. A., Ponomareva L. A., Timashev P. S., Domingues R. M., Stoyanovsky D. A., Greenberger J. S., Mallampalli R. K., Bahar I., Gabrilovich D. I., Bayır H., Kagan V. E., Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 16, 278–290 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Brito Monteiro L., Davanzo G. G., de Aguiar C. F., Correa da Silva F., Andrade J. R., Campos Codo A., Silva Pereira J. A. D., Freitas L. P., Moraes-Vieira P. M., M-CSF- and L929-derived macrophages present distinct metabolic profiles with similar inflammatory outcomes. Immunobiology 225, 151935 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Heap R. E., Marín-Rubio J. L., Peltier J., Heunis T., Dannoura A., Moore A., Trost M., Proteomics characterisation of the L929 cell supernatant and its role in BMDM differentiation. Life Sci. Alliance 4, e202000957 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karki R., Sharma B. R., Tuladhar S., Williams E. P., Zalduondo L., Samir P., Zheng M., Sundaram B., Banoth B., Malireddi R. K. S., Schreiner P., Neale G., Vogel P., Webby R., Jonsson C. B., Kanneganti T.-D., Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168.e17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinberg J. B., Misukonis M. A., Shami P. J., Mason S. N., Sauls D. L., Dittman W. A., Wood E. R., Smith G. K., McDonald B., Bachus K. E., Haney A. F., Granger D. L., Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): Analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 86, 1184–1195 (1995). [PubMed] [Google Scholar]
- 38.Yurdagul A. Jr., Subramanian M., Wang X., Crown S. B., Ilkayeva O. R., Darville L., Kolluru G. K., Rymond C. C., Gerlach B. D., Zheng Z., Kuriakose G., Kevil C. G., Koomen J. M., Cleveland J. L., Muoio D. M., Tabas I., Macrophage metabolism of apoptotic cell-derived arginine promotes continual efferocytosis and resolution of injury. Cell Metab. 31, 518–533.e10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg3505/DC1