

A multilayer hydrogel nanofilm on the cell surface via enzymatic reactions provides a new platform for β cell–based therapy.
Abstract
Pancreatic β cell therapy for type 1 diabetes is limited by low cell survival rate owing to physical stress and aggressive host immune response. In this study, we demonstrate a multilayer hydrogel nanofilm caging strategy capable of protecting cells from high shear stress and reducing immune response by interfering cell-cell interaction. Hydrogel nanofilm is fabricated by monophenol-modified glycol chitosan and hyaluronic acid that cross-link each other to form a nanothin hydrogel film on the cell surface via tyrosinase-mediated reactions. Furthermore, hydrogel nanofilm formation was conducted on mouse β cell spheroids for the islet transplantation application. The cytoprotective effect against physical stress and the immune protective effect were evaluated. Last, caged mouse β cell spheroids were transplanted into the type 1 diabetes mouse model and successfully regulated its blood glucose level. Overall, our enzymatic cross-linking–based hydrogel nanofilm caging method will provide a new platform for clinical applications of cell-based therapies.
INTRODUCTION
Cell transplantation, emerging as a promising strategy in the field of regenerative medicine, consists of delivering live cells or organoids to a patient as a therapeutic drug to repair, reinforce, or replace the biological functions of damaged tissues (1–3). Currently, incurable diseases—such as diabetes, myocardial infarction, osteoarthritis, and spinal cord injuries—have been fundamentally treated via cell transplantation to actively ameliorate disease conditions (1, 4). Despite the benefits, challenges remain including poor viability of injected transplants and vigorous host immune response (4–7). Direct injection of transplants causes mechanical stress against the injection pressure and fluidic shear forces, which led to necrotic and apoptotic cell death by cell membrane rupture (2, 8). Moreover, allogeneic or xenogeneic cell transplantation triggers inflammatory host immune response through direct and indirect immune recognition (9–11). Many efforts have used biomaterials to overcome the low survival rate and vigorous immune rejection of transplantation (1, 2). Among them, encapsulation with biomaterials shed a light to solve the challenges above (12, 13). Working as a protective sacrificial layer and local immunoisolation strategy, encapsulation thereby improves the cell viability against the external stress and host immune system.
Unfortunately, the conventional islet encapsulation methods still have several challenges: large volume ratio of a cell to polymer (14), poor diffusion of oxygen and nutrients (1, 15), heterogeneous coverage due to random trapping (16), and pericapsular fibrotic overgrowth around transplantation site (17). Limitations of these approaches include the fact that durable and stable layers are required to form feasible thickness for transplantation and prevent immune response induced by foreign-body reaction of biomaterials. Therefore, a nanothin, porous, and durable islet encapsulation method—which is sufficiently permeable to nutrient/waste exchange, long-term glycemic control, and immunoisolation effect without the need for systemic immunosuppression—is highly desired.
Here, we fabricated a new concept of the pancreatic β cell spheroid caging system by an enzymatic cross-linking–based hydrogel nanofilm with high stability and cytoprotective property. The hydrogel nanofilm caging system is composed of biocompatible polysaccharide layers of glycol chitosan (GC) and hyaluronic acid (HA). Layers were cross-linked by the monophenol residues conjugated on each polysaccharide, via Streptomyces avermitilis–derived tyrosinase (SA-Ty)–mediated enzymatic reaction. Compared to other tyrosinase, SA-Ty has a flat, wide, and shallow active site that broadens the substrate specificity and shows superior reactivity on long-chain polymers, which accelerates polymer cross-linking (18). We gradually accumulated polysaccharide layers to establish a hydrogel nanofilm on the surface of a single-cell and pancreatic β cell spheroid. In addition, demonstration of our hydrogel nanofilm as a physical barrier against the external environment was validated by the long-term hydrogel nanofilm conservation, endurance against external pressure, and reduction of cell-cell interaction with natural killer cell (NK cell). Last, the hydrogel nanofilm formed by SA-Ty on mouse pancreatic β cell spheroids was able to return type 1 diabetes (T1D)–induced mouse to normoglycemia, compared to other groups. By analyzing long-term blood glucose concentration, intraperitoneal glucose tolerance test (IPGTT), and immunohistochemistry, successful engraftment and therapeutic efficacy of nanofilm-caged spheroids were confirmed.
RESULTS
Synthesis and characterization of biomaterials
To fabricate the multilayer hydrogel nanofilm caging system, two oppositely charged polysaccharides, GC and HA, were chosen as building blocks for layer-by-layer (LbL) assembly (Fig. 1). In addition, tyrosinase-reactive monophenol residues are introduced to each polysaccharide to provide an enzyme-mediated cross-linking reactivity. Through the 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)–N-hydroxysuccinimide (NHS) coupling reaction, monophenol residue–conjugated GC (GC-T) and HA (HA-T) were synthesized, respectively (Fig. 2A). 1H–nuclear magnetic resonance spectra of GC-T showed peaks at 6.791 and 7.100 parts per million (ppm), whereas that of HA-T showed peaks at 6.847, 7.161, 7.272, and 7.447 ppm, which are corresponding to the protons of phenols (fig. S1). The degree of substitution (DS) was calculated as a ratio of integrated areas of a proton of monophenol to three protons of N-acetyl group. DS of GC-T was 10.66%, and that of HA-T was 17.50%. ζ potential of GC-T and HA-T was measured to be 7.962 ± 1.457 and −17.392 ± 3.763 mV, respectively, indicating that both polysaccharides maintained their original charges regardless of monophenol conjugation (Fig. 2B). Then, the enzymatic reactivity of SA-Ty with the modified polysaccharides was measured (Fig. 2C). Oxidation of monophenols rapidly interacts with organic and inorganic substrates via hydrogen bond interaction, Michael addition reaction, and Schiff base reaction (19, 20). The initial oxidation rate, V0, of SA-Ty on GC-T was 2.779 ± 0.046 μM min−1 and HA-T was 1.423 ± 0.023 μM min−1 when SA-Ty (0.0028 U/ml) and 0.1% of each substrate were applied. The reaction solutions turned their color to brown within reaction time. After the full reaction, the Fourier transform infrared (FTIR) spectra were obtained using lyophilized samples (Fig. 2D). The broad peak of the O─H bond at 3100 to 3500 cm−1 increased, and the peak of the aromatic C═C bond decreased at 1500 to 1800 cm−1, indicating that diphenol and o-quinones were increased due to monophenol oxidation mediated by tyrosinase.
Fig. 1. Representative scheme of β cell spheroid transplantation with an enzymatic cross-linking–based LbL hydrogel nanofilm caging system.
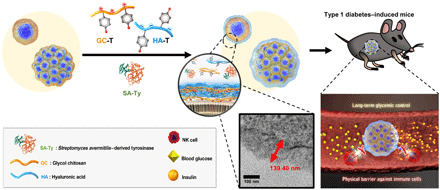
Fig. 2. Synthesis and characterization of GC-T and HA-T, with cross-linking reaction by SA-Ty.
(A) Synthesis of GC-T and HA-T by conjugation of monophenols to GC and HA. (B) ζ potential of 0.1% (w/v) GC-T and 0.1% (w/v) HA-T. (C) Enzymatic reactivity profile of SA-Ty reacted with 0.1% (w/v) GC-T and 0.1% (w/v) HA-T. (D) FTIR spectra of fully oxidized GC-T and HA-T by SA-Ty. (E) Schematic illustration of the enzymatic two-step oxidative reaction mediated by tyrosinase and nonenzymatic cross-linking reactions of o-quinone. (F) Images of 5% (w/v) GC-T, 5% (w/v) HA-T, and 2.5% (w/v) GC-T and 2.5% (w/v) HA-T hydrogel cross-linked by tyrosinase at day 0 and day 1 in PBS. (G) Comparison of swelling ratio between 5% (w/v) GC-T, 5% (w/v) HA-T, and 2.5% (w/v) GC-T and 2.5% (w/v) HA-T hydrogel (N = 6). (H) Schematic illustration of the experimental setup and the diffusion profile of FITC-dextran (20 and 70 kDa) across six layers of GC-T and HA-T (L6) cross-linked by SA-Ty (N = 3). Error bars denote means ± SD. *P < 0.05; **P < 0.01.
The mechanisms of the hydrogel nanofilm formation involve enzymatic oxidative reaction mediated by SA-Ty and cross-linking reactions of o-quinone with an amine, thiol, and other quinones (Fig. 2E). The catalysis of monophenols to o-diphenols (catechol) and o-quinones by the tyrosinase requires two-step oxidations (20). These o-quinones on both polysaccharides form covalent bonds with amines, thiols, and other quinones on cell surface molecules and other polysaccharides (18). To verify such cross-linking by SA-Ty with GC-T and HA-T, we demonstrated bulk hydrogel formation using a high concentration of polysaccharides (Fig. 2F). 5% (w/v) GC-T and 5% (w/v) HA-T individually formed a hydrogel cross-linked by SA-Ty (0.05 U/ml). A half-and-half mixture of GC-T and HA-T [2.5% (w/v) each] formed a more stable hydrogel than the other two individual hydrogels, which aroused from additional electrostatic interactions between GC-T and HA-T. The swelling ratio of each hydrogel represented its polysaccharide characteristic (Fig. 2G). Hydrophilic HA-T–based hydrogel swelled the most; in contrast, GC-T, which is known to be hydrophilic under acidic conditions, did not swell as much in phosphate-buffered saline (PBS), and the hydrogel mixed with both polysaccharides showed the intermediate swelling ratio. Moreover, to analyze the porosity and porous structure of the LbL-formed hydrogel nanofilm, the diffusion profiles of fluorescein isothiocyanate (FITC)–conjugated dextran across the nanofilm were examined. The diffusion test of FITC-dextran molecules (20 and 70 kDa) was conducted in a transwell system with a membrane insert with a six-layered hydrogel nanofilm on it (Fig. 2H). Diffusion profile showed that FITC-dextran (20 kDa) of a lower molecular weight diffused much faster and in larger quantities through the transwell and nanofilm than FITC-dextran (70 kDa) of a higher molecular weight. The amount of diffused FITC-dextran molecules (20 and 70 kDa) across the hydrogel nanofilm was notably less than that of the transwell itself.
Optimization of hydrogel nanofilm formed by the LbL method
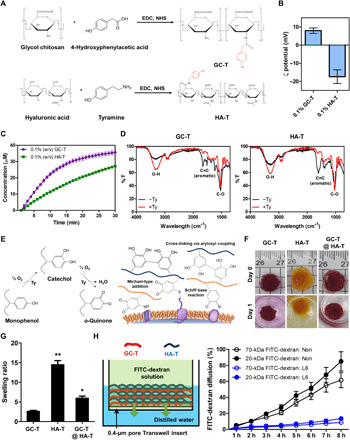
Before forming the hydrogel nanofilm on the cell surface, we verified the biocompatibility of SA-Ty and optimized the hydrogel nanofilm formation (fig. S2A). To optimize the condition for the formation of hydrogel nanofilm on the cell surface, the degree of the coating was investigated with various parameters as follows: the concentration of SA-Ty, reaction time, and the concentration of polysaccharide (fig. S2, B and C). With the optimized SA-Ty treatment, the effect of SA-Ty on cell coating with RITC (Rhodamine B isothiocyanate)–conjugated GC-T (GC-T-RITC) and fluorescein amine isomer I–conjugated HA-T (HA-T-FA) was investigated (Fig. 3A). Fluorescence intensity differs in each polysaccharide as a normal cell surface naturally exposes a negative charge. GC-T with a positive charge is more attractive than negatively charged HA-T. Also, as external GC-T tends to be close to the cell surface, treatment of SA-Ty greatly increased the intensity of GC-T-RITC up to 2.9-fold, while HA-T-FA showed less dependency on SA-Ty. Moreover, the coating efficiency of negatively charged HA-T-FA substantially increased 2.4-fold when GC-T-RITC had been coated before HA-T-FA (Fig. 3B). To inspect the feasibility of the LbL assembly of hydrogel nanofilm, frequency changes were measured in each stacked layer on an O2 plasma–treated Cr/Au electrode by quartz crystal microbalance (Fig. 3C). In the sixth layer, the cumulative mass per area of hydrogel film was 90.16 μg/cm2 without SA-Ty but increased to 148.50 μg/cm2 with SA-Ty (Fig. 3C). The average amount of film deposited increased to 1.6-fold with SA-Ty than without. In addition, layers treated with SA-Ty gradually increased up to 10 layers, but the increasing amount decreased after 6 layers. In contrast, layers formed only by electrostatic force have shown that their mass increment became plateau after six layers. Thus, we designated six layers of polysaccharides as the optimal layers of hydrogel nanofilm on the cell surface. Furthermore, the addition of SA-Ty with the six layers of polysaccharides on MIN6 cells stabilized the nanofilm on the cell surface and did not cause any cytotoxicity (fig. S2, D to F).
Fig. 3. LbL hydrogel nanofilm on the single-cell surface.
(A) The effect of SA-Ty on GC-T and HA-T coating on the cell surface (N = 3). (B) The efficiency difference of HA-T-FA coating on different cell surface charges (N = 3). (C) Quantitative analysis of LbL deposition of GC-T and HA-T on the cell surface. (D) Schematic image of single-cell LbL hydrogel nanofilm formation. (E) Changes in ζ potential of the encapsulated cell surface by the increment of layers (N = 5). (F) Flow cytometry analysis of encapsulated cells based on the increasing numbers of layers. GC-T-RITC and HA-T-FA were detected using the PE channel and FITC channel, respectively. (G) Confocal laser microscopic images of native and L6-encapsulated cells. Scale bars, 10 μm. (H) TEM images of native and L6-encapsulated cell surface. Error bars denote means ± SD.
LbL single-cell caging with hydrogel nanofilm
We then applied the cellular coating by dipping the cell-loaded 30-μm polycarbonate transwell membrane into the coating solutions, medium, and PBS sequentially to minimize cell damage during the typical centrifugation process (Fig. 3D). GC-T was applied for the first and odd-numbered layers, and HA-T was applied for the even-numbered layers. Noncoated cells (termed “native”) and (n)-layer nanofilm-caged cells [termed “L(n)”] were compared for surface charge and stable hydrogel film formation. Cell surface ζ potential switched as we applied GC-T and HA-T alternatively (Fig. 3E). Similarly, fluorescent intensity shifted as we increased the layers with RITC-labeled GC-T (GC-T-RITC) or FITC-labeled HA-T (HA-T-FA) (Fig. 3F). Visualization of nanofilm-caged cells with GC-T-RITC and HA-T-FA was confirmed by a confocal laser microscopy analysis (Fig. 3G). In addition, the cell membranes before and after hydrogel nanofilm coating were visualized by transmission electron microscopy (TEM) images (Fig. 3H). Hydrogel nanofilm was densely fabricated on cell membranes with a thickness of an average 139.40 ± 7.73 nm when six layers were stacked.
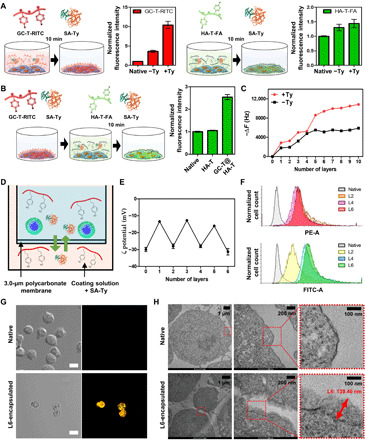
Application of hydrogel nanofilm caging to MIN6 β cell spheroids
LbL cell encapsulation method constructed above for single cells was further applied to β cell spheroids (Fig. 4A). MIN6 β cells were cultured into spheroid form before nanofilm caging since connectivity and cell-cell interactions in β cell clusters are essential for glycemic control. Fabricated β cell spheroids had an average diameter of 350.0 ± 24.5 μm (fig. S3, A to C), and we further applied L6 with SA-Ty. Hydrogel nanofilm on the spheroids was visualized using fluorescence-labeled polysaccharides (Fig. 4A). The fluorescence intensity on the cell surface increased as the number of layers increased with a homogeneous coating of macromolecules (fig. S3D). Furthermore, the surface of native β cell spheroids and L6 nanofilm–caged β cell spheroids was imaged by scanning electron microscopy (SEM) (fig. S3E). Native β cell spheroids displayed a smooth surface without detectable structures, while L6 nanofilm–caged β cell spheroids displayed homogeneous covering of cross-linked macromolecules on the cell surface. We further confirmed the homogeneous covering of hydrogel nanofilm caging on β cell spheroids by confocal image analysis (fig. S3, F to I).
Fig. 4. Encapsulation of β cell spheroids by L6 hydrogel nanofilm.
(A) Schematic and fluorescence images of β cell spheroid encapsulated with hydrogel nanofilm (HA-T-FA in green and GC-T-RITC in red). (B) Live/Dead assay images of native and L6-encapsulated β cell spheroids. Live cells are represented in green, and dead cells are represented in red. Scale bars, 100 μm. (C) The difference in insulin level between native and L6-encapsulated β cell spheroids at low- and high-glucose solutions (N = 4). (D) Comparison of SI of native and L6-encapsulated β cell spheroids calculated by dividing the insulin level at a high-glucose solution by the insulin level at a low-glucose solution. For reference, 25 clusters equate to ~106 cells. Error bars denote means ± SD. *P < 0.05.
Upon application of L6 nanofilm with SA-Ty on β cell spheroids, we assessed the viability of spheroids up to 7 days in vitro and compared it with native β cell spheroids (Fig. 4B). On the basis of the live/dead assay images, most cells were viable in both groups and maintained their spherical shape. To assess whether the L6 hydrogel nanofilm caging altered cellular functionality, glucose-stimulated insulin secretion (GSIS) assay was performed and compared with the native group (Fig. 4C). Both native β cell spheroids and L6 nanofilm–caged β cell spheroids were induced to secrete insulin in response to the low- or high-glucose solution in PBS. Insulin levels of low-glucose solution exposed that the native β cell spheroid and L6 nanofilm–caged β cell spheroid groups were similar. In contrast, L6 nanofilm–caged β cell spheroids secreted a higher amount of insulin compared to the native β cell spheroids when exposed to the high-glucose solution. Stimulation index (SI), calculated by dividing the insulin level at the high-glucose solution by the insulin level at the low-glucose solution, was 4.1-fold higher in the L6 group compared to the native group (Fig. 4D). In addition, we further examined whether the hydrogel nanofilm layering interfered with the insulin secretion and genes related to β cell functions (fig. S4). As we varied the hydrogel nanofilm layering from L2 to L8, our system did not interfere with insulin secretion. In addition, the insulin secretion index was comparable to the native group in both low- and high-glucose solutions. Similarly, the hydrogel nanofilm caging system did not alter the expressions of GLUT2, Ins-1, and Ins-2 genes that are involved in glucose sensing and insulin synthesis.
Hydrogel nanofilm as a physical barrier against the external environment
Cell damage and apoptosis caused by external physical stress such as syringe pressure and blood fluidic shear force are the major challenges in cell transplantation. By the stability and cross-linking advantage of the LbL hydrogel nanofilm driven by SA-Ty, we envisioned that our hydrogel nanofilm can overcome these challenges. To analyze the enhanced sustainability of nanofilm compared to common LbL with polyelectrolytes, L6 nanofilm–caged β cell spheroids, with/without SA-Ty–derived cross-linking, were incubated for 6 days and imaged every 2 days (Fig. 5A). Images represented that the LbL hydrogel nanofilm cross-linked by SA-Ty was able to stably maintain. In contrast, the fluorescence of the LbL formed without SA-Ty dwindled as time went by, indicating that cross-linking with SA-Ty reinforces the interaction between polysaccharides to persist against the exogenous environment. To investigate whether the hydrogel nanofilm protects β cell spheroids from physical stress, both native and L6 groups were repeatedly centrifuged at low speed (81g) and high speed (1073g) (Fig. 5B and fig. S5, A and B). Spheroids in both native and L6 groups maintained their spherical shape at low centrifugal speed; however, at high speed, the native group collapsed while the L6 group withstood the high pressure, proving the durability of the hydrogel nanofilm. Furthermore, the L6 nanofilm–caged β cell spheroid endured the protease attack as we challenged them with trypsin or collagenase (fig. S5, C and D). Native β cell spheroids collapsed within both proteases and dissociated easily with mild pipetting; however, L6 nanofilm–caged β cell spheroids showed less cell detachment and maintained their structure. In addition, to approve hydrogel nanofilm as a physical barrier, inhibition of cell-cell interaction between the spheroids and NK cells was examined, as recognition of peptides or sugars on the cell surface of antigen-presenting cells is a priority to the active immune system in T cells at transplantation (Fig. 5C) (21). Before validating β cell spheroids, we cocultured an L6 nanofilm–caged K562 cell, a human myelogenous leukemia cell line that is easily killed by NK cells, with NK-92 cells, an NK cell line (22). First, verification of single K562 cell coating with GC-T-RITC and HA-T-FA was demonstrated (fig. S6A). Then, green-labeled L6 nanofilm–caged K562 cells were cocultured with blue-labeled NK-92 cells (fig. S6, B and C). A significant decrease in the binding frequency of NK-92 cells was detected in L6 nanofilm–caged K562 cells compared to native K562 cells. On the basis of the successful inhibition effect on K562 single-cell data, L6 β cell spheroids were cocultured with NK-92 cells. Numerous NK-92 cells were detected on the surrounding surface of the native group, but notably, lower coverage was gained in the L6 group (Fig. 5, D and E). By the interaction of NK-92 cell to the β cell spheroids, native β cell spheroid size depleted in time lapse as NK cell dissociated β cells off from the spheroid starting from its surface. However, L6 spheroids conserved their size, representing the reduction of cell-cell interaction with NK cells (Fig. 5F). In addition, coculture with primary splenocytes for 24 hours showed the same loss of cell-cell interaction results in L6 β cell spheroids (fig. S6D). After 24 hours, peripheral β cells on the surface of the native group were observed to protrude outward from the spheroids. In contrast, L6 retained the integrity of the spheroidal form. Thus, with the properties of long-term conservation, endurance against external pressure, and reduction of cell-cell interaction, very attractive and interesting characteristics of the physical barrier were achieved.
Fig. 5. Hydrogel nanofilm as a physical barrier to β cell spheroids.
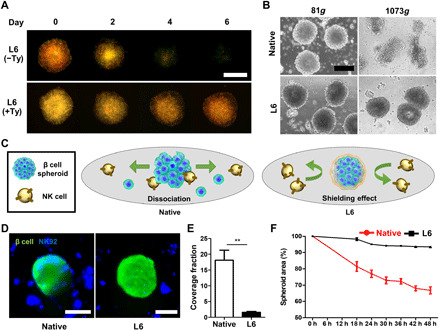
(A) Stability test of hydrogel nanofilm until 1 week. Treatment of SA-Ty prolonged the stability of hydrogel nanofilm. (B) Morphological changes of spheroids with/without hydrogel nanofilm against external stress. (C) Schematic images of immune protection by the L6 hydrogel nanofilm. (D) Confocal microscopic images of β cell spheroids with/without hydrogel nanofilm cocultured with NK-92 cell. (E) Comparison of the coverage fraction of NK cells on each β cell spheroid group (**P < 0.01, N = 3). (F) Percentage reduction of β cell spheroid area by NK cells dissociating β cells from the spheroid (N = 3). Scale bars, 200 μm. Error bars denote means ± SD.
In vivo evaluation of glycemic control in diabetic mice
We further examined the functional effects of the L6 hydrogel nanofilm–caged β cell spheroid on reverse hyperglycemia and regulation of blood glucose levels in a streptozotocin-induced T1D mouse model (Fig. 6A). The 150 L6 spheroids were homogeneously transplanted into the subcapsular membrane of the kidney. For three control groups, sham, native spheroid, and L6 spheroid without SA-Ty–mediated cross-linking were also injected. Transplantation of L6 spheroids with SA-Ty cross-linking efficiently decreased blood glucose level from hyperglycemic condition to normoglycemic level and maintained its state for up to 30 days (Fig. 6B). In contrast, without SA-Ty cross-linking, L6 spheroids did not stably retain blood glucose level to normoglycemic level, as the nanofilm encapsulated by SA-Ty enhanced glucose responsiveness of L6 spheroids. Other control groups showed elevated blood glucose levels throughout the transplant time. Moreover, removal (nephrectomy) of the kidney containing the transplanted spheroids raised the blood glucose level to the hyperglycemic level in the L6 spheroid with SA-Ty cross-linking group, representing that the regulation of blood glucose level in T1D mouse was controlled by the transplanted L6 spheroids only. The weight gain of the treated mice followed the opposite trend of blood glucose level, as the L6 spheroid with SA-Ty cross-linking group gained weight; on the other hand, the other control groups lost their weight because of uncontrolled blood glucose levels (Fig. 6C). After nephrectomy, the mice transplanted with L6 spheroid with SA-Ty cross-linking started to lost weight. In vivo glucose-responsive ability of spheroids was also confirmed by performing IPGTT (Fig. 6D). Sixteen hours after transplantation, the L6 spheroids with SA-Ty cross-linking group sufficiently restored glucose tolerance when challenged with a bolus dose of glucose. Histological analysis and expression of insulin confirmed the stable engraftment and sufficient functionality of transplanted spheroids (Fig. 6E). These results provide evidence that encapsulation of β cell spheroid with L6 hydrogel nanofilm with SA-Ty cross-linking can maintain the regulation function of blood glucose level in vivo and restore euglycemia in diabetic mice.
Fig. 6. β cell spheroids encapsulated with SA-Ty–mediated hydrogel nanofilm sustain normoglycemia in streptozotocin-treated BALB/C mice.
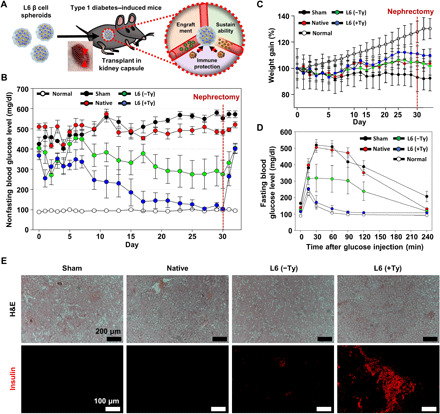
(A) Schematic image of transplantation of β cell spheroids into the kidney capsule of BALB/C mice. Nonfasting blood glucose level (B) and body weight (C) of mice in each group were monitored periodically (N = 4). (D) Measurement of the blood glucose level of mice that were subjected to IPGTT after implantation (N = 4). (E) Histological analysis of implants retrieved 30 days after implantation from the streptozotocin-treated BALB/C mice, represented with hematoxylin and eosin (H&E) staining and immunostaining of insulin in each group. Scale bars, 200 μm (top images) and 100 μm (bottom images). Error bars denote means ± SD.
DISCUSSION
T1D, one of the global epidemic diseases, is an autoimmune disease characterized by the destruction of pancreatic β cells by the patient’s immune system, resulting in high blood glucose levels due to the deficiency of insulin (23–25). Since patients with T1D have few functional pancreatic β cells, an external supply of insulin or the addition of functional pancreatic β cell is required for glycemic control (26). Clinical trials of islet transplantation using biomaterials have increased in number and vindicated its safety and effectiveness for T1D (25). Islets are encapsulated in biocompatible devices or hydrogel microbeads composed of alginate and polyelectrolytes before Langerhans transplantation to allow diffusion of insulin and nutrients necessary for cell function and avoid the need for systemic immunosuppression (12, 23). Even though pancreatic β cell encapsulation within semipermeable hydrogels such as alginate beads represents a functional cure for patients afflicted with T1D, the alginate-based system usually permits ~500-μm-sized beads, which limit the rapid response of glucose sensing and insulin secretion as glucose has to travel several hundred micrometers into the alginate beads.
In this study, we developed an enzymatic cross-linking–based hydrogel nanofilm on the surface of pancreatic β cell spheroids using two kinds of polysaccharides with opposite charges and recombinant tyrosinase enzyme. We envisioned that LbL methods may have an advantage over bulk hydrogel–based encapsulation systems as they may permit the rapid glucose response and insulin secretion as the cells are enclosed within very thin layers of macromolecules. There are many cell-coating strategies, pioneered by M. Matsusaki’s group (27). The LbL assembly of polymers is considered a promising approach for cell encapsulation. However, previous strategies relying on LbL coatings mainly used electrostatic interactions alone. Electrostatic interactions based on LbL coatings have been shown to have a relatively short duration due to their small dissociation energy (28–30). In this study, we report the formation of an LbL hydrogel nanofilm caging system on β cell spheroids with mild and rapid enzymatic cross-linking by SA-Ty. By using chitosan and HA, we were able to form an ultrathin LbL hydrogel on β cell spheroids. In previous studies, 3,4-dihydroxy-l-phenylalanine (dopa)–conjugated macromolecules have been used for adhesive hydrogel due to the oxidation of dopa and formation of o-quinone (31, 32). However, dopa-conjugated macromolecules self–cross-linked to form a hydrogel before forming an LbL hydrogel nanofilm with oppositely charged macromolecules (fig. S7). Compared to the dopa-modified macromolecules, the use of SA-Ty allowed us to initiate the oxidation phenolic moiety on HA or chitosan. Furthermore, it allowed us to cross-link the macromolecules after the LbL hydrogel formation. Thus, by applying SA-Ty on LbL-assembled tyramine-conjugated chitosan and HA, we eliminated any nonspecific cross-linking, reduced the cross-linking time, controlled the cross-linking initiation, and allowed a stable hydrogel nanofilm caging system. Last, covalent bonding by the enzyme enabled stable protection against harsh host environments.
In our study, the LbL hydrogel nanofilm was optimized to perform best in L6-caged β cell spheroids. According to our study, insulin gene expression of nanofilm-caged β cells was improved. Closer cell-cell interaction and cellular condensation have been reported to enhance the glucose responsiveness of β cells (33, 34). Furthermore, transmembrane proteins—including GLUT2 transporter, ATP-sensitive K+ channel (K+ATP), voltage-dependent Na+ channel (VDNaC), voltage-dependent Ca2+ channel (VDCC), voltage-dependent K+ channel (K+V), and gap junction channels—play a pivotal role in the mechanism of glucose-responsive insulin secretion and are well influenced by the environmental factors (35, 36). In our study, the stable hydrogel nanofilm on β cell spheroids may have allowed closer compaction and increased cell-cell contact, leading to the enhanced glucose responsiveness. Along with TEM and confocal images, we confirmed the stable and firm hydrogel nanofilm on the cell membrane. This integral structure on the outer layer of β cell spheroids may have induced β cell function via inducing subtle structural changes on membrane channels without cytotoxicity or functional impairment. Overall, the nanofilm caging of spheroids with the LbL hydrogel nanofilm did not hinder β cell glucose sensitivity. However, detailed molecular analysis and influence on the cellular plasma membrane protein would be desirable.
Stability and diffusivity characterization is an important aspect for cellular nanofilm caging. We observed a stable coating of the first layer of GC-T with the enzymatic reaction on the mammalian cell surface as analyzed using Jurkat cells (fig. S8). We noted that cells are stably coated with 0.1% GC-T-RITC, and they repelled from each other due to electrostatic repulsion, while a lesser amount of GC-T-RITC resulted in cellular clumping. The result confirmed that the positive charges of the GC-T resulted in a homogeneous charge distribution on the cell surface. Furthermore, this homogeneous positive charge distribution on the cell surface by GC-T allowed subsequent homogeneous LbL coating with HA-T. The LbL hydrogel nanofilm acted as a physical barrier against the external environment that endures higher external pressure, reduced acute host immune response based on cell-cell interaction with NK cells, and external cytokine attack. Our nanofilm hydrogel displayed discriminative molecular diffusivity that allowed for free inward and outward diffusion of low–molecular weight nutrients such as glucose, amino acids, and insulin. Moreover, the nanofilm caging system was effective in the cytokine attack. We treated tumor necrosis factor–α (TNF-α; 10 nM) for 48 hours to stimulate cell death and analyzed the apoptotic gene expressions with real-time polymerase chain reaction (PCR). When cells without the hydrogel nanofilm caging system were treated with TNF-α, it elevated significant levels of apoptotic genes such as p53 and Caspase-3. Cells with the hydrogel nanofilm caging system did not respond to the TNF-α treatment, indicating that the nanofilm caging system can prevent cytokine attacks (fig. S9). Furthermore, we have examined that a molecular weight near 20 kDa cannot diffuse across the L6 nanofilm, which supports that the hydrogel nanofilm caging system can protect the cells with cytokine attack while it did not interfere with insulin secretion, which is about 5.8 kDa (Fig. 2H and fig. S4). In addition, the hydrogel nanofilm displayed an impassable barrier against host immune cells that efficiently prevented immune response. By controlling the thickness of the nanofilm, we were able to create a passive barrier between the β cell spheroids and the NK cells. Sufficient thickness that is higher than the cellular arm of the immune system (8 to 22 amino acids in length), which prevents the direct interaction of immune cells to spheroid, was achieved in L6 (37). For these reasons, we confirmed that L6 was the optimal choice of hydrogel nanofilm for our study on β cell spheroid encapsulation as it showed no adverse effect on cell function. Last, for the therapeutic applications, we implanted L6-caged β cell spheroids into T1D mice to regulate blood glucose level in vivo and achieved significantly successful results compared to the noncaged β cell spheroid group. This hydrogel nanofilm formation approach can be effectively used for frequent islet allotransplantation and may be further extended to xenografts in immunocompetent animals over long periods.
MATERIALS AND METHODS
Cell culture
Jurkat cells (Jurkat Clone E6-1, American Type Culture Collection TIB-152) were cultured in 75-cm2 tissue culture flasks with cell culture growth medium at 37°C under humidified atmosphere containing 5% CO2. The cell culture growth medium was RPMI 1640 (Gibco, catalog no. 11995-065) containing 10% fetal bovine serum (FBS; Gibco, catalog no. 16000-044) and 1% penicillin/streptomycin (P/S; Gibco, catalog no. 150710-063). Cells were harvested by centrifugation for subculture or experiments. Media was changed every 2 days. MIN6 cells (provided by Yongsung Hwang laboratory at Soonchunhyang University) were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) containing 15% FBS, 1% P/S, and 55 μM 2-ME (2-Mercaptoethanol) and incubated in 5% CO2 at 37°C. Media was changed every 2 days. K562 cells were cultured in RPMI 1640 media supplemented with 10% FBS and 1% P/S. NK-92 cells were cultured in MEM a media (Gibco, catalog no. 12561-056) supplemented with 12.5% FBS, 12.5% horse serum (Gibco, catalog no. 16050-122), 1% P/S, and human interleukin-2 (20 ng/ml; Peprotech, catalog no. 200-02). All cells were maintained at 37°C with 5% CO2 in a humidified incubator.
Synthesis of GC-T, HA-T, GC-T-RITC, and HA-T-FA
4-Hydroxyphenylacetic acid (HPA) was conjugated to GC via EDC/NHS coupling reaction. First, 200 mg of GC was dissolved in 10 ml of 0.1 M MES buffer (pH 4.7) at 70°C until fully dissolved. HPA (160.82 mg) was dissolved in 10 ml of MES buffer, and 202.64 mg of EDC and 114.76 mg of NHS were added to the solution and stirred for 5 min. Then, two solutions were mixed and reacted overnight at room temperature (RT). Next, the solution was dialyzed (SnakeSkin Dialysis Tubing, Mw cutoff of 1 kDa; Thermo Fisher Scientific) against distilled water for 72 hours and lyophilized for more than 72 hours. In the same manner, HA-T was synthesized by conjugating tyramine hydrochloride to HA. Two hundred milligrams of HA was dissolved in 20 ml of MES buffer, and 197.452 mg of EDC and 111.822 mg of NHS were added and stirred for 5 min. Then, 178.85 mg of tyramine hydrochloride was added and reacted overnight at RT. Subsequently, the final solution was dialyzed and lyophilized. One milliliter of 10 mg/ml of RITC and FA solution, dissolved in N,N-dimethylformamide, was added to the reacting solution of GC-T and HA-T, respectively, to obtain GC-T-RITC and HA-T-FA. To compare the difference between mono- and dihydroxyphenol conjugation, 3,4-dihydroxyphenylacetic acid was conjugated instead of HPA to synthesize GC-dopa.
Expression and purification of recombinant tyrosinase from S. avermitilis
The plasmid was constructed in the previous study (38). Briefly, melC2 gene of tyrosinase (Ty) was extracted from S. avermitilis, and the gene was inserted in MCS2 of pETDuet (Novagen, USA) with His-tag introduced at the C terminus of SA-Ty. For the protein expression and purification of SA-Ty, the plasmid was transformed into Escherichia coli BL21 (DE3) by heat shock and selected on Luria-Bertani (LB) agar plate containing ampicillin (100 μg/ml). A single colony was inoculated into 5 ml of LB broth with ampicillin and cultured overnight in a 37°C shaking incubator at 200 rpm. Then, 2 ml of cell culture was transferred into a 1-liter flask with 200 ml of fresh LB containing ampicillin (100 μg/ml). The cells were grown to an optical density at 600 nm of 0.6, and protein expression was induced by adding 0.2 mM isopropyl-β-d-1-thiogalactopyranoside and 1 mM CuSO4. After 20 hours at 18°C, cells were harvested by centrifugation at 4000 rpm for 10 min at 4°C. Cell pellets were washed once, resuspended in 50 mM tris-HCl buffer (pH 8.0), and disrupted by ultrasonication. Cell lysates were centrifuged at 15,000 rpm for 30 min at 4°C to remove debris. The supernatant was collected and filtered through a sterilized 0.2-μm polyethersulfone membrane (Acrodisc syringe filter with Supor membrane, Pall Life Sciences, USA). The enzyme was purified by the general His-tag purification using Ni-NTA agarose column (QIAGEN). The final enzyme solution was aliquoted and kept at −20°C in tris-HCl buffer (pH 8.0) containing 25% glycerol. The concentration of purified SA-Ty was determined by bicinchoninic acid assay.
Measurement of the enzymatic activity of SA-Ty
SA-Ty (2.5 μM), 5 μM CuSO4, and the substrate [1% (w/v) GC-T, 1% (w/v) HA-T, or 200 μM l-tyrosine] were prepared in a total volume of 200 μl of 50 mM tris-HCl buffer (pH 8.0). The absorbance at 475 nm (εdopachrome = 3600 M−1 cm−1) at 37°C was measured every 1 min for 30 min with a microplate reader (Infinite M200 PRO, TECAN, Switzerland). The initial rate of SA-Ty reaction was defined as the slope of a plot of the product concentration and the reaction time. The enzyme activity is quoted in units per milliliter, where 1 U is the amount of SA-Ty that catalyzes the reaction of 1 μmol of l-tyrosine per minute into dopachrome.
Attenuated total reflection FTIR
A 0.1% GC-T and 0.1% HA-T solution was reacted with SA-Ty (0.05 U/ml) for 30 min at RT. Then, the reaction solution was frozen and lyophilized. The solid sample was directly placed on the zinc selenide attenuated total reflection (ATR) crystal surface of ATR-FTIR (Spectrum 100 FTIR spectrometer, PerkinElmer, USA). The bare ATR crystal was used as a background. The FTIR spectra were obtained by scanning the transmittance at 700 to 4000 cm−1.
Hydrogel formation and swelling ratio
Bulk hydrogels were formed by using a high concentration of polysaccharides. 5% (w/v) GC-T and 5% (w/v) HA-T individually formed a hydrogel cross-linked by adding SA-Ty (0.05 U/ml) for cross-linking. Also, a mixture of 2.5% (w/v) GC-T and HA-T formed a hydrogel by the same procedure. To measure the swelling ratios of the GC-T and HA-T hydrogel, the dry weight of each hydrogel group was measured after freeze-drying. Then, dried hydrogels were incubated in PBS at 37°C for 1 day. The swollen samples were measured after drying the excess solution on the surfaces of the samples. The swelling ratios were calculated by the following equation: swelling ratio (%) = (Ws − Wi)/Wi × 100, where Ws indicates the wet weight of the samples, and Wi indicates the initial dried weight of the samples.
Diffusion test of L6 hydrogel nanofilm
To evaluate the porosity and porous structures of the L6 hydrogel nanofilm, the diffusion test was performed in a transwell system, a common experimental tool for measuring permeability and molecular diffusion. Before L6 hydrogel nanofilm formation, a transwell insert was treated with plasma for a negative charge. Then, the L6 hydrogel nanofilm was formed on a transwell insert (6.5 mm diameter of polycarbonate membrane with 0.4-μm pore size; Corning, USA), and 200 μl of FITC-dextran solutions (0.5 mg/ml; Sigma-Aldrich) with different molecular weights (20 and 70 kDa) was added on top of the transwell insert. Then, the insert was loaded into a 24-well plate containing 800 μl of distilled water. The diffusion profiles of FITC-dextran molecules through the L6 hydrogel nanofilm were determined by collecting the solutions in the 24-well plate at each time point and measuring the fluorescence intensity using a microplate reader (Infinite M200, TECAN, Switzerland).
Cell coating with polysaccharides and SA-Ty
MIN6 β cells were seeded on a tissue culture plate and cultured for 3 days. First, cells were washed twice with PBS. Then, GC-T-RITC solution and SA-Ty were added. After incubation, cells were washed twice with PBS. The degree of the coating was confirmed by measuring the red fluorescence of GC-T-RITC at λex = 543 nm/λem = 580 nm (RITC). Similarly, green fluorescence at λex = 495 nm/λem = 525 nm (FA) was measured to detect HA-T-FA. For suspension cells, cells were collected in a conical tube by centrifugation for every step.
Quartz crystal microbalance
Cr/Au (chromium/gold) crystal (5 MHz, 2.54 cm diameter, AT-cut, plano-plano) was used to deposit GC-T/HA-T layers. Before the deposition, the crystal was treated with piranha solution (H2SO4/H2O2 = 3:1) for 5 min and oxygen plasma for 5 min to clean and set a negative charge on the surface. Next, the crystal was dipped into the 0.1% GC-T solution with SA-Ty (0.05 U/ml). After 10 min, the electrode was rinsed twice with PBS for 1 min. To eliminate the remaining PBS, the crystal was dried with an air blower. LbL deposition proceeded with 0.1% HA-T solution until additional five bilayers were stacked. The amount of hydrogel film deposited on the crystal was analyzed by quartz crystal microbalance (QCM200, Stanford Research Systems, USA) for each layer. The accumulative mass can be calculated by the following equation
where Δf is the observed frequency change in hertz, Δm is the change in mass per unit area in grams per square centimeter, and Cf is the sensitivity factor for the crystal (56.6 Hz μg−1 ∙ cm2 for a 5-MHz AT-cut quartz crystal at RT).
LbL encapsulation of single cells
Before each encapsulation process, the activity of SA-Ty was measured. Lyophilized GC-T and HA-T were dissolved at 10 mg/ml in 0.1% acetic acid and PBS, respectively. After fully dissolved, the solutions were diluted 10 times with PBS, making a final concentration of 1 mg/ml, and filtered through a sterilized 0.2-μm membrane. Jurkat cells were collected, washed twice with PBS, and prepared at the density of 1 × 107 cells per 1 ml of PBS. One hundred microliters of cell suspension was seeded into a 3.0-μm polycarbonate membrane (Transwell 6.5-mm insert, 24-well plate; Corning). After 600 μl of GC-T solution (1 mg/ml) and SA-Ty (0.05 U/ml) were added into a well of a 24-well plate, the cell-containing membrane was dipped for 10 min at RT. During incubation, the plate was tapped every 2 min. After 10 min, the membrane was transferred to the next well with 500 μl of media for 30 s once and 500 μl of PBS for 1 min twice on a shaking incubator. Subsequently, HA-T and GC-T were applied alternatively in the same manner. When the last layer was finished, the cells were dispersed into a cell culture medium.
ζ potential
Cells were fixed with 4% paraformaldehyde (PFA) for 10 min and prepared at a density of 1 × 106 cells per 1 ml of PBS. The ζ potential of native or nanofilm-caged cells was measured with Nano ZS (Malvern Instruments, Germany).
Flow cytometry
Cells were caged with GC-T-RITC and HA-T-FA. After fixing, cells were prepared at 1 × 106 cells per 500 μl of PBS. Fluorescence of RITC and FA was measured by flow cytometry (FACSAria II, BD Biosciences, USA) using lasers of wavelengths of 488 and 633 nm. One-layer–caged cells with GC-T-RITC and HA-T-FA were used as positive controls for gating.
Confocal laser scanning microscopy
After nanofilm caging with GC-T-RITC and HA-T-FA, cells were fixed with 4% PFA for 15 min at RT. Cells were placed on a 20-mm confocal dish and imaged via a confocal microscope (LSM 780, Carl Zeiss, Germany).
Transmission electron microscopy
To confirm the nanothin hydrogel on the cell surface, TEM (Talos L120C, 120 kV, FEI, Czech) image was analyzed. For the preparation of the TEM sample, native and nanofilm-caged cells were fixed with Karnovsky’s fixative. Cells were treated with 1% osmium tetroxide in cacodylate buffer for 1 hour and 0.5% uranyl acetate overnight at 4°C. After dehydrating in ethanol, the samples were embedded in Spurr’s resin. The specimens were sectioned by using an ultramicrotome (EM UC7, Leica, Germany).
Scanning electron microscopy
Images were observed by field-emission SEM (JSM-6701F, JEOL) at 10 μA and 3 to 5 kV. For the preparation of spheroid samples, native and nanofilm-caged spheroids were fixed with 2.5% glutaraldehyde solution. After fixation, spheroids were dehydrated in ethanol and dried using HMDS (hexamethyldisilazane). Before imaging, samples were coated with platinum for 120 s in a vacuum.
LbL encapsulation of pancreatic β cell spheroid
To fabricate β cell spheroids, MIN6 pancreatic β cells were detached from the tissue culture plate with 0.25% trypsin-EDTA. Then, the cells were seeded into an ultralow attachment 96-well plate (Corning, USA) at a density of 5 × 103 cells per well in 100 μl of culture medium and incubated in 5% CO2 at 37°C for 3 days. The assembled spheroids were collected in 15-ml conical tubes and washed twice with PBS. One hundred spheroids were placed on the 3.0-μm polycarbonate membrane, and we proceeded with LbL encapsulation in the same manner as the single-cell encapsulation. After encapsulation, β cell spheroids were transferred to a sterile 35-mm dish in a culture medium. Cell viability was measured by staining the cells with the Live/Dead Viability/Cytotoxicity Kit that contains calcein-AM and ethidium homodimer-1 (EthD-1). After imaging with the fluorescence microscope (EVOS Cell Imaging Systems, Thermo Fisher Scientific), cells were counted in four separate fields. The viability was calculated by dividing the live cell number by the total cell number.
Functional analysis of β cell spheroid
GSIS test was performed to evaluate β cell functionality. β cell spheroids were washed twice with D-PBS and incubated in 500 μl of D-PBS for 1 hour in 5% CO2 at 37°C. After 1 hour, low-glucose solution (3.3 mM glucose in D-PBS) and high-glucose solution (20 mM glucose in D-PBS) were treated to different groups and incubated for 2 hours in a 37°C incubator. The supernatants of the glucose solution, which contain the secreted protein of insulin, were collected from each well. The amount of insulin was measured via mouse insulin enzyme-linked immunosorbent assay (Mouse Insulin ELISA, ALPCO, NH, USA) according to the manufacturer’s instructions. β cell spheroids were lysed with 0.2% Triton X-100 in TE buffer for 30 min with vortexing every 5 min. PicoGreen assay was performed to quantitate double-stranded DNA of the cells in whole. The insulin level was obtained by dividing the total amount of secreted insulin by the amount of DNA. The SI was calculated as the amount of the insulin level under the high-glucose condition divided by the insulin level under the low-glucose condition.
Real-time PCR
Samples were prepared by TRIzol reagent treatment and collected in a 1.8-ml tube. Chloroform was added to the tube and vigorously shaken by hand for 10 s. After a 5-min incubation at RT, the cells were centrifuged (21,055g, 20 min, 4°C). The clear aqueous phase on top was transferred to a new tube. Then, isopropanol was added and inverted several times. After another incubation at RT for 5 min, the cells were centrifuged (21,055g, 20 min, 4°C). The white RNA pellet was collected by eliminating the top aqueous phase, washed with 75% ethanol, dissolved completely in molecular grade water, and denatured for 10 min at 60°C. The RNA concentration was measured, and complementary DNA was prepared by reverse transcription using the EZ006M Kit (Enzynomics, Korea) according to the manufacturer’s instructions. Gene expression levels of pancreatic β cell markers, GLUT-2, insulin-1 (Ins1), and insulin-2 (Ins2) were determined by real-time PCR using SYBR Green PCR Mastermix on a StepOnePlus Real-Time PCR System (Applied Biosystems). Complementary DNA samples were analyzed for the gene of interest and the reference housekeeping gene GAPDH.
Physical stress test
For the physical stress test, β cell spheroids were dispersed in 500 μl of PBS in a 1.7-ml Eppendorf tube after washing twice in PBS. Tabletop centrifuge (MiniSpin Plus, Eppendorf, Germany) was used to centrifuge spheroids at 1100 and 4000 rpm for 5 min. Between each step of centrifugation, clustered spheroids were resuspended without changes in PBS. After three times, residual cell traces were stained with calcein-AM and EthD-1 and imaged with a fluorescence microscope. For the trypsinization test, native and nanofilm-caged spheroids were dispersed in 300 μl of 0.05% trypsin-EDTA and incubated at 37°C with shaking. Image was taken at each time point, and after 30 min, 10 times of pipetting by using a 100-μl volume pipette were held to gently disperse the cells into solution. Type 2 collagenase (Worthington, USA) dissolved in high-glucose DMEM was made at a final concentration of 10 mg/ml. Native and nanofilm-caged spheroids were dispersed in collagenase solution and incubated at 37°C with shaking.
Prevention of cell-cell interaction between L6-encapsulated cell/spheroid and NK cell
After preparation of K562 cell or MIN6 spheroid with L6 encapsulation and labeling with CellTracker Green (Invitrogen, catalog no. C7025), NK-92 cells were washed twice with serum-free media and labeled with 1 μM CellTrace Far Red (Invitrogen, catalog no. C34564) at 37°C for 10 min. Then, the labeled cells were washed with a complete medium containing FBS and used for the experiment. For fluorescence image of the interactions between NK-92 cell and K562 cell, the labeled cells were loaded on 18-μm clean coverslips (NK-92, 0.5 × 106 cells/ml; K562, 0.1 × 106 cells/ml) and incubated for 2 hours at 37°C with 5% CO2 in a humidified incubator. Then, fluorescence images were acquired at that time. For spheroid, each spheroid was loaded on a flat 96-well plate (SPL, Korea). To measure the initial size (t = 0 hour) of the spheroid, fluorescence images were acquired without NK-92 cells. After acquiring the initial images, NK-92 cells (0.15 × 106 cells per well) were added to each well. Then, the flat 96-well plate was mounted on a microscope stage equipped with a Chamlide TC incubator system (Live Cell Instrument, Korea), which maintains a cell culture condition (37°C, 5% CO2). Fluorescence images were acquired every 3 hours for 24 hours. The size of the spheroid was measured through the acquired fluorescence image. A modified Olympus IX 83 epifluorescence microscope with a 10× (UPlanFLN, numerical aperture = 0.30) objective lens and an ANDOR Zyla 4.2 sCMOS camera was used for imaging experiments. A U-LH75XEAPO xenon lamp (75 W, Olympus) and green fluorescent protein (EX BP 470/40, BS 495, and EM BP 525/50) and Cy5 (EX BP 620/60, BS 660, and EM BP 770/75) filter sets were used for fluorescence imaging. The microscope was automatically controlled by Micro-manager. Acquired images were analyzed and processed with ImageJ.
Flow cytometry for NK-92 cytotoxicity (SI)
K562 cells labeled with CellTracker Green (105 cells per well) and NK-92 cells (105 cells per well) were added in 96 wells. After incubating for 4 hours, cell suspension was prepared in PBS containing 2% FBS, 0.1% sodium azide (Sigma-Aldrich, USA), and 1 mM EDTA (Sigma-Aldrich, USA) for staining and flow cytometry analysis. To assess cytotoxicity, dead cells were labeled with CYTOX Red (Invitrogen, S34859) staining. Cytotoxicity was measured by the percentage of CFSE+CYTOX+ cells. Flow cytometry was performed using FACSCanto II (BD Biosciences), and data were analyzed using FlowJo (FlowJo LLC).
Isolation of primary splenocytes
Isolated mouse spleen was washed with PBS twice and placed on a petri dish. Then, the spleen was mashed by the cap of a 1.8-ml tube. After fine grinding, the cap and dish were rinsed, and we gathered the mashed spleen into a 50-ml conical tube. Additional PBS was poured to reach a total volume of 30 ml. We performed centrifugation at 800g for 3 min, discarded the supernatant, and resuspended the pellet with red blood cell lysis buffer (R7757, Sigma-Aldrich) following the manufacturer’s procedure. After the red blood cell lysis, the solution was placed into a 40-μm cell strainer and washed with 30 ml of PBS twice. Cells were counted with a hemocytometer.
Effect of TNF-α to nanofilm-caged β cells
Native and nanofilm-caged spheroids were dispersed in culture media with 10 nM TNF-α added. At day 2, spheroids were collected, and apoptotic gene expressions were analyzed by real-time PCR.
Induction of T1D mellitus in mice
Eight-week-old BALB/C mice were purchased from DaeHan-Bio link (Chungcheongbuk-do, Republic of Korea). All animal procedures were ensured by the Institutional Animal Care and Use Committee (IACUC). Streptozotocin was dissolved in sodium citrate buffer adjusted to pH 4.5 before use. Streptozotocin (80 mg/kg of weight) was administered to BALB/C mice that were fasted overnight. Diabetes was considered to be induced when nonfasting blood glucose levels were maintained over 300 mg/dl for three consecutive days.
β cell transplantation into diabetic mice
MIN6 insulinoma cells were formed into spheroids (5000 cells per spheroid) and transferred to polyethylene tubes before transplantation. BALB/C mice were anesthetized intraperitoneally by a 4:1 mixture of zoletil and rompun. After the removal of hair, a small incision of the skin was made to extrude the kidneys. The subcapsular membrane of the kidney was carefully cut with needles for the entrance of cell-containing tubes. PBS (sham), 150 noncoated spheroids (native), 150 non–cross-linked spheroids (L6 − Ty), or 150 cross-linked spheroids (L6 + Ty) were infused from the polyethylene tubes into the subcapsular membrane of a kidney with a Hamilton syringe. The cut subcapsular membranes of the kidney were closed with a cautery, and the kidneys were returned to the body. After the skin closure with suture, the mice were moved to prewarmed cages. Weight and nonfasting blood glucose levels of the mice were measured daily until day 7 and once every 2 days thereafter until day 30. For nephrectomy on day 30, BALB/C mice were anesthetized intraperitoneally by a 4:1 mixture of zoletil and rompun. After the removal of hair, a small incision of the skin was made to extrude the kidneys. The blood vessels directly connected to the kidneys were tied tightly with threads to prevent bleeding. The kidneys were cut with surgical scissors and fixed in 10% neutral-buffered formalin (NBF). After the skin closure with suture, the mice were moved to prewarmed cages. Weight and nonfasting blood glucose levels of the mice were measured for a couple of days after nephrectomy. The mice were euthanized after the blood glucose levels of all groups increased over 300 mg/dl.
Intraperitoneal glucose tolerance test
The fasting (16-hour) blood glucose levels of MIN6 spheroid–transplanted mice were measured before the administration of glucose. The blood glucose levels were measured at 15, 30, 60, 90, 120, and 240 min after the intraperitoneal injection of 20% d-glucose (2 g/kg of mouse) dissolved in PBS.
Histological evaluation of kidney capsule sections
After fixation in 10% NBF for 3 days, kidneys were transferred to tissue cassettes and rinsed with running tap water. The kidneys were dehydrated and infiltrated with paraffin using a tissue processor. The tissues were embedded in paraffin blocks and sectioned at 5 μm thickness on microscope slides. After the hydration, the antigens of the tissues were unmasked with sodium citrate buffer (pH 6.0) at 100°C. The sections were blocked with goat serum (Abcam, UK) for 1 hour and bound to rabbit anti-insulin primary antibody (catalog no. ab63820, Abcam) overnight at 4°C. After washing, the sections were bound to goat anti-rabbit secondary antibody Alexa Fluor 594 (Invitrogen) in a dark room for 1 hour. The tissues were washed several times and mounted with coverslips. The stained tissues were examined immediately with a fluorescence microscope.
Statistical analysis
Experiments were carried out at least triplicated for statistical analysis. All data are expressed as means ± SD. Statistical significance was determined by paired Student’s t test with *P < 0.05, **P < 0.01, and ***P < 0.005.
Acknowledgments
Funding: This research was financially supported by the Ministry of Science and ICT (NRF-2016R1E1A1A01943393, NRF-2017M3A9C6031786, NRF-2019M3A9G1023840, NRF-2019R1I1A1A01059554, NRF-2019M3A9H1103786, and NRF-2020R1A2C3005834). This research was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (HI18C0453). The Institute of Engineering Research at Seoul National University provided research facilities for this work. This work is also financially supported by the LG Chemical Global Innovation Grant. Author contributions: N.S.H., M.K., and H.K. designed experiments. N.S.H., M.K., and H.K. prepared the paper. M.K., H.K., and Y.-s.L. generated the hydrogel nanofilm caging system on the pancreatic β cell spheroid. S.J. analyzed characteristics of the hydrogel nanofilm. U.-J.L. generated and purified SA-Ty. S.-E.K. and H.K. performed coculture of a spheroid with NK cells. M.K. and S.L. performed in vivo studies. M.K., S.L., and C.-G.P. analyzed in vivo studies. All authors read and approved the final paper. Competing interests: N.H., M.K., and B.-G.K. are inventors on a patent, filed in Korea (10-2021-0042722) and internationally (PCT/KR2021/004203), related to this work filed by Seoul National University, serial number PCT/KR2021/004203, filed on 5 April 2021. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabf7832/DC1
REFERENCES AND NOTES
- 1.Farina M., Alexander J. F., Thekkedath U., Ferrari M., Grattoni A., Cell encapsulation: Overcoming barriers in cell transplantation in diabetes and beyond. Adv. Drug Deliv. Rev. 139, 92–115 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Mitrousis N., Fokina A., Shoichet M. S., Biomaterials for cell transplantation. Nat. Rev. Mater. 3, 441–456 (2018). [Google Scholar]
- 3.Kim H., Shin K., Park O. K., Choi D., Kim H. D., Baik S., Lee S. H., Kwon S. H., Yarema K. J., Hong J., Hyeon T., Hwang N. S., General and facile coating of single cells via mild reduction. J. Am. Chem. Soc. 140, 1199–1202 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Stephan M. T., Irvine D. J., Enhancing cell therapies from the outside in: Cell surface engineering using synthetic nanomaterials. Nano Today 6, 309–325 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hasturk O., Kaplan D. L., Cell armor for protection against environmental stress: Advances, challenges and applications in micro- and nanoencapsulation of mammalian cells. Acta Biomater. 95, 3–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster G. A., Garcia A. J., Bio-synthetic materials for immunomodulation of islet transplants. Adv. Drug Deliv. Rev. 114, 266–271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orive G., Hernández R. M., Gascon A. R., Calafiore R., Chang T. M. S., De Vos P., Hortelano G., Hunkeler D., Lacík I., Shapiro A. M. J., Pedraz J. L., Cell encapsulation: Promise and progress. Nat. Med. 9, 104–107 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Yang J., Yang Y., Kawazoe N., Chen G., Encapsulation of individual living cells with enzyme responsive polymer nanoshell. Biomaterials 197, 317–326 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Stabler C. L., Li Y., Stewart J. M., Keselowsky B. C., Engineering immunomodulatory biomaterials for type 1 diabetes. Nat. Rev. Mater. 4, 429–450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pham-Hua D., Padgett L. E., Xue B., Anderson B., Zeiger M., Barra J. M., Bethea M., Hunter C. S., Kozlovskaya V., Kharlampieva E., Tse H. M., Islet encapsulation with polyphenol coatings decreases pro-inflammatory chemokine synthesis and T cell trafficking. Biomaterials 128, 19–32 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kozlovskaya V., Zavgorodnya O., Chen Y., Ellis K., Tse H. M., Cui W. X., Thompson J. A., Kharlampieva E., Ultrathin polymeric coatings based on hydrogen-bonded polyphenol for protection of pancreatic islet cells. Adv. Funct. Mater. 22, 3389–3398 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vegas A. J., Veiseh O., Gürtler M., Millman J. R., Pagliuca F. W., Bader A. R., Doloff J. C., Li J., Chen M., Olejnik K., Tam H. H., Jhunjhunwala S., Langan E., Aresta-Dasilva S., Gandham S., McGarrigle J. J., Bochenek M. A., Hollister-Lock J., Oberholzer J., Greiner D. L., Weir G. C., Melton D. A., Langer R., Anderson D. G., Long-term glycemic control using polymer-encapsulated human stem cell–derived beta cells in immune-competent mice. Nat. Med. 22, 446–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bose S., Volpatti L. R., Thiono D., Yesilyurt V., McGladrigan C., Tang Y., Facklam A., Wang A., Jhunjhunwala S., Veiseh O., Hollister-Lock J., Bhattacharya C., Weir G. C., Greiner D. L., Langer R., Anderson D. G., A retrievable implant for the long-term encapsulation and survival of therapeutic xenogeneic cells. Nat. Biomed. Eng. 4, 814–826 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mao A. S., Shin J.-W., Utech S., Wang H., Uzun O., Li W., Cooper M., Hu Y., Zhang L., Weitz D. A., Mooney D. J., Deterministic encapsulation of single cells in thin tunable microgels for niche modelling and therapeutic delivery. Nat. Mater. 16, 236–243 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colton C. K., Oxygen supply to encapsulated therapeutic cells. Adv. Drug Deliv. Rev. 67-68, 93–110 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Velasco D., Tumarkin E., Kumacheva E., Microfluidic encapsulation of cells in polymer microgels. Small 8, 1633–1642 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Ryan A. J., O’Neill H. S., Duffy G. P., O’Brien F. J., Advances in polymeric islet cell encapsulation technologies to limit the foreign body response and provide immunoisolation. Curr. Opin. Pharmacol. 36, 66–71 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Kim S. H., Lee S. H., Lee J. E., Park S. J., Kim K., Kim I. S., Lee Y. S., Hwang N. S., Kim B. G., Tissue adhesive, rapid forming, and sprayable ECM hydrogel via recombinant tyrosinase crosslinking. Biomaterials 178, 401–412 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Faure E., Falentin-Daudre C., Jerome C., Lyskawa J., Fournier D., Woisel P., Detrembleur C., Catechols as versatile platforms in polymer chemistry. Prog. Polym. Sci. 38, 236–270 (2013). [Google Scholar]
- 20.Land E. J., Ramsden C. A., Riley P. A., Tyrosinase autoactivation and the chemistry of ortho-quinone amines. Acc. Chem. Res. 36, 300–308 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Kyluik-Price D. L., Scott M. D., Effects of methoxypoly (ethylene glycol) mediated immunocamouflage on leukocyte surface marker detection, cell conjugation, activation and alloproliferation. Biomaterials 74, 167–177 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Choi Y. H., Lim E. J., Kim S. W., Moon Y. W., Park K. S., An H. J., IL-27 enhances IL-15/IL-18-mediated activation of human natural killer cells. J. Immunother. Cancer 7, 168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desai T., Shea L. D., Advances In islet encapsulation technologies. Nat. Rev. Drug Discov. 16, 338–350 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibly R. F., Graham J. G., Luo X., Lowe W. L. Jr., Hering B. J., Shea L. D., Advancing islet transplantation: From engraftment to the immune response. Diabetologia 54, 2494–2505 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapiro A. M. J., Pokrywczynska M., Ricordi C., Clinical pancreatic islet transplantation. Nat. Rev. Endocrinol. 13, 268–277 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Shapiro A. M. J., Ricordi C., Hering B. J., Auchincloss H., Lindblad R., Robertson R. P., Secchi A., Brendel M. D., Berney T., Brennan D. C., Cagliero E., Alejandro R., Ryan E. A., DiMercurio B., Morel P., Polonsky K. S., Reems J.-A., Bretzel R. G., Bertuzzi F., Froud T., Kandaswamy R., Sutherland D. E. R., Eisenbarth G., Segal M., Preiksaitis J., Korbutt G. S., Barton F. B., Viviano L., Seyfert-Margolis V., Bluestone J., Lakey J. R. T., International trial of the edmonton protocol for islet transplantation. N. Eng. J. Med. 355, 1318–1330 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Matsusaki M., Ajiro H., Kida T., Serizawa T., Akashi M., Layer-by-layer assembly through weak interactions and their biomedical applications. Adv. Mater. 24, 454–474 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Bieber T., Meissner W., Kostin S., Niemann A., Elsasser H.-P., Intracellular route and transcriptional competence of polyethylenimine–DNA complexes. J. Control. Release 82, 441–454 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Liu X., Carter S.-S. D., Renes M. J., Kim J., Rojas-Canales D. M., Penko D., Angus C., Beirne S., Drogemuller C. J., Yue Z., Coates P. T., Wallace G. G., Development of a coaxial 3D printing platform for biofabrication of implantable islet-containing constructs. Adv. Healthc. Mater. 8, 1801181 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Wilson J. T., Chaikof E. L., Challenges and emerging technologies in the immunoisolation of cells and tissues. Adv. Drug Deliv. Rev. 60, 124–145 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koivusalo L., Kauppila M., Samanta S., Parihar V. S., Ilmarinen T., Miettinen S., Oommen O. P., Skottman H., Tissue adhesive hyaluronic acid hydrogels for sutureless stem cell delivery and regeneration of corneal epithelium and stroma. Biomaterials 225, 119516 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Zhu W., Iqbal J., Wang D.-A., A DOPA-functionalized chondroitin sulfate-based adhesive hydrogel as a promising multi-functional bioadhesive. J. Mater. Chem. B 7, 1741–1752 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Cavallari G., Zuellig R. A., Lehmann R., Weber M., Moritz W., Rat pancreatic islet size standardization by the “hanging drop” technique. Transplant. Proc. 39, 2018–2020 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Bernard A. B., Lin C.-C., Anseth K. S., A microwell cell culture platform for the aggregation of pancreatic β-cells. Tissue Eng. Part C Methods 18, 583–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Armendariz E. M., Connexin 36, a key element in pancreatic beta cell function. Neuropharmacology 75, 557–566 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Shyr Z. A., Wang Z. Y., York N. W., Nichols C. G., Remedi M. S., The role of membrane excitability in pancreatic β-cell glucotoxicity. Sci. Rep. 9, 6952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gray D. W. R., An overview of the immune system with specific reference to membrane encapsulation and islet transplantation. Ann. N. Y. Acad. Sci. 944, 226–239 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Lee S. H., Baek K., Lee J. E., Kim B. G., Using tyrosinase as a monophenol monooxygenase: A combined strategy for effective inhibition of melanin formation. Biotechnol. Bioeng. 113, 735–743 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabf7832/DC1