

Astrocyte-secreted CCL2 promotes cancer cell extravasation at the brain via chemotaxis and chemokinesis.
Abstract
Although brain metastases are common in cancer patients, little is known about the mechanisms of cancer extravasation across the blood-brain barrier (BBB), a key step in the metastatic cascade that regulates the entry of cancer cells into the brain parenchyma. Here, we show, in a three-dimensional in vitro BBB microvascular model, that astrocytes promote cancer cell transmigration via their secretion of C-C motif chemokine ligand 2 (CCL2). We found that this chemokine, produced primarily by astrocytes, promoted the chemotaxis and chemokinesis of cancer cells via their C-C chemokine receptor type 2 (CCR2), with no notable changes in vascular permeability. These findings were validated in vivo, where CCR2-deficient cancer cells exhibited significantly reduced rates of arrest and transmigration in mouse brain capillaries. Our results reveal that the CCL2-CCR2 astrocyte-cancer cell axis plays a fundamental role in extravasation and, consequently, metastasis to the brain.
INTRODUCTION
An estimated 20% of all cancer patients develop metastatic tumors at the brain (1). Highly lethal with a 30% 5-year survival rate (2), these metastases stem from circulating tumor cells (TCs) in brain capillaries, originating predominantly from lung, breast, melanoma, or colorectal primary cancers (3), that transmigrate into the parenchyma. This occurs despite the fact that the interface between brain capillaries and tissue, also termed blood-brain barrier (BBB), is one of the tightest vascular barriers in humans and is characterized by highly regulated transport mechanisms (4).
The vast majority of studies on the mechanisms of brain metastasis have focused on TC survival and proliferation following transmigration, once cells are lodged in the brain tissue (5–9). For instance, interactions, in the brain parenchyma, between TCs and brain astrocytes (ACs) have been shown to promote the secretion of inflammatory factors by ACs via connexin 43 gap junctions, which, in turn, support tumor growth and chemoresistance (6). Although these findings provide insights into cellular interactions and signaling pathways used by metastatic cancer cells to survive and proliferate in the brain tissue, extravasation precedes growth at the secondary site and is a rate-regulating event in the metastatic cascade, where ~40% of extravasated TCs are able to colonize and successfully establish metastases within the brain (10, 11). Despite the importance of extravasation in the development of metastases and the significant fraction of cancer patients that develop brain metastases, the processes that govern extravasation from blood to brain remain poorly understood.
Difficulties in disentangling the mechanisms responsible for the extravasation step from those promoting colonization and outgrowth in the brain have impeded in-depth understanding of the processes underlying brain metastasis. In addition, conducting high spatiotemporal resolution imaging in vivo has been challenging. To address these limitations, a few studies have used in vitro models to investigate the mechanisms of TC extravasation at the brain. Among these models, two-dimensional (2D) Transwell coculture systems have been and continue to be frequently used, including cocultures of ACs with human umbilical vein endothelial cells (ECs) (12) or brain ECs (13). These high-throughput 2D in vitro extravasation platforms (12, 13) have revealed important insights into the molecular mechanisms mediating extravasation at the brain; however, they cannot recapitulate TC circulation, arrest, and extravasation from three-dimensional (3D) brain capillaries, which occur in vivo (12–14). To overcome the major limitations of current in vivo and in vitro brain extravasation models, we used a 3D platform designed by our group (15) that recapitulates BBB capillaries with microvascular networks (MVNs) composed of induced pluripotent stem cell–derived ECs (iPS-ECs), brain pericytes (PCs), and ACs into which we perfused circulating TCs to study extravasation at the brain.
In addition to the triculture BBB model, we used three variants incorporating different combinations of iPS-ECs and brain stromal cells to dissect the individual roles of PCs, ACs, and their secreted factors on TC extravasation. The all-inclusive triculture model with iPS-ECs, PCs, and ACs (15) was found to be the most robust system, faithfully recapitulating the extravasation microenvironment of the human BBB in vivo.
In this triculture 3D BBB platform, where distinct cellular and molecular interactions governing extravasation can be observed and quantified in real time or by end point analysis, we uncovered a central role for the C-C motif chemokine ligand 2 (CCL2)–C-C chemokine receptor type 2 (CCR2) signaling pathway in brain extravasation, where ACs directly promote TC transmigration through CCL2 secretion. These findings were further validated in vivo, confirming that this moderate-throughput human BBB-on-a-chip system can provide unparalleled strength in dissecting cellular and molecular mechanisms underlying the extravasation of cancer cells at the brain and could serve as a robust, complementary method to in vivo approaches.
RESULTS
Brain stromal cells promote cancer cell extravasation, yet barrier integrity remains intact
To investigate mechanisms governing TC extravasation at the brain and the roles of different stromal cells in this rate-limiting step of brain metastasis, we designed four MVN platforms in a single gel-channel microfluidic device (15–17): (i) a monoculture of iPS-EC, (ii) a coculture of iPS-EC and PC, (iii) a coculture of iPS-EC and AC, and (iv) a triculture of iPS-EC, PC, and AC as our BBB microvascular model (Fig. 1A). Notably, iPS-ECs are used in this assay owing to their superior plasticity and ability to self-assemble into perfusable MVNs in both the presence and absence of PCs and/or ACs compared to other EC lines (18, 19). The sequential addition of PCs and ACs allowed us to isolate their individual contributions on TC extravasation.
Fig. 1. The presence of PCs and ACs in the MVNs promotes TC extravasation but decreases barrier permeability.
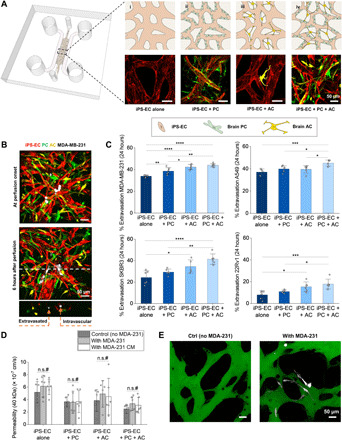
(A) Schematic of the microfluidic chip used in the study to generate four MVN platforms from the sequential addition of PCs and ACs to iPS-ECs. (B) MDA-MB-231 (MDA-231) cells perfused in the triculture MVNs and left to extravasate for 6 hours. Examples of extravasated and intravascular TCs are shown. (C) Extravasation efficiencies of breast MDA-231, breast SKBR3, lung A549, and prostate 22Rv1 in the four MVN platforms at 24 hours after perfusion. (D) MVN permeability to 40 kDa of dextran in the four platforms considered. Permeability is also assessed in the presence of MDA-231 TCs and their conditioned media (CM). (E) Representative images of triculture MVNs perfused with 40-kDa fluorescein isothiocyanate–dextran (green) with and without MDA-231 TCs. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; not significant; n.s.#, not significant across all conditions (pairwise).
The step-wise incorporation of PCs and ACs progressively improved the morphology and barrier permeability of the MVNs (15), and the all-inclusive, triculture MVNs highly recapitulated key morphological and functional properties of the natural BBB, particularly its cellular organization, its expression of junctional proteins, and its deposition of a functional basement membrane and glycocalyx (fig. S1, A to C, and table S1) (20, 21). Notably, the vessel structures of the triculture MVN model closely resembled in vivo brain capillaries (22) compared to all other in vitro BBB models to date (19, 23–26).
To evaluate the robustness of our 3D in vitro BBB model as a surrogate platform to study brain metastasis, we used well-established in vivo and in vitro models from previous studies as our benchmarks (12, 27). In these studies, both parental breast cancer cells (MDA-MB-231, abbreviated here as “MDA-Par” or “MDA-231”) and lung metastatic cells derived from the parental line (MDA-LM2-4175 or “MDA-LM”) (27) exhibited reduced ability to form brain metastases after intracardiac injection, in sharp contrast to the brain metastatic line derived from the parental line (MDA-BrM2a or “MDA-BrM”) (12). In our platforms, while both MDA-BrM and MDA-LM demonstrated superior extravasation ability compared to parental MDA-231 in the monoculture iPS-EC MVNs, this was not the case in the triculture system, where only MDA-BrM cells displayed increased extravasation levels compared to both parental MDA-231 and MDA-LM. This suggests that the triculture MVN model functionally recapitulates other in vivo models of brain metastasis (fig. S2, A and B).
The highly expressed ST6GALNAC5 (“ST6”) in MDA-BrM cells was found to promote cancer cell extravasation in the brain in a 2D in vitro model (12), and knockdown (KD) of ST6 reduced brain metastasis formation after intracardiac injection. To further validate the triculture MVN model and its use as a tumor extravasation platform, we knocked down ST6 in MDA-BrM cells (fig. S2, C, D, F, and G) and validated that their extravasation efficiency was reduced compared to control cells (fig. S2E). However, their reduced extravasation levels remained greater than those of parental MDA-231 cells, as previously found in the in vivo model, but unlike prior observations in 2D in vitro models (12). Our triculture 3D BBB model, therefore, more closely recapitulates the features of in vivo models of brain metastasis compared to 2D in vitro brain extravasation assays, and, importantly, these findings suggest that ST6GALNAC5 is not the only factor affecting TC extravasation at the brain.
Breast cancer, lung cancer, and melanoma are the leading causes of brain metastasis among all cancer patients (3). In addition, brain metastases are also observed in patients with prostate cancer (28). Hence, we used breast cancer cell lines (MDA-231 and SKBR3), lung cancer cell line (A549), and prostate cancer cell line (22Rv1) in this study, aiming to identify general mechanisms of cancer cell extravasation into the brain. Following TC perfusion under transient flow in the MVNs (Fig. 1A) (17), all cells observed to partially or completely cross the endothelium were classified as “extravasated” (Fig. 1B). Despite differences in the baseline extravasation efficiency of each cell line in the iPS-EC alone MVNs (fig. S3A), the sequential addition of brain stromal cells always resulted in significant increases in extravasation after 24 hours (Fig. 1C and fig. S2A). Similar trends were observed 6 hours following perfusion (fig. S3B). These results are in line with prior evidence of the prometastatic role of PCs and ACs when TCs are lodged in the brain tissue after extravasation (5–7, 9).
Next, we sought to assess whether stromal-tumor cell interactions at the brain and the subsequent increase in extravasation were caused by changes in barrier integrity. The latter was evaluated using solute permeability to fluorescent dextran (29, 30). For each of the four MVN platforms considered, the permeability to either 10 or 40-kDa dextrans in MVNs remained unaltered despite the presence of TCs, up to 24 hours after TC perfusion (Fig. 1D and fig. S3C). In addition, the treatment of the MVNs with conditioned media (CM) from MDA-231 for 24 hours resulted in similar observations. This, coupled with a visible lack of focal leaks in the microvasculature following extravasation (Fig. 1E), confirmed that barrier integrity in each of four platforms considered remains intact despite TC transmigration or the presence of TC-secreted factors. This suggests that increased extravasation efficiencies in the presence of PCs and ACs are not caused by disruptions in endothelial junctions, which would increase paracellular transport and result in increased permeability values to low–molecular weight dextrans (29).
Secreted factors from activated stromal cells increase cancer cell extravasation
On the basis of these results, we hypothesized that brain stromal cells act directly on intravascular cancer cells and their ability to transmigrate without needing to alter barrier integrity. To test this, monoculture iPS-EC MVNs were perfused with TCs and treated with conditioned medium from PCs or ACs, cultured either alone (PC and AC CM) or in Transwell systems above MDA-231 [PC (+MDA-231) CM and AC (+MDA-231) CM] (Fig. 2A). The addition of stromal cell CM, particularly when PCs and ACs are activated by the cancer cells in the case of PC (+MDA-231) CM and AC (+MDA-231) CM, significantly increased the extravasation efficiency of TCs (Fig. 2, B and C), suggesting that PC- and AC-secreted factors play important roles in the ability of cancer cells to extravasate.
Fig. 2. PC- and AC-secreted factors increase the extravasation potential of TCs.
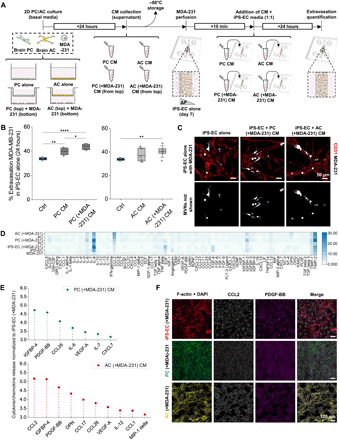
(A) Schematic of the collection of PC and AC CM and subsequent treatment of the MVNs before TC perfusion. (B) Extravasation efficiencies of breast MDA-231 in monoculture MVNs with or without CM treatment at 24 hours after perfusion. (C) Representative images of breast MDA-231 in monoculture MVNs with or without CM treatment at 24 hours after perfusion. (D) Heatmap for cytokine array showing relative magnitudes of secreted factors from MDA-231, iPS-EC, PC, and AC alone, as well as iPS-EC, PC, and AC cultured in Transwell inserts with MDA-231 cells after 24 hours of culture. (E) Relative magnitude of significantly up-regulated cytokines/chemokines by PC or AC cultured in inserts with MDA-231 compared to iPS-EC cultured with MDA-231.(F) Representative images of 2D immunofluorescence staining for F-actin + DAPI, CCL2, and PDGF-BB in iPS-ECs, PCs, and ACs, cultured with MDA-231 in Transwell inserts. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; not significant; n.s.#, not significant across all conditions (pairwise).
Cytokine arrays on conditioned medium from iPS-ECs, PCs, and ACs, in the presence or absence of MDA-231, revealed several highly expressed factors, notably inflammatory and chemotactic cytokines such as interleukin-6 (IL-6), IL-8, and CCL2 (Fig. 2D). Since cancer cell extravasation was significantly increased in the presence of brain stromal cells and their secreted factors compared to monoculture iPS-EC MVNs (Figs. 1C and 2B and fig. S3B), we postulated that up-regulated cytokines by PC (+MDA-231) and AC (+MDA-231) compared to iPS-EC (+MDA-231) could play a role in increasing extravasation (Fig. 2E). Several of these were commonly up-regulated by both PCs and ACs, such as insulin-like growth factor–binding protein 4 (IGFBP-4), platelet-derived growth factor (PDGF)–BB, and CCL26, while some were specific to each brain stromal cell, such as IL-6 for PCs or CCL2 and osteopontin (OPN) for ACs. These findings were validated by immunofluorescence staining for PDGF-BB, CCL2, and IGFBP-4 (Fig. 2F and fig. S4A).
CCL2 was found to be specifically up-regulated by AC (+MDA-231) compared to iPS-EC (+MDA-231) and PC (+MDA-231), whereas PDGF-BB was found to be up-regulated by both stromal cells compared to iPS-ECs (fig. S4B). Similar results were obtained for iPS-ECs, PCs, and ACs cultured in the absence of MDA-231 cancer cells (fig. S4, C and D). Having established that the cytokine profiles of PCs and ACs exhibit notable differences compared to those of iPS-ECs, we next investigated the role of the identified up-regulated factors in cancer cell extravasation.
AC-derived CCL2 promotes cancer cell extravasation from in vitro MVNs
To uncover the key secreted factors responsible for the proextravasation effect, we used a panel of blocking antibodies against candidate cytokines previously identified (Fig. 2E) (31). Only CCL2 blockade significantly reduced TC transmigration in the triculture BBB MVNs (Fig. 3A). As evidenced in media collected from triculture devices, the concentration of CCL2 is the largest of all secreted cytokines (~2700 pg/ml) (fig. S5, A and B) and is primarily expressed by ACs as shown from cytokine array results (Fig. 2D), immunofluorescence staining (Fig. 3B), and quantitative reverse transcription polymerase chain reaction (qRT-PCR) (fig. S5A). In addition, increasing the concentration of blocking antibody for CCL2 from 10 to 50 μg/ml did not affect cancer cell extravasation, suggesting that the concentration used in the blocking antibody panel (10 μg/ml) was sufficient to elicit a response (fig. S5C).
Fig. 3. AC-secreted CCL2 significantly increases the extravasation of TCs via their CCR2 receptor.
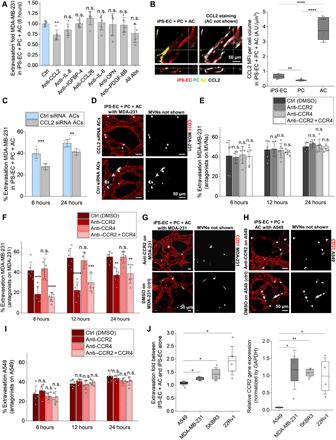
(A) Extravasation efficiency of MDA-231 TCs in triculture MVNs with neutralizing antibodies at 6 hours after perfusion. Abs, antibodies. (B) Representative CCL2 staining in triculture MVNs and quantification of the CCL2 MFI per cell volume in iPS-ECs, PCs, and ACs. (C) Extravasation efficiency of MDA-231 in triculture MVNs with ACs treated with CCL2 small interfering RNA (siRNA) or control siRNA before device seeding. (D) Representative images of MDA-231 in triculture MVNs stained with anti-CD31 at 6 hours after perfusion, in the case of MVNs seeded with ACs treated with CCL2 siRNA or control siRNA. (E) Extravasation efficiency of MDA-231 in triculture MVNs treated with anti-CCR2 and/or anti-CCR4, and the appropriate control at three time points. Significance is assessed with respect to the control [dimethyl sulfoxide (DMSO)] condition. (F) Extravasation efficiency of MDA-231 in triculture MVNs, in the case of MDA-231 treated with anti-CCR2 and/or anti-CCR4, and the appropriate control at three time points. Significance is assessed with respect to the control (DMSO) condition. (G) Representative images of MDA-231 in triculture MVNs stained with anti-CD31, in the case of TCs treated with anti-CCR2 or the appropriate control at 6 hours after perfusion. (H) Representative images of A549 in triculture MVNs stained with anti-CD31, in the case of TCs treated with anti-CCR2 or the appropriate controlat 6 hours after perfusion. (I) Extravasation efficiency of A549 in triculture MVNs, in the case of A549 cells treated with anti-CCR2 and/or anti-CCR4, and the appropriate control at three time points. Significance is assessed with respect to the control (DMSO) condition. (J) Extravasation fold of all four TCs between iPS-EC + AC MVNs and the monoculture iPS-EC MVNs at 24 hours (left) and relative CCR2 gene expression [normalized by glyceraldehyde phosphate dehydrogenase (GAPDH)] for all four TCs measured via qRT-PCR (right). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; not significant; n.s.#, not significant across all conditions (pairwise).
The role of AC-derived CCL2 on transmigration was further validated by treating coculture MVNs of iPS-EC + PC and iPS-EC + AC with blocking antibody for CCL2. Extravasation was significantly reduced only in the case of MVNs cultured with ACs, confirming that these brain stromal cells constitute the main source of CCL2 in the devices (fig. S5D). This was also demonstrated by immunofluorescence staining, whereby the mean fluorescent intensity (MFI) for CCL2 was the highest in ACs in the different MVN platforms compared to PCs and iPS-ECs (fig. S5E). CCL2 levels were significantly higher in ACs in the triculture BBB MVNs compared to the coculture iPS-EC + AC MVNs. This could indicate that cellular interactions among all three cell types, occurring when the human BBB is recapitulated in the in vitro devices, are conducive to increases in CCL2 chemokine production by ACs. It is useful to note that while FGF-2 (fibroblast growth factor 2) was found at an average concentration of ~2900 pg/ml (greater than that of CCL2 in the triculture MVNs), it is a major component of the iPS-EC culture medium used in the devices (present at 5000 pg/ml) and was thus assumed not to be significantly secreted by any of the cells in the BBB MVNs (fig. S5B). Furthermore, KD of CCL2 in ACs before device seeding resulted in significant reductions in TC extravasation in the triculture MVNs at both 6 and 24 hours after perfusion (Fig. 3, C and D, and fig. S5F), highlighting the importance of AC-specific contributions of CCL2 in tumor transmigration at the BBB. Together, these findings suggest that, of all up-regulated cytokines by PCs and ACs compared to iPS-ECs, only AC-secreted CCL2 plays a role in cancer cell extravasation. We thus focused our investigation on this chemokine and its observed effect on the transmigration of circulating TCs at the brain.
ACs signal to cancer cells via their CCR2 receptor to promote their extravasation
CCL2 is known to preferentially bind CCR2 (32). It has also been reported to bind CCR4 on cytotoxic T lymphocytes and regulatory T cells in the context of their recruitment to melanoma tumors, although with lower affinity compared to CCR2 (33, 34). Considering these two receptors, we next evaluated whether blocking the CCL2-CCR2 or CCL2-CCR4 axes would result in any changes in TC extravasation.
Prior evidence suggests that intravascular colon cancer cells secrete CCL2 to activate the endothelium, increase its permeability via the Janus kinase 2–signal transducer and activator of transcription 5 and p38 mitogen-activated protein kinase pathways, up-regulate E-selectin expression, and ultimately promote cancer cell adhesion and extravasation from the vasculature (35). We thus first investigated whether AC-derived CCL2, in our case, could similarly activate the endothelium to promote extravasation. To assess whether AC-derived CCL2 affects the vasculature in the context of extravasation, we treated the triculture MVNs with CCR2 and CCR4 antagonists before TC perfusion. Blocking either receptor or both in the ECs did not result in any changes in the extravasation potential of MDA-231 (Fig. 3E).
Next, we assessed the direct effect of CCR2 and CCR4 on cancer cells by blocking CCR2, CCR4, or both, on the MDA-231 cells, before perfusion. Here, blocking CCR2 or both receptors resulted in a significant decrease in TC extravasation (Fig. 3, F and G). In contrast, CCR4 blockade did not affect MDA-231 extravasation, suggesting that AC-TC interactions are mostly occurring via the CCR2 receptor on TCs.
This was also validated in MDA-BrM cells, where CCR2 blockade alone resulted in greater reductions of extravasation efficiencies compared to KD of ST6 alone (fig. S6, A and B). Similar observations were made in the parental MDA-231 line (fig. S6, C and D); however, blockade of ST6 did not result in significant reductions in extravasation compared to control cells. KD of CCR2 alone in MDA-BrM was sufficient to reduce extravasation levels below those of parental MDA-231, which was not the case when ST6 was knocked down alone (fig. S6A). These findings, coupled with increased CCR2 levels in parental MDA-231 and MDA-BrM (fig. S6, E to H), attest to the significance of AC-secreted CCL2 in promoting their extravasation at the brain through their CCR2 receptor.
To validate our observations on the role of CCR2 in other cell lines, we treated SKBR3, A549, and 22Rv1 cell lines with the same antagonist combinations (CCR2 and/or CCR4). Consistent with our hypothesis, both SKBR3 and 22Rv1 exhibited similar trends as MDA-231 TCs in their extravasation potential (fig. S7, A to C); however, the extravasation efficiencies of A549 were not affected by any of the antagonist treatments (Fig. 3, H and I). Notably, A549 cells exhibited the lowest fold change in extravasation efficiency between iPS-EC + AC and iPS-EC alone compared to other cell lines examined and also expressed the lowest CCR2 levels compared to the three other lines (Figs. 3J and 1C). This is consistent with our hypothesis that the proextravasation effect of ACs is indeed mediated through CCR2 expressed by cancer cells.
Prior evidence suggests that autocrine secretion of CCL2 promotes cancer cell transmigration, invasion, and metastasis in colon cancer (35), prostate cancer, and cervical carcinoma (36, 37). To exclude this possibility, we blocked the endogenous secretion of CCL2 in MDA-231 with the use of small interfering RNA (siRNA) probes, as performed with ACs before device seeding (Fig. 3, C and D). Perfusion in the monoculture or triculture MVNs did not result in any significant differences in the extravasation potential of MDA-231 treated with siRNA for CCL2 or control siRNA (fig. S8, A and B). Consistently, enzyme-linked immunosorbent assay (ELISA) revealed more than 100-fold larger secretions of CCL2 in the triculture MVNs with ACs compared with MDA-231 cells (~15 to 25 pg/ml for MDA-231 compared to ~2700 pg/ml in the triculture BBB, which had comparable numbers of cells at the time of analysis) (figs. S8C and S5B), validating that AC-secreted CCL2 plays a crucial role in TC transmigration in our system.
We next compared CCL2 levels across cancer cell lines. qRT-PCR results showed that A549 expressed the highest levels of CCL2, followed by SKBR3, MDA-231, and 22Rv1 (fig. S8D). We then examined whether blocking TC-secreted CCL2 in SKBR3, A549, and 22Rv1 would give rise to similar results as MDA-231 cells. Treatment with anti-CCL2 before TC perfusion resulted in significant reductions in the transmigration potential of A549 and SKBR3 only (fig. S9E), suggesting that there exists a correlation between autocrine CCL2 levels and TC transmigration at the brain. Given that ACs express significantly larger levels of CCL2 compared to all cancer cells (fig. S8D), endogenous CCL2 in A549 and SKBR3 must be directly affecting their transmigration without having major consequences on the endothelium, contrary to prior findings on colorectal and prostate TC-derived CCL2 on endothelial permeability and extravasation (35, 37).
The CCL2-CCR2 AC-cancer cell axis drives cancer cell chemotaxis and chemokinesis at the BBB
Given its important role on TC transmigration, we next investigated the mechanism of action of AC-derived CCL2. First, we verified that the secreted chemokine could reach the intravascular TC. Immunofluorescence staining of triculture MVNs for CCL2 with and without perfused MDA-231 showed that although the overall CCL2 MFI per region of interest (ROI) remained constant, the presence of TCs resulted in significant decreases in CCL2 MFI per cell volume in ACs only (Fig. 4A and fig. S9A). This could be explained by an exchange of CCL2 between ACs and TCs, ultimately inducing transmigration.
Fig. 4. Increased extravasation at the brain results from chemotactically and chemokinetically driven CCL2-CCR2 interactions between ACs and TCs.
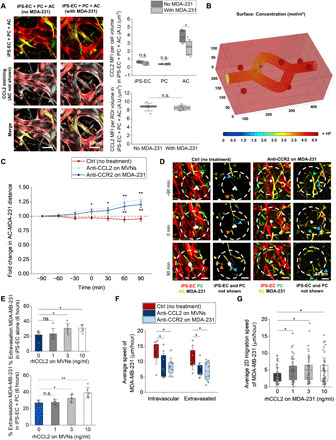
(A) Representative CCL2 staining in triculture MVNs with and without MDA-231 cells and quantification of the CCL2 MFI per cell volume with and without TCs. CCL2 is shown in white, iPS-EC in red, PC in green, and AC in yellow. A.U., arbitrary units. (B) Heatmap of the CCL2 concentration in the 3D MVNs surrounded by ACs with an intravascular TC. Only diffusion is considered here given the absence of luminal fluid flow in the MVNs. (C) AC-TC distance near extravasating TCs over time in triculture MVNs with anti-CCL2 on MVNs or anti-CCR2 on TCs. (D) Representative images of extravasating TCs and its surrounding ACs over time in triculture MVNs with and without anti-CCR2 on TCs. The blue, green, and orange arrows at the different time points refer to individual ACs moving closer to the TC in the control case, or away from it in the anti-CCR2 case. (E) Extravasation efficiency of MDA-231 in monoculture iPS-EC and coculture iPS-EC + PC MVNs at 6 hours after perfusion, in the case of MVNs treated with different recombinant human CCL2 (rhCCL2) concentrations. (F) Average speed of MDA-231 before and after extravasation in the triculture MVNs treated with anti-CCL2 or for the case of TC treatment with anti-CCR2. (G) Average 2D migration speed of MDA-231 with or without rhCCL2 treatment at different concentrations. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; not significant; n.s.#, not significant across all conditions (pairwise).
To validate these results further, we developed a computational model of CCL2 transport from ACs located near an intravascular TC (table S2). Simulation results verified that a constant flux of CCL2 from the ACs led to diffusive transport through the endothelial barrier via paracellular and transcellular pathways, reaching the TC and allowing its local consumption (TC surface concentration of ~4.5 × 108 mol/m3) (Fig. 4B) (38). Given that our MVNs are not subjected to fluid flow as is the case in vivo (39), we validated that the presence of convection (fluid velocity of ~0.5 mm/s) (40), in addition to diffusion (fig. S9B), resulted in CCL2 internalization by the intravascular TC within the same time frame and at similar surface concentration levels (~5 × 108 mol/m3) (fig. S9C). Because astrocytic sources of CCL2 are also located upstream from the cancer cells, CCL2 diffuses into the traveling bulk fluid ensuring that the chemokine is carried via convection to the downstream adhered TC. Thus, in the presence of microvascular convective phenomena, the CCL2 cytokine would still reach its target. It should be noted, however, that this transport analysis disregards the source/consumption kinetics of other cell types present in the in vivo microenvironment, such as neurons, microglia, or blood components (41, 42). Nevertheless, their effects were rendered negligible, given their low consumption rates and greater intercellular distances compared to AC-TC interactions.
Having confirmed that AC-derived CCL2 reaches the TCs, we next evaluated whether blocking the CCL2-CCR2 pathway resulted in any changes in the physical interactions between ACs and TCs. We transduced iPS-ECs, PCs, and ACs to express stable fluorescent tags before seeding the microfluidic chips to visualize cellular interactions in real time with high resolution (fig. S9D). The CCL2-CCR2 axis was blocked using a blocking antibody for CCL2 in the MVNs or an antagonist for CCR2 on TCs. Strikingly, distances between ACs and extravasating MDA-231 were found to increase over time when CCL2 was prevented from reaching the TCs (Fig. 4C and fig. S9E). This was not the case in the control MVNs, where ACs maintained close proximity with extravasating TCs (Fig. 4D). In addition to increased AC-TC distances over time, the number of ACs near MDA-231 was found to decrease when the CCL2-CCR2 pathway was blocked (fig. S9F). Conversely, in the control triculture MVNs, ACs were recruited near TCs during extravasation, attesting to the presence of spatial interactions that are dependent on the CCL2-CCR2 axis between the two cells.
CCL2, also referred to as monocyte chemoattractant protein-1 (MCP-1), is an established chemotactic factor that controls the infiltration of monocytes and macrophages (43). It has also been implicated in improvements in the motility and migration of chondrosarcoma cells, as well as gastric and bladder cancer cells (44–46). We sought to understand whether these mechanisms were also observed at the BBB and whether CCL2-dependent chemotaxis between ACs and TCs was similarly responsible for increases in extravasation. To recapitulate the presence of CCL2 in the extracellular space of the MVNs where ACs are found, mono- and coculture devices with iPS-EC alone and iPS-EC + PC (no ACs) were treated with different concentrations of recombinant human CCL2 (rhCCL2) for 30 min before washing and TC perfusion. Given that CCL2 (~2700 pg/ml) is secreted in the triculture BBB MVNs, mostly by ACs compared to iPS-ECs and PCs (fig. S5A), we subjected the mono- and coculture devices to rhCCL2 [0 ng/ml (control), 1 ng/ml (low), 3 ng/ml (BBB MVN level), and 10 ng/ml (high)]. Washing the MVNs before perfusion removed all intravascular rhCCL2 to establish a chemokine gradient between luminal and interstitial spaces. Extravasation efficiencies of MDA-231 at 6 hours were found to significantly increase in the presence of 3 and 10 ng/ml compared to the control MVNs (Fig. 4E), confirming that the increase in TC transmigration can be partly explained by chemotaxis, where the astrocytic source of CCL2 is present in the extracellular matrix.
The average speed of MDA-231 in the triculture BBB MVNs was significantly lower when the CCL2-CCR2 axis was blocked (from ~11 μm/hour in control MVNs to ~7 μm/hour with anti-CCL2 or anti-CCR2) (Fig. 4F). This was the case for both intravascular and extravasated MDA-231, which were found to travel significantly less in the vessel and surrounding matrix (fig. S10A). These findings suggest that MDA-231 cells respond to chemokinetic cues in the BBB MVNs, whereby their speed is affected by available levels of the CCL2 chemokine (47, 48). The speed of ACs, on the other hand, was not altered when the CCL2-CCR2 axis was blocked, near intravascular or extravasated TCs (fig. S10B). This result was expected, given that MVN treatment with anti-CCL2 solely targets soluble chemokines in the devices without affecting their source (ACs in our case). In BBB MVNs not perfused with MDA-231, ACs also maintained the same average speed (~20 to 25 μm/hour) (fig. S10C), suggesting that their migration patterns are independent of the presence of TCs and their secreted factors.
To further validate the chemokinesis hypothesis, MDA-231 were plated in 2D and subjected to different concentrations of rhCCL2 for 6 hours. Even minute amounts of rhCCL2 (1 ng/ml) resulted in significant increases in the speed of MDA-231, which plateaued at ~5 μm/hour for 3 and 10 ng/ml compared to ~3.3 μm/hour for 0 ng/ml (control) (Fig. 4G). TCs also spanned larger areas throughout their migration in 2D, as evidenced by their x-y displacement tracks over time for the four different treatments (fig. S10D). Together, these results indicate that TCs, via their CCR2 receptor, respond to both chemotactic and chemokinetic cues from AC-derived CCL2 in the MVNs. These cues ultimately trigger TC migration and transmigration from the brain vasculature and into the extracellular space.
CCR2 KD in breast cancer cells decreases brain metastasis formation in vivo
Having established the role of ACs on TC extravasation via the CCL2-CCR2 pathway in an in vitro BBB MVN model, we next sought to validate our findings in vivo by knocking down CCR2 in MDA-231 cells (Fig. 5A). CCR2 KD short hairpin RNA (shRNA) MDA-231 transmigrated significantly less than control shRNA MDA-231 for all time points considered (6, 12, and 24 hours) (Fig. 5B), demonstrating that the shRNA KD can mimic the effects of the CCR2 antagonist applied on MDA-231 in vitro (Fig. 3F).
Fig. 5. CCR2 KD in breast TCs decreases brain metastasis formation in mice.
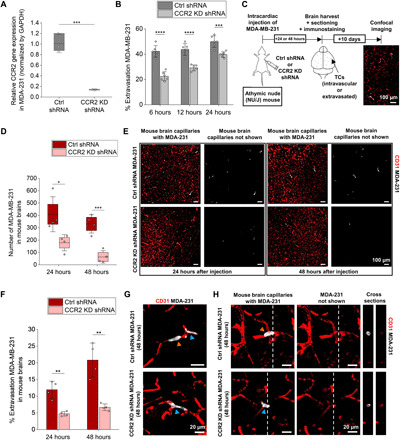
(A) Relative CCR2 gene expression normalized by GAPDH in control shRNA and CCR2 KD shRNA MDA-231 cells as measured by qRT-PCR. (B) Extravasation efficiency of control shRNA and CCR2 KD shRNA MDA-231 in triculture MVNs at three different time points. (C) Schematic of the TC intracardiac injection in mice, followed by brain harvesting, sectioning, immunofluorescence staining, and imaging. (D) Number of control shRNA and CCR2 KD shRNA MDA-231 cells remaining in mouse brains at 24 and 48 hours following intracardiac injection. (E) Representative images of brain sections perfused with MDA-231 (control shRNA and CCR2 KD shRNA) and stained with anti-CD31 at different time points. (F) Extravasation efficiency of control shRNA and CCR2 KD shRNA MDA-231 in mouse brains at 24 and 48 hours following intracardiac injection. (G) Representative images (collapsed z-stacks) of extravasated (orange arrows) and intravascular (blue arrows) MDA-231 (control shRNA and CCR2 KD shRNA) in mouse brains stained with anti-CD31 at 48 hours. (H) Representative images with cross sections of extravasated (orange arrows) and intravascular (blue arrows) MDA-231 (control shRNA and CCR2 KD shRNA) in mouse brains stained with anti-CD31 at 48 hours. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; not significant; n.s.#, not significant across all conditions (pairwise).
Following the in vitro confirmation that CCR2 KD reduces extravasation, CCR2 KD shRNA or control shRNA MDA-231 cells were injected in athymic nude (NU/J) mice via ultrasound-guided intracardiac injections (49). Prior studies suggested that lung and melanoma cancer cells are able to extravasate at the brain as early as 24 hours after injection in mice (11). We thus chose 24 and 48 hours after injection as our time points for this study. At both time points, significantly fewer CCR2 KD shRNA MDA-231 were found in the mouse brain vessels or parenchyma compared to the control shRNA cells (Fig. 5, D and E, and fig. S11A). Given that arrest in the microcirculation is a necessary initial step of the metastatic cascade (10, 50), the decreased number of CCR2 KD shRNA MDA-231 remaining in the mouse brains over time suggests that these cells do not seem to be stimulated at this secondary site, potentially because of their lack of activation/interaction with cellular and molecular cues in the brain microenvironment (50).
We next quantified the transmigration potential of CCR2 KD shRNA and control shRNA MDA-231 in the mouse brain capillaries. The overall extravasation efficiencies of the control shRNA MDA-231 at 24- and 48-hour time points were comparable to previous reports of lung and melanoma cancer cells (11). On the other hand, CCR2 KD significantly impaired MDA-231 extravasation at 24 and 48 hours (Fig. 5F). In addition, while the extravasation potential of control shRNA MDA-231 increased over time (from ~12% at 24 hours to ~21% at 48 hours), that of CCR2 KD shRNA MDA-231 remained relatively constant (~5% at 24 hours and ~7% at 48 hours), consistent with our hypothesis that the latter are not activated in the brain microenvironment. Extravasated control shRNA MDA-231 were found to remain in close proximity with brain capillaries following transmigration (Fig. 5, G and H, and fig. S11B), adopting a perivascular position as formerly observed in the case of lung and melanoma TCs in mouse brains (11). Although early in the metastatic cascade, our results are in agreement with previous findings, suggesting that TCs aim to co-opt the vasculature following transmigration to form successful metastases (11, 51, 52).
DISCUSSION
To investigate one of the rate-limiting steps of brain metastasis formation, namely, extravasation across the BBB, the tightest vascular barrier in the body, we used a 3D in vitro human BBB model to evaluate, in real time, the mechanisms influencing cancer cell extravasation. Progress in this area has previously been hampered by the lack of proper in vivo and in vitro platforms to model TC extravasation and to identify relevant players in this key step of the metastatic cascade. Our in vitro human BBB MVN model overcomes major limitations of currently available models by improving the relevance to human physiology with a 3D cellular architecture and morphology recapitulating brain capillaries, increasing the experimental throughput and spatiotemporal resolution, and allowing for the isolation and identification of cellular and molecular factors directly affecting TC extravasation (15, 53, 54). We also validated our platform as a tumor model assay through a comparison to in vivo and 2D in vitro findings from previous studies (12). MDA-BrM exhibited increased extravasation efficiencies in the triculture BBB MVNs compared to MDA-LM and parental MDA-231, and we verified ST6GALNAC5 to be one of the important factors promoting extravasation, as observed in previous in vivo and in vitro studies (12).
Through the sequential integration of brain stromal cells in the 3D MVNs, we were able to identify some of the key cellular and molecular players in TC transmigration across the BBB. ACs were found to promote extravasation at the brain through secretion of CCL2, which targets the CCR2 receptor expressed by TCs. Via CCL2-driven chemotaxis and chemokinesis, the extravasation and migratory potentials of TCs were considerably enhanced in the BBB MVNs. The KD of AC-secreted CCL2 or tumor-expressing CCR2 resulted in significant decreases in extravasation, attesting to the central role of the CCL2-CCR2 AC-cancer cell axis in metastasis at the brain. CCR2 blockade alone was sufficient to reduce the extravasation levels of MDA-BrM below those of the parental MDA-231 line, which was not the case when ST6GALNAC5 was individually silenced. The significance of the CCL2-CCR2 axis in brain extravasation was validated in vivo, where CCR2 KD in TCs reduced extravasation efficiencies and cancer cell numbers in the brain after intracardiac injection compared to control TCs. These results suggest that the CCL2-CCR2 axis is one of the important mediators of TC extravasation at the brain.
The CCL2-CCR2 axis has been shown to play a role in colorectal cancer cell extravasation at the lung, where endogenous secretion of CCL2 by TCs resulted in altered barrier properties and improved transmigration (35). In addition, up-regulated CCL2 secretions by ACs in response to inflammatory conditions such as brain metastasis (9), glioma (55), epilepsy (56), or autoimmune diseases (57, 58) have been shown to promote trafficking of immune cell, including CCR2-positive T cells, monocytes, and myeloid cells. These studies are consistent with our findings that increased secretion of CCL2 by ACs plays an important role in promoting extravasation at the brain.
However, elevated CCL2 levels at the brain during inflammation (59), particularly in the context of ischemia (60, 61), have been found to disrupt the adherens junctions of the BBB, hence its permeability. Unexpectedly, however, triculture MVNs with PCs and ACs exhibited reduced rather than increased vascular permeabilities compared to vessels composed of iPS-ECs alone. The permeability values in our triculture BBB MVNs (table S1) are comparable to those found in vivo by other groups (61, 62). Even in the presence of extravasating TCs or upon incubation with CM from TCs for up to 24 hours, barrier permeability remained unchanged in all four MVN platforms. This suggests that BBB integrity is not significantly altered during the time that most TCs extravasated and that inflammation of the brain endothelium is not responsible for the increased cancer cell transmigration observed in the triculture system. Rather, the direct interactions between ACs and TCs through the CCL2-CCR2 pathway, via both chemotaxis and chemokinesis, are major contributors to extravasation in our model.
In summary, the in vitro 3D BBB MVN model is a unique platform to functionally interrogate molecular and cellular mechanisms during brain extravasation. With this model, we revealed the central role of AC–cancer cell interactions via the CCL2-CCR2 pathway, in promoting TC extravasation across the BBB. Inhibition of CCR2 in cancer cells resulted in greater reductions of their extravasation efficiencies compared to inhibition of the previously identified ST6GALNAC5 signaling pathway (12). In the future, combinatory inhibitions of both CCR2 and ST6GALNAC5 would be worth exploring as prevention treatments for brain metastasis.
MATERIALS AND METHODS
Experimental design
The objective of this study was to investigate the role of human brain ACs in TC extravasation in an in vitro microvascular BBB model and to validate these findings in vivo. CCL2 was found to be up-regulated by ACs and was found to promote extravasation through CCR2 expressed on TCs, via both chemotaxis and chemokinesis. In addition, ST6GALNAC5, previously found to promote brain metastases in mice, was validated to play a role in extravasation in the in vitro microvascular BBB model. The dual KD of CCR2 and ST6GALNAC5 further reduces extravasation and could be exploited in the clinic. In all in vitro experiments, six devices per condition were used unless otherwise indicated.
These findings were validated in mouse, where CCR2 KD in circulating TCs significantly reduced their transmigration at the brain. In all in vivo mice experiments, four mice per time point and per condition were used. All animal studies were approved by the Massachusetts Institute of Technology (MIT) Institutional Animal Care and Use Committee.
Cell culture and treatments
Human iPS-ECs [Cellular Dynamics International (CDI)], human brain PCs (catalog no. 1200, ScienCell), and human brain ACs (catalog no. 1800, ScienCell) were cultured as described previously (15) and used at passage 5 for all experiments. For time-lapse imaging, iPS-ECs were stably transduced with the LentiBrite GFP Control Lentiviral Biosensor (Millipore Sigma), PCs were transduced with a tagBFP plasmid (Lenti-ef1a-dcas9-vp64-t2a-tagBFP; catalog no. 99371, Addgene; gift from the Jacks laboratory, MIT), and ACs were transduced with a tdTomato plasmid (PGK-Luc-EFS-tdTomato; gift from the Jacks laboratory, MIT).
For the extravasation assays, breast MDA-MB-231 [American Type Culture Collection (ATCC)], brain metastatic variant MDA-BrM2a, and lung metastatic variant MDA-LM2-4175 were obtained from the Massagué group (12, 27) following stable transduction with a triple modality reporter gene, thymidine kinase, green fluorescent protein (GFP), and luciferase (TGL), as previously described (63, 64). The Massagué cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (catalog no. 11995073, Gibco) supplemented with 10% fetal bovine serum (FBS; catalog no. 26140-079, Invitrogen). Breast SKBR3 (ATCC) and lung A549 (ATCC) were stably transduced with the LentiBrite RFP Control Lentiviral Biosensor (Millipore Sigma) and maintained in DMEM supplemented with 10% FBS. Prostate 22Rv1 (ATCC) were maintained in RPMI supplemented with 10% FBS and were stained with the CellTracker Orange CMRA (Invitrogen) at 1 μM in phosphate-buffered saline (PBS) for 10 min at 37°C and 5% CO2. The shRNA-mediated KD of CCR2 was performed on MDA-231 with CCR2 MISSION shRNA bacterial glycerol stock [clone ID: NM_001123041.2-1247s21c1 (human), Millipore Sigma]. Control MDA-231 cells were treated with nontarget shRNA control plasmid DNA MISSION TRC2 pLKO.5-puro (SHC216, Millipore Sigma). Target sequences are shown in table S1.
Microfluidic device fabrication and MVN formation
The 3D microfluidic devices used in this study were fabricated using soft lithography as previously described (16, 17) and comprise a single microchannel for cell seeding and two adjacent media channels with dimensions outlined in detail elsewhere (15). To recapitulate the in vivo organization of the BBB and isolate the effects of the different stromal cells on barrier characteristics and TC extravasation, four MVN platforms were used by culturing different combinations of iPS-ECs, PCs, and ACs in a fibrinogen-thrombin gel (Fig. 1A) (15). For all four platforms, iPS-ECs were resuspended at 5 million cells/ml in the fibrin hydrogel. For the coculture of iPS-EC + AC and the triculture of iPS-EC + PC + AC, ACs were resuspended at 1 million cells/ml and mixed in the fibrinogen-thrombin gel with the iPS-ECs or with both the iPS-ECs and PCs, respectively. For the coculture of iPS-EC + PC, PCs were resuspended at 1 million cells/ml in the fibrin hydrogel and mixed with iPS-ECs. The PC cell density was decreased to 0.5 million cells/ml in the triculture of iPS-EC + PC + AC to ensure a final PC-to-EC ratio on day 7 similar to that found in in vivo mice brains. The triculture iPS-EC + PC + AC model was found to recapitulate the in vivo BBB most closely (table S2). To test the influence of AC-secreted CCL2 on TC extravasation, the KD of CCL2 in ACs was performed before device seeding using a Silencer Select siRNA probe at 10 μM (s12567, Thermo Fisher Scientific) with Lipofectamine RNAiMAX reagent (13778030, Thermo Fisher Scientific) in Opti-MEM reduced serum medium (31985062, Thermo Fisher Scientific). A Silencer Select Negative Control siRNA was used as a control (no. 2, 4390843, Thermo Fisher Scientific). ACs were treated twice more than 48 hours with siRNA probes and left to rest for an additional 24 hours in fresh media before device seeding with iPS-ECs and PCs to form triculture BBB MVNs. Total RNA from ACs was collected at day 0 (MVN seeding) and at day 7 (TC extravasation experiments) from replated ACs following device seeding, as described below. The KD of CCL2 in the ACs was confirmed via qRT-PCR using the protocol and the CCL2 TaqMan Gene Expression Assay mentioned below.
All devices were cultured in VascuLife media (Lifeline Cell Technology) supplemented with 10% FBS and 2% l-glutamine, according to the protocol developed by CDI. For the first 4 days of culture, this media was supplemented with vascular endothelial growth factor–A (VEGF-A) (50 ng/ml) (catalog no. 100-20, PeproTech) in each of the four platforms to promote vasculogenesis following cell seeding. The addition of VEGF-A (50 ng/ml) was performed identically and consistently in all MVN platforms, and the results were compared across platforms.
CM collection
iPS-ECs, PCs, ACs, and MDA-231 cells were plated onto six-well plates in their respective media until 60% confluence, after which media was replaced with VascuLife basal media (without growth factors). Cells were incubated for 24 hours, and CM was collected as previously described (31) and stored at −80°C until use. To test the influence of MDA-231 on the cytokine production of iPS-ECs, PCs, and ACs, a six-well plate with Transwell inserts was used. MDA-231 were added to the bottom compartment, and iPS-ECs, PCs, and ACs were plated onto the insert at a 7:1 ratio with the TCs, as determined via cell quantification in 3D triculture MVNs perfused with MDA-231. Media was replaced with basal VascuLife once the iPS-ECs, PCs, or ACs reached 60% confluence, and cells were incubated for an additional 24 hours before CM collection. The CM of iPS-ECs, PCs, and ACs cultured above MDA-231 was collected exclusively from the inserts and labeled as iPS-EC (+MDA-231), PC (+MDA-231), and AC (+MDA-231).
MVN permeability and dimensions
To assess the MVN permeability, a monolayer of iPS-ECs was added to both media channels of the microfluidic device on day 4 following cell seeding by perfusing 50 μl of iPS-ECs resuspended at 1 million cells/ml in VascuLife (29). Permeability was measured on day 8 using 10- and 40-kDa fluorescein isothiocyanate (FITC)–dextrans at 0.1 mg/ml (FD10S, FD40S, Millipore Sigma), as previously described (29), via imaging using a confocal laser scanning microscope (FV1000, Olympus) with an environmental chamber maintained at 37°C and 5% CO2. Following automatic thresholding and segmentation using the Fiji distribution of ImageJ [National Institutes of Health (NIH)], z-stack images were used to generate a 3D mask of the microvasculature as previously defined (29). At times, MVNs were perfused with MDA-231 on day 7, and the permeability was assessed 24 hours following perfusion on day 8. In addition, CM from MDA-231 was collected as described above and used to treat MVNs on day 7 in a 1:1 ratio with iPS-EC media. The permeability was assessed 24 hours following incubation with CM on day 8. Average vessel diameter and other MVN dimensions (average branch length, average cross-sectional area, surface area of the vessels, and 3D volume of the vessels) were computed as previously described (15, 29) by using the 3D mask generated from the MVN perfusion with dextran in ImageJ.
Immunostaining and quantification
Cells plated in 2D or in Transwell inserts were fixed with 2% paraformaldehyde for 10 min, permeabilized with 0.1% Triton-X for 5 min, and blocked with 4% bovine serum albumin (BSA) and 0.5% goat serum in PBS overnight at 4°C on a shaker. Relevant cytokines were observed by staining the fixed 2D plated cells with anti-CCL2 (MAB279, R&D Systems), anti-IGFBP4 (18500, Proteintech), anti–PDGF-BB (PA5-19524, Invitrogen), anti-CCR2 (711255, Thermo Fisher Scientific), and anti-ST6GALNAC5 (MAB67151, R&D Systems) at 1:200 in PBS overnight at 4°C. Microfluidic chips were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton-X for 5 min, and blocked with 4% BSA and 0.5% goat serum in PBS overnight at 4°C on a shaker. Following a PBS wash, protein visualization was achieved by staining the fixed devices with anti-CD31 (ab3245, Abcam), anti-PDGF receptor b (ab69506, Abcam), anti-GFAP (ab10062, Abcam), and anti–CCL2/MCP-1 (2D8, Invitrogen) at 1:200 in PBS overnight at 4°C on a shaker. After washing, cells and devices were incubated with secondary antibodies at 1:200 in PBS (Alexa Fluor 488 goat anti-rabbit A-11008, 568 goat anti-rabbit A-11011, or 633 goat anti-mouse A-21052; Invitrogen) and 4′,6-diamidino-2-phenylindole (DAPI) (D1306, Invitrogen) at 1:1000 in PBS overnight at 4°C on a shaker. Imaging was performed with a confocal laser scanning microscope (FV1000, Olympus).
Following staining of CCL2 in devices with fluorescently labeled iPS-ECs, PCs, and ACs, MFI per cell volume was computed by collecting stacks covering the full gel thickness containing MVNs using a 20× objective at a resolution of 800 × 800 pixels with a z-spacing of 5 μm. For each channel corresponding to each cell type (iPS-ECs, PCs, or ACs), automatic thresholding and segmentation were performed using the Fiji distribution of ImageJ (NIH) as described previously (29). The 3D masks generated for each cell type were used to compute the total cell volume and determine MFI, when applied on the channel with CCL2 staining. For 2D staining, MFI per cell area was computed by measuring the mean intensity of the antibody of interest per area and normalizing over DAPI intensity.
Mice brains were harvested at two different time points (24 and 48 hours) following ultrasound-guided intracardiac perfusion of TCs and immediately fixed whole with 4% paraformaldehyde (v/v) in PBS at room temperature for 48 hours. Fixing with paraformaldehyde perfusion was avoided to prevent the high flow rates of perfusion from detaching intravascular TCs. Fixed brains were sectioned in 100-μm-thick slices with a vibrating blade microtome (VT1000S, Leica Biosystems) and washed with a wash buffer constituted of 1% Triton X (v/v) in PBS on a shaker. Brain slices were incubated with blocking buffer [10% goat serum (v/v) with 1% Triton X (v/v) in PBS] for 4 hours at room temperature on a shaker. Following washing in wash buffer, samples were incubated in mouse anti-CD31 (553370, BD Biosciences) at 1:100 in wash buffer for 5 days at room temperature on a shaker to visualize the brain microvasculature (65). Brain slices were washed in wash buffer and incubated in secondary antibody (Alexa Fluor 633 goat anti-rat A-21094, Invitrogen) at 1:250 for 5 days at room temperature on a shaker. Samples were washed in wash buffer and placed on a glass coverslip for confocal imaging (FV1000, Olympus). Images were acquired using a 60× oil objective to visualize individual TCs and the brain vasculature.
Cytokine/chemokine/protein measurements
Undiluted CM from MDA-231 and iPS-ECs, PCs, and ACs with or without MDA-231 was assayed using a human 80-cytokine array (AAH-CYT-5, RayBiotech) following the manufacturer’s protocol. Cytokine levels were also evaluated with the Human Cytokine Array/Chemokine Array 42-Plex (HD42, Eve Technologies Corp.) from undiluted media pooled from the media channels of three microfluidic chips per experimental repeat.
ST6GALNAC5 protein levels were measured with a human a-N-acetylgalactosaminide a-2,6-sialyltransferase 5 (ST6GALNAC5) ELISA kit (AMS.E01A1995, Amsbio) from cell lysates obtained from TCs cultured in 2D in 24-well plates. Four wells per TC were used for this assay.
Real-time qRT-PCR
Gene expression was quantified via real-time qRT-PCR. Cells were plated in 2D in wells of a 24-well plate, and total RNA was isolated and purified using the RNeasy Mini Kit (74104, Qiagen) according to the manufacturer’s instructions. The concentration of total RNA was measured using a NanoDrop 1000 spectrophotometer. cDNA was synthesized using the High-Capacity RNA-to-cDNA Kit (4387406, Thermo Fisher Scientific) according to the manufacturer’s protocol. Real-time qRT-PCR was performed on the 7900HT Fast Real-Time PCR System using the TaqMan Fast Advanced Master Mix (4444556, Thermo Fisher Scientific) as previously described (30). Glyceraldehyde phosphate dehydrogenase (GAPDH) was used as a housekeeping gene, and the following genes with respective TaqMan Gene Expression Assays (Thermo Fisher Scientific) were used: GAPDH (Hs01921207_s1), ST6GALNAC5 (Hs05018504_s1), CCR2 (Hs00704702_s1), CCL2 (Hs00234140_m1), CCR4 (Hs00747615_s1), IL-8 (Hs00174103_m1), CXCR1 (Hs01921207_s1), and CXCR2 (Hs01891184_s1).
TC extravasation assays
TC extravasation assays were performed on day 7 once the vasculature was stabilized morphologically (15, 54). TCs were resuspended at 1 million cells/ml and a 20-μl suspension volume was perfused in the MVNs as previously described (17, 31) via a 10-min hydrostatic pressure drop between the two media channels of the device. Media was replenished, and extravasation was quantified as described (66) at different time points following perfusion. TCs observed to cross the endothelium partially or completely were classified as extravasated cells, and the percentage of extravasation or extravasation efficiency was measured as the ratio of extravasated cells to the total number of TCs per device for each time point and MVN condition.
The effect of PC- or AC-secreted factors on TC extravasation was determined by treating monoculture MVNs of iPS-EC alone with PC, PC (+MDA-231), AC, or AC (+MDA-231) CM obtained as described above. TCs were perfused in the MVNs in a 1:1 ratio of CM and iPS-EC media (necessary for MVN stability), and extravasation was quantified at different time points.
The KD of ST6GALNAC5 in the TCs was performed with Silencer Select siRNA probes at 10 μM [s224888 as siRNA #1 (human) and s37827 as siRNA #2 (human); Thermo Fisher Scientific] using Lipofectamine RNAiMAX reagent (13778030, Thermo Fisher Scientific) in Opti-MEM reduced serum medium (31985062, Thermo Fisher Scientific). Silencer Select Negative Control siRNAs were used as controls (nos. 1 and 2; 4390843, Thermo Fisher Scientific). TCs were treated twice more than 48 hours with siRNA probes and left to rest for an additional 24 hours in fresh media before perfusion in the MVNs. KD was validated with qRT-PCR using TaqMan Gene Expression Assays GAPDH (Hs01921207_s1, Thermo Fisher Scientific) as control and ST6GALNAC5 (Hs05018504_s1, Thermo Fisher Scientific).To test the influence of various relevant cytokines on TC extravasation, MVNs were incubated with anti-CCL2 at 10 μg/ml (MAB279), anti–IL-8 at 1 μg/ml (MAB208), anti–IGFBP-4 at 20 μg/ml (MAB8041), anti-CCL26 at 15 μg/ml (MAB653), anti–IL-6 at 3 μg/ml (MAB2061), anti-OPN at 12 μg/ml (AF1433), and anti–PDGF-BB at 3 μg/ml (AF220), either individually or altogether (R&D Systems). Incubation was performed for 30 min at 37°C and 5% CO2 before TC perfusion, without washing. At times, TCs were also incubated with anti-CCL2 at 10 μg/ml (MAB279) for 30 min at 37°C and 5% CO2 followed by a PBS wash and perfusion in untreated MVNs. Extravasation was quantified 6 hours following perfusion. To block CCR2 or CCR4, MVNs or TCs were incubated with a CCR2 antagonist at 50 μM (227016, Millipore Sigma) and/or a CCR4 antagonist at 500 nM (21885, Cayman Chemical). Incubation was performed 24 hours before TC perfusion and repeated 1 hour before perfusion, followed by a PBS wash. Antagonists were resuspended in dimethyl sulfoxide (DMSO) before dilution in MVN or TC media. A DMSO control was used on both MVNs and TCs when quantifying extravasation. TC extravasation was quantified at 6, 12, and 24 hours after perfusion.
To test the influence of CCL2 in the MVNs on TC extravasation, rhCCL2 at various concentrations (279-MC-010, R&D Systems) was used to treat MVNs, followed by a PBS wash before perfusion. The KD of CCL2 in the TCs was also performed with Silencer Select siRNA probes at 10 μM (s12566 and s12567, Thermo Fisher Scientific) using Lipofectamine RNAiMAX reagent (13778030, Thermo Fisher Scientific) in Opti-MEM reduced serum medium (31985062, Thermo Fisher Scientific). Silencer Select Negative Control siRNAs were used as controls (nos. 1 and 2; 4390843, Thermo Fisher Scientific). TCs were treated twice more than 48 hours with siRNA probes and left to rest for an additional 24 hours in fresh media before perfusion in the MVNs. The KD of CCL2 in the TCs was confirmed via ELISA (DCP00, R&D Systems).
Cell migration analysis
To test the influence of CCL2 on migration in 2D, MDA-231 were plated in a 24-well glass-bottom plate and treated with different concentrations of rhCCL2 (279-MC-010, R&D Systems). Time-lapse images of TC migration in 2D were obtained at 15-min intervals for 6 hours following treatment using a confocal microscope (Olympus) with environmental chamber maintained at 37°C and 5% CO2. Resulting TC speeds and migration tracks in 2D (x and y displacements) were graphed for 50 randomly selected TCs per rhCCL2 condition.
To quantify migration of TCs and ACs in 3D, time-lapse images of extravasating TCs were obtained with fluorescently labeled triculture MVN cells at 30-min intervals. At times, MVNs were treated with anti-CCL2 or TCs were treated with an antagonist for CCR2 as defined above before TC perfusion and live imaging with a confocal microscope (Olympus) with environmental chamber maintained at 37°C and 5% CO2.
Over the course of extravasation, distances between the centers of ACs and extravasating TCs were quantified. Given that the average vessel branch length of the triculture MVNs is ~200 μm, ACs located within a 200 μm diameter centered on the TC were classified as “near” the TC and were considered in the distance measurements. The speeds of TCs, ACs near extravasating TCs, and ACs in control MVNs without TCs were also measured over time. Using the 3D time-lapse images of TC extravasation in the triculture MVNs, speeds and AC-TC distances were quantified over time via the Manual Tracking function of the TrackMate plugin in the Fiji distribution of ImageJ (NIH). Briefly, the x-y-z coordinates of the centers of TCs and ACs were tracked at each time point, and their speeds were obtained in micrometers per hour. The AC-TC distances were obtained in micrometer by averaging the distances of all ACs found near extravasating TCs. A time frame of 90 min before and after extravasation was considered to quantify the fold change in AC-TC distance (with respect to the first time point, 90 min before extravasation).
The number of ACs near extravasating TCs was also quantified by counting ACs located within a 200-μm diameter centered on the TC. As for AC-TC distances, this count was presented as a fold change in number of ACs with respect to the first time point 90-min before extravasation.
Computational model of CCL2 transport
A finite element model was developed to evaluate the spatial distribution of CCL2 in the MVNs using the COMSOL Multiphysics software. A bifurcating microvascular geometry similar as previous models from our group (31) was used, with a diameter and branch length corresponding to values measured in this study. A TC was modeled as a solid sphere of similar size adjacent to a wall within the vascular domain. Similarly, ACs were modeled as spheres with the same surface area as ex vivo measurements from electron micrographs (67). A total of seven ACs were distributed across the extracellular domain with an average distance of 10 μm from the vascular walls, both number and distance corresponding to experimental measurements in the MVNs.
CCL2 was modeled as a species with a similar molecular weight (10 kDa) to approximate its diffusion coefficient (D = 150 μm2/s). A constant flux of the species was ascribed to the surface of the modeled ACs, and a surface consumption of 0.5 s−1 × C was applied to the TC, as previously done in studies modeling TC morphogen consumption (68, 69). To account for the transvascular transport of CCL2, a thin diffusion barrier was introduced between the extracellular and vascular domains, through which both paracellular and transcellular transports of the species were considered. The relative flux of the species through this barrier is given by product of the differential concentration across the domains and the species-specific permeability (P) (Nvasc = PCCL2 × ΔC). This permeability value accounts for the diffusive transport between endothelial junctions and CCR2-regulated transcytosis of CCL2 (70). The former was obtained from permeability measurements of 10-kDa FITC-dextran performed in this study. The transcellular contribution was approximated from our previous study in MVNs (29). For the computational models incorporating convection due to fluid flow in the MVNs, an incompressible Stokes flow behavior was applied
where μ is the viscosity, P is the pressure across the system, and u is the velocity vector field. Within the vascular domain, the inlet fluid velocity was set at a physiological value of 0.5 mm/s (40). Mass conservation resulted in the same velocity at the outlet, and a no-slip boundary condition was applied to the internal vascular walls. A symmetry boundary condition (n·N = 0) was applied on the lateral boundaries of the extracellular domain given the periodic pattern of the vessels across the microfluidic device. A no-flux condition was applied at the top and bottom boundaries since they are situated close to the upper and lower walls of the device (n·− D∇C = 0).
A quasi-steady state assumption was made for all the models, given that the diffusion and convection time scales (~1 to 10 s) are two to three orders of magnitude lower than the cellular movement time scales (~1000 s), obtained from measurements in the MVNs. It was thus assumed that the species achieves a steady distribution before the onset of cell migration. The mass conservation and transport equations are reduced to
where N is the flux, R accounts for the source and consumption of the species, and C is the molar concentration. The models were numerically solved using the Transport of Diluted Species module in COMSOL, and the concentration profiles were visualized by generating heatmaps of the species concentration in the MVNs with a lodged TC and ACs in the surrounding matrix.
Mice extravasation assay
All animal studies were approved by the MIT Institutional Animal Care and Use Committee. Athymic nude (NU/J) mice were purchased from the Jackson laboratory. MDA-231 cells were treated with the CCR2 MISSION shRNA Bacterial Glycerol Stock or the nontarget shRNA control plasmid DNA MISSION TRC2 pLKO.5-puro as a control shRNA (Millipore Sigma), and CCR2 gene expression was validated via real-time qRT-PCR. Once CCR2 KD was confirmed, mice were injected with a 100-μl suspension of TCs in PBS at 0.5 million cells/ml via ultrasound-guided intracardiac injections (49). Half of the mice were injected with the CCR2 KD MDA-231, and the other half were injected with the control shRNA MDA-231. Mice were sacked, and their brains were collected at two different time points following intracardiac injection (24 and 48 hours). Whole brains were fixed via immersion in 4% paraformaldehyde in PBS at room temperature for 48 hours. Brains were sliced, stained, and mounted on coverslips for imaging as described above. Eight 100-μm brain slices per mouse were imaged via confocal microscopy using a 10× objective to quantify number of TCs lodged in the brain capillaries or parenchyma. To quantify TC extravasation, confocal imaging was performed using a 60× oil-based objective for adequate visualization of TCs and extravasation events over the entire 100-μm-thick brain slices. Two randomly selected ROIs per brain slice were imaged for each mouse (eight slices total), and extravasation was computed as the ratio of extravasated cells to total number of TCs per mouse, as done in vitro.
Statistical analysis
All data are plotted as means ± SD, unless indicated otherwise. All boxes show quartiles, center lines show means, and whiskers show SDs. Statistical significance was assessed using Student’s t tests performed with the software OriginPro when comparing two conditions/groups. When comparing more than two groups, significance was assessed using one-way analysis of variance (ANOVA) with Tukey post hoc test (if the unequal variance assumption holds, as measured by Levene’s test for homogeneity of variance) or using Brown-Forsythe and Welch ANOVA with Dunnett’s T3 post hoc test (if the unequal variance assumption is violated) with the software GraphPad Prism. Significance is represented as follows: n.s. stands for not significant, n.s.# stands for not significant across all conditions (pairwise), *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. In all in vitro experiments, six devices per condition were used unless otherwise indicated. In all in vivo mice experiments, four mice per time point and per condition were used.
Acknowledgments
We thank M. Gillrie for the help transfecting iPS-ECs with the GFP construct; Y. Chen for the help transfecting MDA-MB-231 with the CCR2 KD and control shRNA constructs; N. Henning, V. Spanoudaki, and the Preclinical Imaging and Testing core facility for the help in mice intracardiac tumor cell injections; A. Burds Connor and the Preclinical Modeling Facility for the help in mice brain harvests; K. Cormier for histology support; and the Swanson Biotechnology Center for the exceptional core facilities. We also thank all the members of the Kamm laboratory for helpful discussions while developing this work. Funding: C.H. is supported by the Ludwig Center for Molecular Oncology Graduate Fellowship and by grant U01 CA202177 from the National Cancer Institute (NCI). Y.S. acknowledges support from Cure Alzheimer’s Fund. J.C.S. is supported by a National Science Foundation (NSF) graduate research fellowship. L.L. was supported by the Swiss National Science Foundation (SNSF) Early and Advanced Postdoc Mobility Fellowships and the Hope Funds Postdoctoral Fellowship. T.J. is a Howard Hughes Medical Institute investigator, a David H. Koch professor of Biology, and a Daniel K. Ludwig scholar. T.J. also acknowledges support from the Ludwig Center for Molecular Oncology and Cancer Center Support Grant (CCSG) P30 CA14501 from the NCI. Author contributions: C.H., Y.S., and R.D.K. designed the study. C.H., Y.S., and L.L. performed the experiments. J.C.S. performed the computational simulations. C.H. and L.L. analyzed the data. T.J. and R.D.K. supervised the work. C.H. and R.D.K. wrote the manuscript, and all authors contributed to its final form. Competing interests: R.D.K. is a cofounder of AIM Biotech that markets microfluidic systems for 3D culture. R.D.K. also receives financial support from Amgen, Biogen, and Gore. T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific. He is also a cofounder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to the design or execution of this study or interpretation of data presented in this manuscript. T.J. laboratory currently also receives funding from the Johnson & Johnson Lung Cancer Initiative and the Lustgarten Foundation for Pancreatic Cancer Research, but this funding did not support the research described in this manuscript. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg8139/DC1
REFERENCES AND NOTES
- 1.Achrol A. S., Rennert R. C., Anders C., Soffietti R., Ahluwalia M. S., Nayak L., Peters S., Arvold N. D., Harsh G. R., Steeg P. S., Chang S. D., Brain metastases. Nat. Rev. Dis. Primers. 5, 5 (2019). [DOI] [PubMed] [Google Scholar]
- 2.D’Andrea G., Palombi L., Minniti G., Pesce A., Marchetti P., Brain metastases: Surgical treatment and overall survival. World Neurosurg. 97, 169–177 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Nayak L., Lee E. Q., Wen P. Y., Epidemiology of brain metastases. Curr. Oncol. Rep. 14, 48–54 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Langen U. H., Ayloo S., Gu C., Development and cell biology of the blood-brain barrier. Annu. Rev. Cell Dev. Biol. 35, 591–613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong X., Hou Z., Endsley M. P., Gronseth E. I., Rarick K. R., Jorns J. M., Yang Q., Du Z., Yan K., Bordas M. L., Gershan J., Deepak P., Geethadevi A., Chaluvally-Raghavan P., Fan Y., Harder D. R., Ramchandran R., Wang L., Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. NPJ Precis. Oncol. 3, 24 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Q., Boire A., Jin X., Valiente M., Er E. E., Lopez-Soto A., S. Jacob L., Patwa R., Shah H., Xu K., Cross J. R., Massagué J., Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 533, 493–498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valiente M., Obenauf A. C., Jin X., Chen Q., Zhang X. H.-F., Lee D. J., Chaft J. E., Kris M. G., Huse J. T., Brogi E., Massagué J., Serpins promote cancer cell survival and vascular cooption in brain metastasis. Cell 156, 1002–1016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiwary S., Morales J. E., Kwiatkowski S. C., Lang F. F., Rao G., McCarty J. H., Metastatic brain tumors disrupt the blood-brain barrier and alter lipid metabolism by inhibiting expression of the endothelial cell fatty acid transporter Mfsd2a. Sci. Rep. 8, 8267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L., Zhang S., Yao J., Lowery F. J., Zhang Q., Huang W.-C., Li P., Li M., Wang X., Zhang C., Wang H., Ellis K., Cheerathodi M., McCarty J. H., Palmieri D., Saunus J., Lakhani S., Huang S., Sahin A. A., Aldape K. D., Steeg P. S., Yu D., Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shibue T., Weinberg R. A., Metastatic colonization: Settlement, adaptation and propagation of tumor cells in a foreign tissue environment. Semin. Cancer Biol. 21, 99–106 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Kienast Y., von Baumgarten L., Fuhrmann M., Klinkert W. E. F., Goldbrunner R., Herms J., Winkler F., Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 16, 116–122 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Bos P. D., Zhang X. H.-F., Nadal C., Shu W., Gomis R. R., Nguyen D. X., Minn A. J., van de Vijver M. J., Gerald W. L., Foekens J. A., Massagué J., Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sevenich L., Bowman R. L., Mason S. D., Quail D. F., Rapaport F., Elie B. T., Brogi E., Brastianos P. K., Hahn W. C., Holsinger L. J., Massagué J., Leslie C. S., Joyce J. A., Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat. Cell Biol. 16, 876–888 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu H., Li Z., Yu Y., Sizdahkhani S., Ho W. S., Yin F., Wang L., Zhu G., Zhang M., Jiang L., Zhuang Z., Qin J., A dynamic in vivo-like organotypic blood-brain barrier model to probe metastatic brain tumors. Sci. Rep. 6, 36670 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campisi M., Shin Y., Osaki T., Hajal C., Chiono V., Kamm R. D., 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials 180, 117–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin Y., Han S., Jeon J. S., Yamamoto K., Zervantonakis I. K., Sudo R., Kamm R. D., Chung S., Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protoc. 7, 1247–1259 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen M. B., Whisler J. A., Fröse J., Yu C., Shin Y., Kamm R. D., On-chip human microvasculature assay for visualization and quantification of tumor cell extravasation dynamics. Nat. Protoc. 12, 865–880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bang S., Lee S.-R., Ko J., Son K., Tahk D., Ahn J., Im C., Jeon N. L., A low permeability microfluidic blood-brain barrier platform with direct contact between perfusable vascular network and astrocytes. Sci. Rep. 7, 8083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S., Chung M., Lee S.-R., Jeon N. L., 3D brain angiogenesis model to reconstitute functional human blood–brain barrier in vitro. Biotechnol. Bioeng. 117, 748–762 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Bonkowski D., Katyshev V., Balabanov R. D., Borisov A., Dore-Duffy P., The CNS microvascular pericyte: Pericyte-astrocyte crosstalk in the regulation of tissue survival. Fluids Barriers CNS 8, 8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank R. N., Dutta S., Mancini M. A., Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Invest. Ophthalmol. Vis. Sci. 28, 1086–1091 (1987). [PubMed] [Google Scholar]
- 22.Wong A. D., Ye M., Levy A. F., Rothstein J. D., Bergles D. E., Searson P. C., The blood-brain barrier: An engineering perspective. Front. Neuroeng. 6, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herland A., van der Meer A. D., FitzGerald E. A., Park T.-E., Sleeboom J. J. F., Ingber D. E., Distinct contributions of astrocytes and pericytes to neuroinflammation identified in a 3D human blood-brain barrier on a chip. PLOS ONE 11, e0150360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown J. A., Pensabene V., Markov D. A., Allwardt V., Diana Neely M., Shi M., Britt C. M., Hoilett O. S., Yang Q., Brewer B. M., Samson P. C., McCawley L. J., May J. M., Webb D. J., Li D., Bowman A. B., Reiserer R. S., Wikswo J. P., Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 9, 054124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park T.-E., Mustafaoglu N., Herland A., Hasselkus R., Mannix R., FitzGerald E. A., Prantil-Baun R., Watters A., Henry O., Benz M., Sanchez H., McCrea H. J., Goumnerova L. C., Song H. W., Palecek S. P., Shusta E., Ingber D. E., Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 10, 2621 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buzhdygan T. P., DeOre B. J., Baldwin-Leclair A., Bullock T. A., McGary H. M., Khan J. A., Razmpour R., Hale J. F., Galie P. A., Potula R., Andrews A. M., Ramirez S. H., The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier. Neurobiol. Dis. 146, 105131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minn A. J., Gupta G. P., Siegel P. M., Bos P. D., Shu W., Giri D. D., Viale A., Olshen A. B., Gerald W. L., Massagué J., Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatzoglou V., Patel G. V., Morris M. J., Curtis K., Zhang Z., Shi W., Huse J., Rosenblum M., Holodny A. I., Young R. J., Brain metastases from prostate cancer: An 11-year analysis in the MRI era with emphasis on imaging characteristics, incidence, and prognosis. J. Neuroimaging 24, 161–166 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Offeddu G. S., Haase K., Gillrie M. R., Li R., Morozova O., Hickman D., Knutson C. G., Kamm R. D., An on-chip model of protein paracellular and transcellular permeability in the microcirculation. Biomaterials 212, 115–125 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Shin Y., Choi S. H., Kim E., Bylykbashi E., Kim J. A., Chung S., Kim D. Y., Kamm R. D., Tanzi R. E., Blood–brain barrier dysfunction in a 3D in vitro model of Alzheimer’s disease. Adv. Sci. 6, 1900962 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen M. B., Hajal C., Benjamin D. C., Yu C., Azizgolshani H., Hynes R. O., Kamm R. D., Inflamed neutrophils sequestered at entrapped tumor cells via chemotactic confinement promote tumor cell extravasation. Proc. Natl. Acad. Sci. U.S.A. 115, 7022–7027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim S. Y., Yuzhalin A. E., Gordon-Weeks A. N., Muschel R. J., Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget 7, 28697–28710 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Craig M. J., Loberg R. D., CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 25, 611–619 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Zhang T., Somasundaram R., Berencsi K., Caputo L., Gimotty P., Rani P., Guerry D. P., Swoboda R., Herlyn D., Migration of cytotoxic T lymphocytes toward melanoma cells in three-dimensional organotypic culture is dependent on CCL2 and CCR4. Eur. J. Immunol. 36, 457–467 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Wolf M. J., Hoos A., Bauer J., Boettcher S., Knust M., Weber A., Simonavicius N., Schneider C., Lang M., Stürzl M., Croner R. S., Konrad A., Manz M. G., Moch H., Aguzzi A., van Loo G., Pasparakis M., Prinz M., Borsig L., Heikenwalder M., Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell 22, 91–105 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Zijlmans H. J. M. A. A., Fleuren G. J., Baelde H. J., Eilers P. H. C., Kenter G. G., Gorter A., The absence of CCL2 expression in cervical carcinoma is associated with increased survival and loss of heterozygosity at 17q11.2. J. Pathol. 208, 507–517 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Lu Y., Cai Z., Galson D. L., Xiao G., Liu Y., George D. E., Melhem M. F., Yao Z., Zhang J., Monocyte chemotactic protein-1 (MCP-1) acts as a paracrine and autocrine factor for prostate cancer growth and invasion. Prostate 66, 1311–1318 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Deshmane S. L., Kremlev S., Amini S., Sawaya B. E., Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 29, 313–326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Follain G., Osmani N., Azevedo A. S., Allio G., Mercier L., Karreman M. A., Solecki G., Garcia Leòn M. J., Lefebvre O., Fekonja N., Hille C., Chabannes V., Dollé G., Metivet T., Hovsepian F. D., Prudhomme C., Pichot A., Paul N., Carapito R., Bahram S., Ruthensteiner B., Kemmling A., Siemonsen S., Schneider T., Fiehler J., Glatzel M., Winkler F., Schwab Y., Pantel K., Harlepp S., Goetz J. G., Hemodynamic forces tune the arrest, adhesion, and extravasation of circulating tumor cells. Dev. Cell 45, 33–52.e12 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Ivanov K. P., Kalinina M. K., Levkovich Y. I., Blood flow velocity in capillaries of brain and muscles and its physiological significance. Microvasc. Res. 22, 143–155 (1981). [DOI] [PubMed] [Google Scholar]
- 41.Abbott N. J., Rönnbäck L., Hansson E., Astrocyte–endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Mestre H., Tithof J., Du T., Song W., Peng W., Sweeney A. M., Olveda G., Thomas J. H., Nedergaard M., Kelley D. H., Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat. Commun. 9, 4878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Siddiqui J. A., Partridge N. C., CCL2/monocyte chemoattractant protein 1 and parathyroid hormone action on bone. Front. Endocrinol. 8, 49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang C.-H., Tsai C.-C., CCL2 increases MMP-9 expression and cell motility in human chondrosarcoma cells via the Ras/Raf/MEK/ERK/NF-κB signaling pathway. Biochem. Pharmacol. 83, 335–344 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Kuroda T., Kitadai Y., Tanaka S., Yang X., Mukaida N., Yoshihara M., Chayama K., Monocyte chemoattractant protein-1 transfection induces angiogenesis and tumorigenesis of gastric carcinoma in nude mice via macrophage recruitment. Clin. Cancer Res. 11, 7629–7636 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Chiu H.-Y., Sun K.-H., Chen S.-Y., Wang H.-H., Lee M.-Y., Tsou Y.-C., Jwo S. C., Sun G. H., Tang S. J., Autocrine CCL2 promotes cell migration and invasion via PKC activation and tyrosine phosphorylation of paxillin in bladder cancer cells. Cytokine 59, 423–432 (2012). [DOI] [PubMed] [Google Scholar]
- 47.D. A. Lauffenburger, J. Linderman, in Receptors: Models for Binding, Trafficking, and Signaling (Oxford Univ. Press, 1993), 376 p. [Google Scholar]
- 48.Lauffenburger D., Rothman C., Zigmond S. H., Measurement of leukocyte motility and chemotaxis parameters with a linear under-agarose migration assay. J. Immunol. 131, 940–947 (1983). [PubMed] [Google Scholar]
- 49.Hebert J. D., Myers S. A., Naba A., Abbruzzese G., Lamar J. M., Carr S. A., Hynes R. O., Proteomic profiling of the ECM of xenograft breast cancer metastases in different organs reveals distinct metastatic niches. Cancer Res. 80, 1475–1485 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Obenauf A. C., Massagué J., Surviving at a distance: Organ-specific metastasis. Trends Cancer 1, 76–91 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bentolila L. A., Prakash R., Mihic-Probst D., Wadehra M., Kleinman H. K., Carmichael T. S., Péault B., Barnhill R. L., Lugassy C., Imaging of angiotropism/vascular co-option in a murine model of brain melanoma: Implications for melanoma progression along extravascular pathways. Sci. Rep. 6, 23834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuczynski E. A., Vermeulen P. B., Pezzella F., Kerbel R. S., Reynolds A. R., Vessel co-option in cancer. Nat. Rev. Clin. Oncol. 16, 469–493 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Hajal C., Campisi M., Mattu C., Chiono V., Kamm R. D., In vitro models of molecular and nano-particle transport across the blood-brain barrier. Biomicrofluidics 12, 042213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Offeddu G. S., Hajal C., Foley C. R., Wan Z., Ibrahim L., Coughlin M. F., Kamm R. D., The cancer glycocalyx mediates intravascular adhesion and extravasation during metastatic dissemination. Commun. Biol. 4, 255 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carrillo-de Sauvage M. A., Gómez A., Ros C. M., Ros-Bernal F., Martín E. D., Perez-Vallés A., Gallego-Sanchez J. M., Fernández-Villalba E., Barcia C. Sr., Barcia C. Jr., Herrero M.-T., CCL2-expressing astrocytes mediate the extravasation of T lymphocytes in the brain. Evidence from patients with glioma and experimental models in vivo. PLOS ONE 7, e30762 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Varvel N. H., Neher J. J., Bosch A., Wang W., Ransohoff R. M., Miller R. J., Dingledine R., Infiltrating monocytes promote brain inflammation and exacerbate neuronal damage after status epilepticus. Proc. Natl. Acad. Sci. U.S.A. 113, E5665–E5674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim R. Y., Hoffman A. S., Itoh N., Ao Y., Spence R., Sofroniew M. V., Voskuhl R. R., Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 274, 53–61 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ponath G., Park C., Pitt D., The role of astrocytes in multiple sclerosis. Front. Immunol. 9, 217 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roberts T. K., Eugenin E. A., Lopez L., Romero I. A., Weksler B. B., Couraud P.-O., Berman J. W., CCL2 disrupts the adherens junction: Implications for neuroinflammation. Lab. Invest. 92, 1213–1233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dimitrijevic O. B., Stamatovic S. M., Keep R. F., Andjelkovic A. V., Effects of the chemokine CCL2 on blood–brain barrier permeability during ischemia–reperfusion injury. J. Cereb. Blood Flow Metab. 26, 797–810 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Shi Y., Zhang L., Pu H., Mao L., Hu X., Jiang X., Xu N., Stetler R. A., Zhang F., Liu X., Leak R. K., Keep R. F., Ji X., Chen J., Rapid endothelial cytoskeletal reorganization enables early blood–brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat. Commun. 7, 10523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan W., Lv Y., Zeng M., Fu B. M., Non-invasive measurement of solute permeability in cerebral microvessels of the rat. Microvasc. Res. 77, 166–173 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Minn A. J., Kang Y., Serganova I., Gupta G. P., Giri D. D., Doubrovin M., Ponomarev V., Gerald W. L., Blasberg R., Massagué J., Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest. 115, 44–55 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ponomarev V., Doubrovin M., Serganova I., Vider J., Shavrin A., Beresten T., Ivanova A., Ageyeva L., Tourkova V., Balatoni J., Bornmann W., Blasberg R., Gelovani Tjuvajev J., A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur. J. Nucl. Med. Mol. Imaging 31, 740–751 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Zukor K. A., Kent D. T., Odelberg S. J., Fluorescent whole-mount method for visualizing three-dimensional relationships in intact and regenerating adult newt spinal cords. Dev. Dyn. 239, 3048–3057 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.G. Offeddu, C. Hajal, C. Foley, Z. Wan, L. Ibrahim, M. Coughlin, R. D. Kamm, Glycocalyx-mediated vascular dissemination of circulating tumor cells. bioRxiv 2020.04.28.066746. 30 April 2020. 10.1101/2020.04.28.066746. [DOI]
- 67.Williams V., Grossman R. G., Edmunds S. M., Volume and surface area estimates of astrocytes in the sensorimotor cortex of the cat. Neuroscience 5, 1151–1159 (1980). [DOI] [PubMed] [Google Scholar]
- 68.Fleury M. E., Boardman K. C., Swartz M. A., Autologous morphogen gradients by subtle interstitial flow and matrix interactions. Biophys. J. 91, 113–121 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Polacheck W. J., Charest J. L., Kamm R. D., Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. U.S.A. 108, 11115–11120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ge S., Song L., Serwanski D. R., Kuziel W. A., Pachter J. S., Transcellular transport of CCL2 across brain microvascular endothelial cells. J. Neurochem. 104, 1219–1232 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg8139/DC1