

Innovations in the American lobster genome provide insight into the ecological success of this iconic marine crustacean.
Abstract
The American lobster, Homarus americanus, is integral to marine ecosystems and supports an important commercial fishery. This iconic species also serves as a valuable model for deciphering neural networks controlling rhythmic motor patterns and olfaction. Here, we report a high-quality draft assembly of the H. americanus genome with 25,284 predicted gene models. Analysis of the neural gene complement revealed extraordinary development of the chemosensory machinery, including a profound diversification of ligand-gated ion channels and secretory molecules. The discovery of a novel class of chimeric receptors coupling pattern recognition and neurotransmitter binding suggests a deep integration between the neural and immune systems. A robust repertoire of genes involved in innate immunity, genome stability, cell survival, chemical defense, and cuticle formation represents a diversity of defense mechanisms essential to thrive in the benthic marine environment. Together, these unique evolutionary adaptations contribute to the longevity and ecological success of this long-lived benthic predator.
INTRODUCTION
The American lobster, Homarus americanus (Milne-Edwards, 1837), is a decapod crustacean of ecological and commercial importance, supporting one of the largest and most valuable fisheries along the east coast of North America (1). Changing environmental conditions have become a concern over the past few decades as elevated temperatures and partial pressure of CO2 levels have been shown to severely affect lobster growth, reproduction, and survival (2–4) and are implicated in increased occurrences of epizootic shell disease (5, 6). Monitoring population structure and dynamics of the American lobster have proven challenging because of limited genomic resources and weak genetic differentiation resulting from large population size and high dispersal rates (7). Nevertheless, single-nucleotide polymorphism genotyping revealed regional-scale genetic structure throughout much of the species’ range and enabled an assessment of how environmental factors such as currents and sea surface temperature shape lobster populations (8, 9). Expanded genomic resources will enhance efforts to investigate population structure and disease susceptibility to support sustainable fisheries management during this time of rapidly changing environmental conditions in the Northwest Atlantic Ocean (10).
In addition to its ecological and commercial importance, the lobsters’ accessible nervous system has provided a powerful model for neuroscience. Studies of lobster heart, stomatogastric, and chemosensory circuits have enabled an understanding of central pattern generations (11), neuromodulation (12), and olfaction (13). Transcriptomic studies have uncovered a complex repertoire of neurotransmitters and their receptors, including numerous neuropeptides (12, 14). However, a complete assessment of the suspected diversification of multiple signaling and integrative components of the lobster nervous system has not been possible without a reference genome.
The American lobster is among the largest benthic invertebrate throughout its range weighing up to 20 kg and reaching a length of more than 1 m (15). Lobsters exhibit indeterminate growth through a stepwise process of molting and exhibit lifelong reproduction suggesting negligible senescence (16). The lack of permanent calcified structures makes accurate age determinations challenging, but H. americanus has been estimated to attain a life span of more than 50 years and perhaps as much as 100 years (15, 16). Similar to other decapod crustaceans, neoplastic diseases have rarely been reported for lobsters, indicating high fidelity of the genome over their long life (16, 17).
Crustaceans are a diverse and ancient group of arthropods noted for variability in their genome size and structure. Crustacean genomes range from 0.12 to 47.88 giga–base pairs (Gbp) (18) with the haploid number of chromosomes ranging from 3 to 188 (19). Polyploidy has been reported in some species (19, 20), and the occurrence of supernumerary or B chromosomes (asynaptic heterochromatic chromosomes with non-Mendelian segregation) has also been reported (19, 21). Total nuclear DNA content of the American lobster has been estimated to be between 6.26 and 9.5 pg (6.12 and 9.28 Gbp) (18, 22) distributed on approximately 136 chromosomes along with two to four supernumerary chromosomes (23). In this study, we report a highly contiguous draft genome for the American lobster and demonstrate a remarkable expansion of the neural gene repertoire and suggested unique interactions between the immune and nervous systems. Together with a robust repertoire of genes associated with innate immunity, genome integrity, cell survival, and chemical defense, the genome reveals adaptations to the benthic marine environment that may contribute to the ecological success and remarkable life history of the lobster.
RESULTS
Sequencing and assembly of the lobster genome
Paired-end and mate pair Illumina libraries were combined with Oxford Nanopore long reads to generate high-quality contigs using the MaSuRCA genome assembler (24). Contigs were scaffolded using proximity ligation data from Dovetail Chicago and Hi-C libraries to yield a genome assembly of 2.29 Gbp. The final assembly consisted of 47,245 scaffolds with 50 and 90% of the genome represented in 242 scaffolds (N50 = 759.6 kb) and 22,569 scaffolds (N90 = 15.2 kb), respectively. Benchmarking Universal Single-Copy Ortholog (BUSCO) analysis with the arthropod (obd9) gene set indicated a high-quality assembly with only 28 missing, 28 fragmented, and 18 duplicated genes of the 1066 examined. Additional sequencing and assembly statistics can be found in Table 1 and table S1.
Table 1. Statistics for the H. americanus draft genome assembly and gene predictions.
| Assembly | |
| Total length | 2.29 Gbp |
| Number of scaffolds | 47,245 |
| N50 (minimum length/number scaffolds) |
759.6 kbp/242 |
| N90 (minimum length/number scaffolds) |
15.2 kbp/22,569 |
| Longest scaffold | 42.52 Mbp |
| Contig N50 | 133.3 kbp |
| Number of gaps | 8350 |
| Percentage of genome in gaps | 0.036% |
| BUSCO (genome assembly) | Arthropod (obd9) gene set |
| Complete | 992 |
| Duplicated | 18 |
| Fragmented | 28 |
| Missing | 28 |
| Gene predictions | |
| No. of genes | 25,284 |
| Average gene length | 9560 bp |
| Average coding sequence length | 1171 bp |
| No. of exons per gene | 5.79 |
| BUSCO (gene predictions) | Arthropod (obd9) gene set |
| Complete | 930 |
| Duplicated | 19 |
| Fragmented | 67 |
| Missing | 50 |
The genome size estimated from k-mer analysis (k = 23) was 3.2 Gbp, which is within the range of values previously reported for the 1C nuclear DNA content of H. americanus (3.06 to 4.64 Gbp) (18, 22) but indicates that our assembly is missing about 28% of the genome. This discrepancy could be explained by DNA missed by sequencing bias or challenges of assembling highly repetitive regions and is consistent with other reported crustacean genomes that, with the exception of the marbled crayfish, are missing between 22 and 47% of the predicted genome size (table S2).
Repeat analysis revealed that 52.9% of the lobster genome assembly was made up of repetitive sequences. A total of 1302 repeat families were identified, 433 of which have been previously reported in other species, while the remaining 869 families were unknown (table S3). Of the previously characterized repeat families, long interspersed nuclear element retrotransposons were the most common, occurring 398,238 times, making up 10.2% of the genome. Short interspersed nuclear element retrotransposons made up 0.4% of the genome, while long terminal repeat (LTR) elements and DNA elements accounted for 4.5 and 4.8%, respectively. Gypsy-like sequences were the most common LTR elements (3.28% of genome), and TcMar-Tigger were the most common DNA elements (1.28%). Simple repeats represented 1,709,134 occurrences making up 6.3% of the genome (table S3). A comparison of the occurrence of repetitive elements between H. americanus and four other crustacean genomes shows a high level of variability across species (table S4).
A de novo assembled transcriptome from stage IV and stage V larval lobsters consisted of 478,211 transcript contigs and served as evidence for the MAKER2 gene prediction pipeline (25) that identified 25,284 protein-coding gene models (table S5). The structure of predicted genes for H. americanus was similar to that of other previously sequenced crustacean genomes (table S6) (20, 26–28). BUSCO analysis on the predicted gene models demonstrated 949 complete (single, 930; duplicated, 19), 67 fragmented, and 50 missing (Table 1). A total of 12,598 gene models had high homology to validated proteins in Swiss-Prot, an additional 3513 gene models were assigned putative annotations based on homology to proteins in the nr database or known Pfam domains, and the remaining 9173 were annotated as uncharacterized (table S5).
Ortholog analysis of genes from eight arthropod species including H. americanus, Daphnia magna, Litopenaeus vannamei, Cherax quadricarinatus, Procambarus virginalis, Parhyale hawaiensis, Hyalella azteca, and Drosophila melanogaster identified 23,238 gene family clusters. We constructed a phylogeny using 264 single-copy orthologs and analyzed the expansion and contraction of all ortholog families at each node (Fig. 1). In the H. americanus lineage, 821 gene families were expanded with significant enrichment in genes associated with neuronal function (52 families), cuticle structure and remodeling (34 families), and transposons (25 families) (Fig. 1 and table S7). The gene ontology (GO) terms associated with the most highly expanded families include structural constituent of cuticle and chitin metabolic process, as well as ligand-gated ion channel (LGIC) activity and ionotropic glutamate receptor (iGluR/IR) activity (table S7). For the 2445 contracted gene families in H. americanus, the three most represented GO terms were protein binding, protein kinase activity, and nucleic acid binding (Fig. 1 and table S7). Of the contracted gene families, 2210 gene families were lost in the lobster lineage. These included genes with GO terms associated with protein binding (244 families), nucleic acid/DNA binding (211 families), and adenosine 5′-triphosphate binding (200 families).
Fig. 1. The American lobster, arthropod phylogenetic tree, and gene family expansion/contraction.
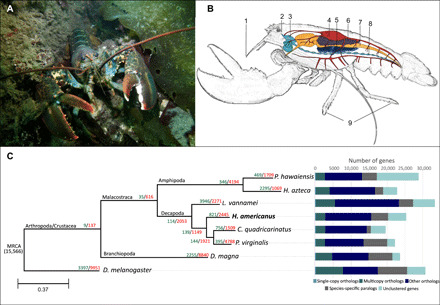
(A) The American lobster, H. americanus, in its natural habitat in the Gulf of Maine, USA. Photo credit: J.M. Polinski (B) Schematic representation of lobster anatomy, indicating the tissues types used for this study: 1, antennular lateral flagella; 2, eye stalk ganglion; 3, brain (supraesophogeal ganglion); 4, heart; 5, hepatopancreas; 6, gonads/testis; 7, intestine; 8, abdominal ganglion; 9, walking leg dactyls. (C) Phylogenetic tree inferred from 263 single-copy orthologs among eight arthropod species. Numbers of expanded gene families are marked in green, and numbers of contracted gene families are marked in red. The number below the MRCA (most recent common ancestor) represents the total number of orthologs from OrthoMCL analysis used as input for CAFE expansion/contraction analysis. Single-copy orthologs are defined as orthologs that were present as a single-copy gene in all eight species. Multiple-copy orthologs represent the gene groups present in all species with a gene number of >1 in at least one species. Species-specific paralogs represent genes uniquely present in only one species. Other types of orthologs represent the gene groups that are absent in some species and not species-specific paralogs.
The expanded gene families involved in cuticle formation, and remodeling included 189 genes encoding cuticular proteins, chitin binding proteins, crustacyanins, and chitinases (table S7). The cuticle provides mechanical support for the body and protects the lobster from invading parasites, pathogens, and physical injuries. To grow, the cuticle is shed through the molting process, and a new, larger cuticle is formed. The organic matrix of the cuticle is secreted by the epithelial cell layer and is composed of chitin polymers and associated proteins that are mineralized by the deposition of calcium minerals (29). Structural studies have shown that the H. americanus cuticle is more complex than that of other arthropods, with interconnected chitin-protein fibers bending around a well-developed pore canal system to form a planar honeycomb-like structure (29). This structural complexity may account for the expansion of cuticle-related gene families and equip the lobster with an effective means of physical defense.
Many gene families associated with transposable elements were expanded in the lobster genome. In total, there were 972 gene models with Swiss-Prot annotations as transposable element-derived, and the vast majority of these (917) showed expression in lobster developmental stages and/or adult tissues (table S8). This suggests that transposable elements are active in the lobster, although it is possible that some have been domesticated for other functions. Products encoded by members of the Tc1/mariner superfamily of DNA transposons represented the majority (80%) of expressed transposable element genes, while 16% were products of retrotransposons (table S8).
Neural signaling complexity and neuroimmune interactions in H. americanus
For more than 50 years, H. americanus has been a powerful model in neuroscience, especially for the identification of neural circuits controlling rhythmic behaviors (11) and olfaction (13). Results from the gene family expansion and contraction analysis revealed an expansion of many families associated with neuronal function, and targeted examination of the lobster neural gene complement identified an extensive repertoire of genes encoding sensory signaling components within neural circuits (>900 genes; table S9). The most remarkable features include a marked expansion of LGICs compared to other crustaceans and arthropods, a putative nitrite/nitrate sensor previously not characterized in metazoans, a complex secretome with 52 neuropeptide prohormones, and an unusual family of ligand-gated channels with lectin-binding domains suggesting novel neuroimmune interactions. In addition, essential components for functional deciphering of the cellular basis of behavior include genes encoding 16 gap junction proteins (pannexins/innexins) critical for central pattern generations and genes responsible for neuromuscular organization (table S9). Last, we found an expanded complement of 483 genes encoding ion channels (Fig. 2A), including vertebrate-type rapid voltage-gated sodium (Nav) channels (fig. S1 and table S9), pH-gated and pH-sensitive components, and genes associated with photosensitivity and circadian rhythm critical for lobster ecology and behavior (table S9).
Fig. 2. Diversity of ion channels encoded in the H. americanus genome.
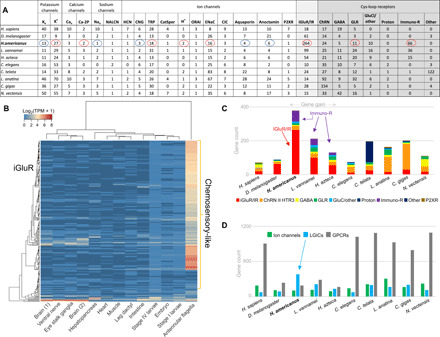
(A) Number of homologs encoding ion channels, including voltage-gated ion channels/LGICs and related receptors, identified in the lobster genome compared to other arthropod and metazoan species. (B) Heatmap of normalized expression levels of the 264 iGluR/IRs in developmental stages and adult tissues of H. americanus represented as a gradient of log2 TPM (transcripts per million). Dendrograms on the left and top sides indicate the hierarchical clustering of homologs and tissues, respectively. (C) Number of homologs encoding iGluR/IRs, cys-loop receptors, and purinoceptors (P2XR) in H. americanus compared to other arthropod and nonarthropod species. (D) Number of homologs encoding ion channels, LGICs, and G protein–coupled receptors (GPCRs) in H. americanus compared to other arthropod and nonarthropod species. LGIC, ligand-gated ion channel; GPCR, G protein-coupled receptor; TRP, transient receptor potential channel; HCN, hyperpolarization-activated cyclic nucleotide-gated channel; CNG, cyclic nucleotide-gated channel; ChRN, nicotinic acetylcholine receptor; GluCl, glutamate-gated chloride receptor; GLR, glycine receptor.
As an opportunistic predator, the lobster has diversified sensory systems, where olfaction is the most dominant trait. Genes encoding the chemosensory machinery represent one of the most developed molecular toolkits of H. americanus with at least 264 iGluR/IRs versus 99 in the shrimp (L. vannamei) or 61 in D. melanogaster (Fig. 2, A and C). The iGluR/IRs were predominantly expressed in antennules, the major chemosensory organs of the lobster (Fig. 2B and table S9). This gene expression pattern implies the recruitment of iGluR/IRs in olfactory processing as one of the key ecologically relevant adaptations of this species.
Unexpectedly, expansion of the iGluR’s family correlates with a relatively smaller complement of 188 G protein–coupled receptors (GPCRs; belonging to the seven-transmembrane superfamily with predicted relatively slow kinetics) compared to other species (Fig. 2D and table S9). This finding represents an uncommon molecular shift, possibly related to the predatory lifestyle, which requires relatively fast responses to olfactory cues (via LGICs) and, therefore, increased speed of information processing. To support this hypothesis, we identified 18 transient receptor potential ion channels (table S9), some of which were highly expressed in the antennules and might be responsible for the detection of broad senses (including temperature, pH, and mechanosensation) together with a piezo mechanosensitive channel, which was also abundantly expressed in the antennular lateral flagella (table S9).
Transcriptome analysis illuminated the involvement of cyclic nucleotide–gated and hyperpolarization-activated channels in the chemosensation of lobster (table S9). This pathway frequently works together with the gaseous nitric oxide (NO)–mediated signaling machinery in chemosensory and neuronal processes (30, 31). Further genomic deciphering of these NO-dependent pathways and, in particular, the quest for NO-reception complement, led to the discovery and identification of very unusual guanylate cyclases with unique nitrate- and nitrite-sensing (NIT) domains (32) in the lobster genome. These NIT domains were previously characterized in prokaryotes (e.g., bacteria and cyanobacteria) (33). This unusual molecular architecture implies the existence in animals of previously unidentified nitrite or nitrate sensing (34), which might have broad implications for environmental biology. NO and products of NO oxidation might also contribute to innate immunity and be a part of the nitrogen cycle in the marine environment. Unexpectedly, as an expansion of these NO-nitrite pathways, we identified the primarily neuronal expression of ammonium transporter–encoding genes (table S9). These genes are also expressed in the embryo and larval stages.
There were 116 predicted lobster proteins with a cys-loop neurotransmitter-gated ion-channel ligand-binding domain (PF02931) and membrane domain (PF02932) (Fig. 3A and table S9). Of these, 50 were the canonical class of LGIC composed of 24 nicotinic type acetylcholine receptors, 10 glutamate/histamine-gated chloride channels, and 16 glycine/γ-aminobutyric acid (GABA)–like receptors (Fig. 3, A and B, and table S9). However, there were 66 predicted LGIC receptors with unknown functions. Besides the PF02931/PF02932 domains, this novel class of channels had several unique domains, including the C-type lectin or carbohydrate-recognition domain (CLECT), the pentaxin C-reactive domain (PF00354), and the low-density lipoprotein receptor domain class A (LDLa; PF00057) (Fig. 3A and table S9). Gene expression analysis showed that some of these predicted LGIC receptors were highly expressed in the brain and the antennules (Fig. 3C). To the best of our knowledge, this is the first demonstration of cys-loop receptors with immune-related domains and may represent a previously unknown link between immunity and neural signaling in lobsters. Notably, we did not find these “chimeric” immune-neurotransmitter binding transmembrane receptors in the sequenced genomes of Drosophila and many other lophotrochozoans, cnidarians, and vertebrate models. It appears that this type of innovation is unique to the lobster and related crustaceans. The presence and expansion of this type of pentaxin-lectin-LGIC receptors suggest mechanistic links between immune and neural systems leading to rapid pattern recognition responses and their modulation of neurotransmitters.
Fig. 3. Novel cys-loop receptors with immune-related domains in H. americanus.
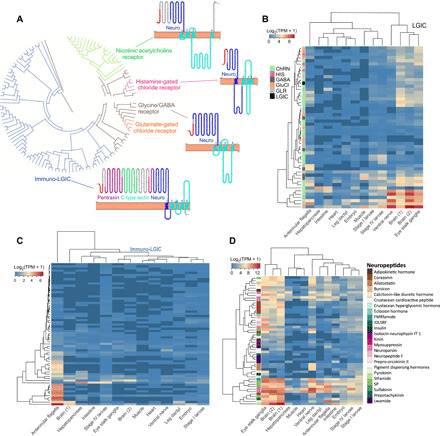
(A) There are 116 predicted homologs encoding proteins with a cys-loop neurotransmitter-gated ion-channel ligand-binding domain (PF02931) and membrane domain (PF02932) in the lobster genome. Of these, 50 are the class of LGIC composed of 24 nicotinic type acetylcholine receptors (ChRN), 10 glutamate-gated chloride receptors (GluCl)/histamine-gated chloride receptors (HIS), and 16 glycine/GABA-like receptors (GLR/GABA) and one unknown that is a root in the phylogenetic tree. There are 66 predicted LGIC receptors with unknown function. Besides the PF02931/ PF02932 domains, these channels have the CLECT, the pentaxin C-reactive domain (PF00354) and the LDLa (PF00057). Phylogenetically, the different classes all form unique clusters. (B) Heatmap of expression of canonical LGIC receptors in developmental stages and adult tissues of H. americanus. (C) Heatmap of expression levels of the immune-LGIC receptors in developmental stages and adult tissues of H. americanus. (D) Heatmap of expression levels of the predicted secretory prohormones, with many showing high expression in the brain, chemoreceptory structures, and eyestalk. All heatmaps show normalized expression levels log2 TPM with dendrograms on the left and top sides, indicating the hierarchical clustering of homologs and tissues, respectively.
Despite intensive and systematic studies related to the identification of 35 neuropeptide precursors in the lobster (12, 35), we found several previously unidentified prohormones, totaling 52 genes encoding secretory signaling peptides (Fig. 3D and table S9). Embryos, larvae, and adult peripheral tissues had notably less expression of predicted secretory molecules, suggesting that most of the identified prohormones are nervous system derived (Fig. 3D and table S9). Most of these were highly expressed in the brain (e.g., SIFamide, allatostatin C, IDLSRF-like, and a new peptide as the most abundant) (Fig. 3D and table S9). Their functions are diverse with both effector and modulatory actions and might also include control of nociception and immunity by analogy with Drosophila (36). Another part of the central nervous system, the ventral nerve cord, had a different pattern of prohormone expression, with the highest expression of neuroparsins and novel peptides (Fig. 3D and table S9). There were also distinct profiles of neuropeptides in chemosensory structures with orcokinin as the top candidate for chemosensory modulation and processing (table S9).
Only 2 of 52 neuropeptide prohormones have no detectable expression in the lobster eye stalk ganglia (Fig. 3D and table S9), suggesting complex modulatory capabilities in photoreception. In the eyes, we detected high levels of expression for these systemic action peptides as crustacean hyperglycemic, eclosion, and pigment-dispersing prohormones. Neuroparsins, involved in energetics, were most dominantly expressed in the heart, and we identified 27 neuropeptide prohormones that may be involved in the modulation of skeletal muscle innervation and activity (table S9), greatly exceeding the level of modulation found in mammals.
Highlighted immune genes in the H. americanus genome
A robust innate immune system is paramount to long-term survival in the benthic marine environment, and a wide variety of genes associated with pathogen recognition and other immune effectors were identified in the lobster genome (table S10). These include eight Toll-like receptors and 11 isoforms of its binding partner spatzle (37). A variety of pathogen recognition receptors were found including three β-1,3-glucan–binding proteins and four peptidoglycan-recognition proteins. There were also 82 lectins with similarity to CLECT, techylectins, ficolins, and mannose-binding proteins. The Down syndrome cell adhesion molecule (DSCAM) gene was also identified; however, it is located across two scaffolds and will require additional directed sequencing for full characterization. Antimicrobial peptides (AMPs) are a critical part of the American lobster’s immune repertoire, and 37 homologs encoding AMPs were identified in the genome including 9 antilipopolysaccharide factors (ALFs), 23 crustins/carcinins, and 5 β-defensin–like panusins. Gene family expansion/contraction analysis suggested an expansion of CLECT, crustin, proclotting factor, and complement factor H genes relative to the other arthropods (table S7). Additional gains were noted for DSCAM, but this is a result of the gene annotation program finding several separate sections of the large DSCAM gene and erroneously reporting them as multiple genes.
ALFs are primarily cationic peptides that bind to lipid A or lipoteichoic acid from Gram-negative and Gram-positive bacteria (38) and β-glucan from fungi (39). Nine transcribed ALFs were found in the H. americanus genome. Isoelectric point (pI) predictions (40) of the mature peptide of these ALFs found that all except one would be positively charged in lobster hemolymph. Phylogenetic predictions separate most of the ALFs into two major clades (Fig. 4A). There were two groups of three ALF genes that cluster together in the genome. ALF gene cluster 1 contains ALF-like2, ALF-like4, and ALF-like3 separated more than ~10 kbp, while ALF gene cluster 2 contains ALF-like6, ALF-like9, and ALF-like7 separated more than ~9 kbp. Both outer ALF genes in each cluster are more similar to each other than to the middle ALF gene, but there is more intracluster similarity than intercluster similarity. This indicates a long period of divergent evolution even if ALF gene cluster 2 is a result of a cluster duplication from ALF cluster 1. The ALF genes, ALF-like1, ALF-like2, and ALF-like3 correspond to ALFHa-2, ALFHa-4, and ALFHa-6, genes that have been previously shown to be up-regulated during bacterial (41), protist (42), and viral (43) pathogen challenges (44).
Fig. 4. AMPs encoded in the genome of H. americanus.

Phylogenetic comparison of the predicted mature peptide sequences of expressed ALFs (A) and crustins (B) generated by Multiple Alignment using Fast Fourier Transform (MAFFT) multisequence alignment and maximum-likelihood tree generation. Bootstrap values are present at nodes with scale bars representing the number of substitutions per site. Peptides highlighted with blue are positive, while those highlighted with red are negative, at a lobster’s physiological pH (7.7 to 7.9). Gray circles denote crustin peptides lacking recognizable WAP domains. (C) ALF, crustin, and panusin expression represented as a gradient of log2 TPM with hierarchical clustering of both AMPs and tissue. Color bars along the sides of the heatmap represent gene groups (i.e., crustin, panusin, and ALF).
Crustins/carcinins can be either cationic or anionic peptides expressed primarily in hemocytes and contain three to four disulfide bonds and essential whey-acidic protein (WAP) domains (45). Twenty-three transcribed crustin genes were located in the lobster genome (Fig. 4B). Their mature protein pIs indicate that they are a mix of positively (8) and negatively charged (14) peptides in lobster hemolymph. Six of the peptides lacked predicted WAP domains, although they shared similarity to the WAP domain–containing peptides in their N terminus. Three of these crustins (Crustin-like18, Crustin-like1, and Crustin-like20) had the traditional eight cysteines of WAP domains, although they were not annotated as such. Two crustins, Crustin-like22 and Crustin-like23, have identical mature peptide protein sequences, although they are on different scaffolds. The predicted transcript sequence of Crustin-like23 is identical to the 1010 nucleotides from the 3′ end of Crustin-like22, and comparison to expressed transcripts suggests that the predicted 5′ transcript sequence of Crustin-like22 is not exonic sequence. Therefore, these two crustins have identical transcript sequences. Phylogenetic analysis separated the crustins into several clades, which is common in crustin genes, but they did not all group with the four distinct types of crustins (45). The sequence for only one H. americanus crustin, Hoa-crustin (46), has previously been described, and its protein sequence is identical to the predicted Crustin-like2.
Defensins are a family of disulfide-rich peptides expressed by a variety of tissues in vertebrates and nonarthropod invertebrates (47). Five genes encoding β-defensin–like panusin AMPs were found in the lobster genome. This is significant because only two β-defensin–like AMPs have previously been found in Ecdysozoa, one in Panurilus spp. (48) and one in H. americanus (49). These five genes encode three different proteins where Panusin-like4 and Panusin-like3 encode an identical protein, while Panusin-like2 and Panusin-like5 encode another identical protein. The only difference between these latter two proteins is a substitution of F37S38 with S37F38. The final homolog, Panusin-like1, corresponds to the previously identified Hoa-D1 isolated from H. americanus granulocytic hemocytes (49).
The expression of the AMPs varied greatly over the tissues analyzed with a group of seven being constitutively expressed, while the remaining 30 were expressed in several samples (Fig. 4C and table S10). The panusins were all expressed in relatively few samples with the exception of Panusin-like1, which was constitutively expressed. Its constitutive expression is likely the reason why it was the only H. americanus panusin previously identified. The differential expression of AMPs in different tissues illustrates that tight control of AMP expression depends on the life stage, tissue, or the presence of pathogens and may also reflect differing levels of hemocytes in the tissue preparations. Clustering of the samples provided no clear rationale for expression except to note that similar expression was observed in lobster embryos and stage I larval tissues. We noted that some AMPs were highly expressed in the brain, ventral nerve cord, and antennules at levels comparable to canonical neuropeptides (Fig. 4C and table S10) suggesting potential interactions between the immune and nervous systems.
Genes associated with genome stability and cell survival in H. americanus
Maintaining fidelity of the genome is critical to support longevity and to impede the formation of neoplastic disease; therefore, the H. americanus genome was surveyed for genes associated with genome stability. Inspection of the expanded gene families in the lobster compared to short-lived arthropods revealed several that play a role in maintaining genome integrity and cell survival such as members of the Argonaute (AGO) protein family and inhibitors of apoptosis. To investigate this in more detail, a list of 326 genes was assembled including those known to play a role in DNA repair, cell cycle regulation, tumor suppression, apoptosis, chromosome structure, and transposon silencing (50, 51). Of these 326 genes, 237 had at least one homolog identified in the lobster genome, and more than 75% of the surveyed genes associated with DNA repair, tumor suppression, cell cycle, and chromosome structure were identified (table S11). Among these were three homologs of TP53 (tumor protein p53) and an expansion of the AGO gene family and DNA repair genes, particularly those with a role in double-strand break repair. In contrast, only 50% of the genes associated with apoptosis were identified.
Identification of three homologs encoding the transcription factor p53 was particularly intriguing in light of its central role in safeguarding genome integrity through modulating expression of genes involved in cell cycle regulation, DNA repair, and apoptosis (52). Analysis of lobster transcriptomes from developmental stages and adult tissues confirmed that all three p53 homologs were actively expressed and the corresponding transcripts were used to map the exon structure of the TP53 genes (Fig. 5A). All three lobster homologs have the transactivation, DNA binding, and tetramerization domains characteristic of p53 family members (Fig. 5A and table S12). One homolog also contained the sterile alpha-motif (SAM) domain and showed the highest similarity to family member p63 from molluscs, echinoderms, and humans (Fig. 5C and table S12). Pairwise comparisons between the three lobster p53/p63 homologs showed limited similarity (24.5 to 29.4%), indicating a high level of divergence (table S12). Lobster homologs, p63-like and p53-like2, were expressed across all developmental stages and adult tissues surveyed, with p53-like2 showing a higher level of expression in tissues of the nervous system (Fig. 5B). The other p53 homolog (p53-like1) had a more restricted expression pattern with low levels in developmental stages and high levels of expression in the intestine and hepatopancreas (Fig. 5B and table S11). The observation that p53 participates in the regulation of many different metabolic, physiological, and developmental processes in addition to its role as “guardian of the genome” (53) may explain these different expression patterns.
Fig. 5. Gene structure, expression and phylogeny of p53 family members in the H. americanus genome.

(A) Predicted gene and transcript structure of the three H. americanus p53 family members with the transactivation domain (TAD) shown in blue, the DNA binding domain (DBD) shown in pink, the tetramerization domain (4D) in yellow, and the SAM domain in green. (B) Heatmap of normalized expression levels (TPM) of the p53 homologs in developmental stages and adult tissues of H. americanus. Dendrograms on the left and top sides indicate hierarchical clustering of homologs and tissues, respectively. (C) Phylogenetic relationships, based on amino acid sequence similarity, between predicted protein sequences of p53 family members from H. americanus and selected arthropod and nonarthropod species. (D) p53 upstream regulator and downstream effector genes in the H. americanus genome. p53 activation occurs as a result of DNA damage or other cellular stress–induced signaling pathways with selected upstream activators depicted as ovals. Activation of p53 induces transcriptional up-regulation of one or more target genes that mediate the cellular processes shown in the green boxes. The total number of lobster homologs identified per number of target genes investigated is shown above each green box. Selected target genes are listed for each cellular process with homologs identified in the lobster genome shown in blue, while those not found are shown in red. Gene names in capital letters represent human homologs (54), and those in lower-case letters are D. melanogaster homologs (55). A complete list of p53 target genes investigated is shown in table S13 and full gene names are shown in table S11.
A survey of the lobster gene predictions showed that several upstream regulators and downstream targets of p53 were present in the genome (Fig. 5D and table S13). In total, 366 validated p53 target genes from mammals and Drosophila were investigated within the categories of cell cycle, DNA repair, apoptosis, and metabolism (table S13) (54, 55). A notable observation was that homologs for only 9 of 46 p53-dependent activators of apoptosis were found in the lobster genome (Fig. 5D and table S13). Extending this analysis to include p53-independent apoptosis genes showed only 29 of 61 homologs were identified in the lobster genome (table S11 and fig. S2). Proapoptotic factors identified in the lobster genome include two homologs for apoptosis inducing factor and 11 homologs encoding caspases, while homologs for several well-characterized activators of apoptosis in Drosophila and mammals were absent (e.g., BAX, BAK, BID, hid, egr, rpr, and grim) (table S11 and fig. S2). Prosurvival homologs present in the lobster genome include two homologs of Bclx-like and two homologs of inhibitor of apoptosis (IAP) and 13 homologs of death-associated inhibitor of apoptosis (Diap2) (table S11 and fig. S2). Autophagic programmed cell death is an alternative to apoptosis that has been well characterized in Drosophila development (56), and three homologs of death-associated protein kinase (DAPk), a key regulator of this process, were identified in the lobster genome (table S11).
While less than 20% of p53 targets involved in apoptosis were identified in the lobster genome, more than 80% of known p53-dependent DNA repair genes were found (table S13). Extending this to include p53-independent DNA repair genes indicated that 56 of 70 genes surveyed were present (table S11). There was an additional copy of excision repair cross-complementing group 1 (ERCC1) relative to other crustaceans and an expanded complement of genes involved in DNA double-strand break repair including two homologs of each RAD50 and RAD52, four homologs of RAD51 (in addition to DMC1), seven homologs of RAD54, and seven homologs of PARP (table S11).
Heterochromatin formation through DNA or histone methylation plays an important role in the detection and repair of DNA lesions and also serves as a protective mechanism to suppress transposition and aberrant recombination between repetitive DNA elements (57). Several families of methyltransferases were expanded in the lobster genome relative to other crustaceans (table S7) with the lobster genome containing 31 homologs encoding histone-lysine N-methyltransferases and 11 members of the arthropod-specific SET gene family of histone modifiers, SmydA (58) (table S11).
The AGO protein family plays an essential role in maintaining heterochromatin structure and genomic stability. There were 20 homologs belonging to this family identified in the lobster genome, which is more than twice the number found in other arthropods and higher animals (Fig. 6A and table S11). Through interaction with noncoding RNA (ncRNA), this family of proteins facilitates regulation of transcription, translation, heterochromatin formation, and transposon silencing (59). Among the 20 lobster homologs, three share similarity to the P element–induced wimpy testis (PIWI) subfamily, while 17 were more similar to the AGO subfamily (Fig. 6A and table S11). In addition to PIWI and AGO genes, the lobster genome encoded a full complement of genes for the biogenesis of small ncRNAs including Dicer, TARBP2, Drosha, and DGCR8 (Fig. 6A and table S11). Examination of lobster transcriptomes revealed that the PIWI and AGO variants and the ncRNA biogenesis genes were expressed at very different levels in embryonic, larval, and adult tissues (Fig. 6B and table S11). One PIWI homolog (PIWI-like3) was preferentially expressed in early life stages (embryos and larvae) as well as hepatopancreas and intestine, while the other PIWI homologs were mainly expressed in tissues of the nervous system (brain, eye stalk ganglia, and antennular lateral flagella) (Fig. 6B). Many of the AGO family members and the ncRNA biogenesis genes showed widespread expression with highest levels in tissues of the nervous system (e.g., brain, eye stalk ganglia, and antennular lateral flagella) suggesting a role in neurosensory function.
Fig. 6. Analysis of the PIWI/AGO family members in the H. americanus genome.
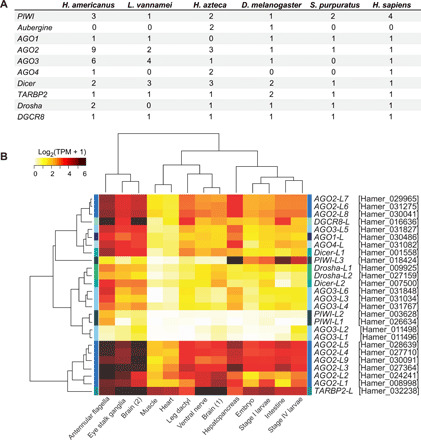
(A) Number of gene predictions encoding members of the PIWI/AGO family in the lobster genome compared to humans and other animals. (B) Heatmap of expression levels represented as a gradient of log2 TPM of the PIWI/AGO family members in developmental stages and adult tissues of H. americanus. Dendrograms on the left and top sides indicate hierarchical clustering of homologs and tissues, respectively.
Genes associated with cellular defense and protection from environmental toxicants in H. americanus
Examination of the expanded gene families in lobster relative to other arthropods revealed several that play a role in cellular defense and protection from environmental toxicants. These include expansion of certain cytochrome P450 monooxygenases (CYP), sulfotransferases, organic cation and anion transporters (OCT and OAT), heat shock proteins (HSPs), glutathione peroxidase (GPX), and superoxide dismutase (SOD) (table S7). We extended this analysis to investigate a comprehensive list of genes associated with the “chemical defensome” (60) and found that the lobster genome contains a robust repertoire of defense genes encoding biotransformation enzymes that transform chemicals to excretable metabolites, efflux transporters that actively export substrates from cells, antioxidant proteins that protect against endogenous or exogenously generated reactive oxygen species (ROS), metal detoxification proteins, and HSPs (table S14). Of particular note were 21 genes encoding well-characterized CYP enzymes involved in detoxification of lipophilic compounds (CYP2, CYP3, and CYP4) and 21 members of the CYP6 gene family. The CYP6 family is specific to arthropods, and increased expression of CYP6 has been associated with pesticide resistance in Drosophila and mosquitoes (61). Additional biotransformation enzymes expanded in the lobster genome include several families of sulfotransferases that are involved in sulfate conjugation of xenobiotic molecules (table S14). The lobster genome contains 57 genes encoding members of the OAT and OCT families that play a role in the efflux of a variety of endogenous organic anions and cations, as well as a wide array of drugs and environmental toxins. HSPs respond to a range of stressors such as ultraviolet radiation, temperature, oxidative stress, hypoxia, and osmotic stress and thus act as a fundamental cellular defense mechanism for maintaining protein homeostasis. Expanded in the lobster genome were members of the HSP70 family (13 gene predictions) and its cochaperone HSP40/DNAJ (48 gene predictions) (table S14). Antioxidant enzymes safeguard the cell against the harmful effects of ROS, and the lobster exhibited an extra copy of two key antioxidant enzymes relative to other arthropods (SOD and GPX), for a total of five homologs for cytosolic Cu-Zn SOD, two homologs for Mn SOD, one homolog for extracellular SOD, in addition to seven homologs of GPX (tables S7 and S14). The extensive repertoire of chemical defense mechanisms in the lobster make it well equipped to cope with the stresses of the benthic marine environment.
DISCUSSION
Despite the scientific, ecological, and economic importance of crustaceans, there are very few genomic resources available. To date, only seven crustacean genomes have been published, and none have achieved chromosome length assemblies that have been reported for other taxa. The challenge appears to be related to the highly repetitive nature of crustacean genomes that may contain as much as 78% repetitive sequences as has been reported for the Pacific white shrimp, L. vannamei (26). Here, we have used a hybrid approach involving a combination of short- and long-read sequencing and scaffolding based on Chicago and Hi-C libraries to assemble a high-quality genome of the American lobster, H. americanus, and yet, our assembly falls short of the complete genome size. The assembly is composed of 52.9% repetitive elements, and given the good assembly statistics and completeness of the BUSCO gene set, it is likely that our assembly is missing additional copies of repetitive elements.
The highly repetitive nature of crustacean genomes poses considerable challenges for maintaining genome integrity as tracts of interspersed nonallelic homologous sequence have the potential to interact during meiotic recombination and DNA repair, resulting in deletions, duplications, and large-scale chromosomal rearrangements. Uncontrolled transposition can create additional aberrant genomic alterations. The American lobster is reported to be a long-lived animal with few incidences of neoplastic disease (16, 17), and maintaining genome fidelity is critical to support these characteristics. Comparison of the lobster genome to that of short-lived arthropods has provided insights into the remarkable life history of the lobster with an expansion of genes involved in heterochromatin formation, DNA repair, and cell survival. Regulating heterochromatin is essential for preventing aberrant recombination, ensuring proper chromosome segregation and DNA repair, and suppressing transposon activity (62). The lobster genome shows an expansion of the AGO/PIWI family and histone methyltransferases that play a key role in regulating heterochromatin formation and have been shown to be important for transposon silencing in both the soma and the germline of other arthropods (59).
Previous studies in Drosophila suggest that an ancestral function of p53 may be related to restricting the movement of transposable elements during meiosis through interaction with the AGO pathway (63). In light of this, it is interesting that three homologs encoding p53 were identified in the lobster genome. p53 plays a key role in maintaining genome integrity through modulating expression of genes involved in cell cycle regulation, DNA repair, and apoptosis. Proper regulation of apoptosis is essential for normal tissue formation and homeostasis and also for elimination of damaged or abnormal cells. The absence of key apoptotic activators and expansion of inhibitors of apoptosis in the lobster suggests unique mechanisms for regulating apoptosis. It has been suggested that a strong antiapoptotic system represents a mechanism to avoid premature or unnecessary apoptosis for enhanced environment resilience in other marine organisms (64). Members of the p53 family participate in the regulation of many different metabolic, physiological, and developmental processes beyond their role in safeguarding genome integrity (53), and future studies are needed to discern the role of the lobster p53 homologs in its biology, longevity, and resistance to neoplastic disease.
The complement of predicted neural genes in the lobster genome notably expanded the known repertoire of molecular players involved in sensory, motor, and integrative processes compared to well-studied insects. The most notable feature was the remarkable diversity of ionotropic glutamate-type receptors, which serve as important mediators of information transduction. This diversity is also greater than that found in the shrimp genome (26) and might be associated with the predatory lifestyle of the lobster and its dependence on olfaction.
The functional role of multiple pH-dependent channels, together with the expansion of the list of neurosecretory precursors to 52 and more than hundreds of receptors, provides an opportunity to decipher novel signal transduction pathways in this advanced neurobiological model. The possible redundancy of multiple sensory and motor pathways is a little-explored phenomenon in neuronal operation, homeostasis, and integrative activity, as well as learning and memory mechanisms to be explored in the lobster.
The potential link between the immune and neural systems, as suggested by the coupling of cys-loop receptors with immune-related domains, may give rise to a new form of cellular communication in lobsters. Neuroimmune interactions have not been explored in crustaceans; however, in Drosophila, sensory neurons comprise a major part of the larval hematopoietic niche suggesting an interface where sensory inputs regulate blood cell homeostasis or immune responses (65). The robust immune system and cellular defense mechanisms of the lobster no doubt play an essential role in its longevity and ecological success, and further investigation of the lobster genome provides an opportunity to expand our understanding of the cellular and organismal adaptations that have enabled the success of this benthic predator. The genome presented here will serve as a valuable resource for fisheries, ecology, and biomedical research, especially in deciphering complex traits including susceptibility to disease and ability to cope with environmental change.
MATERIALS AND METHODS
Sequencing the H. americanus genome with Illumina, Dovetail, and Oxford Nanopore
Wild-caught lobsters purchased live from Steve Connolly Seafood Company (Gloucester, MA) were sent to Dovetail Genomics LLC (Santa Cruz, CA) for DNA extractions and sequencing library preparation. Genomic DNA (gDNA) was isolated from heart tissue using a cetyltrimethylammonium bromide (CTAB) extraction procedure (66), and paired-end (mean insert size, ~500 bp) and mate pair (insert sizes, 3.4 to 8.7 kb) libraries were constructed using Illumina reagents (Illumina Inc., San Diego, CA). In total, we generated 283 million read pairs (2 × 250 bp) totaling 142 Gbp, and 113 million mate pairs (2 × 100 bp and 2× 75 bp), totaling 22 Gbp (table S1).
Chicago libraries were prepared by Dovetail Genomics LLC. Briefly, for each library, 500 ng of high-molecular-weight gDNA, isolated from heart tissue, (mean fragment length, ~50 kbp for libraries 1 to 3 and ~150 kbp for library 4) was reconstituted into chromatin in vitro and fixed with formaldehyde. Fixed chromatin was digested with Mbo I, the 5′ overhangs were filled in with biotinylated nucleotides, and then free blunt ends were ligated. After ligation, cross-links were reversed, and the DNA was purified from protein. Purified DNA was treated to remove biotin that was not internal to ligated fragments. The DNA was then sheared to ~350-bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters (New England Biolabs, Ipswich, MA). Biotin-containing fragments were isolated using streptavidin beads before polymerase chain reaction (PCR) enrichment of each library. The libraries were sequenced on an Illumina HiSeq2500 (rapid run mode). A total of 123 Gbp of data were generated, composed of 404 million 2 × 100–bp read pairs and 143 million 2 × 150–bp read pairs (table S1).
Dovetail Hi-C libraries were prepared by Dovetail Genomics LLC using lobster heart tissue. Briefly, for each library, chromatin was fixed in place with formaldehyde in the nucleus. Extracted fixed chromatin was digested with Dpn II, the 5′ overhangs were filled in with biotinylated nucleotides, and then free blunt ends were ligated. After ligation, cross-links were reversed, and the DNA was purified from protein. Purified DNA was treated to remove biotin that was not internal to ligated fragments. DNA was then sheared to ~350-bp mean fragment size, and sequencing libraries were generated using NEBNext Ultra enzymes and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The libraries were sequenced on an Illumina NextSeq, generating 1 billion 2 × 150–bp read pairs totaling 312 Gbp (table S1).
High–molecular-weight DNA was isolated from lobster heart tissue for Oxford Nanopore long-read sequencing. gDNA prepared by the CTAB extraction method (66) was sheared to a target length of 10 kb using the Megaruptor 2 (Diagenode Inc., Denville, NJ). Nanopore sequencing libraries were prepared using approximately 200 fmol of sheared DNA input into the LSK-109 Kit from Oxford Nanopore Technologies (Oxford, UK) and following the manufacturer’s protocol. Approximately 50 fmol of the sequencing library was loaded onto each PromethION flow cell. In total, we generated 15 million reads with N50 length of 7521 bp, totaling 55.7 Gbp of sequence (table S1). All sequence data generated and the genome assembly reported in this study are available in the National Center for Biotechnology Information (NCBI) GenBank under BioProject PRJNA655509.
Genome assembly
An initial de novo assembly of the lobster genome was conducted by Dovetail Genomics LLC, using the Illumina paired-end and mate pair sequencing data and Meraculous 2 v2.0.2 (67) with a k-mer size of 61 yielding an N50 value of 24 kbp. The contiguity of the assembly was then improved using the data from Chicago and Dovetail Hi-C proximity ligation methods and scaffolded using Dovetail’s HiRise software (table S15). This approach yielded a final assembly size of 1.071 Gbp, less than one-third of what we expected on the basis of previously reported nuclear DNA content for H. americanus (18, 22). Oxford Nanopore long-read sequencing was then undertaken. Assembly of the Nanopore reads was attempted using Flye (68), wtdbg2 (69), and MaSuRCA (24) assemblers, but only MaSuRCA v3.3.3 was successful in completing the assembly. MaSuRCA is a hybrid assembler that uses Illumina and Nanopore data at the same time to correct the long high-error Nanopore reads into highly accurate mega-reads. The mega-reads are then assembled using a modified version of CABOG assembler distributed with MaSuRCA.
The initial assembly produced using MaSuRCA with Illumina and Oxford Nanopore data yielded 55,595 contigs with a contig N50 of 133.3 kb (table S15). The “chromosome scaffolder” tool distributed with MaSuRCA was used to scaffold the resulting contigs using the scaffolds from the Hi-C assembly produced by Dovetail Genomics as “reference.” This yielded the final assembly with scaffold N50 of 759.6 kbp containing 2.29 Gbp of sequence in 47,245 scaffolds. To improve the final sequence quality, the assembly was polished using Illumina data with POLCA software (70). POLCA fixed 2,134,547 substitution errors and 4,422,552 insertion/deletion errors in scaffolds yielding the final estimated assembly consensus quality of at least 99.99% or less than 1 error per 10,000 bases.
Genome size estimation
The genome size was estimated using k-mers in Illumina reads. The original paired-end and mate pair library reads did not generate a reliable estimate due to the low coverage and the noise in the read data. Hence, we generated an additional Illumina paired-end dataset with higher coverage to use for k-mer analysis. For this, a 2 × 150–bp paired-end library was generated from lobster heart tissue using the Kapa HyperPrep DNA Kit with Illumina TruSeq single-index adapters and sequenced on a S1 flowcell of the NovaSeq 6000 at the Molecular Biology Core Facility at the Dana-Farber Cancer Institute to yield 1.99 billion read pairs (598 Gbp). MaSuRCA error correction was used to eliminate erroneous k-mers, and Jellyfish (71) was used to count 23-mers in Illumina reads; the histogram of the 23-mer counts is shown in fig. S3, where the x axis shows how many times the k-mer was encountered in the Illumina data, and the y axis shows the number of k-mers with count x. Two well-defined distinct peaks were observed in the black curve at coordinate 80 and 162. The peak at x = 80 represents k-mers that occur on a single chromosome, and the peak at 162 represents k-mers that are shared between two copies of the chromosome, thus double the count. The genome size was estimated from the k-mer distribution. The distribution suggests that the k-mer coverage for the single chromosome was about 80× and the average read length was 150 bp. For k = 23, each read yields 128 k-mers; thus, the read coverage can be estimated as 80 × 150/128 = ~94×. Total sequence in reads was 598 Gbp. Therefore, the total genome size is 598/94 = 6.36 Gbp, and the 1-N genome size is approximately 3.18 Gbp.
Genomic repeat annotation
Repetitive elements in the lobster genome assembly were identified and classified using the program RepeatModeler v1.0.11 using default parameters. The consensus sequences in the custom repeat library generated with RepeatModeler were used as input for RepeatMasker v4.0.9, which identified and masked all repetitive elements.
Transcriptome sequencing and assembly
Stage IV and stage V lobster larvae, obtained from the Coastal Zone Research Institute–Homarus Inc. (Shippagan, NB, Canada), and adult tissues (hepatopancreas and intestine) from lobsters obtained from Clearwater Seafoods (Bedford, NS, Canada) were preserved frozen in RNAlater. Larval lobsters and tissues were homogenized in 1 ml of RNA preservation reagent [1.4 M guanidine isothiocyanate, 38% phenol (pH 4.5), 5% glycerol, and 0.1 M sodium acetate] using an Omni homogenizer (Omni International, Kennesaw, GA), followed by the addition of 200 μl of chloroform, vigorous inversion, and a 3-min incubation at room temperature. The homogenate was centrifuged at 15,000g for 15 min at 4°C. The resulting aqueous phase was removed and mixed, via inversion, with an equal volume of 100% ethanol. RNA was isolated from the resulting solution using RNeasy spin columns (QIAGEN, Valencia, CA) using the manufacturer’s instructions including the on-column deoxyribonuclease I digestion. RNA was quantified using spectrophotometry (NanoDrop ND-1000, Thermo Fisher Scientific, Waltham, MA), and its quality was assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA) and the RNA 6000 nano cartridge. RNA samples were stored at −80°C until ready for use.
High-quality RNA was sent to Genome Quebec (Montreal, PQ, Canada) for Illumina TruSeq RNA library preparation according to the manufacturer’s instructions. Paired-end 100-bp sequencing was performed on an Illumina HiSeq2000 instrument, and sequencing reads were filtered by removal of all reads with overall quality scores of <36. Raw sequence reads were generated using the Illumina CASAVA pipeline with base quality encoded in phred 33. Trimmomatic v0.36.3 was used to remove Illumina sequence adapters and bases with phred scores of 30 or less. Minimal read length for sequences was set at 32 nucleotides. Trimmed reads for the stage IV and stage V larval lobsters were normalized using Trinity normalization (50×) utility before the de novo generation of the H. americanus transcriptome using the Trinity assembler v2.8.5.
Gene prediction and annotation
Protein-coding gene models were predicted from the genomic sequence using iterative implementations of the Maker2 pipeline (25). The first iteration of Maker2 v2.31.10 inferred gene predictions directly from RNA sequencing (RNA-seq) and protein evidence. The stage IV and stage V larvae transcriptome described above and the Swiss-Prot protein database were used as evidence. The resulting gene models were used to train the ab initio gene prediction programs SNAP (72) and Augustus (73). Ab initio gene predictions were then generated using SNAP, Augustus v3.3.2, and GeneMark-ES v4 algorithms run through Maker2, generating a new set of evidence-based gene annotations. The process of retraining SNAP and Augustus using improved output and rerunning the Maker2 pipeline was performed three times, at which point BUSCO scores indicated no further improvements in gene model completeness. The full set of predicted proteins were then searched for Pfam domains using InterProScan v3.38, and the final gene set was quality-filtered to include only gene models supported by evidence (annotation edit distance, AED, ≤ 0.75) or that encoded a Pfam domain. The resulting set of 25,284 gene models was annotated using a BLASTP search against the Swiss-Prot curated protein database, with an E value cutoff of 1 × 10−6. A BLASTP search against the GenBank nr protein database, with an E value cutoff of 1 × 10−6, was also performed to assign additional annotation information.
Gene expression analysis
To assess gene expression patterns of the predicted gene models, transcripts from RNA-seq data generated in this study were combined with additional datasets representing multiple tissue types. RNA-seq data from stage IV larvae, hepatopancreas, and intestine were generated as described above for the transcriptomic dataset. Additional data from abdominal muscle, heart with neurosecretory pericardial organ, abdominal nerve cord, and supraesophogeal ganglion (brain) previously published by McGrath et al. (14), as well as data from NCBI Sequence Read Archive including: stage I larvae (accession nos. SRX5509373, SRX5509368, and SRX5509367), whole embryo (SRX5509372, SRX5509371, and SRX5509374), antennular lateral flagella (SRX7543840), brain (SRX7543842), walking leg dactyl (SRX7543841), and eye stalk ganglia (SRX2068867, SRX2068868, SRX2068869, and SRX2068870) were included. All datasets from the same tissue type were combined as a single sample, with the exception of the brain datasets generated separately. Sequences were quality trimmed with Trimmomatic v0.38 using a sliding window approach (LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50). Data were then mapped to the genome with STAR v2.7.3 using default parameters. Feature counts were extracted from the mapping file using edgeR and normalized to transcripts per million (TPM) based on total reads per sample and gene length.
Gene family analyses and phylogenic tree construction
Gene family analysis was performed on protein-coding genes from H. americanus and seven additional arthropods: D. melanogaster, D. magna, L. vannamei, C. quadricarinatus, P. virginalis, P. hawaiensis, and H. azteca (table S7). Results from an all-to-all BLASTP search were used as input in OrthoMCL v2.0.9 (74) to generate gene family clusters.
A total of 263 single-copy orthologs, defined here as orthologs that were present as a single-copy gene in all eight species, were identified in the OrthoMCL results and used for phylogenetic tree construction. For each ortholog group, proteins were aligned with MUSCLE v3.8.21. The 263 protein alignments were used to generate a phylogenetic tree with RAxML v8.2.12 under the DAYHOFF model with 1000 bootstrap repetitions. D. melanogaster was set as the outgroup, as it was the only noncrustacean arthropod included.
The phylogenetic tree from RAxML was converted to an ultrametric tree with ETE2. The ultrametric tree and gene family count data generated by OrthoMCL analysis were used as input into CAFE v4.2.1 (75) to identify significant gene family expansions and contractions within the phylogeny.
Targeted analysis of genes associated with neurology, immunity, genome stability, and chemical defense
For targeted gene homology searches, lists of candidate genes were compiled from previous studies in genome stability/DNA metabolism/apoptosis (50, 51), cellular chemical defense (60), immunity (76), and neuronal function (77–79). For each of the candidate gene lists, custom BLAST databases were generated. A minimum of two high-quality reference sequences were selected from the Swiss-Prot curated protein database for each of the genes in the genome stability/DNA metabolism and chemical defense candidate lists for inclusion in a custom BLAST protein database. The custom database for the immune-related homology search was generated with all immune-related transcripts from Clark and Greenwood (76) and the protein sequence of H. americanus panusin (49). The custom database for the neuronal gene complement included reference sequences for the pannexin/innexin family previously described in Moroz and Kohn (78); phenylalanine hydroxylase, tryptophan hydroxylase, tyrosine hydroxylase, carnitine palmitoyltransferase, choline acetyltransferase, sodium channel (SCNN1/ASSC), embryonic lethal abnormal vision mRNA processing (ELAV), and CUGBP-ELAV reference sequences previously described in Moroz and Kohn (77); and the C. elegans neuronal gene complement (79).
The final set of gene models from the Maker2 pipeline was used as query sequences against the custom databases in BLAST homology searches (E value cutoff of 1 × 10−5). All similar gene models, as well as any annotated as a target gene previously but not identified in the custom search, were retained for further inspection. Genes of particular interest, including TP53, PIWI, AGO, crustin, ALF, and panusin, were visualized using IGV v2.7.2 with additional tracks for viewing HISAT2 v2.1.0 based RNA-seq read alignments. Intron-exon junctions were manually inspected to confirm that the predicted protein coding sequences matched the mapped transcript reads. The predicted gene model sequences were amended when they did not match with transcript sequences. Only genes with supporting gene expression data and sufficient homology (E value of ≤1 × 10−5) were retained for subsequent analysis.
Acknowledgments
We thank C. Lawley, Scientific Advisory Board member at GMGI, for input throughout the project and on the manuscript; P. Havlak, L. Shiue, S. McWilson, M. Hartley, T. Dickinson, B. Rice, and M. Vierra from Dovetail Genomics for conducting the Chicago and Hi-C analysis; A. Tai in the Department of Immunology at Tufts University School of Medicine for assistance with the bioinformatics analysis on the preliminary draft genome; C. Munkholm for precuring the first lobsters; and Steve Connolly Seafood Company of Gloucester, MA for supplying the lobsters used in this study. Funding: We gratefully acknowledge the philanthropic support from many generous donors to GMGI in support of the research. This work was supported in part by: NSERC Discovery Grant (RGPIN-2018-05894 to K.F.C.), NSERC Discovery Grant Accelerator (RGPAS-2018-522733 to K.F.C.), PEI-Atlantic Shrimp Corp. Inc. grants (12-LSC-35, 14-LSC-037 to S.J.G. & K.F.C.), NSERC-Strategic Projects Grant (STPGP 463277-14 to S.J.G.), Human Frontiers Science Program (RGP0060/2017 to L.L.M.), National Science Foundation (1557923 and 1645219 to L.L.M.), National Institutes of Health (R01NS114491 to L.L.M.), and Russian Ministry of Science and High Education (agreement 075-15-2020-801 to D.R.). Author contributions: J.M.P. conducted all bioinformatic analyses on the final lobster genome assembly, conducted TPM analysis of the lobster transcriptomes, and conducted analysis of the genome stability and chemical defense genes. A.V.Z. constructed the final assembly of the genome and conducted k-mer analysis. K.F.C. generated RNA-seq data from larval lobsters, intestine, and hepatopancreas and investigated the immune system gene repertoire. L.L.M., A.B.K., D.Y.R., and P.W. conducted analyses of the nervous system gene repertoire. W.T. and N.S. conducted long-read sequencing. P.K. conducted initial bioinformatic analysis on the preliminary draft genome. A.P. conducted additional bioinformatic analysis on the preliminary draft genome. S.J.G. provided RNA-seq data for transcriptome analyses. D.R.W. conceived and initiated the study and provided input throughout its progress. A.G.B. coordinated the activities of all collaborators, led the analysis of genome stability and chemical defense genes, and wrote the manuscript. All authors contributed to editing the manuscript. Competing interests: D.R.W. is the scientific founder of Illumina and holds equity in the company. W.T. has two patents (8,748,091 and 8,394,584) licensed to Oxford Nanopore Technologies. The other authors declare that they have no competing interests. Data and materials availability: All sequence data generated and the genome assembly reported in this study are available in the NCBI GenBank under BioProject PRJNA655509; www.ncbi.nlm.nih.gov/bioproject/PRJNA655509. All additional data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabe8290/DC1
REFERENCES AND NOTES
- 1.Le Bris A., Mills K. E., Wahle R. A., Chen Y., Alexander M. A., Allyn A. J., Schuetz J. G., Scott J. D., Pershing A. J., Climate vulnerability and resilience in the most valuable North American fishery. Proc. Natl. Acad. Sci. 115, 1831–1836 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearce J., Balcom N., The 1999 long island sound lobster mortality event: Findings of the comprehensive research initiative. J. Shellfish. Res. 24, 691–697 (2005). [Google Scholar]
- 3.Klymasz-Swartz A. K., Allen G. J. P., Treberg J. R., Yoon G. R., Tripp A., Quijada-Rodriguez A. R., Weihrauch D., Impact of climate change on the American lobster (Homarus americanus): Physiological responses to combined exposure of elevated temperature and pCO2. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 235, 202–210 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Waller J. D., Wahle R. A., McVeigh H., Fields D. M., Linking rising pCO2 and temperature to the larval development and physiology of the American lobster (Homarus americanus). ICES J. Mar. Sci. 74, 1210–1219 (2017). [Google Scholar]
- 5.Glenn R. P., Pugh T. L., Epizootic shell disease in american lobster (Homarus americanus) in Massachusetts coastal waters: Interactions of temperature, maturity, and intermolt duration. J. Crustacean Biol. 26, 639–645 (2006). [Google Scholar]
- 6.McLean E. L., Katenka N., Seibel B., Decreased growth and increased shell disease in early benthic phase Homarus americanus in response to elevated CO2. Mar. Ecol. Prog. Ser. 596, 113–126 (2018). [Google Scholar]
- 7.Dorant Y., Benestan L., Rougemont Q., Normandeau E., Boyle B., Rochette R., Bernatchez L., Comparing Pool-seq, Rapture, and GBS genotyping for inferring weak population structure: The American lobster (Homarus americanus) as a case study. Ecol. Evol. 9, 6606–6623 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benestan L., Gosselin T., Perrier C., Sainte-Marie B., Rochette R., Bernatchez L., RAD genotyping reveals fine-scale genetic structuring and provides powerful population assignment in a widely distributed marine species, the American lobster (Homarus americanus). Mol. Ecol. 24, 3299–3315 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Benestan L., Quinn B. K., Maaroufi H., Laporte M., Clark F. K., Greenwood S. J., Rochette R., Bernatchez L., Seascape genomics provides evidence for thermal adaptation and current-mediated population structure in American lobster (Homarus americanus). Mol. Ecol. 25, 5073–5092 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Pershing A. J., Alexander M. A., Hernandez C. M., Kerr L. A., Le Bris A., Mills K. E., Nye J. A., Record N. R., Scannell H. A., Scott J. D., Sherwood G. D., Thomas A. C., Slow adaptation in the face of rapid warming leads to collapse of the Gulf of Maine cod fishery. Science 350, 809–812 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Marder E., Bucher D., Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu. Rev. Physiol. 69, 291–316 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Christie A. E., Chi M., Lameyer T. J., Pascual M. G., Shea D. N., Stanhope M. E., Schulz D. J., Dickinson P. S., Neuropeptidergic signaling in the American lobster Homarus americanus: New insights from high-throughput nucleotide sequencing. PLOS ONE 10, e0145964 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ache B. W., Young J. M., Olfaction: Diverse species, conserved principles. Neuron 48, 417–430 (2005). [DOI] [PubMed] [Google Scholar]
- 14.McGrath L. L., Vollmer S. V., Kaluziak S. T., Ayers J., De novo transcriptome assembly for the lobster Homarus americanus and characterization of differential gene expression across nervous system tissues. BMC Genomics 17, 63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolff T., Maximum size of lobsters (Homarus) (Decapoda, Nephropidae). Crustaceana 34, 1–14 (1978). [Google Scholar]
- 16.Vogt G., Ageing and longevity in the Decapoda (Crustacea): A review. Zoologischer Anzeiger A J. Comp. Zool. 251, 1–25 (2012). [Google Scholar]
- 17.Vogt G., How to minimize formation and growth of tumours: Potential benefits of decapod crustaceans for cancer research. Int. J. Cancer 123, 2727–2734 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Jeffery N. W., Gregory T. R., Genome size estimates for crustaceans using Feulgen image analysis densitometry of ethanol-preserved tissues. Cytometry A 85, 862–868 (2014). [DOI] [PubMed] [Google Scholar]
- 19.LÉCher P., Defaye D., Noel P., Chromosomes and nuclear DNA of Crustacea. Invertebrate Reproduct. Develop. 27, 85–114 (1995). [Google Scholar]
- 20.Gutekunst J., Andriantsoa R., Falckenhayn C., Hanna K., Stein W., Rasamy J., Lyko F., Clonal genome evolution and rapid invasive spread of the marbled crayfish. Nat. Ecol. & Evol. 2, 567–573 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Coluccia E., Cannas R., Cau A., Deiana A. M., Salvadori S., B chromosomes in Crustacea Decapoda. Cytogenet. Genome Res. 106, 215–221 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Jimenez A. G., Kinsey S. T., Dillaman R. M., Kapraun D. F., Nuclear DNA content variation associated with muscle fiber hypertrophic growth in decapod crustaceans. Genome 53, 161–171 (2010). [DOI] [PubMed] [Google Scholar]
- 23.E. Coluccia, A. Cau, R. Cannas, A. Milia, S. Salvadori, A. M. Deiana, in Mitotic and Meiotic Chromosomes of the American Lobster Homarus americanus (Nephropidae, Decapoda) (Springer, 2001), vol. 449, pp. 149–152. [Google Scholar]
- 24.Zimin A. V., Puiu D., Luo M. C., Zhu T., Koren S., Marçais G., Yorke J. A., Dvořák J., Salzberg S. L., Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27, 787–792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holt C., Yandell M., MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X., Yuan J., Sun Y., Li S., Gao Y., Yu Y., Liu C., Wang Q., Lv X., Zhang X., Ma K. Y., Wang X., Lin W., Wang L., Zhu X., Zhang C., Zhang J., Jin S., Yu K., Kong J., Xu P., Chen J., Zhang H., Sorgeloos P., Sagi A., Alcivar-Warren A., Liu Z., Wang L., Ruan J., Chu K. H., Liu B., Li F., Xiang J., Penaeid shrimp genome provides insights into benthic adaptation and frequent molting. Nat. Commun. 10, 356 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao D., Lai A. G., Stamataki E., Rosic S., Konstantinides N., Jarvis E., Di Donfrancesco A., Pouchkina-Stancheva N., Sémon M., Grillo M., Bruce H., Kumar S., Siwanowicz I., Le A., Lemire A., Eisen M. B., Extavour C., Browne W. E., Wolff C., Averof M., Patel N. H., Sarkies P., Pavlopoulos A., Aboobaker A., The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. eLife 5, e20062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colbourne J. K., Pfrender M. E., Gilbert D., Thomas W. K., Tucker A., Oakley T. H., Tokishita S., Aerts A., Arnold G. J., Basu M. K., Bauer D. J., Cáceres C. E., Carmel L., Casola C., Choi J.-H., Detter J. C., Dong Q., Dusheyko S., Eads B. D., Fröhlich T., Geiler-Samerotte K. A., Gerlach D., Hatcher P., Jogdeo S., Krijgsveld J., Kriventseva E. V., Kültz D., Laforsch C., Lindquist E., Lopez J., Manak J. R., Muller J., Pangilinan J., Patwardhan R. P., Pitluck S., Pritham E. J., Rechtsteiner A., Rho M., Rogozin I. B., Sakarya O., Salamov A., Schaack S., Shapiro H., Shiga Y., Skalitzky C., Smith Z., Souvorov A., Sung W., Tang Z., Tsuchiya D., Tu H., Vos H., Wang M., Wolf Y. I., Yamagata H., Yamada T., Ye Y., Shaw J. R., Andrews J., Crease T. J., Tang H., Lucas S. M., Robertson H. M., Bork P., Koonin E. V., Zdobnov E. M., Grigoriev I. V., Lynch M., Boore J. L., The ecoresponsive genome of Daphnia pulex. Science 331, 555–561 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romano P., Fabritius H., Raabe D., The exoskeleton of the lobster Homarus americanus as an example of a smart anisotropic biological material. Acta Biomater. 3, 301–309 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Benton J. L., Sandeman D. C., Beltz B. S., Nitric oxide in the crustacean brain: Regulation of neurogenesis and morphogenesis in the developing olfactory pathway. Dev. Dyn. 236, 3047–3060 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moroz L. L., Kohn A. B., Parallel evolution of nitric oxide signaling: Diversity of synthesis and memory pathways. Front. Biosci. 16, 2008–2051 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oulavallickal T., Brewster J. L., McKellar J. L. O., Fairhurst M. J., Tenci N. A., Gerth M. L., The Pseudomonas syringae pv. actinidiae chemoreceptor protein F (PscF) periplasmic sensor domain: Cloning, purification and x-ray crystallographic analysis. Acta Crystallogr. F Struct. Biol. Commun. 73, 701–705 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shu C. J., Ulrich L. E., Zhulin I. B., The NIT domain: A predicted nitrate-responsive module in bacterial sensory receptors. Trends Biochem. Sci. 28, 121–124 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Moroz L. L., Romanova D. Y., Nikitin M. A., Sohn D., Kohn A. B., Neveu E., Varoqueaux F., Fasshauer D., The diversification and lineage-specific expansion of nitric oxide signaling in Placozoa: Insights in the evolution of gaseous transmission. Sci. Rep. 10, 13020 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nusbaum M. P., Blitz D. M., Marder E., Functional consequences of neuropeptide and small-molecule co-transmission. Nat. Rev. Neurosci. 18, 389–403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachtel N. D., Hovsepian G. A., Nixon D. F., Eleftherianos I., Allatostatin C modulates nociception and immunity in Drosophila. Sci. Rep. 8, 7501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneider D. S., Jin Y., Morisato D., Anderson K. V., A processed form of the Spatzle protein defines dorsal-ventral polarity in the Drosophila embryo. Development 120, 1243–1250 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Tanaka S., Nakamura T., Morita T., Iwanaga S., Limulus anti-LPS factor: An anticoagulant which inhibits the endotoxin mediated activation of Limulus coagulation system. Biochem. Biophys. Res. Commun. 105, 717–723 (1982). [DOI] [PubMed] [Google Scholar]
- 39.Sun C., Xu W. T., Zhang H. W., Dong L. P., Zhang T., Zhao X. F., Wang J. X., An anti-lipopolysaccharide factor from red swamp crayfish, Procambarus clarkii, exhibited antimicrobial activities in vitro and in vivo. Fish Shellfish Immunol. 30, 295–303 (2011). [DOI] [PubMed] [Google Scholar]
- 40.de Castro E., Sigrist C. J., Gattiker A., Bulliard V., Langendijk-Genevaux P. S., Gasteiger E., Bairoch A., Hulo N., ScanProsite: Detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark K. F., Acorn A. R., Greenwood S. J., Differential expression of American lobster (Homarus americanus) immune related genes during infection of Aerococcus viridans var. homari, the causative agent of Gaffkemia. J. Invertebr. Pathol. 112, 192–202 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Clark K. F., Acorn A. R., Greenwood S. J., A transcriptomic analysis of American lobster (Homarus americanus) immune response during infection with the bumper car parasite Anophryoides haemophila. Dev. Comp. Immunol. 40, 112–122 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Clark K. F., Greenwood S. J., Acorn A. R., Byrne P. J., Molecular immune response of the American lobster (Homarus americanus) to the white spot syndrome virus. J. Invertebr. Pathol. 114, 298–308 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Clark K. F., Characterization and functional classification of American lobster (Homarus americanus) immune factor transcripts. Fish Shellfish Immunol. 41, 12–26 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Tassanakajon A., Somboonwiwat K., Amparyup P., Sequence diversity and evolution of antimicrobial peptides in invertebrates. Dev. Comp. Immunol. 48, 324–341 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Christie A. E., Rus S., Goiney C. C., Smith C. M., Towle D. W., Dickinson P. S., Identification and characterization of a cDNA encoding a crustin-like, putative antibacterial protein from the American lobster Homarus americanus. Mol. Immunol. 44, 3333–3337 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Gerdol M., Schmitt P., Venier P., Rocha G., Rosa R. D., Destoumieux-Garzón D., Functional insights from the evolutionary diversification of big defensins. Front. Immunol. 11, 758 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montero-Alejo V., Corzo G., Porro-Suardíaz J., Pardo-Ruiz Z., Perera E., Rodríguez-Viera L., Sánchez-Díaz G., Hernández-Rodríguez E. W., Álvarez C., Peigneur S., Tytgat J., Perdomo-Morales R., Panusin represents a new family of β-defensin-like peptides in invertebrates. Dev. Comp. Immunol. 67, 310–321 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Vu G. H., Do D., Rivera C. D., Dickinson P. S., Christie A. E., Stemmler E. A., Characterization of the mature form of a β-defensin-like peptide, Hoa-D1, in the lobster Homarus americanus. Mol. Immunol. 101, 329–343 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Robertson A. J., Croce J., Carbonneau S., Voronina E., Miranda E., McClay D. R., Coffman J. A., The genomic underpinnings of apoptosis in Strongylocentrotus purpuratus. Dev. Biol. 300, 321–334 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Fernandez-Guerra A., Aze A., Morales J., Mulner-Lorillon O., Cosson B., Cormier P., Bradham C., Adams N., Robertson A. J., Marzluff W. F., Coffman J. A., Genevière A.-M., The genomic repertoire for cell cycle control and DNA metabolism in S. purpuratus. Dev. Biol. 300, 238–251 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Eischen C. M., Genome stability requires p53. Cold Spring Harb. Perspect. Med. 6, a026096 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ingaramo M. C., Sánchez J. A., Dekanty A., Regulation and function of p53: A perspective from Drosophila studies. Mech. Dev. 154, 82–90 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Fischer M., Census and evaluation of p53 target genes. Oncogene 36, 3943–3956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akdemir F., Christich A., Sogame N., Chapo J., Abrams J. M., p53 directs focused genomic responses in Drosophila. Oncogene 26, 5184–5193 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Denton D., Aung-Htut M. T., Kumar S., Developmentally programmed cell death in Drosophila. Biochim. Biophys. Acta 1833, 3499–3506 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Nair N., Shoaib M., Sørensen C. S., Chromatin dynamics in genome stability: Roles in suppressing endogenous DNA damage and facilitating DNA repair. Int. J. Mol. Sci. 18, 1486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang F., Liu Q., Wang Y., Zhang J., Wang H., Song T., Yang M., Wang X., Kang L., Comparative genomic analysis of SET domain family reveals the origin, expansion, and putative function of the arthropod-specific SmydA genes as histone modifiers in insects. GigaScience 6, 1–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis S. H., Quarles K. A., Yang Y., Tanguy M., Frézal L., Smith S. A., Sharma P. P., Cordaux R., Gilbert C., Giraud I., Collins D. H., Zamore P. D., Miska E. A., Sarkies P., Jiggins F. M., Pan-arthropod analysis reveals somatic piRNAs as an ancestral defence against transposable elements. Nat. Ecol. Evol. 2, 174–181 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goldstone J. V., Hamdoun A., Cole B. J., Howard-Ashby M., Nebert D. W., Scally M., Dean M., Epel D., Hahn M. E., Stegeman J. J., The chemical defensome: Environmental sensing and response genes in the Strongylocentrotus purpuratus genome. Dev. Biol. 300, 366–384 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edi C. V., Djogbénou L., Jenkins A. M., Regna K., Muskavitch M. A. T., Poupardin R., Jones C. M., Essandoh J., Kétoh G. K., Paine M. J. I., Koudou B. G., Donnelly M. J., Ranson H., Weetman D., CYP6 P450 enzymes and ACE-1 duplication produce extreme and multiple insecticide resistance in the malaria mosquito Anopheles gambiae. PLOS Genet. 10, e1004236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janssen A., Colmenares S. U., Karpen G. H., Heterochromatin: Guardian of the genome. Annu. Rev. Cell Dev. Biol. 34, 265–288 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Wylie A., Jones A. E., D’Brot A., Lu W.-J., Kurtz P., Moran J. V., Rakheja D., Chen K. S., Hammer R. E., Comerford S. A., Amatruda J. F., Abrams J. M., p53 genes function to restrain mobile elements. Genes Dev. 30, 64–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song H., Guo X., Sun L., Wang Q., Han F., Wang H., Wray G. A., Davidson P., Wang Q., Hu Z., Zhou C., Yu Z., Yang M., Feng J., Shi P., Zhou Y., Zhang L., Zhang T., The hard clam genome reveals massive expansion and diversification of inhibitors of apoptosis in Bivalvia. BMC Biol. 19, 15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makhijani K., Brückner K., Of blood cells and the nervous system: Hematopoiesis in the Drosophila larva. Fly 6, 254–260 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Doyle J. J., Doyle L. L., A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bullet. 19, 11–15 (1987). [Google Scholar]
- 67.Chapman J. A., Ho I., Sunkara S., Luo S., Schroth G. P., Rokhsar D. S., Meraculous: De novo genome assembly with short paired-end reads. PLOS ONE 6, e23501 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kolmogorov M., Yuan J., Lin Y., Pevzner P. A., Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019). [DOI] [PubMed] [Google Scholar]
- 69.Ruan J., Li H., Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimin A. V., Salzberg S. L., The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLOS Comput. Biol. 16, e1007981 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marçais G., Kingsford C., A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korf I., Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stanke M., Keller O., Gunduz I., Hayes A., Waack S., Morgenstern B., AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li L., Stoeckert C. J. Jr., Roos D. S., OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han M. V., Thomas G. W., Lugo-Martinez J., Hahn M. W., Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Clark K. F., Greenwood S. J., Next-generation sequencing and the Crustacean immune system: The need for alternatives in immune gene annotation. Integr. Comp. Biol. 56, 1113–1130 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Moroz L. L., Kohn A. B., Unbiased view of synaptic and neuronal gene complement in ctenophores: Are there pan-neuronal and pan-synaptic genes across metazoa? Integr. Comp. Biol. 55, 1028–1049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moroz L. L., Kohn A. B., Independent origins of neurons and synapses: Insights from ctenophores. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371, 20150041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O. Hobert, The neuronal genome of Caenorhabditis elegans, in WormBook, The C. elegans Research Community, Ed. (WormBook, 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berman H., Henrick K., Nakamura H., Announcing the worldwide protein data bank. Nat. Struct. Mol. Biol. 10, 980 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Kelley L. A., Mezulis S., Yates C. M., Sternberg M. J., The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabe8290/DC1