

Nevi (moles) harbor resident immune cells that can be activated to eradicate moles and, potentially, to prevent melanoma.
Abstract
Melanoma and melanocytic nevi harbor shared lineage-specific antigens and oncogenic mutations. Yet, the relationship between the immune system and melanocytic nevi is unclear. Using a patient-derived xenograft (PDX) model, we found that 81.8% of the transplanted nevi underwent spontaneous regression, while peripheral skin remained intact. Nevus-resident CD4+ T helper 1 cells, which exhibited a massive clonal expansion to melanocyte-specific antigens, were responsible for nevus rejection. Boosting regulatory T cell suppressive function with low-dose exogenous human interleukin-2 injection or treatment with a human leukocyte antigen (HLA) class II–blocking antibody prevented nevus rejection. Notably, mice with rejected nevus PDXs were protected from melanoma tumor growth. We detected a parallel CD4+ T cell–dominant immunity in clinically regressing melanocytic nevi. These findings reveal a mechanistic explanation for spontaneous nevus regression in humans and posit the activation of nevus-resident CD4+ effector T cells as a novel strategy for melanoma immunoprevention and treatment.
INTRODUCTION
Melanoma is composed of malignant melanocytes, which are targeted by T cell immunity directed at melanocyte lineage–specific and mutated antigens (1–3). In contrast, a benign melanocytic nevus is composed of nests of melanocytes with low mutational burden (4, 5). Despite their stable nature, attributed to oncogene-induced senescence (6–9), melanocytic nevi can undergo spontaneous regression or transformation into melanoma (4). Approximately 33%, reported ranges 4 to 72%, of melanomas have an associated or preexisting melanocytic nevus (10–13). Yet, the role of the immune system in the homeostasis of a melanocytic nevus is unknown. Considering that melanocyte lineage–specific antigens including gp100 and melanoma antigen recognized by T cells 1 (MART-1) are shared between melanocytic nevi and melanoma (1, 3, 14), the immune response to nevi could be harnessed to prevent and treat melanoma.
Melanocytic nevus nests develop because of a transient hyperproliferative state driven by constitutive mitogen-activated protein kinase (MAPK) signaling pathway activation, which ultimately results in a benign and relatively stable neoplasm in the skin (5, 15). While melanoma has, on average, 49.1 mutations per megabase, melanocytic nevi on highly sun-exposed skin harbor 3.9 mutations per megabase (12, 13, 16). The most common shared mutation between nevi and melanoma affects BRAF, a cell-signaling protein that regulates the MAPK pathway (5, 15). The BRAF-V600E mutation is detected in up to 95% of nevi and 50% of cutaneous melanomas (4, 5, 15). The shared cell lineage and the oncogenic relationship between nevi and melanoma further highlight the potential for leveraging the immunobiology of benign nevi as a means to prevent and treat melanoma.
Compared to melanoma, far fewer T and antigen-presenting cell (APC) populations are found in the microenvironment of common melanocytic nevi (17–19). However, clinical evidence exists for inherent immune surveillance and control of these lesions. Halo nevi and vitiligo are marked by autoreactive CD8+ T cells targeting melanocytes for destruction within the melanocytic nevus nests and the surrounding skin (20, 21). In addition, there are well-documented cases of spontaneous nevus regression in humans and the loss of common melanocytic nevi in the elderly, which are distinct from the halo nevi/vitiligo phenomenon (17, 22, 23). It has been reported that the median number of nevi peaks in the age range 20 to 29 and decreases through age 80 (22). Understanding the interplay between the immune system and melanocytic nevi can explain these fundamental clinical observations and provide critical insights into the immune response to early melanomagenesis.
To determine the impact of the host immune response on the biology of benign melanocytic nevi, we generated a patient-derived xenograft (PDX) model in which common melanocytic nevi from participants were transplanted onto the immunodeficient nonobese diabetic (NOD) severe combined immunodeficient gamma (NSG) mice. Unexpectedly, we found that most of the transplanted nevi underwent spontaneous regression, which was mediated by the activation of CD4+ effector memory T cell resident in the nevi. The underlying mechanism of nevus PDX rejection not only explains the long withstanding observations that benign melanocytic nevi can undergo spontaneous regression and are lost with age but also provides fundamental insights into the mechanisms of peripheral tolerance, spontaneous regression of melanoma, and immunity in advanced disease.
RESULTS
Melanocytic nevi regress in immunodeficient hosts
To determine the impact of the host’s immune response on the homeostasis of melanocytic nevi, we developed a nevus PDX model by transplanting clinically benign, acquired melanocytic nevi from cutaneous oncology patients onto NSG mice. Two melanocytic nevi and a normal skin sample were collected from the participants’ trunk or upper extremities by shave biopsy. Nevus I and the normal skin were transplanted onto immunodeficient NSG mice, while nevus II was used for direct histological and molecular studies (fig. S1A). A total of 81.8% of the transplanted nevi underwent spontaneous regression, while 18.2% remained stable over time with no apparent changes in size or shape (Fig. 1, A and B). During the nevus regression, we observed an inflammatory reaction at the site of the nevus that progressed to crust formation, while surrounding skin remained intact in all PDXs (Fig. 1B). No significant change in the total size of the transplanted tissue was observed in stable or regressed nevus PDXs over time (Fig. 1B). Stable nevus PDXs were further treated with ultraviolet B (UVB, 302 to 312 nm) (100 mJ/cm2) three times weekly for over 35 weeks. UVB treatment did not change the size or shape of the stable nevi, which retained the histological features of benign melanocytic proliferation at harvest (fig. S1B). Among participant demographics, including age, gender, cutaneous versus ocular melanoma, cancer stage, systemic treatment, size, anatomical site of the nevi, and the histological diagnosis of nevus II, only increased age was significantly correlated with the regression phenotype (Fig. 1C). The degree of nevus sun exposure (upper arm/chest/upper back versus abdomen/lower back) did not predict nevus regression in NSG mice (Fig. 1C). Consistent with the findings in this observational cohort, one of two nevus PDXs generated from participants with advanced basal cell carcinoma, two of two nevus PDXs from participants with cutaneous melanoma stage IB, and six of nine nevus PDXs from participants with ocular melanoma underwent spontaneous regression in our follow-up studies.
Fig. 1. Most of the melanocytic nevus PDXs undergo regression, which is associated with increased participant age.
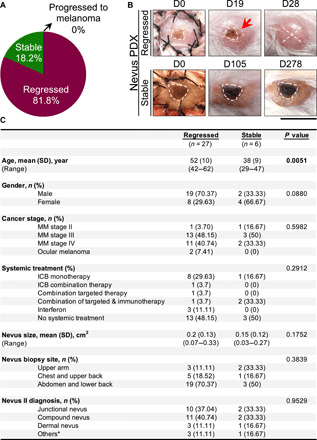
(A) Distribution of nevus PDX outcomes (regressed, stable, or progressed to melanoma, n = 33 mice). (B) Representative images of regressed nevus PDX at days 0, 19, and 28 and stable nevus PDX at days 0, 105, and 278 after transplant. Dashed lines outline the melanocytic nevus on the human skin graft, “X” signifies nevus rejection, and the red arrow points to the inflammation in the regressing nevus; scale bar, 1 cm. (C) Participant demographic information associated with regressed and stable nevus PDXs (n = 33). Mann-Whitney U test is used for age and size variables, and Pearson’s χ2 tests are used for other categorical variables. *Others, benign melanocytic proliferations; MM, malignant melanoma; ICB, immune checkpoint blockade. Photo credit: Erik B. Schiferle, Massachusetts General Hospital.
Cytotoxic immunity is responsible for nevus rejection in NSG mice
We detected a massive accumulation of human T cells, which were predominantly CD4+ T cells, in the regressed nevus PDXs compared with the participant-matched nevus II and stable nevus PDXs (Fig. 2A and fig. S1C). To determine whether T cell expansion occurred only at the site of the nevus, we generated a series of nevus PDXs in which normal skin was cotransplanted with the nevus onto the same NSG mouse (fig. S1D). At the site of the regressed nevi, we detected a massive expansion of T cells, which did not affect the normal human skin graft cotransplanted on the back of the same mouse or the peripheral recipient mouse skin (fig. S1E). We did not detect human B, natural killer (NK), or mast cells in the regressed nevi (fig. S1, F to H). The marked expansion of nevus-resident T cells in the regressed nevus PDXs suggests that melanocytic nevi in NSG mice undergo a T cell–mediated rejection.
Fig. 2. Melanocytic nevus PDX regression is associated with a massive expansion of human CD4+ T cells in the regressed nevi, the apoptosis of nevus melanocytes, and expansion of nevus-derived human T cells in the lymph node and spleen of the transplanted mice.
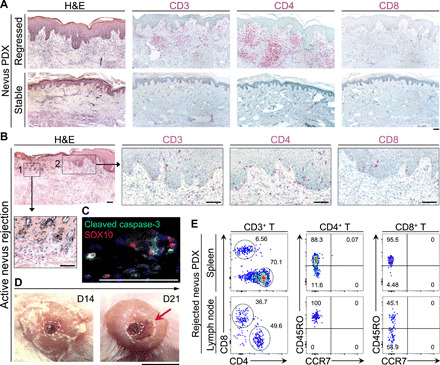
(A) Representative images of hematoxylin and eosin (H&E), human CD3, CD4, and CD8 stained histological sections of regressed and stable nevus PDXs. Scale bar, 100 μm. (B) Representative images from H&E- and CD3/CD4/CD8-stained sections of actively rejecting nevus PDX. Insets show (1) the site of active nevus nest rejection and (2) immune cell infiltration. Scale bars, 100 μm. (C) Representative immunofluorescence (IF) image of cleaved caspase-3 (green) and SOX10 (red) in an actively rejecting nevus PDX at day 14 after transplant. Scale bar, 100 μm. (D) Representative macroscopic images of actively rejecting nevus PDX at days 14 and 21 after transplant. Dashed lines outline the area of nevus before rejection on the human skin, and arrow points to the inflammation and scabbing. Scale bar, 1 cm. (E) Representative flow cytometry plots of human CD4+ and CD8+ T cells in the spleen and lymph node of the rejected nevus PDX. CD45RO versus CCR7 expression shows the “effector memory” status of the rejected nevus-derived human T cells in the lymphoid organs of the NSG mice; numbers on the plots represent the percent cells within each gate. Photo credit: Erik B. Schiferle, Massachusetts General Hospital.
Active nevus rejection, which took place between days 14 and 28 after transplant, coincided with the expansion of CD4+ T cells and the apoptosis of SOX10+ melanocytes in the nevus nests (Fig. 2, B to D, and fig. S2A). Nevus-derived CD45RO+ CCR7− effector memory CD4+ and CD8+ T cells were readily detectable in the spleen and skin-draining lymph nodes of the rejected nevus PDXs (Fig. 2E) (24). We did not detect human CD4+ T cells in the spleen or lymph nodes of stable nevus PDXs (fig. S2B). Further, CD4+ T cells in the stable nevi expressed the p16INK4A senescence marker (fig. S2C). To establish whether nevus rejection in NSG mice was immune-mediated, we treated a cohort of nevus PDXs with a T cell–suppressing agent, cyclosporine (25, 26), at 30 mg/kg daily dose injected subcutaneously from days 2 to 30 after transplant. Cyclosporine treatment prevented nevus rejection in NSG mice (Fig. 3, A and B). Nonetheless, five of six nevus PDXs underwent rejection after cyclosporine treatment cessation (Fig. 3, A and B). In contrast to a CD8+ T cell–mediated halo nevus phenomenon, the immune-mediated rejection of nevi in NSG mice did not affect the normal melanocytes in the surrounding skin of the rejected nevus PDX (fig. S2D). Multiplex immunostaining for T cell markers revealed a significant expansion of CD4+ T, CD8+ T, and T-bet+ CD4+ T helper 1 (TH1) cells and a markedly increased TH1/regulatory T cell (Treg) ratio in the rejected nevus compared with its participant-matched nevus II (Fig. 3, C to G, and fig. S2, E and F). The CD8+ T/Treg ratio, GATA3+ TH2, and RORγt+ TH17 cell counts were not significantly increased in rejected nevus PDXs compared with participant-matched nevus II samples (Fig. 3H and fig. S2, G to J). We found a significant increase in granzyme B+ CD4+ T cells in the rejected nevus PDXs compared with their participant-matched nevus II samples (fig. S2, K and L). Stable nevus PDXs showed no increase in any T cell subtype compared with their participant-matched nevus II samples (Fig. 3, C to H, and fig. S2, G, I, and K). These findings demonstrate that cytotoxic immunity rejects nevi in NSG mice, which is dominated by CD4+ TH1 cells specifically targeting the melanocytes in the nevus.
Fig. 3. Melanocytic nevi are rejected by nevus-resident immune cells, which are primarily CD4+ TH1 cells.
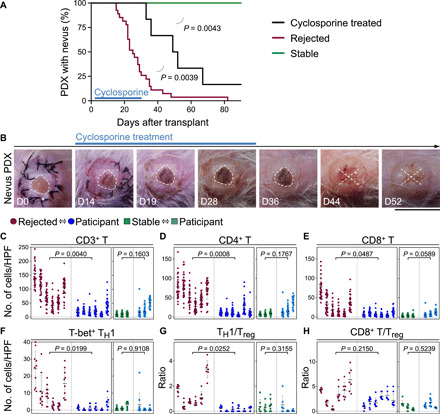
(A) Time to nevus PDX rejection in cyclosporine-treated mice (black, n = 6) compared with the rejected (red, n = 27, P = 0.0039) and stable (green, n = 6, P = 0.0043) nevus PDXs (log-rank test). Note that rejected and stable nevus PDXs are shown in Fig. 1, A and C. (B) Representative images of nevus PDX in cyclosporine-treated mice at days 0, 14, 19, 28, 36, 44, and 52 after transplant. Dashed lines outline the area of nevus before rejection on the human skin, and X signifies nevus rejection. Scale bar, 1 cm. (C to H) Immune cell quantification in the rejected and stable nevus PDXs compared with their participant-matched nevus II samples. CD3+ T (C), CD4+ T (D), CD8+ T (E), and T-bet+ TH1 (F) cell counts and TH1/Treg (G) and CD8+ T/Treg (H) ratios are shown (nested t test). Cells were counted in 10 random high-power field (HPF) images per rejected nevus PDX (n = 6), participant-matched nevus II (n = 6), stable nevus PDX (n = 3), and participant-matched nevus II (n = 3) samples. Photo credit: Erik B. Schiferle, Massachusetts General Hospital.
TH1 cell activation mediates rejection of nevi in NSG mice
Tregs are dependent on exogenous interleukin-2 (IL-2) and lose their suppressive function when transferred from humans to NSG mice (27). To determine whether Treg function in newly transplanted nevi could be bolstered to suppress nevus-resident effector T cells, we treated a cohort of nevus PDXs with a low-dose (80,000 U/kg) human IL-2 from 2 to 30 days after transplant (27). Unlike high-dose IL-2 as a therapy for melanoma, low-dose IL-2 is used for the treatment of autoimmune diseases (28). While on treatment, five of six IL-2–treated nevus PDXs remained stable, while six of seven nevus PDXs treated with phosphate-buffered saline (PBS, carrier control) underwent rejection (Fig. 4A). Following the treatment cessation, nevi in the IL-2–treated cohort underwent rejection at a similar ratio to those in the PBS-treated group (fig. S3A). In an attempt to determine whether Treg depletion in stable nevus PDXs could result in the induction of nevus rejection, we intradermally injected stable nevus PDXs with an anti-CD25 antibody, basiliximab. Although basiliximab has shown efficacy for Treg depletion (29), we did not observe any changes in stable nevus PDXs treated with basiliximab (fig. S3B). Collectively, these data suggest that waning Treg-mediated peripheral tolerance during aging may be a reason for nevus regression in the elderly (22, 30). This hypothesis is supported by the significantly higher age of the patients without detectable melanocytic nevi on their skin compared with those who had nevi in our study (P < 0.0001; fig. S3C).
Fig. 4. Break in peripheral tolerance leads to the rejection of nevus PDX.
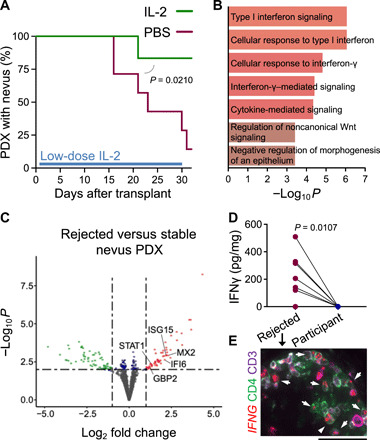
(A) Time to nevus PDX rejection in mice treated with low-dose IL-2 (green, n = 6) compared with PBS-treated controls (red, n = 7, log-rank test). (B) Gene Ontology pathway enrichment analysis of up-regulated genes in the rejected compared with stable nevus PDXs (n = 3 per group). (C) Volcano plot of significantly up-regulated (red dots) and down-regulated (green dots) genes in the rejected compared with stable nevus PDXs (n = 3, log2 fold change > |1| and −log10 of P > 2 is considered significant). (D) IFNγ protein levels in the rejected nevus PDXs versus participant-matched nevus II samples (n = 7 per group, paired t test). (E) Representative IF image of IFNG RNA in situ hybridization combined with CD4 (green) and CD3 (purple) antibody staining in the rejected nevus PDX. Arrowheads point to IFNG-expressing CD4+ T cells. Scale bar, 50 μm.
To determine which factor(s) contributed to nevus rejection in NSG mice, we examined the transcriptional profiles of rejected and stable nevus PDXs. Mouse-specific transcripts were computationally removed to enable this analysis (fig. S4, A and B). The transcriptome of the rejected nevus PDXs clustered away from stable nevus PDXs and participant-matched nevus II samples (fig. S4, C and D). Type I interferon (IFN), IFNγ signaling, and the cellular response to IFN were the highest up-regulated pathways in the rejected versus stable nevus PDXs (Fig. 4B). Signal transducer and activator of transcription 1 (STAT1), MX2, IFI6, ISG15, and GBP2 were significantly up-regulated in the rejected versus stable nevus PDXs (Fig. 4C). At the protein level, IFNγ was markedly elevated in the rejected nevus PDXs compared with participant-matched nevus II samples (Fig. 4D and fig. S4E). At the RNA level, IFNG-expressing CD4+ T cells were readily detectable in the rejected nevus PDXs (Fig. 4E and fig. S4F). In addition to IFNγ, IL-6, C-X-C motif chemokine ligand 8 (CXCL8), and CXCL10 were detectable in the rejected nevus PDXs, while IL-4, IL-10, and IL-17A were largely undetectable (fig. S4G).
Next, we investigated the role of APCs in CD4+ T cell activation in the rejected nevus PDXs. We found a significant expansion of human leukocyte antigen class II (HLA II)–expressing dermal APCs in the rejected nevi compared with stable nevi and participant-matched nevus II samples (Fig. 5A and fig. S5A). Most of the APCs in the rejected nevus PDXs were activated CD80+ CD68+ macrophages (Fig. 5, B to D, and fig. S5, B to E). In addition to macrophages, HLA II expression was detectable in the epidermal keratinocytes at the site of nevus rejection (fig. S5F). In contrast to the massive expansion of the activated APCs, very few programmed cell death ligand-1 (PD-L1)–expressing dermal cells were detected in the rejected nevi (fig. S5G). In addition, PD-L1 was not expressed by the melanocytes in the rejected or stable nevus PDXs (fig. S5H).
Fig. 5. HLA II blockade inhibits the CD4+ T cell–mediated rejection of nevus PDX.
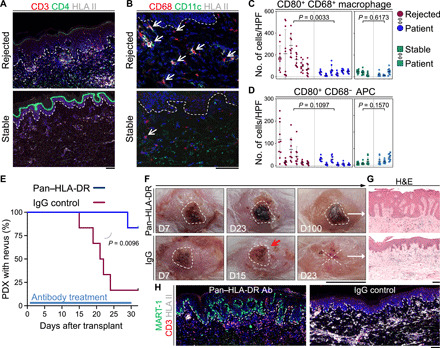
(A) Representative IF images of CD3-stained (red), CD4-stained (green), and HLA II–stained (light gray) sections of the rejected and stable nevus PDXs. Dashed lines highlight the epidermal basement membrane. Scale bar, 100 μm. (B) Representative IF images of CD68-stained (red), CD11c-stained (green), and HLA II–stained (light gray) sections of the rejected and stable nevus PDXs. Dashed lines highlight epidermal basement membrane, and arrows point to CD68+ macrophages. Scale bar, 100 μm. (C and D) Quantification of CD80+ CD68+ macrophages (C) and CD80+ CD68− APCs (D) in 10 random HPFs per sample of rejected nevus PDXs (n = 6), their participant-matched nevus II (n = 6), stable nevus PDXs (n = 3), and their participant-matched nevus II (n = 3) samples (nested t test). (E) Time to nevus PDX rejection comparing pan–HLA-DR antibody–treated (blue, n = 6) to IgG control–treated (red, n = 6) PDX mice (log-rank test). (F) Representative images of pan–HLA-DR antibody–treated nevus PDX at days 7, 23, and 100 after transplant and IgG control–treated nevus PDX at days 7, 15, and 23 after transplant. Dashed lines outline the area of the nevus on the human skin, X signifies nevus rejection, and red arrow points to the inflammation at the site of the nevus PDX. Scale bar, 1 cm. (G) Representative images of H&E-stained sections from pan–HLA-DR antibody– and IgG control–treated nevus PDXs. Scale bar, 100 μm. (H) Representative IF images of MART-1–stained (green), CD3-stained (red), and HLA II–stained (light gray) sections of pan–HLA-DR antibody (Ab)– and IgG control–treated nevus PDXs. Dashed lines indicate epidermal basement membrane. Scale bar, 100 μm. Photo credit: Erik B. Schiferle, Massachusetts General Hospital.
To examine the requirement of T cells to reject melanocytic nevi in NSG mice, we attempted to deplete CD4+ T or CD8+ T cells in the newly transplanted animals using anti-human CD4 (OKT4) or CD8 (OKT8) antibodies, respectively. However, these antibody injections did not deplete T cells in PDXs likely because of the inability of mouse complements to bind human antibodies. To examine the requirement of CD4+ T effector function to reject melanocytic nevi in NSG mice, we treated newly transplanted animals with a pan–HLA-DR antibody [L243 (31)] that blocks HLA-DR/T cell receptor (TCR) interaction (31–33). Pan–HLA-DR antibody treatment of nevus PDXs at a dose of 12 μg/g via intraperitoneal injection biweekly from days 2 to 30 after transplant led to a significant increase in the percentage of mice with stable nevi compared with immunoglobulin G (IgG)–treated nevus PDXs (P = 0.0096; Fig. 5E). Size, shape, and the melanocyte nests of the nevus PDXs treated with the pan–HLA-DR antibody remained intact compared with IgG-treated controls (Fig. 5, F to H). Although we observed a reduction in HLA II–expressing APCs, T cell accumulation persisted in pan–HLA-DR antibody–treated nevus PDXs similar to IgG-treated controls (Fig. 5H). These findings demonstrate that CD4+ effector T cells are responsible for melanocytic nevus rejection in the NSG mice.
Expanded CD4+ T cell clones target melanocyte lineage–specific antigens in the nevus
To determine whether T cells within the rejected nevus PDXs underwent clonal expansion, we performed immunoSEQ high-throughput DNA sequencing of the TCR B gene (TCRB) on rejected nevus PDXs and their participant-matched nevus II samples (34). T cell populations were exceptionally clonal in seven of seven rejected nevus samples analyzed. Three-dimensional plots of TCRBV versus TCRBJ regions of top clones in rejected samples illustrated that single clone count frequencies were as high as 39.1% (Fig. 6A). Productive frequencies for the top 10 clones in the rejected nevi accounted for 29.2 to 81.8% of the total T cells compared with 8.5 to 21.2% for nevus II samples (fig. S6A). Likewise, clonality was significantly increased and entropy, a measure of TCR rearrangement diversity, was significantly decreased when comparing rejected nevus PDXs to participant-matched nevus II samples (Fig. 6, B and C). Many of the T cell clones that expanded in the rejected nevus PDXs were also found in their participant-matched nevus II samples (fig. S6, B and C). Investigation of shared V-D-J gene names across rejected nevus samples revealed seven gene names that were shared among the seven rejected nevus samples examined (Fig. 6D and fig. S6D). Aligning the productive frequencies of the T cell clones that significantly expanded in the rejected nevus PDXs versus participant-matched nevus II samples with CD4+ and CD8+ T cell percentages in the rejected nevus PDXs revealed that many of the top expanded clones were CD4+ T cells, as the total CD8+ T cell percentage was less than the frequency of the top T cell clone in the PDX (Fig. 6E and fig. S6E).
Fig. 6. CD4+ T cells undergo clonal expansion in the rejected nevus PDXs.
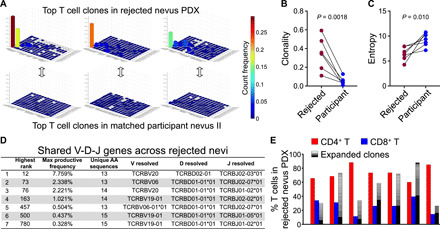
(A) Three-dimensional bar graphs of TCRB sequencing data in three rejected nevus PDXs and their participant-matched nevus II samples (x axis = TCRBV, y axis = TCRBJ, and z axis = count frequency). (B and C) T cell clonality (B) and entropy (C) in the rejected nevus PDXs (n = 7) and their participant-matched nevus II samples based on CDR3β.AA sequences (n = 7, paired t test). (D) Shared V-D-J gene names across the seven rejected nevus PDXs. “Highest rank” refers to the rank of the maximum productive frequency observed in one sample from the list that contains the total number of sequences (18,536) across 14 rejected nevus PDXs and their participant-matched nevus II samples sorted by decreasing productive frequencies. “Max productive frequency” refers to the value of rank above. “Unique amino acid (AA) sequences” refers to the number of unique CDR3β.AA sequences shared across the seven rejected samples that are observed for the respective V-D-J gene name. (E) Grouped bar graph of % CD4+ T (red), CD8+ T (blue), and significantly expanded T cell clones (black-white shading) of total T cells in the rejected nevus PDXs (n = 7). Note that CD4+ and CD8+ T cell frequencies were obtained using HALO software to count cells across the entirety of immunohistochemical-stained nevus PDX sections.
To investigate whether the expanded CD4+ T cell clones were targeting melanocyte lineage–specific antigens, we stained CD4+ T cells from PDX nevi with a DRB1*0401 gp10044–59 tetramer (35) and a DRB1*0101 MART-151–65 dextramer (14) versus control peptide containing tetramer and dextramer. gp100-specific CD4+ T cells were significantly expanded in the rejected compared with stable nevus PDXs with HLA DRB1*04 (Fig. 7A and fig. S7, A to C). Notably, in one of the rejected nevus PDXs, 27% of total CD4+ T cells were labeled with the DRB1*0401 gp10044–59 tetramer (Fig. 7B). We did not detect any DRB1*0101 MART-151–65 dextramer–positive CD4+ T cells in the analyzed PDXs (Fig. 7B and fig. S7C). We examined whether the differential expression of gp100 and MART-1 in nevus versus normal skin melanocytes could explain the presence of gp100- but not MART-1–specific CD4+ T cells in the rejected nevus PDXs (36, 37). In contrast to MART-1, gp100+ melanocyte percentage was markedly higher in the nevus compared with the adjacent normal skin (Fig. 7, C and D). This finding demonstrates that gp100 antigen expression is more restricted to nevus melanocytes compared with MART-1. Gating off gp100-specific CD4+ T cells revealed that most of these cells were CD45RO+ CCR7− and also expressed tissue-resident memory (TRM) marker CD103 (fig. S7, D and E). Immunofluorescence staining supported the expansion of CD103+ CD69+ TRM cells in the rejected compared with stable nevus PDXs (fig. S7F). Collectively, these findings reveal a critical role for nevus-resident melanocyte antigen–specific CD4+ T cells in rejecting melanocytic nevi in NSG mice.
Fig. 7. Clonally expanded CD4+ T cells target melanocyte lineage–specific antigens in the rejected nevus PDXs.
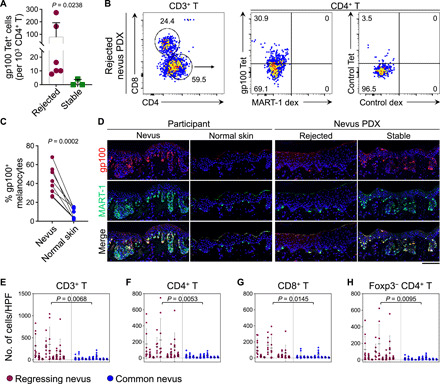
(A) gp10044–59 tetramer–positive CD4+ T cell counts in the rejected (n = 6) versus stable nevus PDXs (n = 3, Mann-Whitney U test). (B) Representative flow cytometry plots of human T cells isolated from a rejected nevus PDX. gp10044–59 tetramer– versus MART-151–65 dextramer–labeled CD4+ T cells are compared with control peptide tetramer- and dextramer-stained CD4+ T cells from the same nevus PDX. Numbers on the plots represent the % cells within each gate. (C) Percentage of gp100+ melanocytes in nevus II samples compared with the adjacent normal skin. Note that MART-1 is used as the marker to identify melanocytes in the nevi and normal skin (paired t test). (D) Representative image of gp100 and MART-1–stained nevus II and adjacent normal skin from participants, rejected, and stable nevus PDXs. Note that a large number of melanocytes in nevus II and stable nevus PDX express gp100. Scale bar, 100 μm. (E to H) Immune cell quantification in clinically regressing nevi compared with common melanocytic nevi. CD3+ T cells (E), CD4+ T (F), CD8+ T (G), and Foxp3− CD4+ effector T (H) cell counts are shown (nested t test). Cells were counted in 10 random HPF images per sample of the regressing (n = 9) and common nevi (n = 10).
CD4+ T cell immunity is characteristic of spontaneously regressing nevi in humans
To determine whether a similar CD4+ T cell immunity is involved in the nevus regression phenomenon observed in humans, we compared a collection of clinically regressing nevi with common melanocytic nevi. All regressing nevi were marked by a massive T cell infiltrate, which were predominantly CD4+ effector T cells, compared with common nevi (Fig. 7, E to H and fig. S8, A to C). CD4+ T cell infiltrates were accompanied by a large expansion of dermal APCs in the regressing nevi (fig. S8C). Unlike CD8+ T cell–mediated melanocyte destruction in halo nevi, the MART-1+ melanocytes in the normal skin surrounding the regressing nevi were intact (fig. S8C). The parallels observed between the rejected nevus PDX and clinically regressing nevi highlight a mechanistic explanation for nevus regression in humans.
Mice with rejected nevus are protected from melanoma
To determine whether T cell immunity against melanocytic nevi can protect against melanoma, we subcutaneously implanted SKMEL30, a pigmented human melanoma cell line (38), into the human skin graft of the rejected nevus (test) and participant-matched normal skin (control) PDXs. SKMEL30 tumor growth was significantly blunted when tumor cells were implanted in the rejected nevus site compared with normal human skin PDXs and the dorsal skin of NSG mice without a graft (fig. S9, A to D). In addition, the metastasis of melanoma from the subcutaneous implantation site to mouse lymph nodes was significantly decreased in the rejected nevus PDXs (fig. S9B). Likewise, the SKMEL30 subcutaneous tumor growth rate was lowest in the rejected nevus compared with normal skin PDXs and NSG mice without a graft (fig. S9E). Three stable nevus PDXs that subcutaneously received SKMEL30 cells in the nevus graft site did not experience any protection from melanoma growth compared with normal skin PDXs (fig. S9E). Tumor-infiltrating CD4+ T and CD8+ T cells were markedly higher in the rejected nevus compared with normal skin PDXs (fig. S9, F and G). These findings suggest that antinevus immunity may provide cross-protection against melanoma.
DISCUSSION
Translation of fundamental mechanisms governing T cell tolerance and exhaustion have led to modern immunotherapeutics, which are mainstay treatments in the fight against multiple cancers, including melanoma. Although it is known that nearly 33% of melanomas have an associated or preexisting melanocytic nevus (10–13), the role that T cells play in the homeostasis and transformation of benign melanocytic nevi has remained largely unexplored. Using a novel nevus PDX model, we demonstrate that human nevus-resident CD4+ TH1 cells mediate spontaneous rejection of benign acquired melanocytic nevi. Nevus rejection is blocked by treating newly transplanted nevus PDXs with low-dose human IL-2 to boost Tregs or with an HLA II–blocking antibody. The introduction of human-derived pigmented melanoma cells to the rejected nevus PDXs illustrates the potential of antinevus immunity to protect from melanoma growth. These findings posit an explanation for the long withstanding observations that melanocytic nevi can undergo spontaneous regression and are lost with age (22). Furthermore, the activation of CD4+ effector T cells present in common melanocytic nevi can be used for melanoma immunoprevention and treatment.
The loss of Treg suppressive function in our PDX model has revealed that melanocytic nevi harbor melanocyte antigen–specific CD4+ TH1 cells, which can be activated in situ to reject the nevus. The deletion of self-reactive T cells occurs as part of the process of thymic selection (39). However, Treg-mediated peripheral tolerance provides an additional layer of defense against self-reactive T cells that escape thymic selection (40, 41). This mechanism is particularly critical for CD4+ T cell tolerance to tissue-restricted “self” antigens (42). Melanocyte antigen–specific memory CD4+ T cells have been detected in human blood, which further supports the critical role for peripheral tolerance in suppressing their activation (35). In light of the loss of Treg suppressive function in PDX models (27, 43), melanocyte antigen–specific CD4+ effector T cells are unleashed to cause nevus rejection in PDXs. We find gp100 antigen to be more restricted to nevus melanocytes compared with MART-1, which is broadly expressed by nevus and normal skin melanocytes. Thus, we speculate that gp100-specific CD4+ effector T cells may be more likely to escape central tolerance mechanisms and be controlled through peripheral tolerance in the nevus microenvironment with age. Functional assays with isolated CD4+ T cell clones are required to fully assess the antigen specificities of CD4+ T cells in nevi. Considering the increased risk of break in peripheral tolerance and autoimmunity with age, our findings provide an explanation for the clinical observation that spontaneous nevus regression occurs in individuals with increased age. Future studies are warranted to further examine the immune basis of spontaneous nevus regression in humans with age.
The field of cancer immunology is dominated by studies on CD8+ cytotoxic T cells in the context of advanced cancers, including melanoma (44). However, recent studies have provided evidence that CD4+ T cells can broaden antitumor responses and overcome obstacles to current cancer immunotherapies (45, 46). Specifically, neoantigen-specific CD4+ T cells are shown to be critical for the optimal cancer control and the efficacy of immune checkpoint inhibitor therapy (47). Likewise, intratumoral cytotoxic CD4+ T cells have been found in human bladder cancer, which play a critical role in response to immune checkpoint inhibitor therapy in patients (48). CD4+ T cells are upstream activators of adaptive immunity and release a broad array of cytokines that recruit and regulate the activity of other immune cell types. In particular, TH1 cells produce IFNγ to directly target malignant cells for apoptosis and to recruit and activate CD8+ T and NK cells against cancer (49, 50). Thus, the activation of melanocyte antigen–specific CD4+ TH1 cells can generate a potent immunity against melanoma development and progression. Our data demonstrate that CD4+ TH1 cells, with high IFNγ expression and potential for direct cytotoxicity, are responsible for nevus rejection in NSG mice. CD4+ TH1 cells’ induction of melanocytes’ death likely incorporates direct cytotoxicity (48), cytokine-mediated [e.g., IFNγ (51, 52)] and ligand-mediated [e.g., Fas ligand (53)] cytotoxicity, and downstream activation of CD8+ cytotoxic T cells (47). The contribution of each of these effector mechanisms downstream of CD4+ TH1 activation to nevus rejection will be examined in future studies. In sum, the activation of TH1 cells, which are already present in the microenvironment of benign melanocytic nevi, highlights a novel therapeutic strategy to eliminate melanocytic nevi and may provide an effective strategy for melanoma immunoprevention.
MATERIALS AND METHODS
Study approval
The clinical study was approved by the Massachusetts General Hospital Institutional Review Board. All participants provided written informed consent to participate in the study. The clinical study was conceived, designed, initiated, and performed by the academic investigators. Animal studies were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee (IACUC).
Participants
Patients with cancer with Fitzpatrick skin types II and III were recruited into the study as they presented to the cutaneous oncology clinics at Massachusetts General Hospital. Eligibility criteria included age of at least 18 years and the presence of at least two clinically benign acquired melanocytic nevi (brown macules with regular shape and pigment) on the trunk or upper extremities. All the target melanocytic nevi were evaluated by a board-certified dermatologist who deemed the lesions to be clinically consistent with benign nevi and that would otherwise not be excised to rule out a melanoma diagnosis. Three skin shave biopsy sites, including two nevi and a normal skin region, were selected. Photographs of the nevi and normal skin were taken. Biopsy sites were sterilized and anesthetized, and a shave biopsy was performed. Biopsies were stored in RPMI 1640 (Life Technologies, Carlsbad, CA, catalog no. 21870076) at 4°C until transplantation onto NSG mice within 2 hours of collection. Three-digit unique identifiers were used to identify all biopsies, photographs, and demographic data, which included the site of the biopsy, age/gender of the participant, the presence or absence of a history of melanoma, melanoma stage, and treatment. Nevus PDXs with a histological diagnosis other than benign melanocytic proliferation (e.g., lentigo and seborrheic keratosis) on their participant-matched nevus II samples were excluded from the analysis.
Mice
Male and female 6- to 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG, the Jackson laboratory, Bar Harbor, ME, strain code: 614, Research Resource Identifiers (RRID): BCBC_4142) mice were used for all PDXs. All mice were housed under pathogen-free conditions in the animal facility at Massachusetts General Hospital, adhering to all animal care regulations. All procedures were approved by the IACUC at Massachusetts General Hospital.
Nevus transplantation
Anesthesia, ketamine [10 mg/ml; KetaVed, Boehringer, Ingelheim Vetmedica, Fort Dodge, IA, New Animal Drug Application (NADA) no. 045-290] combined with xylazine (0.5 mg/ml; AnaSed, Akorn, Lake Fortes, IL, NADA no. 139-236) diluted in PBS (Life Technologies, Thermo Fisher Scientific, Grand Island, NY, catalog no. 14190144) under sterile conditions, was administered to mice by subcutaneous injection at 4.5 μl of working solution per gram of body weight. Mice were also administered 3% isoflurane (Baxter Healthcare Corporation, Deerfield, IL, catalog no. 1001936040) as needed during the procedure. Anesthetized mice were placed on a heating pad, and back skin was treated with 70% ethanol to sterilize the surgical site. A circular incision was made in mouse back skin with sterile surgical scissors to remove roughly twice the area of the biopsied human nevus. 1× PBS was added to the surgical area as needed to keep tissue from drying out. Using a 6-0 silk suture (Ethicon, San Lorenzo, Puerto Rico, catalog no. K889H), 8 to 10 surgical knots were tied equidistant around the biopsied skin to the recipient skin. The transplanted nevus/skin was photographed. Triple antibiotic ointment (Medline Industries, Northfield, IL, catalog no. CUR001231) was placed on the transplant area. A 2 cm by 2 cm patch of Vaseline gauze (Covidien, Mansfield, MA, catalog no. 884413605) was placed over the transplant area. The gauze was secured with 4-0 silk surgical sutures (Ethicon, catalog no. K871) and athletic tape (Tensoplast, BSN medical, Charlotte, NC, catalog no. 02115-00). All mice received sulfadiazine/trimethoprim antibiotics (Equisul-SDT Oral Suspension, Aurora Pharmaceuticals, Santa Cruz Animal Health, Dallas, TX, catalog no. sc-395895Rx) following transplantation to prevent infection. Bandages were removed 5 to 7 days after surgery. Graft sutures were removed by day 14 days after surgery. PDXs were monitored daily and photographed every 1 to 7 days. ImageJ (version 1.52a) was used to measure graft size.
Tissue harvesting
Nevus II samples were cut in half, parallel to the shortest perceived diameter of the nevus, using a razor blade. Half of the biopsy was placed in optimum cutting temperature compound (OCT compound) (Thermo Fisher Scientific, Waltham, MA, catalog no. 23-730-571), frozen on dry ice, and then moved to −80°C for long-term storage. The other half was treated with 4% paraformaldehyde (PFA; Sigma-Aldrich, St. Louis, MO, P6148) in 1× PBS at 4°C for 16 hours. The tissue was then dehydrated in an ethanol series, processed, and embedded in paraffin. To harvest PDX, mice were euthanatized by CO2 inhalation. Hair near the transplant site was shaved. The graft and adjacent recipient skin were simultaneously excised using surgical scissors. The tissues were processed for histology (in PFA and OCT), flow cytometry, and molecular studies.
Flow cytometry
Dorsal skin-draining lymph nodes were harvested and placed in 1 ml of digestion buffer [collagenase IV (200 U/ml; Thermo Fisher Scientific, catalog no. 17104019) in RPMI 1640] in an Eppendorf tube (Eppendorf, Hamburg, Germany) and incubated at 37°C for 30 min. A total of 10 μl of 0.5 M EDTA (Boston BioProducts, Ashland, MA, catalog no. BM-150-K-100) was added to the digestion tube and incubated for 5 min at room temperature (RT). Then, single-cell suspensions of spleen and digested lymph nodes were prepared by passing the tissue through 70-μm filters in RPMI 1640 and centrifugation at 300g for 5 min. The spleen samples were resuspended in 1 ml of 1X RBC Lysis Buffer (RBC lysis buffer 10×, BioLegend, San Diego, CA, catalog no. 420301) and incubated at RT for 5 min. Samples were washed once with RPMI 1640 and stained with antibodies (table S1) suspended in 100 μl of PBA [500 ml PBS, 25 ml of newborn calf serum (Invitrogen, Carlsbad, CA, catalog no. 26010074), and 1 ml of 10% sodium azide (Sigma-Aldrich, catalog no. S2002-100G)] for 30 min at 4°C. Samples were washed with PBA and assayed on a BD LSRFortessa X-20 flow cytometer (BD Bioscience, Billerica, MA) or BD FACSCanto (BD Bioscience). Single cells were first identified by forward scatter area (FSC-A) versus FSC-H. Then, lymphocytes were gated from FSC-A versus side scatter area (SSC-A). T cells were then identified as CD45+ CD3+. No CD56+ NK cells or CD19+ B cells were detected in the PDX flow analyses. Data were analyzed using FlowJo software version 10 (Tree Star, Ashland, OR).
Tetramer and dextramer experiments
The DRB1*0401 gp10044–59 phycoerythrin (PE)–conjugated tetramer was generated by the National Institutes of Health (NIH) tetramer core facility at Emory University, on the basis of a previous publication (35). A DRB1*0401 control peptide (PVSKMRMATPLLMQA) PE-conjugated tetramer was also provided by the NIH. The DRB1*0101 Mart151–65 fluorescein isothiocyanate (FITC)–conjugated dextramer was purchased from Immudex (Copenhagen, Denmark), on the basis of a previous report (14). A DRB1*0101 control peptide dextramer (PVSKMRMATPLLMQA) FITC-conjugated dextramer was also acquired from Immudex. Tissue samples were minced with a razor blade and digested with 10 ml of RPMI 1640 medium, containing 10% fetal bovine serum (FBS; Corning, Corning, NY, catalog no. 35-010-CV), 1% penicillin-streptomycin (Invitrogen, catalog no. 10378016), 1% l-glutamine (Sigma-Aldrich, catalog no. G7513-20ML), and collagenase P (1 mg/ml; Sigma-Aldrich, catalog no. 11213865001), for 6 hours at 37°C (54). The tissue was homogenized through a 70-μm strainer, washed with 37°C RPMI 1640 medium, and centrifuged at 1200 rpm for 5 min. On the basis of the instructions provided by the NIH tetramer core facility and Immudex, up to 1 million cells per sample were treated with 46 nM test and control tetramers and 0.64 nM test and control dextramers for 3 hours at 37°C. Cells were washed with 37°C RPMI 1640 medium and centrifuged at 1200 rpm for 5 min. Cells were stained for other markers and analyzed as described above.
LEGENDplex and enzyme-linked immunosorbent assay
Using a microtome, 20 μm by 50 μm sections were cut from OCT-frozen blocks and dissolved in 1 ml of sterile water. After centrifugation at 300g for 5 min, the supernatant was discarded, and the remaining pellet was suspended in 575 μl of 0.1% (v/v) Tween 20 in 1× PBS with 25 μl of Pierce EDTA-free proteinase inhibitor (Thermo Fisher Scientific, Waltham, MA, catalog no. A32955). Tissues were homogenized with a Qiagen TissueLyser (Qiagen, Hilden, Germany) at 30 Hz for 6 min. The homogenate was flash-frozen with liquid nitrogen and thawed at 37°C for 5 min. After sonication using Sentry Ultrasonic for 15 s, the samples were centrifuged at 13,000g for 5 min. Protein concentration was determined using a Bicinchoninic acid assay (BCA) kit (Thermo Fisher Scientific, catalog no. 23225) according to the manufacturer’s instructions. Quantifications of IL-4, IL-6, IL-10, IL-17A, CXCL8, and CXCL10 were obtained using LEGENDplex kits (BioLegend, catalog nos. 740933, 740935, 740939, 740940, 740941, 740904, 740932, 740931, and 740368) following the manufacturer’s instructions for analysis on the BD LSRFortessa X-20 flow cytometer. Enzyme-linked immunosorbent assay–based quantification of IFNγ (Biolegend, catalog no. 430107) was performed according to the manufacturer’s instructions.
RNA sequencing and analysis
OCT-frozen tissue sections were prepared in 10 μl of TCL buffer (Qiagen, catalog no. 1031576) supplemented with 1% β-mercaptoethanol (Thermo Fisher Scientific, catalog no. 21-985-023) and added into a 96-well Eppendorf twin-tec barcoded plate provided by the Broad Institute (Cambridge, MA). Modified SmartSeq2 complementary DNA and Illumina Nextera XT library construction and sequencing were conducted at the Broad Institute using the Illumina NextSeq 500 System. The quality of FASTQ files was examined by using FastQC-0.11.5. To estimate the proportion of human and mouse sequences, the FASTQ files were analyzed by using FastQ Screen-0.14.0 (55). Any sequences specifically matched to the mouse genome were removed from PDX samples using FastQ Screen with filter 03 inverse options, where 0 denotes “Read does not map” with respect to human, 3 means “Read maps (one or more times)” with respect to the mouse, and inverse option indicates complementary sets. The trimmed sequences were then aligned to the human genome/GRCh38 and transcriptome/ensGene98 using STAR-2.7.0e RNA sequence aligner (56). The quality of the alignment was examined with Qualimap-2.2.1 (57) and NOISeq-2.28.0 (58). Sequences located at transcripts were quantified by using RSEM-1.3.1 (59). Differentially expressed genes were analyzed using DESEq2-1.24.0 (60). Only protein-coding RNAs were included for further analyses. Pathway enrichment analysis of genes used Gene Ontology library. The heatmap was generated using Heatmapper with default settings (www.heatmapper.ca/expression/). Original data are available at the National Center for Biotechnology Information Gene Expression Omnibus, accession number: GSE153399.
TCR sequencing and analysis
DNA isolation from OCT-frozen blocks was performed using the Qiagen DNeasy Blood and Tissue Kit (Qiagen, catalog no. 69504) according to the manufacturer’s instructions. Briefly, tissue was lysed, DNA was bound to a spin column using chaotropic salts and isopropanol, the column was washed to remove impurities, and then the DNA was eluted from the column. A NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE, catalog no. ND-1000) was used to determine DNA quality and concentration. Adaptive biotechnologies (Seattle, WA) performed high-throughput sequencing of the provided DNA, produced the raw data, and generated the XY scatter plot and differential abundance graphs in fig. S6 (B and E). The remaining graphs were generated using MATLAB (MathWorks, Natick, MA).
Histology
For paraffin-embedded tissues, sections were cut at 4 μm and baked at 60°C for 1 hour. Slides were dewaxed in xylenes and then rehydrated using 100/90/70% ethanol series, followed by either PBS or tris-buffered saline (TBS) solutions, at the recommendation of the manufacturer’s instructions. For immunofluorescence studies, sections were permeated with 0.2% Triton X-100 (Thermo Fisher Scientific, catalog no. BP151) in 1× PBS or TBS for 5 min. Antigen retrieval was performed using a Cuisinart pressure cooker for 20 min at high pressure in antigen unmasking solution (Vector Laboratories, Burlingame, CA, catalog no. H-3300). Slides were washed three times 3 min each in 1× PBS or TBS supplemented with 0.1% Tween 20 (Sigma-Aldrich, catalog no. P1379). Nonspecific protein binding was blocked with 5% bovine serum albumin (Thermo Fisher Scientific, catalog no. BP1600) and 5% goat serum (Sigma-Aldrich, catalog no. G9023) in 1× PBS or TBS. Sections were stained overnight at 4°C with primary antibodies (table S2) diluted in the blocking buffer. For stains that required signals to be increased or had an overlap in primary antibody species, the Alexa Fluor 594 Tyramide SuperBoost Kit, goat anti-rabbit IgG (Thermo Fisher Scientific, catalog no. B40944) was used according to the manufacturer’s instructions, followed by blocking and primary incubation as above. Following primary antibody application, slides were washed as above and incubated in secondary antibodies (table S2) diluted in blocking buffer for 2 hours at RT. Slides were washed as above and stained with a 1:4000 dilution of 4′,6-diamidino-2-phenylindole (Invitrogen, catalog no. D3571) in 1× PBS or TBS for 10 min at RT. Slides were washed as above and mounted with ProLong Gold Antifade Reagent (Invitrogen, catalog no. P36930). Ten randomly selected fields of view at ×200-magnification (high-power field) were obtained for each section using a Zeiss Axio Scan (Zeiss, Oberkochen, Germany). Manual counting was performed using the ZEN Blue Software (Zeiss). Slides stained for hematoxylin and eosin were cut and rehydrated as described above, stained according to standard procedures, and mounted with Cytoseal XYL (Thermo Fisher Scientific, catalog no. 8312-4). Mast cell stains were performed using Toluidine Blue O (Sigma-Aldrich, catalog no. T3260-25 g) at pH 2.0 for 2.5 min. Slides were washed in water three times, dehydrated with xylene, and mounted with Cytoseal XYL. Immunohistochemistry (IHC) staining was performed using a Ventana Ultra automated immunostainer (Ventana Medical Systems, Tucson, AZ; table S2) according to the manufacturer’s instructions. Whole-slide imaging was performed using a Zeiss Axio Scan.Z1 and NanoZoomer S60 Digital slide scanner (Hamamatsu, Japan) and analyzed with NDP-view2 software (Hamamatsu). For IHC slides, HALO image analysis (Indica Labs, Albuquerque, NM) was also used to count cells. This was accomplished by creating user-modified counting algorithms that were built off positive cells defined by manually counted images.
RNA in situ hybridization
RNA in situ hybridization was performed on PFA-fixed paraffin-embedded tissue sections using the RNAscope 2.5 HD detection reagent protocol (Advanced Cell Diagnostics, Newark, CA) with accommodation to simultaneously stain for CD3 and CD4 proteins. The 4-μm sections were baked at 60°C for 30 min. Slides were treated with xylene, followed by 100% ethanol, and allowed to dry. Slides were treated with hydrogen peroxide at RT for 10 min and then washed with deionized (DI) water. Samples were treated with RNAscope 1× Target Retrieval Reagent (Advanced Cell Diagnostics, catalog no. 322000) at 102°C for 15 min and then washed. Slides were incubated with deoxyribonuclease I (bovine, Sigma-Aldrich, catalog no. D5319-500UG), to remove DNA fragments, at 37°C for 30 min in a HybEZ Oven II (Advanced Cell Diagnostics, catalog no. 321720) and then washed. RNAscope Protease Plus (Advanced Cell Diagnostics, catalog no. 322331) treatment was applied at 40°C for 30 min and then washed. After target probe amplification and hybridization steps, sections were stained with Fast RED reagent (RNAscope 2.5 HD Detection Reagents–RED, Advanced Cell Diagnostics, catalog no. 322360). Slides were washed with DI water and then PBST [18 g of NaCl and 3 ml of Triton X-100 (Thermo Fisher Scientific, catalog no. BP151-500) to 1 liter of 0.2 M PBS and 1 liter of distilled water]. Slides were blocked with 5% serum in PBST for 1 hour at RT. Tissue sections were then stained according to the immunofluorescence protocol described above.
PDX treatments
Cyclosporine (BioVision, Milpitas, CA, catalog no. 1522-1G), dosed at 0.75 mg suspended in 10 μl of dimethyl sulfoxide (Corning, catalog no. 25-950-CQC) and 90 μl of olive oil, was injected subcutaneously at the base of the mouse neck. The protocol was adapted from the publication of Klaus and Kunkl (25), but the dosing was halved to reduce the mouse fatality rate. Mice were treated with 250 μg of L243 (pan–HLA-DR, mouse IgG2a) or isotype control (mouse IgG2a, BioXCell, catalog no. BE0085) antibody in 100 μl of PBS biweekly by intraperitoneal injection (31). L243 (pan–HLA-DR) was a gift from J. Moon, Massachusetts General Hospital/Harvard Medical School, Boston, MA, USA. Mice receiving IL-2 (BioLegend, catalog no. 589104) treatment were administered 80,000 U/kg suspended in PBS by intraperitoneal injection. Mice were treated with UVB (302 to 312 nm) (100 mJ/cm2) three times weekly for up to 42 weeks using a UVP Blak-Ray Lamp UVB (VWR, Radnor, PA, catalog no. 36575-052) to simulate 25 to 50 min of sun exposure in Florida at midday in the summer (61). Mice receiving basiliximab were administered a single injection of 50 μg suspended in 100 μl of PBS intradermally and were monitored for at least 90 days after injection.
SKMEL30 cell culture and mouse injection
SKMEL30 cells were cultured in RPMI 1640 media, supplemented with 20 mM glutamine, 10% FBS, and 1% penicillin-streptomycin at 37°C and 5% CO2. Cells were passaged with 0.25% (w/v) trypsin–0.53 mM EDTA solution (Sigma-Aldrich, catalog no. T4049). The cell line was a gift from D. Fisher, Massachusetts General Hospital/Harvard Medical School, Boston, MA, USA. A total of 1 × 106 SKMEL30 cells suspended in 100 μl of RPMI 1640 were injected subcutaneously into nevus, skin grafts, or into the dorsal skin of NSG mice with no graft.
DRB1*04 typing
PDX DNA isolation, concentration, and purity determination were performed as above. DRB1*04 typing experiments were performed according to the manufacturer’s instructions of Olerup SSP (CareDx, Brisbane, CA, catalog no. 101.114-03u). Briefly, DNA, master mix, and Klentaq LA (DNA Polymerase Technologies, St. Louis, MO, catalog no. 110) were added to appropriate primer cocktails. The reaction mix was placed on a thermocycler; samples were run on 1% agarose (Denville Scientific, Metuchen, NJ, catalog no. GR140-500) gels and then imaged. The kit typed for all phenotypically different DRB1*04 alleles, DRB1*04:01-DRB1*04:241, using a total of 47 reactions and 1 control reaction. It is important to note that a positive band could indicate other DRB alleles, such as DRB1*14:57. Therefore, the data are analyzed in Olerup software to confirm DRB1*04 status.
Statistical analysis
Graphs show means ± SD. The number of mice per group used in each experiment is annotated in the corresponding figure legends. Graphs and statistical analyses were performed using GraphPad Prism 8 (Prism, La Jolla, CA), Visual Basic Analysis (Microsoft, Redmond, WA), or MATLAB (MathWorks). All tumor quantifications and cell counts were performed blindly. A log-rank test was used for all Kaplan-Meier graphs. Nested t tests were used for immune cell counts. A paired t test was used for protein, TCR sequencing–related experiments and % gp100+ melanocytes comparisons. An unpaired t test was used for comparison of participants with or without nevi. A Mann-Whitney U test was used for analysis of patient demographic information “age” and “size” variables as well as for tetramer- and SKMEL30-related experiments. Pearson’s χ2 tests are used for remaining patient demographic categorical variables. A P value of less than 0.05 was considered significant.
Acknowledgments
We thank D. E. Fisher for providing SKMEL30 cells. We thank T. Cunningham and K. Ngo for technical assistance. Funding: S.D. holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund. E.B.S., S.Y.C., H.G.S., J.L.M., and S.D. were supported by grants from the Burroughs Wellcome Fund, Sidney Kimmel Foundation, and NIH (DP5OD021353 and R01AR076013). Author contributions: S.D. conceived the study. K.T.F., R.J.S., and S.D. designed the clinical study. E.B.S. and S.D. designed animal experiments. D.P.L., J.V.C., K.T.F., R.J.S., and S.D. recruited participants and conducted the clinical study. E.B.S., S.Y.C., S.H., H.G.S., and J.L.M. performed the experiments and analyzed the data. J.J.M. provided expertise and reagents for human CD4+ T cell suppression studies. C.G.L. provided expertise on nevi pathology and contributed regressing nevi samples. E.B.S. and S.D. interpreted the data and wrote the manuscript. Competing interests: R.J.S. received research funding from Merck and Amgen. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg4498/DC1
REFERENCES AND NOTES
- 1.Bakker A. B., Schreurs M. W., de Boer A. J., Kawakami Y., Rosenberg S. A., Adema G. J., Figdor C. G., Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J. Exp. Med. 179, 1005–1009 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ott P. A., Hu Z., Keskin D. B., Shukla S. A., Sun J., Bozym D. J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., Chen C., Olive O., Carter T. A., Li S., Lieb D. J., Eisenhaure T., Gjini E., Stevens J., Lane W. J., Javeri I., Nellaiappan K., Salazar A. M., Daley H., Seaman M., Buchbinder E. I., Yoon C. H., Harden M., Lennon N., Gabriel S., Rodig S. J., Barouch D. H., Aster J. C., Getz G., Wucherpfennig K., Neuberg D., Ritz J., Lander E. S., Fritsch E. F., Hacohen N., Wu C. J., An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin J. Y., Fisher D. E., Melanocyte biology and skin pigmentation. Nature 445, 843–850 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Damsky W. E., Bosenberg M., Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 36, 5771–5792 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shain A. H., Yeh I., Kovalyshyn I., Sriharan A., Talevich E., Gagnon A., Dummer R., North J., Pincus L., Ruben B., Rickaby W., D’Arrigo C., Robson A., Bastian B. C., The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 373, 1926–1936 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Michaloglou C., Vredeveld L. C., Soengas M. S., Denoyelle C., Kuilman T., van der Horst C. M., Majoor D. M., Shay J. W., Mooi W. J., Peeper D. S., BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720–724 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Liu X.-L., Ding J., Meng L.-h., Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 39, 1553–1558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhomen N., Reis-Filho J. S., da Rocha Dias S., Hayward R., Savage K., Delmas V., Larue L., Pritchard C., Marais R., Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell 15, 294–303 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Courtois-Cox S., Jones S. L., Cichowski K., Many roads lead to oncogene-induced senescence. Oncogene 27, 2801–2809 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Tsao H., Bevona C., Goggins W., Quinn T., The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: A population-based estimate. Arch. Dermatol. 139, 282–288 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Shitara D., Tell-Martí G., Badenas C., Enokihara M. M. S. S., Alós L., Larque A. B., Michalany N., Puig-Butille J. A., Carrera C., Malvehy J., Puig S., Bagatin E., Mutational status of naevus-associated melanomas. Br. J. Dermatol. 173, 671–680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stark M. S., Tan J.-M., Tom L., Jagirdar K., Lambie D., Schaider H., Soyer H. P., Sturm R. A., Whole-exome sequencing of acquired nevi identifies mechanisms for development and maintenance of benign neoplasms. J. Invest. Dermatol. 138, 1636–1644 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Wilmott J. S., Wilmott J. S., Johansson P. A., Newell F., Waddell N., Ferguson P., Quek C., Patch A.-M., Nones K., Shang P., Pritchard A. L., Kazakoff S., Holmes O., Leonard C., Wood S., Xu Q., Saw R. P. M., Spillane A. J., Stretch J. R., Shannon K. F., Kefford R. F., Menzies A. M., Long G. V., Thompson J. F., Pearson J. V., Mann G. J., Hayward N. K., Scolyer R. A., Whole genome sequencing of melanomas in adolescent and young adults reveals distinct mutation landscapes and the potential role of germline variants in disease susceptibility. Int. J. Cancer 144, 1049–1060 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Zarour H. M., Kirkwood J. M., Kierstead L. S., Herr W., Brusic V., Slingluff C. L. Jr., Sidney J., Sette A., Storkus W. J., Melan-A/MART-151-73 represents an immunogenic HLA-DR4-restricted epitope recognized by melanoma-reactive CD4+T cells. Proc. Natl. Acad. Sci. U.S.A. 97, 400–405 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shain A. H., Joseph N. M., Yu R., Benhamida J., Liu S., Prow T., Ruben B., North J., Pincus L., Yeh I., Judson R., Bastian B. C., Genomic and transcriptomic analysis reveals incremental disruption of key signaling pathways during melanoma evolution. Cancer Cell 34, 45–55.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayward N. K., Wilmott J. S., Waddell N., Johansson P. A., Field M. A., Nones K., Patch A.-M., Kakavand H., Alexandrov L. B., Burke H., Jakrot V., Kazakoff S., Holmes O., Leonard C., Sabarinathan R., Mularoni L., Wood S., Xu Q., Waddell N., Tembe V., Pupo G. M., De Paoli-Iseppi R., Vilain R. E., Shang P., Lau L. M. S., Dagg R. A., Schramm S.-J., Pritchard A., Dutton-Regester K., Newell F., Fitzgerald A., Shang C. A., Grimmond S. M., Pickett H. A., Yang J. Y., Stretch J. R., Behren A., Kefford R. F., Hersey P., Long G. V., Cebon J., Shackleton M., Spillane A. J., Saw R. P. M., López-Bigas N., Pearson J. V., Thompson J. F., Scolyer R. A., Mann G. J., Whole-genome landscapes of major melanoma subtypes. Nature 545, 175–180 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Speeckaert R., van Geel N., Luiten R. M., van Gele M., Speeckaert M., Lambert J., Vermaelen K., Tjin E. P. M., Brochez L., Melanocyte-specific immune response in a patient with multiple regressing nevi and a history of melanoma. Anticancer Res 31, 3697–3703 (2011). [PubMed] [Google Scholar]
- 18.Yan B. Y., Garcet S., Gulati N., Kiecker F., Fuentes-Duculan J., Gilleaudeau P., Sullivan-Whalen M., Shemer A., Mitsui H., Krueger J. G., Novel immune signatures associated with dysplastic naevi and primary cutaneous melanoma in human skin. Exp. Dermatol. 28, 35–44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitsui H., Kiecker F., Shemer A., Cannizzaro M. V., Wang C. Q. F., Gulati N., Ohmatsu H., Shah K. R., Gilleaudeau P., Sullivan-Whalen M., Cueto I., McNutt N. S., Suárez-Fariñas M., Krueger J. G., Discrimination of dysplastic nevi from common melanocytic nevi by cellular and molecular criteria. J. Invest. Dermatol. 136, 2030–2040 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Riding R. L., Harris J. E., The role of memory CD8+T cells in vitiligo. J. Immunol. 203, 11–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeff R. A., Freitag A., Grin C. M., Grant-Kels J. M., The immune response in halo nevi. J. Am. Acad. Dermatol. 37, 620–624 (1997). [DOI] [PubMed] [Google Scholar]
- 22.MacKie R. M., English J., Aitchison T. C., Fitzsimons C. P., Wilson P., The number and distribution of benign pigmented moles (melanocytic naevi) in a healthy British population. Br. J. Dermatol. 113, 167–174 (1985). [DOI] [PubMed] [Google Scholar]
- 23.Martin J. M., Pinazo I., Jordá E., Monteagudo C., Differential clinicopathological features in spontaneous regression of melanomas and melanocytic naevi. Acta Derm. Venereol. 97, 692–697 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Watanabe R., Gehad A., Yang C., Scott L. L., Teague J. E., Schlapbach C., Elco C. P., Huang V., Matos T. R., Kupper T. S., Clark R. A., Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med. 7, 279ra239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klaus G. G., Kunkl A., Effects of cyclosporine on the immune system of the mouse. II. Cyclosporine inhibits the effector function of primary T helper cells, but not helper cell priming. Transplantation 36, 80–84 (1983). [DOI] [PubMed] [Google Scholar]
- 26.Gad S. C., Cassidy C. D., Aubert N., Spainhour B., Robbe H., Nonclinical vehicle use in studies by multiple routes in multiple species. Int. J. Toxicol. 25, 499–521 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Goettel J. A., Kotlarz D., Emani R., Canavan J. B., Konnikova L., Illig D., Frei S. M., Field M., Kowalik M., Peng K., Gringauz J., Mitsialis V., Wall S. M., Tsou A., Griffith A. E., Huang Y., Friedman J. R., Towne J. E., Plevy S. E., O’Hara Hall A., Snapper S. B., Low-dose interleukin-2 ameliorates colitis in a preclinical humanized mouse model. Cell. Mol. Gastroenterol. Hepatol. 8, 193–195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klatzmann D., Abbas A. K., The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 15, 283–294 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Okita R., Yamaguchi Y., Ohara M., Hironaka K., Okawaki M., Nagamine I., Ikeda T., Emi A., Hihara J., Okada M., Targeting of CD4+CD25high cells while preserving CD4+CD25low cells with low-dose chimeric anti-CD25 antibody in adoptive immunotherapy of cancer. Int. J. Oncol. 34, 563–572 (2009). [PubMed] [Google Scholar]
- 30.Jagger A., Shimojima Y., Goronzy J. J., Weyand C. M., Regulatory T cells and the immune aging process: A mini-review. Gerontology 60, 130–137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Besneux M., Greenshields-Watson A., Scurr M. J., MacLachlan B. J., Christian A., Davies M. M., Hargest R., Phillips S., Godkin A., Gallimore A., The nature of the human T cell response to the cancer antigen 5T4 is determined by the balance of regulatory and inflammatory T cells of the same antigen-specificity: Implications for vaccine design. Cancer Immunol. Immunother. 68, 247–256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gehring A. J., Rojas R. E., Canaday D. H., Lakey D. L., Harding C. V., Boom W. H., The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and FcγR1 on human macrophages through Toll-like receptor 2. Infect. Immun. 71, 4487–4497 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Racle J., Michaux J., Rockinger G. A., Arnaud M., Bobisse S., Chong C., Guillaume P., Coukos G., Harari A., Jandus C., Bassani-Sternberg M., Gfeller D., Robust prediction of HLA class II epitopes by deep motif deconvolution of immunopeptidomes. Nat. Biotechnol. 37, 1283–1286 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Emerson R. O., DeWitt W. S., Vignali M., Gravley J., Hu J. K., Osborne E. J., Desmarais C., Klinger M., Carlson C. S., Hansen J. A., Rieder M., Robins H. S., Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat. Genet. 49, 659–665 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Su L. F., Kidd B. A., Han A., Kotzin J. J., Davis M. M., Virus-specific CD4+ memory-phenotype T cells are abundant in unexposed adults. Immunity 38, 373–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagner S. N., Wagner C., Schultewolter T., Goos M., Analysis of Pmel17/gp100 expression in primary human tissue specimens: Implications for melanoma immuno- and gene-therapy. Cancer Immunol. Immunother. 44, 239–247 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smoller B. R., McNutt N. S., Hsu A., HMB-45 recognizes stimulated melanocytes. J. Cutan. Pathol. 16, 49–53 (1989). [DOI] [PubMed] [Google Scholar]
- 38.Joshi S. S., Jiang S., Unni E., Goding S. R., Fan T., Antony P. A., Hornyak T. J., 17-AAG inhibits vemurafenib-associated MAP kinase activation and is synergistic with cellular immunotherapy in a murine melanoma model. PLOS ONE 13, e0191264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein L., Robey E. A., Hsieh C.-S., Central CD4+T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 19, 7–18 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Mueller D. L., Mechanisms maintaining peripheral tolerance. Nat. Immunol. 11, 21–27 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Walker L. S. K., Abbas A. K., The enemy within: Keeping self-reactive T cells at bay in the periphery. Nat. Rev. Immunol. 2, 11–19 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Legoux F. P., Lim J. B., Cauley A. W., Dikiy S., Ertelt J., Mariani T. J., Sparwasser T., Way S. S., Moon J. J., CD4+ T cell tolerance to tissue-restricted self antigens is mediated by antigen-specific regulatory T cells rather than deletion. Immunity 43, 896–908 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boroughs A. C., Larson R. C., Choi B. D., Bouffard A. A., Riley L. S., Schiferle E., Kulkarni A. S., Cetrulo C. L., Ting D., Blazar B. R., Demehri S., Maus M. V., Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight 5, e126194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Waldman A. D., Fritz J. M., Lenardo M. J., A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 20, 651–668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tay R. E., Richardson E. K., Toh H. C., Revisiting the role of CD4+T cells in cancer immunotherapy––New insights into old paradigms. Cancer Gene Ther. 28, 5–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borst J., Ahrends T., Bąbała N., Melief C. J. M., Kastenmüller W., CD4+T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18, 635–647 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Alspach E., Lussier D. M., Miceli A. P., Kizhvatov I., DuPage M., Luoma A. M., Meng W., Lichti C. F., Esaulova E., Vomund A. N., Runci D., Ward J. P., Gubin M. M., Medrano R. F. V., Arthur C. D., White J. M., Sheehan K. C. F., Chen A., Wucherpfennig K. W., Jacks T., Unanue E. R., Artyomov M. N., Schreiber R. D., MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh D. Y., Kwek S. S., Raju S. S., Li T., McCarthy E., Chow E., Aran D., Ilano A., Pai C.-C. S., Rancan C., Allaire K., Burra A., Sun Y., Spitzer M. H., Mangul S., Porten S., Meng M. V., Friedlander T. W., Ye C. J., Fong L., Intratumoral CD4+T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 181, 1612–1625.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kennedy R., Celis E., Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol. Rev. 222, 129–144 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Braumüller H., Wieder T., Brenner E., Aßmann S., Hahn M., Alkhaled M., Schilbach K., Essmann F., Kneilling M., Griessinger C., Ranta F., Ullrich S., Mocikat R., Braungart K., Mehra T., Fehrenbacher B., Berdel J., Niessner H., Meier F., van den Broek M., Häring H.-U., Handgretinger R., Quintanilla-Martinez L., Fend F., Pesic M., Bauer J., Zender L., Schaller M., Schulze-Osthoff K., Röcken M., T-helper-1-cell cytokines drive cancer into senescence. Nature 494, 361–365 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Wang W., Green M., Choi J. E., Gijón M., Kennedy P. D., Johnson J. K., Liao P., Lang X., Kryczek I., Sell A., Xia H., Zhou J., Li G., Li J., Li W., Wei S., Vatan L., Zhang H., Szeliga W., Gu W., Liu R., Lawrence T. S., Lamb C., Tanno Y., Cieslik M., Stone E., Georgiou G., Chan T. A., Chinnaiyan A., Zou W., CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su Q., Wang F., Dong Z., Chen M., Cao R., IFN‑γ induces apoptosis in human melanocytes by activating the JAK1/STAT1 signaling pathway. Mol. Med. Rep. 22, 3111–3116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hahn S., Gehri R., Erb P., Mechanism and biological significance of CD4-mediated cytotoxicity. Immunol. Rev. 146, 57–79 (1995). [DOI] [PubMed] [Google Scholar]
- 54.Salimi M., Subramaniam S., Selvakumar T., Wang X., Zemenides S., Johnson D., Ogg G., Enhanced isolation of lymphoid cells from human skin. Clin. Exp. Dermatol. 41, 552–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wingett S. W., Andrews S., FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 7, 1338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Okonechnikov K., Conesa A., García-Alcalde F., Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tarazona S., Furió-Tarí P., Turrà D., Pietro A. D., Nueda M. J., Ferrer A., Conesa A., Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 43, e140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D’Orazio J. A., Nobuhisa T., Cui R., Arya M., Spry M., Wakamatsu K., Igras V., Kunisada T., Granter S. R., Nishimura E. K., Ito S., Fisher D. E., Topical drug rescue strategy and skin protection based on the role of Mc1r in UV-induced tanning. Nature 443, 340–344 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabg4498/DC1