

Pseudorabies virus interacts with active regions in host genome to efficiently transcribe viral genes.
Abstract
Like most DNA viruses, herpesviruses precisely deliver their genomes into the sophisticatedly organized nuclei of the infected host cells to initiate subsequent transcription and replication. However, it remains elusive how the viral genome specifically interacts with the host genome and hijacks host transcription machinery. Using pseudorabies virus (PRV) as model virus, we performed chromosome conformation capture assays to demonstrate a genome-wide specific trans-species chromatin interaction between the virus and host. Our data show that the PRV genome is delivered by the host DNA binding protein RUNX1 into the open chromatin and active transcription zone. This facilitates virus hijacking host RNAPII to efficiently transcribe viral genes, which is significantly inhibited by either a RUNX1 inhibitor or RNA interference. Together, these findings provide insights into the chromatin interaction between viral and host genomes and identify new areas of research to advance the understanding of herpesvirus genome transcription.
INTRODUCTION
Mammalian cell nuclei are elegantly occupied with different chromosomes in discrete territories for spatially and temporally coordinated gene expression (1). Most eukaryotic DNA viruses, such as herpesvirus can precisely deliver their genomes into the cell nuclei and hijack the host machinery for the maintenance, replication, and expression of their genome (2, 3). Herpesvirus represents a large virus family that is capable of infecting a wide range of vertebrate hosts, including human. For example, herpes simplex virus (HSV) infects up to 85% of the human population (4), and Epstein-Barr virus (EBV) can be oncogenic in human (5). Herpesviruses have double-stranded DNA, which is maintained as a chromatinized episome in the host cell nucleus (6). The chromosome conformation capture (3C) techniques have revealed the remote chromatin interaction between different chromosomal sites in the nucleus (1). Emerging evidence suggests that the viral episome can also take part in this transchromosome interaction (7–11).
It has been reported that hepatitis B virus and adenovirus type-5 (Ad5) target transcription start sites and CpG islands regions, which may provide an environment propitious to their own replication and transcription (12). Whereas, EBV virus preferentially associates with Epstein-Barr nuclear antigen 1 (EBNA1) binding genomic regions enriched with repressive histone mark H3K9me3, which is functionally linked to viral latency (13). The genome of EBV can move from repressive heterochromatin region to active euchromatin region upon reactivation from latency (14). However, it is largely unknown how DNA viral genome specifically interacts with the host genome and hijacks host transcription machinery.
Pseudorabies virus (PRV), an alphaherpesvirus of swine, has been used as a model virus to study alphaherpesvirus in all aspects of molecular biology, pathogenesis, and neuron virulence (15, 16). Herpesvirus genes are transcribed by host RNA polymerase II (RNAPII) in a temporally coordinated order of immediate early (IE), early (E or β), and late (L or γ) genes (17). However, the underlying mechanism of how herpesviruses recruit host RNAPII to transcribe their genome remains elusive. Here, we used PRV as a model virus to explore trans-species chromatin interaction between the virus and host and elucidate the processes underlying recruitment of host transcription machinery.
RESULTS AND DISCUSSION
Detection of the direct chromatin interaction between PRV and host cells
First, we performed a multiple circular 3C (4C) assay in PRV-infected PK15 cells, a porcine kidney cell line. After cross-linking with formaldehyde to fix the in situ chromatin complex structure, the chromatin complex was subjected to Bam HI digestion, proximity ligation, Alu I digestion, and self-ligation (Fig. 1A). Thirty-two pairs of primers were designed on the loci of the PRV genome flanking Bam HI and Alu I enzyme sites and used for polymerase chain reaction (PCR) amplification of the ligated chromatins independently, of which the products were pooled together for 4C library construction (fig. S1 and table S1). With this approach, the chimeric DNA fragments coupling the genetic information from PRV and host cell genomes could be used to decipher the chromatin interaction between PRV and the host cell. Sanger sequencing of the 4C fragment demonstrated that the chimeric DNA fragments were indeed coupled with genetic information from both PRV and host genomes (fig. S2A).
Fig. 1. Detection of chromatin interaction between PRV and host cell.
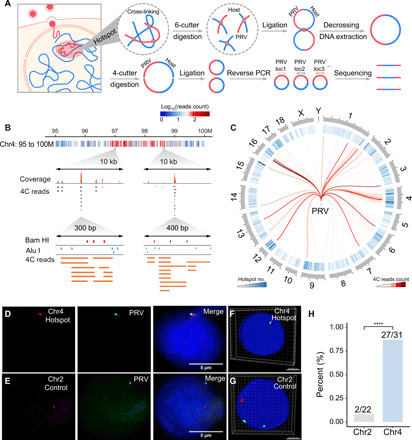
(A) Schematic illustration of multiple 4C procedure, including attachment, penetration, and chromatin interaction between PRV and the host cell. The chromatin complexes were cross-linked and then digested by Bam HI. After proximity ligation and DNA purification, the DNA fragments were digested by Alu I and then self-ligated for reverse PCR, of which the products were subjected to high-throughput sequencing. (B) Distribution of PRV-host 4C reads in the 95 to 100M region on chromosome 4. Red dots represent Bam HI sites, and blue dots represent Alu I sites. (C) Circos plot of high frequent PRV-host interaction hotspots distributed on each chromosome. (D) DNA hybridization chain reaction (HCR) validation of the colocalization between PRV and hotspot region in Chr4. (E) No colocalization was observed between PRV and noninteracting regions in Chr2. (F and G) Three-dimensional (3D) reconstruction of DNA HCR images of the interaction site (Chr4, hotspot) and noninteraction site (Chr2, control). Green, PRV signals; red, host genome site signal. (H) Quantification of the colocalization ratio of the interaction sites (Chr4, hotspot) and the noninteraction sites (Chr2, control). ****P < 0.0001 from Fisher’s exact test.
Next, the 4C PCR products were subjected to high-throughput sequencing and analyzed by a custom script (fig. S2B). A total of 838,863 uniquely mapping reads were obtained and allocated to the host genome (fig. S2C). The hotspot interaction sites in the 95 to 100M region of chromosome 4 were demonstrated with different resolutions. At 1-kb resolution, we observed a cluster of stacked fragments with Bam HI and Alu I sites as an interaction hotspot (Fig. 1B). Next, the adjacent 4C reads were combined as clusters. Figure S2D shows that clusters with read counts less than 6 were too short in span and probably were noise without sufficient recurrence. Then, the clusters with read counts equal to or greater than 6 were retained for further analysis. Figure 1C shows the genome-wide cluster distribution with their contact frequency.
Validation of virus-host chromatin interactions
To validate the 4C result, we performed another independent multi-4C experiment for enriched interaction sites on the PRV genome. As shown in fig. S2E, 93 to 100% of the virus-host interaction (VHI) sites with high-frequency 4C read counts (6 or more) overlapped between two replicates. Next, we used 3C-PCR to further validate the VHI sites using the primers from the 4C paired-end reads sequences, with one side from the PRV genome and another side from the host genome. Figure S2F demonstrated that 11 of 13 randomly selected VHI sites (84.6%) from the 4C assay could be validated by 3C-PCR, indicating the integrity of the 4C data and the specificity of VHI.
To further confirm the specificity of VHI, one high frequent “hotspot” interaction site in chromosome 4 and one negative control site without VHI were selected for in situ DNA hybridization chain reaction (HCR) assay. As shown in Fig. 1D, green fluorescence dots representing the PRV genome were colocalized with red hotspot interaction sites in the genome of PK15 cells (87.1%), while such colocalization was hardly (9.1%) observed in the negative control sites (Fig. 1E). We also reconstructed three-dimensional (3D) images of the interaction and noninteraction sites, which further supported spatial colocalization of the PRV genome and hotspot sites in the host genome (Fig. 1, F and G). Notably, this colocalization was observed in 27 of 31 cells examined (Fig. 1H). Together, the 3C, 4C, and in situ HCR results unambiguously demonstrate that PRV specifically interacts with the host cell genome.
PRV preferentially interacts with the open chromatin regions in the host genome
The mammal genome is folded into a highly compacted structure with a portion of the genome maintaining an open and loose chromatin state (18). It is conceivable that VHI might tend to occur in the accessible open and loose regions in the genome. To test this hypothesis, we performed assay for transposase-accessible chromatin using sequencing (ATAC-seq) on PRV-infected PK15 cells. The 2D density plot demonstrated a strong positive correlation between ATAC-seq peak and VHI 4C read counts (Fig. 2A and fig. S3A). Next, the average chromatin accessibility at VHI hotspots ± 50 kb was calculated, which indicated that the accessibility of genome regions in the interaction hotspots was significantly higher than that of the regions outside the hotspots (fig. S3B). Our ATAC-seq data showed that the open regions of the host genome remained largely unaltered upon PRV infection (fig. S3B), indicating that the PRV preferentially interacted swine genome regions were mostly pre-open.
Fig. 2. PRV preferentially interacts with open and active chromatin regions in the swine genome.
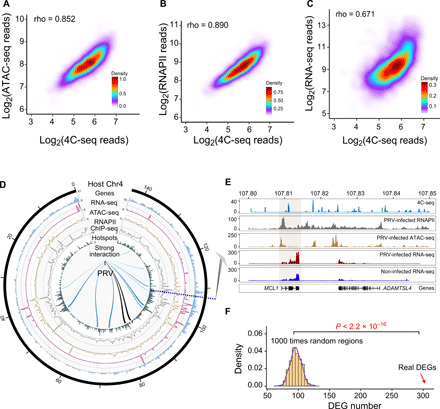
(A) Density plot demonstrating the positive correlation between ATAC-seq reads and PRV-host 4C reads in PRV-infected cells. (B) Density plot showing the positive correlation between RNAPII ChIP-seq reads and PRV-host 4C reads. (C) Density plot showing the positive correlation between RNA sequencing (RNA-seq) reads and PRV-host 4C reads. (D) Circos plot of the integrated 4C-seq data, ATAC-seq, RNAPII ChIP-seq, and RNA-seq data on chromosome 4. (E) Integrated sequencing tracks for the 4C-seq data, ATAC-seq, RNAPII ChIP-seq, and RNA-seq data of the MCL1 gene. (F) Number of differentially expressed genes (DEGs) related to the random regions and interaction regions, respectively, indicating that DEGs are enriched in the interaction hotspots. P < 2.2 × 10−16 from the Student’s t test.
As the open regions in the genome are often actively associated with transcription protein complexes, we hypothesized that PRV delivers its genome to these regions to hijack the host active transcription machinery to transcribe viral genes in an efficient and timely manner. In this scenario, it would be expected that the VHI 4C reads frequency should be positively correlated to the enrichment of RNAPII in the interaction sites. To ascertain whether this was the case, we performed RNAPII chromatin immunoprecipitation sequencing (ChIP-seq) experiments in PK15 cells infected with PRV. As shown in Fig. 2B and fig. S3C, there was a positive correlation between RNAPII ChIP read counts and VHI 4C read counts. Analysis of the average RNAPII enrichment around PRV-host interaction hotspots ± 50 kb demonstrated that the RNAPII was highly enriched in the interaction hotspots, and the RNAPII enrichment did not change in the host genome after the runt related transcription factor 1 (RUNX1) interference (fig. S3D). To further analyze the distribution of the interaction hotspots in A/B compartments, we used the Hi-C data from pig embryonic fibroblasts (mouse embryonic fibroblast) as a reference. The integrated multi-omics analysis demonstrated the VHI hotspots, ATAC-seq peaks, and RNAPII enrichment were mainly enriched in the A compartment (fig. S3E).
Next, we performed RNA sequencing (RNA-seq) to evaluate the correlation between RNA-seq read counts and VHI 4C read counts. As shown in Fig. 2C, the RNA-seq reads were positively correlated with PRV-host 4C read counts. The results of 4C, ATAC-seq, RNA-seq, and RNAPII ChIP-seq integrated multi-omics analysis demonstrated a strong correlation of VHI 4C peaks with ATAC-seq peaks, RNA-seq peaks, and RNAPII enrichment (Fig. 2D). One typical example of a PRV interaction site is illustrated in detail in Fig. 2E, indicating that PRV interacts with the myeloid cell leukemia-1 (MCL1) gene region with high levels of gene expression, RNAPII, and ATAC-seq peaks. Collectively, these data suggest that PRV has the tendency to load its genome in the open regions and in transcriptional active hubs of the host genome, potentially to efficiently use the on-site host active gene transcription machinery such as RNAPII for its own viral gene transcription. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that the differentially expressed genes (DEGs) were enriched in several pathways such as the “regulation of inflammatory response,” “GABAergic, cholinergic, and glutamatergic synapse,” etc., (fig. S3, F to H), suggesting that PRV may cause neurological symptoms and inflammation response.
As the PRV genome preferentially interacts with transcriptional active regions, we tested whether the transcription of the host genes in these loci was modulated by VHI. Thus, genes whose expression was significantly different following PRV infection were plotted to the genome regions with and without VHI. As shown in Fig. 2F, the distribution of DEGs in the VHI regions was much higher than that of the random regions, implying that the interaction of the PRV genome might influence the transcription of genes inside the interaction regions. Consistent with this observation, previous studies have identified that EBV can likely reset the host cell transcription program by remodeling chromatin architecture (19). Moreover, the DNA virus minute virus of mice established replication at cellular DNA damage sites, where enriched with replication and expression machineries (20). Human papillomavirus (HPV) E2 protein has been found associated with actively transcribing genes and active chromatin, presumably to facilitate expression of HPV through hijacking the host transcriptional machinery at these sites (21). It would be of great interest to further investigate whether this VHI could remodel the host chromatin structure and subsequently reprogram gene transcription in the corresponding regions.
DNA binding protein RUNX1 mediates the PRV-host chromatin interaction
As DNA binding proteins play crucial roles in the maintenance of the 3D genome structure (13, 17, 22), we hypothesize that VHI might also be mediated by DNA binding proteins. Thus, we performed motif analysis to screen the conserved DNA motifs in the interaction regions and the corresponding putative DNA binding proteins. As shown in Fig. 3A, we identified a conserved RCCACAGY motif enriched in the VHI regions in both host and PRV genomes. The distribution of this motif in the VHI regions was significantly higher than that in the random regions (Fig. 3B). One typical example of a PRV interaction site is presented in Fig. 3C, indicating that PRV interacts with the Centromere Protein A (CENPA) gene region with a high level of gene expression, RUNX1 motif binding site, ATAC-seq, and RNAPII peak.
Fig. 3. DNA binding protein RUNX1 mediates the PRV-host chromatin interaction.
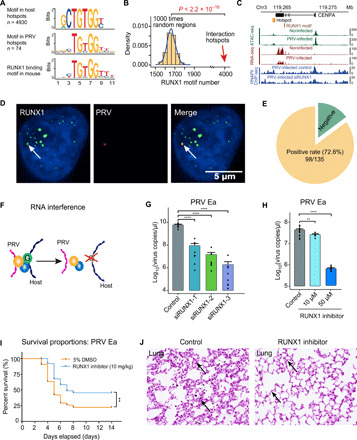
(A) Significant protein binding motifs in both PRV and host cell interaction hotspots and the potential DNA binding protein RUNX1. (B) Number of RUNX1 motifs in random regions and interaction hotspots, respectively. (C) Integrated sequencing tracks for the RUNX1 motif, 4C-seq data, ATAC-seq, RNA-seq data, and RNAPII ChIP-seq in CENPA gene locus. (D) Colocalization of the RUNX1 protein immunofluorescence signal and PRV genome DNA FISH (HCR) signal. (E) PRV genome and RUNX1 protein colocalized in 72.6% (98 of 135) of the tested cells. (F) Schematic illustration of the RUNX1 RNA interference effect on PRV-host chromatin interaction. (G) Significant decrease of PRV copy number after RUNX1 interference in PRV-infected PK15 cells. (H) PRV copy number in PRV-infected PK15 cells treated with different concentrations of RUNX1 inhibitor (Ro 5-3335). (I) Survival curves of the PRV-infected C57BL/6 mice treated with Ro 5-3335 and 5% dimethyl sulfoxide (DMSO; control); n = 42 per group. (J) Histological staining of the lung with or without the RUNX1 inhibitor Ro 5-3335 treatment after PRV Ea infection, respectively. Arrows show the thickened alveolar septum in control group. **P < 0.01 and ****P < 0.0001 from Student’s t test. Error bars represent SD from eight independent experiments.
RUNX family members are host nucleus proteins that have been shown to be involved in virus replication regulation such as HIV-1 (23). As we identified RUNX1 as the putative DNA binding protein of this motif, the localization of RUNX1 protein and the PRV genome was subsequently investigated by simultaneous immunostaining and DNA fluorescence in situ hybridization (FISH). As shown in Fig. 3D and fig. S4A, the PRV genome is indeed colocalized with RUNX1 protein in host cells. This colocalization was observed in 72.6% of tested cells (98 of 135 cells; Fig. 3E), supporting that RUNX1 represents a potential candidate protein bridging VHI. Next, we knocked down RUNX1 by RNA interference to further validate its function in VHI (Fig. 3F and fig. S4B). As shown in Fig. 3G, PRV copy number was significantly decreased upon RUNX1 knockdown. Notably, this reduction of viral titer was likely due to the direct effect of RUNX1 knockdown, as there was no notable difference in cell growth, proliferation, or morphology between RUNX1 knockdown cells and control cells (fig. S4, C and D). Moreover, the treatment of RUNX1 inhibitor Ro 5-3335 also decreased PRV copy number in PK15 cells (Fig. 3H). Notably, no substantial difference in cell growth, proliferation, or morphology was observed between RUNX1-inhibited cells and control cells (fig. S4E).
To further validate this phenomenon in vivo, we inoculated mice with PRV by the intramuscular route and treated half of them with RUNX1 inhibitor Ro 5-3335 and another half with the vehicle only. As shown in Fig. 3I, 75.7% of infected mice succumbed to infection with PRV in the vehicle control group, whereas treatment with Ro 5-3335 significantly improved the survival of PRV-infected mice. Histological analyses demonstrated an obviously thickened alveolar septum in PRV-infected mice receiving vehicle control group, while the alveolus was enlarged and distended in the Ro 5-3335–treated group (Fig. 3J and fig. S4F). Moreover, we also observed an obvious coagulative necrosis around the hepatic lobules in PRV-infected mice receiving vehicle control group, while this damage was much attenuated in the Ro 5-3335–treated group (fig. S4G).
As PRV is often used as a model virus for the family Herpesviridae (16), we explored the RUNX1 motif sequences in the genomes of different viruses in this family such as HSV, EBV, and equine herpesvirus (EHV). Through motif scanning, we found that the RUNX1 binding motif is conserved in the family Alphaherpesvirinae, especially in HSV (fig. S5A). On the basis of this observation, we speculate that RUNX1 may also affect HSV replication via a similar mechanism. Thus, we treated HSV129–green fluorescent protein (GFP) infected with human embryonic kidney (HEK) 293T and Vero cells with RUNX1 inhibitor for 6 hours and checked the florescence signal. Figure S5B demonstrates that the GFP signals were significantly decreased in the inhibitor-treated group. Next, we further checked the expression of different viral genes in different time points after RUNX1 inhibitor treatment. As shown in fig. S5C, our data showed that inhibition of RUNX1 can indeed repress HSV129 viral gene transcription and subsequent replication.
A recent study showed that RUNX1 highly expressed in dorsal root ganglion and also binds to HSV-1 genome but negatively regulates transcription of viral genes (24). It could be possible that this protein may play distinct regulatory roles in different cells and different stages such as latency stage, of which the molecular mechanism needs to further investigate. Therefore, RUNX1 might represent a general mediator of chromatin interaction between alphaherpesviruses and the host genome, and Ro 5-3335 should be further evaluated as a potential drug candidate for the treatment of alphaherpesvirus infections.
PRV hijacks host RNAPII to facilitate viral gene transcription
As the PRV genome is enriched with RUNX1 binding motif, we performed ChIP–quantitative PCR (qPCR) to validate the recruitment of RUNX1 with the PRV genome. Figure S6A showed that RUNX1 can precipitate both swine genome hotspot loci and PRV genome interaction sites. Furthermore, as shown in Fig. 4A, RUNX1 was indeed bound to the PRV genome, with enrichment in hotspots significantly higher that of the non-hotspots. To further test whether the VHI could facilitate virus hijacking host transcription machinery, we analyzed the recruitment of RNAPII with the PRV genome. Figure 4B shows that RNAPII is significantly enriched in the VHI regions of the PRV genome, among which 80.43% of the RNAPII peaks were located around the RUNX1 binding motif (<2 kb; Fig. 4C). To validate the role of RUNX1 in VHI and RNAPII recruitment, we knocked down RUNX1 and performed RNAPII ChIP-seq. As shown in Fig. 4 (D and E), RNAPII enrichment in the PRV genome was significantly reduced following RNA interference.
Fig. 4. PRV hijacks host RNAPII via RUNX1 for its transcription and replication.
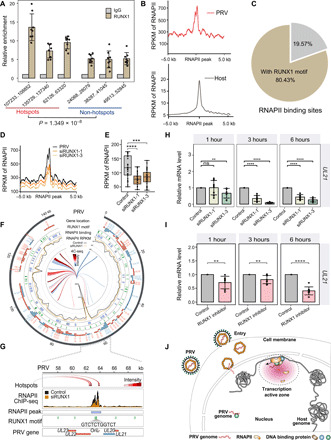
(A) RUNX1 ChIP-qPCR assay demonstrated the enrichment of RUNX1 in the hotspot and non-hotspot regions in the PRV genome [normalized with immunoglobulin G (IgG) control and input]. (B) Average read coverage of RNAPII peaks in the PRV and host genomes. RPKM, reads per kilobase per million mapped reads. (C) 80.43% of the RNAPII peaks were located around the RUNX1 binding motif in PRV genome (<2 kb). (D) Average read coverage of RNAPII peaks in the PRV genome with/without RUNX1 RNA interference. (E) Box plot of RNAPII occupancy on PRV genome with/without RUNX1 RNA interference. (F) Distribution of RNAPII binding sites, RNAPII occupancy, RUNX1 motif, and genes in the PRV genome with/without RUNX1 RNA interference. (G) Distribution of RNAPII binding sites, RNAPII occupancy, RUNX1 motifs, and genes in OriL region (around UL21 locus) of PRV genome with/without RUNX1 RNA interference. (H) Expression levels of UL21 at 1, 3, and 6 hours with/without RUNX1 RNA interference. (I) Expression levels of UL21 at 1, 3, and 6 hours following treatment with RUNX1 inhibitor Ro 5-3335. (J) Model of chromatin interaction between pseudorabies viral genome and host cell genome. ns, not significant (P > 0.05); **P < 0.01, ***P < 0.001, and ****P < 0.0001 from the Student’s t test. Error bars represent SD from eight independent experiments.
We observed marked RNAPII enrichment at RUNX1 motifs in OriL sites in the PRV genome, which were significantly reduced upon RUNX1 knockdown (Fig. 4, F and G). As OriL is the origin for viral replication, further investigation of the role of RNAPII in viral replication is warranted. It has been reported that the RUNX1 can bind to the site near the polyomavirus replication origin (25, 26). This structure is conducive to the large T antigen of the replication origin close to the replication factory. Together with our data showing that RUNX1 can bind to both host and viral genome, this evidence suggests that RUNX1 is an important protein in the complex, which may mediate the viral and host DNA chromatin interaction. It would be of great interest to further explore the detailed components and their molecular functions in this viral host complex, especially their role in viral replication.
To confirm the multi-omics data supporting a role for RUNX1 in the regulation of PRV viral gene transcription, we analyzed RNAPII enrichment around several immediate early genes, early genes, and late genes including IE180, UL54, UL18, and UL21. Figure 4G and fig. S6 (B to D) show that RUNX1 interference significantly decreased RNAPII enrichment around these genes. Next, PK15 cells with or without RUNX1 knockdown were infected with PRV for different time periods and then subjected to qPCR analysis. We found that RUNX1 interference could significantly decrease the expression of these genes at multiple time points after infection (Fig. 4H and fig. S6, E to G). This phenomenon was additionally observed in cells treated with RUNX1 inhibitors (Fig. 4I and fig. S6, H to J). To further validate the role of RUNX1 in virus-host chromatin interaction, we used Hi-C to capture all the chromatin interaction in the PK15 cells. As shown in fig. S7 (A to C), the VHIs reduced 27.8% following 6 hours of RUNX1 inhibitor treatment, while the overall chromatin interaction was unaltered, supporting the role of RUNX1 in mediating VHIs. These data support a role of RUNX1 in directing the PRV genome to transcriptional active zones, subsequently facilitating the recruitment of RNAPII and promoting viral gene transcription and replication.
Collectively, our findings demonstrate genome-wide specific trans-species chromatin interaction between viral and host genomes using PRV as a model virus. We propose a model whereby PRV exploits the host DNA binding protein RUNX1 to deliver its genome to the open chromatin and active transcription zones, facilitating the virus hijacking host transcription machinery such as RNAPII, to ultimately transcribe the viral gene in a timely and efficient manner (Fig. 4I). We show that the RUNX1 inhibitor Ro 5-3335 repressed the proliferation of the PRV in the host cells, which could represent a potential drug candidate for pseudorabies and other herpesvirus-induced diseases. These findings provide insights into the cross-talk between viral and host genomes and elucidate the molecular mechanisms of viral gene preferential transcription. Furthermore, they support new areas of research to advance our understanding of herpesvirus gene expression and contribute to further efforts to characterize the detailed parasitic viral life cycle in host cells.
METHODS
Cell lines and plasmids construction
The porcine kidney cell line PK15 (ATCCR CCL-33), HEK cells 293 (HEK293T cells, ATCCR CRL-3216), and Vero cells (ATCCR CCL81.4) were all grown in 25-cm2 culture flasks (Corning, Sigma-Aldrich) containing Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), l-glutamine (2 mM), and penicillin (100 U/ml) and streptomycin (100 U/ml) (P/S) in a 5% CO2 incubator in a humidified atmosphere at 37°C. PK15, HEK293T, and Vero cells were subcultured into new flasks containing fresh culture media thrice weekly.
Adeno-associated virus (AAV) viral packaging and preparation
Plasmids for pAAV-U6-si-scramble-CMV-mCherry and pAAV-U6-siRUNX1-CMV-mCherry were generated from pAAV-U6-siRNA-CMV-mCherry and packaged in 293T cells. 293T cells were cotransfected with pAAV-U6-siRNA-CMV-mCherry (control, siRUNX1), pDJ, and pAAV-Helper by standard polyethylenimine (PEI) transfection. Cells were harvested 72 hours after transfection, and the cell pellets were stored at −80°C. The virus preparation was performed as previously described (27). The titer of rAAV-U6-si-scramble-CMV-mCherry was 3 × 1012 viral particles/ml and rAAV-U6-siRUNX1-CMV-mCherry was approximately 2 × 1012 viral particles/ml.
Virus infection
The PRV strain PRV Ea was used in this study (a gift from Z. Liu). PRV Ea was amplified in PK15 cells. Virus-containing medium was collected in DMEM supplemented with 2% FBS and 1% P/S. The virus was collected at 72 hours postinfection (hpi). After three freeze-thaws in liquid nitrogen, the virus-containing material was clarified by centrifugation at 10,000g for 15 min at 4°C and stored at −80°C. The virus titer [50% tissue culture infectious doses (TCID50)] was determined by cytopathic effect.
The HSV129-GFP used in this study was a gift from M. Luo. HSV129-GFP was amplified in Vero cells. Virus-containing medium was collected in DMEM supplemented with 2% FBS and 1% P/S. The virus was collected at 72 hpi.
TCID50 assay
Serial 10-fold dilutions (from 10−1 to 10−10) of the original PRV Ea virus were made in DMEM. Diluted virus was inoculated in each plate well, with 100 μl of each dilution in eight wells. PK15 cell suspension (100 μl) was added into each well to achieve a final concentration of 2 × 105 cells/ml. The plate was incubated for 7 days at 37°C in CO2 (5%), and the cytopathic effect was observed and recorded daily. All titers were calculated by the method of Reed and Muench. The TCID50 can be converted to plaque-forming units (PFU), with a conversion factor of PFU = 0.7 × TCID50. The multiplicity of infection (MOI) is reported as PFU number per cell.
The virus titer (TCID50) of HSV129-GFP was calculated using the dilution method of counting as mentioned above. The results were observed under a fluorescence microscope, and the number of the cells with green fluorescence was counted. All titers were calculated by the method of Reed and Muench.
In vitro HSV129-GFP infection
HEK293T cells were plated into 12-well plates at a density of 200,000 cells per well containing DMEM supplemented with 10% FBS, l-glutamine (2 mM), and penicillin (100 U/ml) and streptomycin (100 U/ml) (P/S) in a 5% CO2 incubator in a humidified atmosphere at 37°C. HEK293T cells were first treated with RUNX1 inhibitor Ro 5-3335 for 24 hours. Next, the cells were infected with HSV129-GFP at a MOI of 3. The cells were harvested at 3 and 6 hours for qPCR detection.
RNA-seq library preparation
PK15 cells were infected with PRV Ea at a MOI of 0.1. After 1 hour of adsorption at 37°C, inoculum was removed and replaced with cell maintenance solution (RPMI with 2% FBS) after a single rinse with 1× phosphate-buffered saline (PBS). The infected cells were harvested 8 hpi and resuspended in RNAiso Plus (Takara). mRNA Capture Beads (VAHTS mRNA-seq v2 Library Prep Kit for Illumina, Vazyme) were used to extract mRNA from total RNA. The PrimeScript Double Strand cDNA Synthesis Kit (Takara) was used to synthesize double-stranded cDNA from the purified polyadenylated mRNA templates. The TruePrep DNA Library Prep Kit V2 for Illumina (Takara) was used to prepare cDNA libraries for Illumina sequencing.
Multiple 4C assay
The initial steps of the 3C assays were performed as described previously (28, 29). Briefly, the nuclei were fixed by 1% paraformaldehyde and then subjected to Bam HI overnight digestion at 37°C at a concentration of 100 ng/μl. The restriction enzyme was inactivated at 65°C for 25 min. The samples were then transferred to a 50-ml tube for overnight proximity ligation at 16°C (150 μl of sample in 14 ml using 400 U of T4 ligase). The ligated products were added with 2 M NaAC, glycogen, and 100% ethanol and stored at −80°C until completely frozen. DNA samples were precipitated by ethanol, after which the supernatant was removed and the pellets were briefly dried at room temperature. The pellets were dissolved in 150 μl of 10 mM tris (pH 7.5) at 37°C and then digested with 50 U of Alu I restriction enzyme at 37°C for 3 hours. These digested products were purified again and subjected to self-ligation by T4 ligase.
Next, these DNA fragments were purified and amplified by a set of inverse PCRs. Primers were designed according to the PRV genome sequence flanking the Bam HI and Alu I sites as shown in table S1. A typical 4C-PCR protocol was as follows: one cycle of 95°C for 8 min followed by 21 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 1 min and one cycle of 72°C for 10 min and 16°C for 2 min. The 200– to 500–base pair (bp) amplified fragments were purified using Agarose Gel Extraction Kit and then ligated with Illumina P5 and P7 adapters by T4 ligase (Takara) at 16°C overnight. These ligation products were amplified 12 cycles and then subjected to Illumina sequencing.
ATAC-seq library preparation
PK15 cells were infected with PRV Ea at a MOI of 0.1, with infected cells harvested at 2 hpi. Cells were immediately resuspended in 1 ml of nuclei permeabilization buffer and rotated for 15 min at 4°C, and then, 200,000 cells were spun down at 4000g for 5 min at 4°C. Supernatant was discarded, and the remaining cells were subjected to immediate transposition reaction. The composition of nuclei permeabilization buffer is as follows: 5% bovine serum albumin (BSA), 0.2% (m/v) IGEPAL CA-630, and 1 mM cOmplete EDTA-free protease inhibitor.
The TruePrep DNA Library Prep Kit V2 for Illumina (TD501, Vazyme, China) was used to prepare DNA libraries for next-generation sequencing. Briefly, the nuclei were resuspended in the transposition reaction mix [10 μl of 5× TruePrep Tagment Buffer L (TTBL), 5 μl of TruePrep Tagment Enzyme Mix (TTE Mix) V50, and 50 ng of DNA] and then incubated at 55°C for 10 min. DNA fragments were immediately purified using a MinElute PCR purification kit (QIAGEN). To amplify the transposed DNA fragments, the following components were added in a sterile PCR tube: 24 μl of DNA fragments, 10 μl of 5× TruePrep Amplify Buffer (TAB), 5 μl of PCR Primer Mix (PPM), 5 μl of N5, 5 μl of N7, 1 μl of tris-acetate-EDTA, and ddH2O. The amplified libraries were purified with 1× VAHTS DNA Clean Beads (N411-02, Vazyme) separated and viewed on a 1% agarose gel.
Chromosome conformation capture–PCR
3C-PCR analysis was performed as previously described (29). Briefly, PRV-infected PK15 cells were fixed with 2% formaldehyde. Chromatin was digested using Bam HI and then proximity ligated by T4 DNA ligase under diluted condition. Ligated DNA was then decross-linked overnight at 65°C, purified by phenol extraction procedures, and then subjected to PCR using primers as shown in table S2. The noninteraction site was used as the negative control for the 3C assays.
3D in situ DNA HCR analysis
PK15 cells were infected with PRV Ea at a MOI of 3. The cells were harvested at 6 hpi. Fixation, permeabilization, and denaturation of DNA were performed as described previously (30). DNA HCR hairpins were designed and synthesized according to previous studies (31). The DNA probes are 61 nucleotides (nt) long (36-nt initiator, 5-nt spacer, and 20-nt mRNA recognition sequence), which are shown in table S3. The detection stage and amplification stage were performed according to the research of Choi et al. (31). Briefly, the cells were lysed with 0.56% KCl solution at 37°C for 30 min to release the nuclei. The nuclei were resuspended in methanol acetic acid fixing solution (3:1) for prefixation and final fixation. The fixed nuclei were dropped onto slides and dehydrated through a 70, 85, and 100% graded ethanol series. The 70% formamide was used to degenerate the DNA on the slides for 10 min at 75°C. To prevent DNA renaturation, the slides were rapidly dehydrated through a 70, 85, and 100% graded precold ethanol series. The nuclei were prehybridized with 350 μl of probe hybridization buffer [50% formamide, 5× SSC, 9 mM citric acid (pH 6.0), 0.1% Tween 20, heparin (50 μg/ml), 1× Denhardt’s solution, and 10% dextran sulfate] for 30 min at 45°C. After removing the prehybridization solution, the probe solution (500 μl of probe hybridization buffer containing 1 pmol of each probe) was added and incubated for 16 hours at 45°C. The sequences of the probes to detect the swine genome and PRV in PK15 cells are shown in table S3. Excess probe was removed by washing the slides with a series of 75, 50, and 25% gradient probe wash buffer [50% formamide, 5× SSC, 9 mM citric acid (pH 6.0), 0.1% Tween 20, and heparin (50 μg/ml)]. Sequences of the cy3-labeled hairpins for the swine genome (Chr2, Chr4) and the Alexa Fluor 488–labeled hairpins for PRV are shown in table S3. The hairpin solution was prepared by adding all the snap-cooled hairpins (30 pmol of each fluorescently labeled hairpin in 10 μl of 5× SSC buffer, heated at 95°C for 90 s and then reduced stepwise to 25°C at a rate of 0.1°C/s) to 500 μl of amplification buffer (5 × SSC, 0.1% Tween 20, and 10% dextran sulfate) at room temperature. Nuclei were preamplified with 350 μl of amplification buffer for 30 min at room temperature. After removing the preamplification solution, the hairpin solution was added. Last, the slides were incubated overnight at room temperature. Slides were washed five times with 5× SSCT (5× SSC and 0.1% Tween 20). After washing, slides were stained with 4′,6-diamidino-2-phenylindole (DAPI; Life Technologies) and analyzed under a fluorescence microscope.
Virus titer experiment
PK15 cells were first infected with RUNX1 and scramble control small interfering RNA (siRNA) AAV virus for 48 hours. Next, the cells were infected with PRV Ea at a MOI of 3 for 24 hours and then harvested 72 hours after the initial transfection. The siRNA sequences are shown in table S4; the online website Invitrogen was used to set the siRNA primer: http://rnaidesigner.thermofisher.com/rnaiexpress/sort.do.
PK15 cells were first treated with RUNX1 inhibitor Ro 5-3335 for 24 hours. Next, the cells were infected with PRV Ea at a MOI of 3 for 24 hours and then harvested 48 hours after the initial RUNX1 blocking.
Detection of PRV copy number by real-time PCR
To establish a real-time PCR method for PRV titer detecting, a pair of specific primer was designed according to the sequence of gI/gE gene in PRV. gI/gE primers: (forward) 5′-GGTGTTTGCATAATTTTGTGGGTGG-3′ and (reverse) 5′-GAAAGGGCCGCATGGTCTCA-3′. A series diluted PRV bacterial artificial chromosome plasmid containing PRV genome was used to obtain the standard curve and melt curve, which showed a fine linear relationship between threshold cycle and template concentration. The melt curve was specific, and the correlation coefficient was 0.991. The copy number formula is as follows: copies/μl = concentration (ng/μl) × 10−9 × 6.02 × 1023/molecular weight [length of template (bp) × 648].
ChIP-qPCR and ChIP-seq assay
RUNX1 ChIP-qPCR assays were performed as previously described (32). Briefly, PK15 cells (106 cells) were infected with PRV Ea at a MOI of 3 for 6 hours. The cells were harvested and fixed with 1% formaldehyde (Sigma-Aldrich) for 20 min to cross-link DNA and proteins. Fixation was quenched by adding glycine. Chromatin was sheared using Qsonica (Q125) (five cycles of sonication: 10 s ON; 30 s OFF; and AMPL, 30%). Chromatin (2 μg) was then incubated with RUNX1 antibody (ChIP Grade; ab23980, Abcam) and immunoglobulin G (IgG) antibody (negative control; CST#2729, Cell Signaling Technology), respectively, and immunoprecipitated by protein G magnetic beads (20 μl). After extensive washing [RIPA-LS (low salt), RIPA-HS (high salt), RIPA-LiCl, and 10 mM tris (pH 8)], the beads were gently resuspended in tagmentation reaction using the TruePrep DNA Library Prep Kit V2 for Illumina (TD501, Vazyme, China) according to the manufacturer’s instructions, incubated for 10 min at 37°C, and then cooled on ice. The beads were washed with RIPA-LS and tris-EDTA (TE) buffer. After washing, protein-DNA complexes were eluted, and cross-linking was reversed by adding 6 μl of 5 M NaCl and 2 μl proteinase K and incubated for 2 hours at 65°C. DNA was purified and subjected to qPCR for DNA enrichment detection for which the primers are listed in table S5. The results showed as follows: 2% × 2(CTinput−CTRUNX1)/[2% × 2(CTinput−CTIgG)]. The purified DNA was subjected to DNA library construction using the TruePrep DNA Library Prep Kit V2 for Illumina (TD501, Vazyme, China) according to the manufacturer’s instructions.
RNAPII ChIP assays were performed using the SimpleChIP Plus Sonication Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer’s instructions. Briefly, PK15 cells (107 cells) were transfected with siRNA-RUNX1 and siRNA-scramble for 48 hours and then infected with PRV Ea at a 3 MOI for 6 hours. The cells were fixed with 1% formaldehyde (Sigma-Aldrich) for 20 min to cross-link DNA and proteins, respectively. Fixation was quenched by adding glycine. Chromatin was sheared using Bioruptor Picoruptor (Diagenode; eight cycles of sonication: time on, 30 s and time off, 30 s). The chromatin (10 μg) was then incubated with RNAPII antibody (C-terminal repeat domain repeat YSPTSPS antibody; ChIP Grade; ab817, Abcam), histone H3 antibody (positive control), and IgG antibody (negative control), respectively, and immunoprecipitated by protein G magnetic beads (30 μl). After extensive washing, protein-DNA complexes were eluted and cross-linking was reversed by adding 6 μl of 5 M NaCl and 2 μl of proteinase K and incubated for 2 hours at 65°C. DNA was purified and subjected to qPCR for DNA enrichment detection for which the primers are listed in table S5. The purified DNA was subjected to DNA library construction.
DNA library construction
Hieff NGS Fast-Pace End Repair/dA-Tailing Module Kit (YEASEN) and GeneRead adapters were used for end repair of the DNA fragment, A-addition, and adapter ligation. First, the dA tailing and end repair buffer containing 20 μl of fragmented DNA, 3 μl of End Repair/dA-Tailing Buffer, 2 μl of End Repair/dA-Tailing Enzyme, and 5 μl of ddH2O were added in a sterile PCR tube for the dA tailing and end repair. The thermal cycler was programmed to incubate the reaction mixer for 30 min at 20°C, followed by 20 min at 65°C. Then, the Y adapters were ligated to the end-repaired DNA fragments by adding the following components: 2 μl of T4 DNA ligase, 2 μl of Y adapter, 4 μl of T4 ligase buffer, 30 μl of DNA fragments, and 2 μl of ddH2O and then incubated at room temperature for 1 hour. Ligated DNA fragments were purified with 1.2× VAHTS DNA Clean Beads. The adapter-ligated DNA libraries were amplified using Phanta Max Super-Fidelity DNA Polymerase. The PCR amplification program was as follows: 1 cycle of 98°C for 1 min followed by 16 cycles of 98°C for 10 s, 57°C for 30 s, and 72°C for 30 s and 1 cycle of 72°C for 5 min and 16°C for 2 min. The amplified DNA libraries were purified with 1.2× VAHTS DNA Clean Beads before Illumina sequencing.
Digestion-Ligation-Only (DLO) Hi-C assay
DLO Hi-C assay was performed as previously described in our laboratory (33). Briefly, PK15 cells (107 cells) were treated with RUNX1 inhibitor Ro 5-3335 for 24 hours and then infected with PRV Ea at a 3 MOI for 6 hours. The cells were harvested and fixed with 1% formaldehyde (Sigma-Aldrich) for 10 min to cross-link DNA and proteins. Fixation was quenched by adding glycine. The cross-linked cells were subsequently lysed in lysis buffer. Nuclei were then gently resuspended in 100 μl of 4% SDS, incubated at 55°C for 5 min, and placed on ice immediately. After incubation and centrifugation, 20 μl of 20% Triton X-100, 30 μl of Mse I [100 U/μl; New England Biolabs (NEB)], and 40 μl of 1.3× NEB Buffer 2.1 were added to the 310 μl of nuclei, and the samples were incubated for 4 hours at 37°C with metal bath at 1000 rpm. After restriction enzyme, the Mse I–bio-linker was ligated to the digested chromatin. The 400 μl of nuclei, 30 μl of Mse I–bio-linker, and 40 μl of T4 ligase buffer (Thermo Fisher Scientific) were added to the tubes and incubated for 10 min; then, 20 μl of T4 ligase were added, and the tubes were incubated for 1 hour at room temperature with rotation at 15 rpm. After Mse I–bio-linker ligation, chromatin DNA-protein complexes were centrifuged at 16°C for 5 min at 2000 rpm, and the supernatant was discarded again. The chromatin DNA-protein complexes were resuspended with 170 μl of ddH2O. The 170 μl of chromatin DNA-protein complexes, 10 μl of T4 PNK (NEB), and 20 μl of T4 buffer (Thermo Fisher Scientific) were added to the tubes and incubated for 30 min at 37°C. The samples were then transferred to a 1.5-ml tube for overnight proximity ligation at 16°C (200 μl of sample in 500 μl using 25 μl of T4 ligase). Next, the tubes were incubated for 1 hour at room temperature with rotation at 15 rpm. The samples were digested with 100 μl of 10% SDS and 100 μl of proteinase K (10 mg/ml; Sigma-Aldrich) at 65°C for 4 hours to release DNA. After incubation, an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1) was added to the sample to precipitate the DNA. A total of 6 μl of 10× CutSmart buffer, 3 μl of S-adenosyl-methionine (SAM) (NEB), and 3 μl of Mme I (2 U/μl; NEB) was added to the 30 μl of DNA sample and digested at 37°C for 1.5 hours. The digested DNA sample was subjected to electrophoresis in native polyacrylamide gel electrophoresis gels. The Oligo Clean & Concentrator (Zymo Research) was used to purify DNA. A total of 2 μl of T4 ligase, 2 μl of T4 buffer, 2.5 μl of MGI adapter A, and 2.5 μl of MGI adapter B were added to the 11 μl of sample and incubated at room temperature for 20 min. Next, the products were purified with 1.6× DNA Clean Beads. The eluted DNA was repaired using PreCR Repair Mix (NEB) for 30 min at 37°C in a final volume of 20 μl. Repaired DNA (10 μl) was used as a template and amplified for 15 cycles.
Real-time qPCR
Total RNA from PRV-infected PK15 cells and HSV129-infected HEK293T cells were both purified using RNAiso Plus reagent (Takara). After DNase treatment, reverse transcription was performed using a ReverTra Ace qPCR RT Master Mix kit (TOYOBO, Shanghai, China). Primers for qPCR analyses are shown in table S6. Measurements of the target genes were normalized using glyceraldehyde-3-phosphate dehydrogenase mRNA levels as an internal control for each sample.
Simultaneous immunohistochemistry and DNA HCR assay
PK15 cells were cultured in 96-well plate (PerkinElmer) and infected with PRV Ea at a MOI of 3. The cells were fixed by 4% paraformaldehyde and washed thrice with ice-cold PBS at 6 hpi. The samples were incubated in blocking solution (5% BSA and 0.1% Triton X-100 in PBS) at room temperature for 1 hour and then incubated with primary antibody (1:200; rabbit anti-RUNX1, Cell Signaling Technology) in primary dilution buffer (1% BSA and 0.3% Triton X-100 in PBS) at 4°C overnight. After washing four times for 5 min with PBS at room temperature, the samples were incubated with secondary antibody (1:1000; goat anti-rabbit, Alexa Fluor 488) and DAPI (1:1000) in blocking solution at 37°C for 2 hours. After washing four times for 5 min with PBS at room temperature, the samples were analyzed with microscopy for the first round imaging using the Opera Phenix (PE Corporation).
Subsequently, the DNA HCR assay was performed on the same sample analyzed by immunohistochemistry. PK15 cells were postfixed by 4% paraformaldehyde and washed thrice with PBS. The cells were permeabilized in 100% methyl alcohol for 5 min at room temperature and washed thrice with PBS. The cells were incubated in 0.2 M HCl (pH 7.4) for 5 min at room temperature and washed thrice with PBS. Next, cells were incubated with ribonuclease A (100 μg/ml) at 37°C for 10 min and washed thrice with PBS. The cells were subjected to DNA HCR assay as described above. The samples were analyzed with microscopy for the second round imaging. The images were aligned and then analyzed by Adobe Illustrator CC 2017.
Animal experiments and Ro 5-3335 treatment
All mice (female C57BL/6 mice, 6 weeks old, 20 ± 2 g) used in this study were purchased from Beijing Vital River Laboratory Animal Technology. Animal experiments were approved by the Scientific Ethics Committee of Huazhong Agricultural University (HZAUMO-2019-023). To generate the pseudorabies model, mice were injected intramuscularly with 3.5 × 104.5 TCID50/100μl PRV Ea (titer TCID50 107.5). The RUNX1 inhibitor Ro 5-3335 (10 mg/kg) in 5% dimethyl sulfoxide (DMSO) and 5% DMSO control were injected intraperitoneally 5 hours after PRV infection, respectively. All the mice were observed, and the survival rates were calculated. Mice were anesthetized and transcardially perfused with 4% paraformaldehyde in PBS. Liver was fixed in 4% buffered paraformaldehyde solution 8 days after treatment for histopathological examination.
Confocal microscopy
Fixed cells were imaged by a confocal laser scanning microscope (ZEISS LSM 800) using a 63× oil immersion with 1.4 optical aperture. A diode laser (λ = 405 nm) was used for excitation of DAPI counterstain. A helium-neon (λ = 543 nm) laser and an argon (λ = 488 nm) laser were used for spectrum cy3 probes and spectrum green excitation (Alexa Fluor 488 probes), respectively. 3D image stacks with an image format of 1024 × 1024 pixels and constant voxel sizes of 0.058 μm by 0.058 μm by 0.204 μm were acquired. The number of z-stacks was adjusted according to the height of cell nuclei, resulting in an average amount of 20 2D images for each cell nucleus. The DAPI-stained nucleus, swine genome probe, and PRV genome probe signals were acquired in parallel at a constant scanning speed of 1000 Hz. Imaris software was used to render the image.
Analysis of ATAC-seq, ChIP-seq, and RNA-seq data
The quality of the raw paired-end reads from ATAC-seq, ChIP-seq, and RNA-seq were evaluated with FASTQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/). Low-quality reads and contamination were filtered with Trimmomatic (34). The cleaned reads were aligned to the swine genome (Sscrofa10.2) using Burrows-Wheeler Aligner (BWA) (35) and TopHat (36) with default parameters, respectively. Unmapped reads and non-uniquely mapped reads (mapping quality < 30) were removed, while PCR duplicate reads were removed using SAMtools (37). To comprehensively analyze the multi-omics data from 4C, ATAC-seq, ChIP-seq, and RNA-seq, deepTools2 (38) was used to count the coverage through whole genome with bin size of 1 Mb. The samples were normalized by reads per kilobase per million mapped reads. MACS (39) was used to call RNAPII binding sites. The overlap of reads with genes was estimated using HTSeq (40). The normalization of counts and detection of DEGs were performed by DESeq2 (the absolute value of fold change ≥ 2 and P < 0.05) (41). Gene Ontology analysis was executed using DAVID (42). To describe the intensity of RNAPII occupancy in the PRV genome, PCR duplicate reads were removed, after which mapped reads were used for calling peak and downstream processing.
Analysis of multiple 4C sequencing data
Paired-end reads from multiple 4C sequencing were first filtered by 32 PRV primers using a custom JavaScript. Under the condition of allowing two mismatches, the script can divide the sequenced reads into 32 groups and each group of reads has a specific pair of primers from PRV genome. The reads without paired primers are removed from further analysis. The filtered reads in each group with distinct pair of PRV primers were used for downstream processing separately. To obtain clean reads from the swine genome, sequences from the PRV genome in the remaining sequences were removed to the extent possible. We cut the part of the sequences that originated from the PRV genome, which are theoretically the sequences from the primers to their nearest restriction sites (Bam HI and Alu I). The clean reads were aligned to the swine genome (Sscrofa10.2) using BWA (35) with default parameters. The nonredundant and uniquely aligned reads were used for subsequent analysis. BEDTools (43) was used to extract 4C read counts, and a custom Perl script was used to analyze interaction hotspots between the PRV genome and the swine genome. Briefly, the 4C reads were sorted according to their loci. The adjacent 4C read counts that the span of 4C read counts less than 250 bp were combined to form a cluster. The 4C read counts in the cluster were counted as the intensity of interaction hotspots. In theory, the ends of each interacted DNA fragments contain a pair of Bam HI and Alu I restriction sites. Thus, to obtain the interaction hotspots of the swine genome that PRV prefers to interact, we first extracted fragments with Bam HI and Alu I restriction sites at the end of the cluster. The clusters with pet number equal to one indicate only one interaction between the virus and the host genome, which are likely to be noise. Moreover, the length of these noise fragments was mostly less than 100 bp. To obtain reliable interaction hotspots between virus and host genome, we removed these noises as much as possible. As shown in fig. S2D, the 4C reads in the cluster with count number less than six contain much more noise fragments of which the length is less than 100 bp. Therefore, the 4C read in the cluster with count number equal to or greater than six were chosen as candidate interaction hotspots. Gene Ontology analysis was executed using DAVID (42).
DLO Hi-C data analysis
DLO Hi-C data were processed using the DLO Hi-C Tool (44). In detail, the linkers were filtered first. The clean reads were mapped to one new merged genome that contained the swine genome (v 10.2) and PRV genome. The self-ligation and religation reads were removed using the DLO Hi-C Tool (44). The iterative correction method (45) was performed to normalize interaction matrixes at different resolutions to remove the bias of raw matrices. For each sample, we merged all valid pairs from biological replicates for further analysis after confirming that all replicates were highly correlated. We counted the number of DLO Hi-C reads aligned to the swine genome at one end and aligned to the PRV genome at the other end as the trans-species interactions.
DNA binding motif screening
To detect DNA binding motifs enriched in the interaction hotspots, the genomic sequences of the interaction hotspots were submitted to a meme web server (46) to identify known or similar motifs (E value < 0.0005). To test whether the RUNX1 motif was significantly enriched in interaction hotspots, we randomly selected the same number and similar length range as hotspots in the host genome and counted the number of RUNX1 motifs in these random regions. The random procedure was repeated 1000 times.
Statistical analysis
All statistical tests were executed using the R statistical package (www.r-project.org/). All data were presented as means ± SEM of n independent measurements. Statistical analysis of the survival curves was performed with GraphPad Prism 5, and statistical comparisons between two groups were made by the Fisher’s exact test, Wilcoxon test, and Student’s t test. Significance levels were set at P = 0.05.
Acknowledgments
We are thankful to Z. Hu (State Key Laboratory of Agricultural Microbiology Core Facility), Q. Zhang (Platform of National Key Laboratory of Crop Genetic Improvement), and Z. Zhang (Spatial FISH Co. Ltd.) for technical help and suggestions. We are grateful to H. Chen, Z. Liu, and M. Luo for providing PRV Ea and HSV-GFP. Funding: This work was supported by the National Key Research and Development Project of China (grant 2017YFD0500300 to G. Cao), the National Natural Science Foundation of China (grants 31941014 to G. Cao, 31970590 and 31771402 to G.L., 31702196 to K.X., and 31941005 to J.D.), the Key Research and Development Program of Guangdong Province (grant 2019B020211003 to G.C.), Fundamental Research Funds for the Central Universities (grants 2662018PY025 and 2662019YJ004 to G. Cao), and the China Postdoctoral Science Foundation (grant 2019M662676 to K.X.). Author contributions: G. Cao and G.L. conceived the study with the help of Y.R. and Z.F. K.X. performed all the experiments with the help of G.Ch., J.Y., Y.L., K.C., A.G., L.Z., Y.X., J.D., K.Y., and X.Hu. D.X. analyzed all the data with the help of Q.X., X.Hua., and K.C. G. Cao, G.L., K.X., and D.X. wrote the manuscript. All authors read and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The raw sequencing data from this study have been deposited in the Genome Sequence Archive in BIG Data Center (https://bigd.big.ac.cn/), Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under the accession number: CRA002718 (https://bigd.big.ac.cn/gsa/s/S5XR6Cw9). All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabf8962/DC1
REFERENCES AND NOTES
- 1.McCord R. P., Kaplan N., Giorgetti L., Chromosome conformation capture and beyond: Toward an integrative view of chromosome structure and function. Mol. Cell 77, 688–708 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roy C. R., Mocarski E. S., Pathogen subversion of cell-intrinsic innate immunity. Nat. Immunol. 8, 1179–1187 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Marsh M., Helenius A., Virus entry: Open sesame. Cell 124, 729–740 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karasneh G. A., Shukla D., Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 8, 481 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farrell P. J., Epstein–Barr virus and cancer. Annu. Rev. Pathol. 14, 29–53 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Aranda A. M., Epstein A. L., Herpes simplex virus type 1 latency and reactivation: An update. Med. Sci. 31, 506–514 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Washington S. D., Singh P., Johns R. N., Edwards T. G., Mariani M., Frietze S., Bloom D. C., Neumann D. M., The CCCTC binding factor, CTRL2, modulates heterochromatin deposition and the establishment of herpes simplex virus 1 latency in vivo. J. Virol. 93, e00415–e00419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu H., Zheng Y., Role of ND10 nuclear bodies in the chromatin repression of HSV-1. Virol. J. 13, 62–69 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hellert J., Weidner-Glunde M., Krausze J., Lünsdorf H., Ritter C., Schulz T. F., Lührs T., The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc. Natl. Acad. Sci. U.S.A. 112, 6694–6699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H., Deng Z., Vladimirova O., Wiedmer A., Lu F., Lieberman P. M., Patel D. J., Structural basis underlying viral hijacking of a histone chaperone complex. Nat. Commun. 7, 12707–12716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tempera I., Klichinsky M., Lieberman P. M., EBV latency types adopt alternative chromatin conformations. PLOS Pathog. 7, e1002180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreau P., Cournac A., Palumbo G. A., Marbouty M., Mortaza S., Thierry A., Cairo S., Lavigne M., Koszul R., Neuveut C., Tridimensional infiltration of DNA viruses into the host genome shows preferential contact with active chromatin. Nat. Commun. 9, 4268–4281 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim K.-D., Tanizawa H., de Leo A., Vladimirova O., Kossenkov A., Lu F., Showe L. C., Noma K.-i., Lieberman P. M., Epigenetic specifications of host chromosome docking sites for latent Epstein-Barr virus. Nat. Commun. 11, 877 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moquin S. A., Thomas S., Whalen S., Warburton A., Fernandez S. G., McBride A. A., Pollard K. S., Miranda J. J. L., The Epstein-Barr virus episome maneuvers between nuclear chromatin compartments during reactivation. J. Virol. 92, e01413–e01417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koyuncu O. O., Hogue I. B., Enquist L. W., Virus infections in the nervous system. Cell Host Microbe 13, 379–393 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pomeranz L. E., Reynolds A. E., Hengartner C. J., Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 69, 462–500 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dremel S. E., DeLuca N. A., Genome replication affects transcription factor binding mediating the cascade of herpes simplex virus transcription. Proc. Natl. Acad. Sci. U.S.A. 116, 3734–3739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fraser J., Williamson I., Bickmore W. A., Dostie J., An overview of genome organization and how we got there: From FISH to Hi-C. Microbiol. Mol. Biol. Rev. 79, 347–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coppotelli G., Mughal N., Callegari S., Sompallae R., Caja L., Luijsterburg M. S., Dantuma N. P., Moustakas A., Masucci M. G., The Epstein–Barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group A proteins. Nucleic Acids Res. 41, 2950–2962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majumder K., Wang J., Boftsi M., Fuller M. S., Rede J. E., Joshi T., Pintel D. J., Parvovirus minute virus of mice interacts with sites of cellular DNA damage to establish and amplify its lytic infection. eLife 7, e37750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang M. K., Kwon D., McBride A. A., Papillomavirus E2 proteins and the host Brd4 protein associate with transcriptionally active cellular chromatin. J. Virol. 83, 2592–2600 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng X. Q., Gao Y., Zhang Q., Liu Y., Peng Y., Fu M., Ji Y., Identification of transcription factor AML-1 binding site upstream of human cytomegalovirus UL111A gene. PLOS ONE 10, e0117773 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klase Z., Yedavalli V. S. R. K., Houzet L., Perkins M., Maldarelli F., Brenchley J., Strebel K., Liu P., Jeang K.-T., Activation of HIV-1 from latent infection via synergy of RUNX1 Inhibitor Ro5-3335 and SAHA. PLOS Pathog. 10, e1003997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim D. J., Khoury-Hanold W., Jain P. C., Klein J., Kong Y., Pope S. D., Ge W., Medzhitov R., Iwasaki A., RUNX binding sites are enriched in herpesvirus genomes and RUNX1 overexpression leads to HSV-1 suppression. J. Virol., e00943-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murakami Y., Chen L.-F., Sanechika N., Kohzaki H., Ito Y., Transcription factor Runx1 recruits the polyomavirus replication origin to replication factories. J. Cell. Biochem. 100, 1313–1323 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Ito Y., RUNX genes in development and cancer: Regulation of viral gene expression and the discovery of RUNX family genes. Adv. Cancer Res. 99, 33–76 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Zolotukhin S., Byrne B. J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R. J., Muzyczka N., Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6, 973–985 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Splinter E., Grosveld F., de Laat W., 3C technology: Analyzing the spatial organization of genomic loci in vivo. Methods Enzymol. 375, 493–507 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Hagège H., Klous P., Braem C., Splinter E., Dekker J., Cathala G., de Laat W., Forné T., Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat. Protoc. 2, 1722–1733 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Bacher C. P., Guggiari M., Brors B., Augui S., Clerc P., Avner P., Eils R., Heard E., Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat. Cell Biol. 8, 293–299 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Choi H. M. T., Beck V. A., Pierce N. A., Next-generation in situ hybridization chain reaction: Higher gain, lower cost, greater durability. ACS Nano 8, 4284–4294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidl C., Rendeiro A. F., Sheffield N. C., Bock C., ChIPmentation: Fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 12, 963–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin D., Hong P., Zhang S., Xu W., Jamal M., Yan K., Lei Y., Li L., Ruan Y., Fu Z. F., Li G., Cao G., Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture. Nat. Genet. 50, 754–763 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Durbin R., Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trapnell C., Pachter L., Salzberg S. L., TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang D. W., Sherman B. T., Lempicki R. A., Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong P., Jiang H., Xu W., Lin D., Xu Q., Cao G., Li G., The DLO Hi-C tool for digestion-ligation-only Hi-C chromosome conformation capture data analysis. Genes 11, 289 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Imakaev M., Fudenberg G., McCord R. P., Naumova N., Goloborodko A., Lajoie B. R., Dekker J., Mirny L. A., Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bailey T. L., Williams N., Misleh C., Li W. W., MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 34, W369–W373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tombácz D., Tóth J. S., Petrovszki P., Boldogkői Z., Whole-genome analysis of pseudorabies virus gene expression by real-time quantitative rt-pcr assay. BMC Genomics 10, 491–490 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/26/eabf8962/DC1