

Abstract
Background:
It is important to identify patients with monogenic IBD as management may differ from classical IBD. In this position statement we formulate recommendations for the use of genomics in evaluating potential monogenic causes of IBD across age groups.
Methods:
The consensus included paediatric IBD specialists from the Paediatric IBD Porto group of the European Society of Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) and specialists from several monogenic IBD research consortia. We defined key topics and performed a systematic literature review to cover indications, technologies (targeted panel, exome and genome sequencing), gene panel setup, cost-effectiveness of genetic screening, and requirements for the clinical care setting. We developed recommendations that were voted upon by all authors and Porto group members (32 voting specialists).
Results:
We recommend next-generation DNA-sequencing technologies to diagnose monogenic causes of IBD in routine clinical practice embedded in a setting of multidisciplinary patient care. Routine genetic screening is not recommended for all IBD patients. Genetic testing should be considered depending on age of IBD-onset (infantile IBD, very early-onset IBD, paediatric or young adult IBD), and further criteria, such as family history, relevant comorbidities, and extraintestinal manifestations. Genetic testing is also recommended in advance of hematopoietic stem cell transplantation. We developed a diagnostic algorithm that includes a gene panel of 75 monogenic IBD genes. Considerations are provided also for low resource countries.
Conclusions:
Genomic technologies should be considered an integral part of patient care to investigate patients at risk for monogenic forms of IBD.
Keywords: Crohn’s disease, exome sequencing, genetics, primary immunodeficiency, ulcerative colitis, very early-onset inflammatory bowel disease
INTRODUCTION AND BACKGROUND
Genetic technologies have revolutionized the understanding of the genetic basis and subsequent functional understanding of immune-mediated disorders, such as inflammatory bowel diseases (IBD), which encompasses Crohn’s disease, ulcerative colitis, and IBD unclassified (IBDU) (1,2). The genetics of IBD is complex with 3 major areas arising: complex genetics based on hundreds of common polygenic risk variants, rare monogenic IBD genetics, and pharmacogenetics. In most patients with IBD, a large number of common genetic variants (>1% allelic frequency in the general population) contribute to disease susceptibility in a polygenic setting (3–8).
Use of genomic sequencing technologies allows identification of previously undiagnosed disorders in patients with multiple conditions, including gastrointestinal, immunological, and rheumatologic diseases (9). A growing number of rare monogenic disorders presenting with IBD-like intestinal inflammation have been identified (10–14). In these patients, IBD is caused by high penetrance genetic variants in a single gene (monogenic IBD). The group of monogenic IBD defects includes a large number of primary immunodeficiencies as well as intestinal epithelial cell defects.
It can be challenging to distinguish monogenic IBD from classical IBD based on clinical phenotype alone. Whereas some of these monogenic disorders present with almost pathognomonic features (such as immunodysregulation polyendocrinopathy enteropathy X-linked syndrome or Trichohepatoenteric syndrome), others do not. Monogenic IBDs have a higher morbidity compared with classical IBD (15,16). Incomplete penetrance of most causative genes suggests that additional factors, such as environmental factors, the microbiome, and additional genetic risk factors contribute to the phenotype of monogenic IBD (17,18).
Next-generation sequencing technologies including targeted panel sequencing, exome and genome sequencing are increasingly used in the research and in the clinical setting to screen for monogenic disorders associated with IBD (10,11,19). The technologies and analytic approaches are standardised to meet clinical diagnostic needs (20). Panel sequencing is a cost-effective technology to screen for variants in a limited number of genes (typically 10s to few hundred genes). This technology results comparably in high coverage of the sequencing reads and the analysis is relatively simple compared with an exome-wide or genome-wide analysis. Exome sequencing aims to investigate the whole range of protein-coding variants in approximately 20,000 genes (20). The technology does have limitations in the diagnostic setting because of read coverage deficiencies in some regions but newer exon capture assays compensate for uneven coverage. Genome sequencing investigates the entire genome of approximately 3 billion base pairs of which most are biallelic (20). In addition to the protein-coding regions, it allows analysis of regulatory elements and intronic regions. Due to a more even sequencing coverage of the sequence reads compared with exome sequencing, it allows to investigate copy number variation (CNV, deletions or duplications). Nevertheless, genome sequencing has not yet fulfilled its full potential for clinical genomics as current information of the noncoding elements in IBD is still restricted and its analysis is complex (21). The diagnostic yield of exome and genome sequencing is similar or slightly higher in genome sequencing (22,23).
We present a position statement on the application of next-generation sequencing for diagnosis of monogenic IBD in clinical practice. We outline indications for genetic testing and discuss the diagnostic yield in different settings. Focusing on next-generation sequencing for monogenic IBD diagnosis, we will not discuss emerging genetic applications, such as polygenic IBD risk scores (3) or IBD-related pharmacogenetics (24–28).
METHODS
Definition of Topics and Summary of Evidence
Following an open call to the members of the Paediatric IBD Porto and interest group of ESPGHAN, international specialists were selected based on content expertise. In addition to Porto group members, external paediatric and adult IBD as well as immunology specialists were also invited to participate. These included specialists from several research consortia with a focus on monogenic IBD including the Genius working group of ESPGHAN, the ESPGHAN supported COLORS in IBD research network and the VEOIBD consortium (www.veoibd.org).
Initially, the steering committee identified 7 key topics as part of an online iterative discussion process (Box 1). Subsequently, these topics were discussed within working groups, literature was assessed using a PubMed search, and position statements were developed.
Box 1. Key themes identified to assess utilisation of clinical genomics for diagnosis of monogenic inflammatory bowel disease.
Is there evidence to support the clinical use of genomic sequencing technologies for diagnosis of monogenic forms of inflammatory bowel disease (IBD)?
Which patients should be investigated by genomic technologies?
What Mendelian disorders (genes) should be included in a screening panel?
Are there preferred genomic sequencing technologies (targeted panel sequencing, whole exome and whole genome sequencing)?
What is the role of functional validation to establish causality?
What is the role of a multidisciplinary team in the care of patients with suspected monogenic IBD (Paediatric/adult gastroenterology, immunology, clinical genetics, other disciplines)?
Is there evidence of cost-effectiveness for genomic screening technologies for diagnosis of IBD?
Literature Search Strategy and Eligibility Criteria
To assess the use for next-generation sequencing technologies for the diagnosis of monogenic IBD in patient cohorts, a systematic literature review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-analyses statement (PRISMA; http://www.prisma-statement.org). The electronic database PubMed was searched for studies (updated June 2020). To achieve the maximum sensitivity of the search strategy in title and abstract, we combined terms that describe the IBD population (monogenic inflammatory bowel disease, n = 8 articles and very early-onset inflammatory bowel disease, n =110 articles) with search for IBD-related sequencing technologies (Inflammatory Bowel Disease AND Exome Sequencing n = 107 articles or Inflammatory Bowel Disease and targeted panel sequencing n = 21 articles). Patient cohort studies published between 2010 to 2020 were included when the study was published as original article in a peer-reviewed journal, it was written in English, the study population consisted of at least 10 patients diagnosed with IBD, the aim was to screen for a spectrum of monogenic IBD disorders using targeted panel sequencing, and/or exome and/or genome sequencing. Case reports, conference presentations, reviews, editorials, and expert opinions were excluded. The reference lists of relevant articles and reviews on the topic were screened for additional studies that meet the eligibility criteria. From each selected study, the following information was extracted: author name and year of publication, number of patients, cohort description, age at IBD diagnosis, region where participants were recruited and ethnic descent of IBD patients investigated, sequencing technology used, the number of monogenic IBD genes screened and the number of patients with monogenic IBD identified, whether functional validation was performed and whether the results had therapeutic consequences. The selected studies were assessed for diagnostic costs and health economic data.
To assess clinical features that are significantly associated with the group of monogenic IBD, we selected studies that compared clinical features between cohorts of patients with monogenic IBD (including diverse genetic defects) and control IBD patients in whom no monogenic defect was identified. Significantly associated features were reported according to the statistical analysis described in the respective article.
To select an extensive list of genes associated with monogenic IBD, we summarised the gene list discussed in 4 extensive specialist review articles that cover the biology and clinical implications of monogenic IBD (10–13). To reduce selection bias, we included reviews from specialists of different institutions. On the basis of this summary, a consensus list was generated by representatives of the research consortia based on published evidence.
We assessed different diagnostic algorithms to identify patients with monogenic IBD (11,12,29–31).
Definitions
In accordance with previous definitions, we define neonatal IBD as onset within first 28 days of age, infantile (and toddler) onset of IBD (IOIBD, less than 2 years of onset), very early-onset IBD (VEOIBD; less than 6 years of age IBD-onset), early-onset IBD (EOIBD; A1a Paris Classification; less than 10 years of age), paediatric IBD less than 17 years of age-onset (A1a and A1b Paris Classification) as well as adult-onset IBD 17 years and older (29,32).
To define the pathogenicity of genetic variants, we follow the American College of Medical Genetics (ACMG) 5-tier classification system (33). According to this classification: (1) pathogenic and (2) likely pathogenic variants directly contribute to the development of disease or are very likely causative, (3) variants of uncertain significance lack evidence to support a more definitive classification, whereas (4) likely benign, or (5) benign variants do unlikely or not cause disease (33).
Statement Voting
Position statements were developed based on iterative e-mail and conference call group discussions. The writing group voted on all statements while adding specific comments using a web-based voting platform. A second round of electronic voting and revisions was done, including all members of the Paediatric IBD Porto group of ESPGHAN. In total, 32 paediatric IBD specialists voted anonymously on all statements: 23 Porto group members (of whom 9 were authors; Appendix 1, http://links.lww.com/MPG/C127 names of all Porto group IBD specialists) and 9 non-Porto group authors. All statements were supported by at least 80% of the group.
RESULTS
Next-generation Sequencing for Diagnosis of Monogenic Inflammatory Bowel Disease
The initial literature search resulted in 185 studies. The use of next-generation sequencing for diagnosis of diverse monogenic IBD disorders has been described in 18 cohort studies (Table 1). Five studies identified significant clinical and laboratory features of monogenic IBD patients compared with IBD patients in whom no monogenic cause was identified (Table 2). The results of the studies are influenced by several factors including the geographical representation of the cohorts, the ethnic background of the study groups, or the preselection of the study population in respect to clinical phenotype (unselected cohorts, cohorts with undiagnosed immune defects or congenital diarrhea, cohorts with or without clinical suspicion of monogenic IBD before genetic tests). Further factors that influence the diagnostic yield are the age of IBD-onset of the study population (ranging from neonatal-onset to adult-onset IBD), the use of sequencing technologies (targeted panel sequencing, exome and genome sequencing, with and without prior Sanger sequencing), the study setup (single or multicentre studies, analysis focused on singleton sequencing in most studies vs family trio analysis in 1), and the degree of pathogenic classification and functional validation of the genetic variants (Tables 1 and 2). The heterogeneity of these studies has so far precluded a formal meta-analysis to establish a hierarchy of factors. Even in the absence of a multifactorial analysis, however, current data allow to draw conclusions about the patient populations that will benefit most from genetic screening, and how to optimise the diagnostic pathway in the clinical care setting.
TABLE 1.
Inflammatory bowel disease cohort studies to investigate a spectrum of monogenic Inflammatory bowel disease using next-generation sequencing
| Publication | Year | Cohort description Study setup Patient selection Ethnicity | Age-at-IBD diagnosis—median or mean (range; in years) | Sequencing technology / Number of candidate genes | Number of patients Total (n)/monogenic IBD (n) / Functional validation, yes/no | Therapeutic consequences of genetic findings and Comment |
|---|---|---|---|---|---|---|
| Taylor et al (21) | 2015 | Single centre, UK IBD-onset <7 years | WGS 40 genes | Total 15 Monogenic IBD 0 |
N/A | |
| Kammermeier et al (47) | 2014 | Single tertiary referral centre; UK Extensive disease (pancolitis or panenteritis) Diagnosis within the first 36 months of life Caucasian n =11, Asian n =14 |
Median 0.58 years (0.1–1.6) | Sanger sequencing TGPS n = 25 WES n = 20 40 genes | Total 25 Monogenic IBD 7/25 IO-IBD 19% (4/21) |
HSCT assessment initiated and performed |
| Kelsen et al (48) | 2015 | Single tertiary referral centre; USA IBD-onset under 5 years of age |
Range 0.06 to 5 | WES 400 genes |
Total 125 Monogenic 0 Hypomorphic variants Yes |
N/A |
| Ashton et al (49) | 2016 | Single tertiary centre; UK Age <18 years | Median age at diagnosis 12.2 years Median age at onset 11.04 years |
WES n =147 51 genes |
Total 147 Unclear† No |
|
| Ostrowski et al (50) | 2016 | Multicentre; Poland Paediatric IBD No family history of IBD | 88 patients with IBD, 43 under 6 years of age, and 45 more than 40 years of age VEO-IBD: age range 1 to 5; median 3 |
WES n = 43 VEO-IBD and n = 45 after 40 years old 40 genes |
Total 88 Unclear† 2 homozygote variants (NCF4 and WAS) were found in 2 affected adults and one child, no functional validation |
|
| Xiao et al (51) | 2016 | Single tertiary centre, China VEO-IBD Chinese n = 13 |
mean age 0.5 range: 0 to 3 years | TGPS 10 genes, including susceptibility genes | Total 13 Monogenic IBD 3 Others not clear IO-IBD 23% (3/13) No |
|
| Petersen et al (52) | 2017 | Multicentre; international Early-onset IBD or chronic diarrhea Age at diagnosis <10 years of life Caucasian n = 47, Arab n = 9, Turkish n = 5, Other n = 10 |
Average 3 years | Total 71 TGPS n = 46 TGPS+WES n = 25 28 genes, including 5 susceptibility genes |
Total 71 Monogenic IBD 5 IO-IBD 13% (4/31) VEOIBD 9.26% (5/54) Yes |
HSCT initiated and performed |
| Suzuki et al (53) | 2017 | Multicentre; Japan 35 patients age <16 years, among whom 27 patients under the age of 6 Japanese n = 33, Japanese-Chinese n = 1, Japanese-Brazilian n = 1 |
Mean 4.50 years | TGPS n = 35 55 genes |
Total 35 Monogenic IBD 5, IO-IBD 22% (2/9) Paris A1a 13.3% (4/30) Paris A1b 20% (1/5) Yes |
HSCT initiated and performed |
| Kammermeier et al (16) | 2017 | Single centre, tertiary referral, UK IBD-onset <2 years 52% were White Europeans, 16% were Middle Eastern/Arab States, 8% Pakistani, 8% Indian, 6% Bangladeshi, 5% African, and 5% of mixed ethnic origin; 29% were offspring from consanguineous unions; 18% had a positive family history of IBD |
Median 0.25 years (0.1–0.9) | TGPS only n = 17 WES only n = 37 Sanger only n = 8 40 genes |
Total 62 Monogenic IBD 19, IO-IBD 31% (19/62) Partial |
HSCT initiated and performed |
| Quaranta et al (37) | 2018 | Single centre, tertiary referral, UK Age at IBD diagnosis 7–40 years Severe disease (need for intestinal surgery and/or therapy progression to biologics) |
WES 59 genes |
Total 503 Monogenic IBD 1, Paris A1a/b 0.19% (1/503) Yes |
HSCT initiated and performed | |
| Charbit-Henrion et al (31) | 2018 | Multicentre, international Clinical presentation of severe VEO-IBD (n = 185) and congenital diarrhea; History suggestive of monogenic disorder (n = 22) European n = 200, Asian n = 2 African n = 3, Australia n = 2 |
<2 years n = 144; > 6 years n = 22 | TNGS n = 167 WES n = 51 66 genes |
Total 207 Monogenic IBD 66, IO-IBD 41% (59/144) VEO-IBD 33.5% (62/185) 6 years 18% (4/22) Yes | |
| Amininejad et al (38) | 2018 | 660 early-onset/familial cases among the 2390 cases with Crohn’s disease | NGS 23 PID-genes | Hypomorphic variant in XIAP | no | |
| Fang et al (34) | 2018 | Single centre, China IBD-onset before 6 months of age or VEO-IBD accompanied with severe perianal disease, severe malnutrition or growth failure, or resistance to conventional treatment median age of disease-onset was 14 mo (IQR: 0–72 mo) among 54 patients with VEO-IBD Chinese n = 54 |
Median 2.9 years (0.25–14.4) |
TGPS n = 12 WES n = 6 TGPS and WES n = 2 4503 genes |
Total Monogenic IBD 9 IO-IBD 19.3% (6/31) VEOIBD 16.6% (9/54) Yes |
HSCT assessment initiated |
| Lega et al (35) | 2019 | Multicentre, Italy VEO-IBD and patients with early-onset IBD with severe/atypical phenotypes* | Median Monogenic IBD 2.25 years (0.83–4); Nonmonogenic IBD 2 years (0.66–4) | Candidate gene n = 47 TGPS n = 69 WES n = 16 TrioWES n = 5 Candidate genes: WES n = 400 TGPS A n = 30 TGPS A n = 43 | Total 93 Monogenic IBD 12; IO-IBD 14.5% (8/55) VEO-IBD 11.5% (10/87) > 6 years 17% (2/6) Yes | HSCT n = 7 Liver transplant n = 1 |
| Crowley et al (36) | 2020 | Single tertiary centre IBD-onset Canada under 18 years IBD-onset European/Caucasian 566, East Asia n = 19, South Asia n = 104, Africa n = 29, Mixed n = 63, American n = 65, Asian n = 35, West Asian n = 21, Unclassified n = 103 | Median 12.0 years (0–18) | WES (trio analysis) 67 genes | Total 1005 Monogenic IBD 31 Yes IO-IBD 13.8% (4/29) VEO-IBD 6.2% (7/112) 6 to 9.9 year 1.7% (3/179) 10 to 17.9 year 2.5% (17/ 684) | HSCT initiated and performed |
| Ashton et al (54) | 2020 | Single tertiary centre; UK Age <18 years | Median 11.9 years (1.3–17.4) | WES n = 401 68 genes | Total 401 Unclear† No |
|
| Serra et al (8) | 2020 | Multicentre, international Severe IBD disease course (previous surgery or need for biological therapy) no suspicion of monogenic IBD Caucasian n = 99 African n = 2, Asian n = 21 Jewish n = 1 Others/unknown n = 22 |
Median 3.5 years (4–6.8) | WES 67 genes | Total 145 Monogenic IBD 4 VEO-IBD 2.75% (4/145) Mosaicism n = 1 (CYBB) Yes |
HSCT assessment initiated in several patients |
| Uchida et al JPGN | 2020 | Multicentre, Japan Age <17 years, early-onset diarrhea, refractory to conventional therapies Japanese n = 107, Japanese- Laotian n = 1 |
Median age at onset 3.82 (IQR 2.50) years | TGPS 193 genes | Total 108 Monogenic IBD 15 VEO-IBD 9.9% (8/81) IO-IBD 17.1% (7/41) 6 to 10 years 38.4% (5/13) 10 to 17 years 14.3% (2/14) Yes |
HSCT = hematopoietic stem cell transplantation;TPS = Targeted panel sequencing, WES = Exome sequencing; WGS = genome sequencing.
Severe perianal disease, recurrent/atypical infections, skin/annexes abnormalities, abnormal immune status, associated multiple/severe autoimmunity,
history of macrophage activation syndrome or hemophagocytic lymphohistiocytosis, intestinal atresia, or early development of tumors.
In these articles, no functional validation of novel variants was performed. Many variants are found at unexpectedly high allele frequencies (ExAC and gnomAD databases), thus pathogenicity and inheritance pattern is unclear.
TABLE 2.
Clinical features associated with monogenic inflammatory bowel disease in cohort studies
| Author | Year | Patient numbers (n) | Genetic defects | Age of onset | Features significantly associated with monogenic IBD |
|---|---|---|---|---|---|
| Kammermeier et al (16) | 2017 | Monogenic IBD: 19 Control IBD: 43 |
EPCAM (n = 3) IL10 (n = 2) IL10RA (n = 1) IL10RB (n = 2) FOXP3 (n = 3) LRBA (n = 1) SKIV2L (n = 2) TTC37 (n = 2) TTC7A (n = 3) |
Mean age of onset 2 months monogenic 8.3 months control |
Consanguinity Disease-onset <6 months Height-for-age z-score <−3 Extensive disease Epithelial abnormality Parenteral nutrition required |
| Fang et al (34) | 2018 | Monogenic IBD: 9 Control IBD: 45 |
IL10R (n = 5) CYBB (n = 2) XIAP (n = 1) CVID with TNFRSF13B (n = 1) |
Median age of onset 1 months monogenic, 19.5 months control |
Incidence of perianal disease Use of mesalazine Death |
| Kim et al (15) | 2018 | Monogenic IBD: 18 (not all genes specified) Control IBD: 212 |
CGD (n = 3) IPEX (n = 2) GSD (n = 1) Congenital neutropenia (n = 2) Hyper immunoglobulin (Ig)M syndrome (n = 1) Hypogammaglobulinemia (n = 1) IL-10 (n = 8) |
Mean age of diagnosis 1.6-year monogenic, 11.7-year control |
Incidence of surgery per year Hospitalization per year Height less than third percentile Weight less than third percentile IBD-U |
| Lega et al (35) | 2019 | Monogenic IBD: 12 Control IBD: 81 |
XIAP (n = 2) WAS (n = 3) TTC37 (n = 1) DKC1 (n = 1) CD40L (n = 2) CYBA (n = 1) CYBB (n = 1) FOXP3 (n = 1) |
Mean age of onset 27-month monogenic, 24-month control |
Males (n) Extraintestinal findings Disease-onset </ = 1 month Disease location (colon only) Disease location (colon + other location) Extraintestinal: infections Extraintestinal: HLH/MAS Extraintestinal: skin disease Low platelets Low immunoglobulin Lymphocyte subset abnormalities |
| Crowley et al (36) | 2020 | Monogenic IBD: 31 Control IBD: 974 |
ALPI (n = 1) COL7A1 (n = 1) GUCY2C (n = 2) SLCO2A1 (n = 1) TTC7A (n = 1) ARPC1B (n = 2) BTK (n = 1) DKC1 (n = 1) DOCK8 (n = 3) LRBA (n = 2) STAT 1 (n = 1) HPS1 (n = 1) PIK3CD (n = 1) SH2D1A (n = 1) XIAP (n = 5) CYBB (n = 1) CTLA4 (n = 1) FOXP3 (n = 2) IL10RB (n = 1) HSPA1L (n = 1) MASP2 (n = 1) |
Mean age of onset 9.69 years monogenic | Age at diagnosis <2 years family history of autoimmune disease Any extraintestinal manifestation > 1 extraintestinal manifestation Progression to surgical therapy |
Indications for Genetic Testing of Monogenic Inflammatory Bowel Disease Informed by Clinical Phenotypes and Therapeutic Implications
Establishing a genetic diagnosis is of benefit to patients and their families as it can help to predict the disease course, to anticipate complications, to facilitate genetic counselling and to consider specific treatments, most notably allogeneic hematopoietic stem cell transplantation for some underlying primary immunodeficiencies (10–12) (Table 1). As monogenic IBD is associated with a number of demographic features including age of IBD-onset, the family history, clinical phenotypes (in particular, comorbidities and extraintestinal manifestations), as well as abnormal laboratory findings (Table 2), those features can inform the need for genetic investigations as well as the clinical need to progress with informed treatment options (Box2).
Box 2. Summary of clinical features that should prompt considering a monogenic inflammatory bowel disease workup (Red flag signs).
Age of inflammatory bowel disease (IBD) presentation
<2 years IBD symptom onset
<6 years IBD symptom onset in particular when other red flag signs are present
Family history
Affected family member with a suspected monogenic disorder
Consanguinity
Multiple family members with early-onset IBD
Comorbidity and extraintestinal manifestations are particularly relevant for monogenic IBD diagnostic considerations when rare or atypical for patient age irrespective of organ manifestation
Recurrent severe infections or atypical infections consistent with diagnostic criteria of a primary immunodeficiency
Hemophagocytic lymphohistiocytosis
Autoimmune features in particular features of Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome
Malignancies
Multiple intestinal atresias
Age of Inflammatory Bowel Disease Onset as a Predictor of Monogenic Inflammatory Bowel Disease
The age of onset of IBD is a strong predictor for the risk of monogenic IBD (Tables 1 and 2). The majority of patients with monogenic IBD present in the first 6 years of life (ie, very early-onset IBD [VEOIBD]) (16,31,34,35). The age of onset, however, forms a spectrum and across a large number of gene defects and individual patients do present in paediatric care beyond 6 years of age (29).
Studies in patients with infantile-onset IBD (ie, <2 years of age at disease-onset) identified a monogenic cause in 13% to 41%, in patients with VEOIBD in 0% to 33%, and in patients with IBD-onset between 6 and 18 years in 0% to 38% depending on the preselection of the patients investigated (Table 1). Among all paediatric IBD patients ages <18 years in a single centre, monogenic IBD was identified in 3% (36).
Diagnosing a monogenic cause of IBD during adulthood is exceptional. Sequencing studies in adult-onset IBD populations have not identified monogenic IBD (37,38). Among the exceptional rare gene defects that are associated regularly with adult-onset IBD is congenital diarrhea because of GUCY2C defects (39). Adult age-onset IBD is a rare but consistent finding in patients with XIAP defects, who were either previously symptomatic because of immunodeficiency or even nonsymptomatic (40). Hypomorphic variants in XIAP without clinical consequence for the patients are more common in patients with IBD-onset >6 years of IBD-onset, suggesting a role as modifier variant and a spectrum of pathogenicity (38).
Family History as Predictor of Monogenic Inflammatory Bowel Disease
Consanguinity, a family history of autoimmune disease, and family history of suspected or confirmed monogenic disorders are associated with monogenic IBD (16,36) (Table 2). Male predominance is a sign of X-linked disorders, such as XIAP deficiency or IPEX syndrome (41). Suspicion is highest if similar disease phenotypes are observed in several family members. A positive family history is, however, not specific as multiple affected family members can also be found in classical IBD, males predominate in paediatric-onset Crohn’s disease overall, and a quasi Mendelian inheritance pattern has been described in families with NOD2 variants (42,43). On the other hand, monogenic IBD disorders, such as LRBA deficiency (44,45), or CTLA4 haploinsufficiency (46) can present with quite diverse phenotypes reflecting variable expression and incomplete penetrance. In those defects, a Mendelian inheritance pattern can be clouded even if several family members are affected.
Clinical Features of Monogenic Inflammatory Bowel Disease Including Comorbidities and Extraintestinal Manifestations
Current data suggest a limited diagnostic value of endoscopic IBD classification for the diagnosis of monogenic IBD (Tables 1 and 2) whereas some histologic features may raise the suspicion for monogenic IBD (Table 3).
TABLE 3.
Phenotypic features of monogenic inflammatory bowel disease (exemplars)
| Phenotypic features | Exemplar disorder and gene defect | |
|---|---|---|
| Infection | Recurrent typical (eg, Staphylococcus aureus) or single/recurrent atypical infections (eg, mycobacterial, fungal or cytomegalovirus in patients without immunosuppressive therapy | Primary immunodeficiency eg, chronic granulomatous disease |
| Immune activation with and without infection | Hemophagocytic lymphohistiocytosis (HLH) | XIAP (55) and STXBP2 (56). HLH is not specific since a known complication of cytomegalovirus and Epstein-Barr virus infection in patients receiving immunomodulatory medications including thiopurines (57) |
| Autoimmune features | Immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX or IPEX-like) syndrome | FOXP (41), LRBA (45), CTLA4 (58), STAT3 (59), and STAT1 (60). |
| Dermatological features | Oral leukoplakia Ectodermal dysplasia with dysplastic nails and conical teeth |
Telomeropathies (61) NF kappa B essential modulator (NEMO) IKBKG defects (62) |
| Tumours | Woolly hair with trichorrhexis nodosa B cell lymphomas Gastric adenocarcinomas |
Trichoenterohepatic syndrome because of TTC37A (63) or SKIVL2 (64) IL-10 signalling defects (65) CTLA4 (46) and LRBA (66) |
| Intestine Endoscopic and histology features |
Multiple intestinal atresia Complex perianal fistulizing disease accompanying luminal inflammation, especially if manifest in the first year of life Intestinal epithelial cell apoptosis Tissue eosinophilia Enteropathy with villous flattening, similar to celiac disease Germinal cell hypoplasia Granulomas and pigmented macrophages |
TTC7A (67) IL10, IL10RA, and IL10RB (68), TGFB1 (69), or XIAP (40,70). TTC7A, LRBA, XIAP, SH2D1A, ARPC1B, or COL7A1 (29,67,71) IPEX, IPEX-like syndrome or WAS (72) IPEX, or IPEX-like syndrome and CVID (41,44,58) Defect in humoral immunity such as in ICOS or BTK deficiency (73,74) Chronic granulomatous disease |
Comorbidities and extraintestinal features are significantly associated with monogenic IBD (Table 2). We use the terms ‘‘comorbidities and extraintestinal manifestations’’ aligned to acknowledge the fact that potentially unrelated comorbidities in patients with suspected monogenic IBD (pre-test definition) are in fact often extraintestinal manifestations in accordance with the broader phenotypic disease spectrum of individual gene defects (postdiagnosis definition). Several reviews have provided an overview of extraintestinal features of the diverse immunodeficiency and epithelial cell disorders that can present with intestinal inflammation (10,12,29). Those features include recurrent infections, hemophagocytic lymphohistiocytosis (HLH), autoimmune and dermatological features as well as development of malignancy (Table 3).
It is important to note that not all rare extraintestinal manifestations and comorbidities are a consequence of monogenic IBD and that intestinal inflammation in a rare Mendelian disease is not always caused by the gene defect. For instance, hemophagocytic lymphohistiocytosis can be a medication-induced side effect of thiopurines used to treat IBD (Table 3). De novo intestinal inflammation in patients with Mendelian disorders can be a consequence of treatment of malignancy by immune checkpoint inhibitor (anti-CTLA4 or anti-PD1) or mofetil mycophenolate treatment after solid organ transplantation (75).
Laboratory Features Associated With Immune Dysfunction in Monogenic Inflammatory Bowel Disease
Abnormal immune cell numbers and function and abnormal immunoglobulin levels in a patient with intestinal inflammation can suggest a primary immunodeficiency (Table 4). A pragmatic basic workup for patients with IBD and suspected monogenic IBD includes a limited number of essential laboratory tests (Table 4). There are no data in support of more extensive laboratory studies in routine clinical practice. The infection history, auto-immune phenotype, and abnormal immunological laboratory features may, however, trigger more specialized investigations as part of an interdisciplinary effort.
TABLE 4.
Inflammatory markers, immunology, and infectious diseases workup
| Test | Indication and examples of monogenic IBD disease groups were abnormal results expected | |
|---|---|---|
| Blood work essential | Complete blood count (Neutrophils, Thrombocytes, lymphocytes) | Autoimmune neutropenia Neutrophilia (leucocyte adhesion deficiency) Autoimmune thrombocytopenia Congenital neutropenia Inflammation activity—nonspecific |
| Basic immune blood work | Inflammatory markers (CRP, ESR) Immunoglobulin classes (IgA, IgG, IgM, IgE; age-specific normal range) Lymphocyte subsets DHR testing |
CVID Agammaglobulinemia CVID, Agammaglobulinemia Chronic granulomatous disease |
| Blood tests to consider depending on presentation | TREC/TCR repertoire Vaccine antibodies (vaccination history) Autoantibodies coeliac screen Anti-enterocyte antibodies Thyroid function tests, Liver function test Type 1 diabetes autoantibodies Metabolic workup FOXP3 or XIAP-expression* MDP-monocyte stimulation assay IL10-induced phospho-STAT3 or LPS/IL-10 suppression-assay |
Hypomorphic SCID Infection susceptibility CVID, Agammaglobulinemia Exclude coeliac disease Autoimmune enteropathy IPEX and IPEX-like Autoimmune hepatitis, thyroiditis, diabetes, and so forth Glycogen storage disease IB Niemann Pick Type C IPEX, XIAP deficiency XIAP deficiency IL-10 signalling defects |
| Basic infection screen | TB Elispot assay HIV serology |
Exclude infections |
| Stool tests | Microbiology to exclude bacterial and parasitic enteric infections Calprotectin stool |
Exclude infections Inflammation activity—nonspecific |
CVID = combined variable immunodeficiency; DHR = dihydrorhodamin test; HIV = human immunodeficiency virus; IPEX = TB tuberculosis; MDP = muramyl dipeptide; TREC/TCR = T-cell Receptor Excision Circles/T- cell receptor oligoclonal expansion.
A normal expression does not exclude FOXP3 or XIAP deficiency but a substantial proportion of patients can be detected.
Genetic Screening in Advance of Interventions Associated With High Morbidity and Mortality
Genetic investigations to establish monogenic IBD can guide appropriate therapies and inform on risk benefit of therapies associated with high morbidity and mortality (Table 1).
Results of genetic investigations in patients with suspected monogenic IBD guide the application of allogeneic hemopoietic stem cell transplantation in several aspects. Allogeneic hemopoietic stem cell transplantation has developed into a de facto standard of care for several monogenic IBD disorders associated with primary immunodeficiencies, in particular, IL10-signalling defects or regulatory T-cell defects (76–77). For other monogenic IBD disorders with epithelial defects, such as TTC7A or IKBKG defects, allogeneic haematopoietic stem cell transplantation is less or not at all effective (78,79). This means that for some patients with monogenic IBD defects, the likely therapeutic benefit clearly outweighs the transplant-associated mortality and morbidity (such as graft-vs-host disease, medication toxicity, infections) whereas patients with epithelial conditions will unlikely benefit while still experiencing potential complications. Genetic screening can identify patients with pure epithelial defects and prevent progression to haematopoietic stem cell transplantation (47). In patients with monogenic IBD defects that affect both hematopoietic and nonhematopoietic (epithelial) cell lineages (eg, CASP8, IKBKG, or RIPK1), weighing risks and benefits of allogeneic HSCT remains challenging (80–82).
Patients with IL10-signalling defect have an increased susceptibility for lymphoma (83). Whereas in patients with many forms of lymphoma, chemotherapy and autologous hematopoietic stem cell transplantation is a standard of care, autologous transplantation does not correct the underlying genetic driver in IL10-signalling defect. Genetic analysis can, therefore, prevent autologous haematopoietic stem cell transplantation in patients with monogenic IBD defects and inform progression to allogeneic haematopoietic stem cell transplantation (83).
In some patients with monogenic severe therapy refractory Crohn’s disease, multiple resections can cause short gut syndrome. Establishing the genetic diagnosis of XIAP deficiency in a patient with short gut syndrome by exome sequencing resulted in change of clinical management from a proposed small bowel transplantation to allogeneic HSCT (37). Establishing a genetic diagnosis early in the course of the disease may prevent surgery by progressing with curative allogeneic HSCT.
A Genetic Diagnosis of Monogenic Inflammatory Bowel Disease Can Inform on Pharmacological Treatment Options
Preliminary data suggest that patients with distinct monogenic disorders might benefit from specific pharmacologic interventions that are currently not standard of care in patients with classical IBD. Case reports or noncontrolled small case series suggest that patients with IL10-signalling defects and mevalonate kinase deficiency might benefit from IL-1-targeting therapies (84,85), patients with NLRC4 defects from IL-18 or IL-1-targeting treatments (86,87), whereas patients with CTLA4 and LRBA defects can benefit from CTLA4 fusion protein abatacept (88,58).
Prenatal Testing in Families With History of Infantile Inflammatory Bowel Disease
Families with children affected by severe genetic disorders may ask whether predictive prenatal testing can inform recurrence of the disorder in subsequent pregnancies. A recent survey among clinicians in tertiary centres of 10 countries reported referrals for prenatal genetic testing for IL10-signalling defects, that is, a form of monogenic IBD with severe phenotype and complete penetrance in 4 countries (89). Prenatal testing in families with known IL10RA defects was performed as targeted preimplantation test after in-vitro fertilization or as targeted genetics after intrauterine amniocentesis/chorion villus sampling. Prenatal diagnostics requires specific clinical genetic counselling and poses great ethical challenges because of the potential implications of embryo selection or termination of a pregnancy (89).
What Monogenic Disorders Should Be Included in Genetic Sequencing Panels?
Gene sets for monogenic IBD disorders have been discussed in recent literature reviews (10–13). Among those, there is considerable heterogeneity; 32 genes are discussed in all 4 reviews (Table 5). Similarly, there are differences in the gene panel setup of commercial, clinical, and research-focused targeted gene panel assays aiming to screen patients with monogenic IBD, infantile IBD, and/or congenital diarrhoea to include between 21 and 160 genes (Table 1, Supplemental Digital Content, http://links.lww.com/MPG/C125). Those differences can be explained by several factors: an increasing number of candidate genes is identified over time, focus on slightly different patient cohorts (ie, focus on immunodeficiency genes and/or congenital diarrhoea depending on the referral population), different definitions on what defines a causative monogenic IBD gene, and inclusion of genes that are (currently) of research interest but not established as monogenic cause of disease, some panels include genes informed by polygenic risk loci. Acknowledging those factors, we reviewed the gene list and agreed on a consensus list of 75 monogenic IBD genes (Table 5). Depending on the phenotype and the family history, a variable set of candidate genes can be prioritized for the individual patient. The emerging number of newly described genetic causes and the process of variant validation over time means that this list of monogenic IBD genes will evolve and needs updating. The vast majority of these genes has been recognized as disease-causing by an independent international expert committee of the international union of immunological societies (90).
TABLE 5.
Monogenic inflammatory bowel disease gene panel suggested by specialist reviews and consensus (75 genes)
| Gene | Disorder | Inheritance AR/AD/XL | Uhlig and Muise 2017 | Ouahed et al, 2020 | Kelsen et al, 2019 | Pazmandi et al, 2019 | Consensus |
|---|---|---|---|---|---|---|---|
| ADA | Atypical SCID | AR | + | + | + | + | |
| ADAM17 | Inflammatory skin and bowel disease, neonatal | AR | + | + | + | + | + |
| AICDA | Immunodeficiency with hyper-IgM | AR | + | + | + | + | + |
| ALPI | Intestinal Alkaline Phosphatase deficiency | AR | + | + | |||
| ARPC1B | Wiskott-Aldrich syndrome-like | AR | + | + | + | ||
| BTK | Agammaglobulinemia, X-linked 1 | XL | + | + | + | + | + |
| CASP8 | Caspase-8 deficiency | AR | + | + | + | ||
| CD3G | Atypical SCID | AR | + | + | + | ||
| CD40LG | Immunodeficiency, X-linked, with hyper-IgM | XL | + | + | + | + | + |
| CD55 | CHAPLE syndrome | AR | + | + | + | ||
| COL7A1 | Dystrophic epidermolysis bullosa | AR | + | + | + | ||
| CTLA4 | Autoimmune lymphoproliferative syndrome, type V | AD | + | + | + | + | + |
| CYBA | Chronic granulomatous disease | AR | + | + | + | + | + |
| CYBB | Chronic granulomatous disease | XL | + | + | + | + | + |
| DCLRE1C | Omenn syndrome | AR | + | + | + | + | |
| DKC1 | Dyskeratosis congenita—Hoyeraal Hreidarsson Syndrome | XL | + | + | + | + | + |
| DOCK8 | Dedicator of Cytokinesis 8 (DOCK8) deficiency | AR | + | + | |||
| FERMT1 | Kindler syndrome | AR | + | + | + | + | |
| FOXP3 | Immunodysregulation polyendocrinopathy enteropathy X-linked syndrome | XL | + | + | + | + | + |
| G6PC3 | Congenital neutropenia | AR | + | + | + | + | |
| GUCY2C | Familial diarrhea | AD | + | + | + | + | |
| HPS1 | Hermansky-Pudlak syndrome (type 1) | AR | + | + | + | ||
| HPS4 | Hermansky-Pudlak syndrome (type 4) | AR | + | + | + | + | |
| HPS6 | Hermansky-Pudlak syndrome (type 6) | AR | + | + | + | ||
| ICOS | ICOS deficiency | AR | + | + | + | + | + |
| IKBKG | X-linked ectodermal dysplasia, anhidrotic and immunodeficiency | XL | + | + | + | + | |
| IL10 | IL10 deficiency | AR | + | + | + | + | + |
| IL10RA | IL10 receptor deficiency | AR | + | + | + | + | + |
| IL10RB | IL10 receptor deficiency | AR | + | + | + | + | + |
| IL21 | IL21 deficiency (Combined variable immunodeficiency-like 11) | AR | + | + | + | + | |
| IL2RA | IPEX-like (Immunodeficiency with lymphoproliferation and autoimmunity) | AR | + | + | + | + | + |
| IL2RB | IL2RB Immune Dysregulation | AR | + | + | |||
| IL2RG | Atypical SCID | XL | + | + | + | + | |
| ITCH | ITCH deficiency | AR | + | + | + | ||
| ITGB2 | Leukocyte adhesion deficiency 1 | AR | + | + | + | + | + |
| LIG4 | Atypical SCID | AR | + | + | + | + | + |
| LRBA | Combined variable immunodeficiency (CVID 8) | AR | + | + | + | + | + |
| MALT1 | MALT1 deficiency (IPEX-like) | AR | + | + | + | ||
| MASP2 | Mannan Binding Lectin Serine Peptidase 2defect | AR | + | + | + | ||
| MVK | Mevalonate kinase deficiency | AR | + | + | + | + | + |
| NCF1 | Chronic granulomatous disease | AR | + | + | + | + | |
| NCF2 | Chronic granulomatous disease | AR | + | + | + | + | + |
| NCF4 | Chronic granulomatous disease | AR | + | + | + | + | + |
| NLRC4 | Autoinflammation with infantile enterocolitis | AD | + | + | + | + | |
| NPC1 | Niemann-Pick type C disease | AR | + | + | + | + | |
| PIK3CD | PIK3CD deficiency and PI3K activation syndrome | AR&AD | + | + | + | + | + |
| PIK3R1 | Agammaglobulinemia type 7 and activated PI3K syndrome | AR&AD | + | + | + | + | + |
| PLCG2 | Autoinflammation, antibody deficiency, and immune dysregulation syndrome | AD | + | + | + | + | + |
| POLA1 | PDR syndrome (pigmentary disorder, reticulate, with systemic manifestation) | XL | + | + | + | ||
| RAG1 | Atypical SCID | AR | + | + | + | ||
| RAG2 | Atypical SCID | AR | + | + | + | + | |
| RIPK1 | RIPK1 deficiency | AR | + | + | |||
| RTEL1 | Dyskeratosis congenita – Hoyeraal Hreidarsson Syndrome | AR/AD | + | + | + | + | + |
| SH2D1A | X-linked lymphoproliferative syndrome 1 (XLP1) | XL | + | + | + | ||
| SKIV2L | Trichohepatoenteric syndrome 2 | AR | + | + | + | + | + |
| SLC37A4 | Glycogen storage disease type 1b | AR | + | + | + | ||
| SLC9A3 | Congenital diarrhea | AR | + | + | + | + | |
| SLCO2A1 | Prostaglandin transporter deficiency | AR | + | + | + | + | |
| STAT1 | IPEX-like | AD | + | + | + | + | |
| STAT3 | Autoimmune disease, multisystem, infantile-onset, 1 | AD | + | + | + | + | + |
| STIM1 | STIM1 deficiency | AR | + | + | + | ||
| STXBP2 | Familial hemophagoytic lymphohistiocytosis type 5 | AR | + | + | + | + | + |
| STXBP3 | Syntaxin binding protein 3 defect | AD/AR | + | + | |||
| TGFB1 | TGFB1 deficiency | AR | + | + | |||
| TGFBR1 | Loeys-Dietz syndrome 1 | AD | + | + | + | ||
| TGFBR2 | Loeys-Dietz syndrome 2 | AD | + | + | + | ||
| TNFAIP3 | Autoinflammatory syndrome, familial, Behcet-like Syndrome | AD | + | + | |||
| TRIM22 | TRIM22 defect | AR | + | + | + | + | |
| TRNT1 | SIFD (sideroblastic anemia, immunodeficiency, periodic fevers and developmental delay) | AR | + | + | |||
| TTC37 | Trichohepatoenteric syndrome 1 | AR | + | + | + | + | + |
| TTC7A | TTC7A deficiency | AR | + | + | + | + | + |
| WAS | Wiskott-Aldrich syndrome | XL | + | + | + | + | + |
| XIAP | X-linked lymphoproliferative syndrome 2 (XLP2) | XL | + | + | + | + | + |
| ZAP70 | Atypical SCID | AR | + | + | + | + | |
| ZBTB24 | Immunodeficiency, centromeric instability and facial anomalies (ICF) syndrome | AR | + | + | + |
Are There Preferred Sequencing Technologies?
Due to the increasing number of candidate genes, the use of classical Sanger sequencing of candidate genes has been replaced by parallel sequencing technologies. Sanger sequencing is still an effective, fast, and economic way to confirm a suspected genetic diagnosis in a family with a known genetic variant or in a patient with a pathognomonic phenotype. There is now ample evidence that panel sequencing (47,52), exome sequencing (8,36,37,47,48,52), as well as genome sequencing (21,91,92) are excellent tools to investigate genetic variants in patients with suspected monogenic IBD. Comparative studies in patients with IBD have confirmed that sequencing read coverage and diagnostic accuracy of panel sequencing is higher than exome sequencing (47,52). Several clinical genetic laboratories and additional commercial companies offer panel sequencing assays aimed for early-onset IBD (Table 1, Supplemental Digital Content, http://links.lww.com/MPG/C125). It is in the nature of the assay that updating the panel of genes is required over time and retesting of patient samples with a high suspicion of a monogenic IBD is required either with an updated panel or subsequent progression with exome or genome sequencing. Since the first description of exome sequencing in a patient with IBD and XIAP deficiency (70), exome sequencing has demonstrated its clinical utility and potential for discovery (11,19). Using exome sequencing, a specified ‘‘virtual’’ monogenic IBD panel can be used for the initial screening, and exome-wide analysis can be performed as part of a subsequent analysis. Genome sequencing has not yet fulfilled its full potential for clinical genomics as current information of the noncoding elements in IBD is still restricted and its analysis is complex (21). Genome sequencing care pathways for infantile onset IBD are currently being evaluated in a formal trial of the National Genomic program in England (https://www.genomicsengland.co.uk). The genetic DNA sequencing technologies might be complemented by copy number variation analysis via multiplex ligation-dependent probe amplification. Other variants might require validation via RNA sequencing to confirm relevant splice variants.
Which sequencing technology to use in clinical practice depends on the clinical setting and the resources available. For example, regional or national health care services may opt for well-designed panel sequencing approaches as a first-line strategy, whereas academic centres (single centres or national research hubs) may prefer standardized exome or genome sequencing platforms.
In the context of exome-wide sequencing, analysis of patient and parents (family trios) has a higher discovery rate than singleton patients sequencing (20,22). Indeed, the use of trio analysis might explain in part the higher diagnostic rate in a paediatric IBD population compared with previous studies (36).
Analysis and interpretation of genomic data should be performed by specialists trained in clinical genomic medicine. All variants within each gene should be classified as either ‘‘pathogenic,’’ ‘‘likely pathogenic,’’ or ‘‘variants of unknown significance.’’ Databases, such as Clinvar, ClinGen, or The Human Gene Mutation Database can help to assess variant phenotype relations (93,94), although there is currently no single repository of classified monogenic IBD gene variants. Deposition of variants of unknown significance via Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/) or sharing variants via Matchmaker (https://www.matchmakerexchange.org) can initiate communication with interested research groups and facilitate characterization. In some patients, in particular, in consanguineous families, occasionally more than 1 plausible monogenic defect is identified. As annotation and assembly of the human genome is an ongoing project and computational algorithms are constantly being improved, the identification of genetic variants is also subject to evolution.
Is a Functional Validation of Genetic Variants Required Before Genetic Variants Can Be Regarded as Pathogenic?
The causal relation of a genetic finding in a patient with potential monogenic IBD needs to be established on a gene as well as variant level. Relying solely on genetic screening can be misleading as computational variant prediction can either fail to detect functional damaging variants (false-negative) or predict damaging effects in neutral variants (false-positive). It is, therefore, important that novel variants in known genes are functionally validated and assessment of variants in novel candidate genes typically requires further research. In principle, functional studies are required to show that genetic variants cause disease through loss-of-function, or gain-of-function via altered protein function or expression, or localization of the gene product.
It is a limitation that in routine diagnostic laboratories, functional tests are currently available only for a fraction of genes associated with monogenic IBD. Among those tests used in the routine clinical immunology labs, the dihydrorhodamine test is standardised to detect defective neutrophil NADPH oxidase activity in patients with chronic granulomatous disease. Other functional tests assess IL10 signalling (function of the IL10 receptors—IL10RA and IL10RB deficiency), or IL10 secretion in response to LPS, and muramyl dipeptide-induced signalling (XIAP deficiency) (29). Flow cytometry studies for XIAP, SLAM-associated protein (SAP), CTLA4, LRBA, and FOXP3 protein expression will identify a large proportion of patients who harbour gene defects with deletion, stop codon, or frameshift variants in those genes (95,96). In patients with suspected IPEX or IPEX-like syndrome measurement of a range of autoantibodies including antithyroid antibodies, type 1 diabetes-specific antibodies, and anti-enterocyte antibodies can be adequate (41).
Some genetic defects show cell-type specific effects, some variants affect isoforms, some variants only affect a fraction of cells, that is, mosaicism (8). With increasing use of sequencing technologies, it becomes clear that there are many hypomorphic defects and it can be challenging to assess whether a gene variant is a pathogenic variant or a risk factor [NADPH oxidase complex (97); XIAP (38,98)]. Functional validation usually requires in-depth studies either based on primary patient-derived cells or cell lines that are modified to express patient-derived mutations. To assess compound heterozygosity, the effects of both genetic variants should be tested. For a large number of genes, functional validation assays are available in the research setting only. Complex in-vitro models based on organoids or induced pluripotent stem cells (iPS cells) may be employed to study cell types, such as epithelial cells or macrophages (99,100). In-vivo models include animals, such as zebrafish or mice (eg, humanised mouse models) (101). As the clinical analysis depends on research findings, translational research studies must adhere to clear standards when reporting variants and associated pathogenicity. Without performing functional validation studies, research results are potentially misleading (49,54).
Are Genomic Screening Technologies for Diagnosis of Monogenic Inflammatory Bowel Disease Cost-effective?
Formal studies on the overall diagnostic costs of genomic diagnostics in IBD are lacking. Among the next-generation sequencing technologies, the technical sequencing costs are lowest with panel sequencing and highest with genome sequencing (102). Whereas the technical sequencing costs drop over time aiming for costs less than $1000 per genome and less than $500 per exome, the overall costs of the genetic analysis are still substantial. A systematic literature review that included studies until the year 2016 estimated the cost for a single test of exome sequencing from $555 to $5169 and for genome sequencing from $1906 to $24,810 (102). Another study calculated the per-sample costs €1669 for genome sequencing, €792 for exome, and €333 for targeted panel sequencing (103). A microcosting analysis of genome sequencing in a UK National Health Service laboratory processing 399 samples per year estimated the true costs of analysis several times higher than the technical sequencing costs (104). Targeted gene panel sequencing has emerged as a diagnostically accurate and cost-effective technology with approximately 25% the sequencing cost of exome sequencing and less analytic complexity. Petersen et al (52) performed targeted panel sequencing in a cohort of 71 patients with early onset IBD or early onset chronic diarrhoea and compared the findings to exome sequencing performed in 25 of these patients. Costs can differ in different health care settings. Many centres see the trade-off between costs and the expected clinical utility currently in favour of performing targeted panel or exome sequencing as first-line analysis.
Health economic studies are currently not available to assess how genomic diagnostic costs in different care pathways compare long term in patients with suspected monogenic IBD. Although not related to monogenic IBD, a recent prospective study demonstrating cost-effectiveness of early exome sequencing in relation to all diagnosis-related investigations including Sanger sequencing in a group of 40 children with a range of suspected Mendelian disorders (105). An early genetic diagnosis may prevent a diagnostic odyssey with associated diagnostic costs, treatments, operations, and hospitalization in those patients. Case reports, case series, and cohort studies suggest a long diagnostic delay in some patients and confirm that a genetic diagnosis after performance of genomic screening technologies does lead to fundamental change in clinical management, years to even decades after the onset of intestinal inflammation (8,36,37,91).
Multidisciplinary Care Pathways and Clinical Genomics in Monogenic Inflammatory Bowel Disease
In light of the individual benefit for patients and their families, targeted panel sequencing and exome sequencing strategies for patients with suspected monogenic IBD have been implemented in routine clinical care by health care providers in several countries (eg, Switzerland, UK, Israel, USA).
Several diagnostic algorithms for monogenic IBD have been proposed (Figure 1 and Figure 1, Supplemental Digital Content, http://links.lww.com/MPG/C126). The use of genetic technologies complement the diagnostic algorithms for the diagnosis of paediatric Crohn’s disease, UC, and IBDU to establish the extent and activity of the intestinal inflammation (32,106). Due to the continuously increasing number of Mendelian disorders that can cause IBD, the prediction of a single candidate gene based on clinical presentation is unreliable even to the most experienced clinicians. Next-generation sequencing technologies are therefore key to help establish the diagnosis of monogenic diseases associated with IBD.
FIGURE 1.
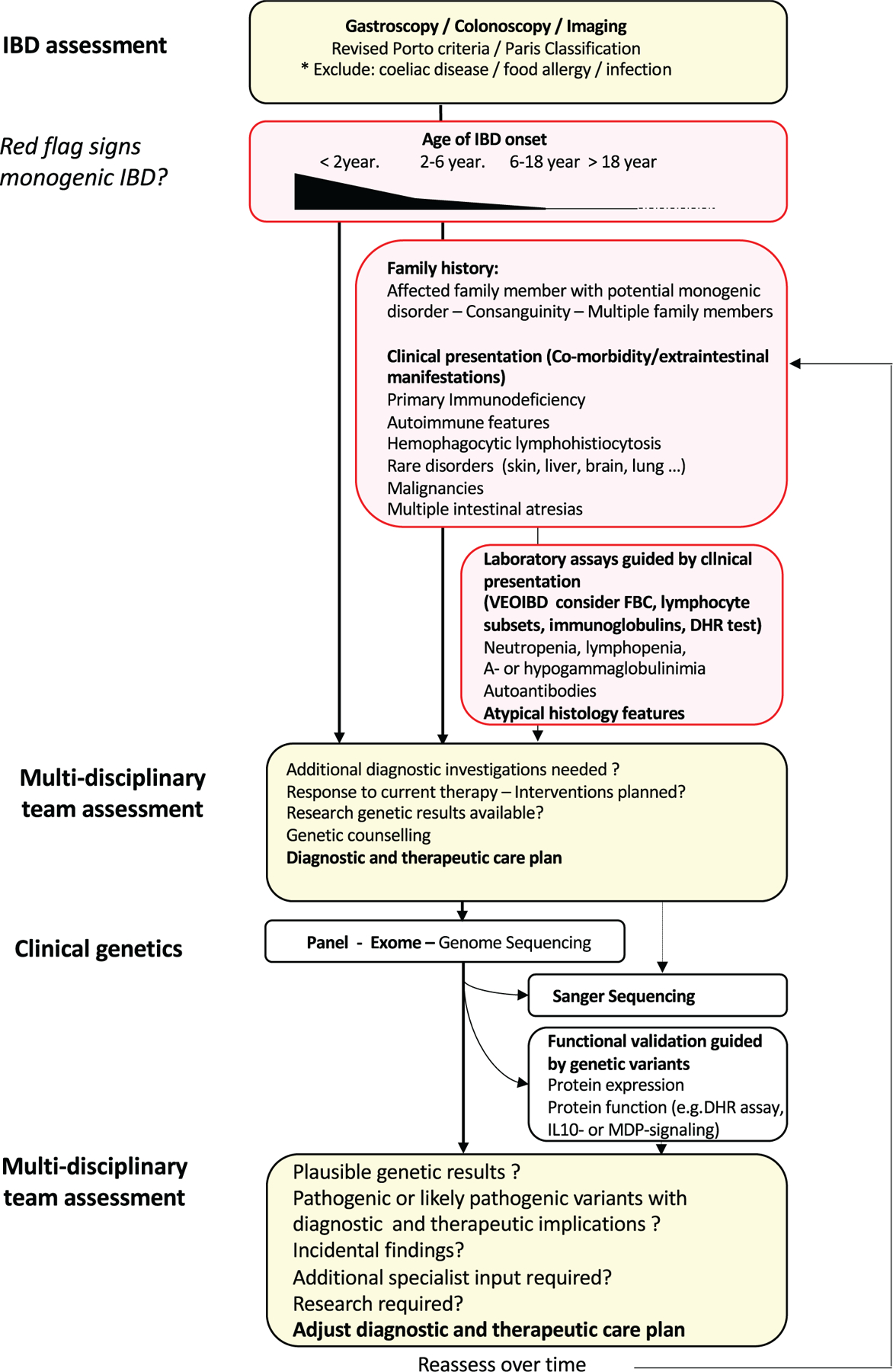
Diagnostic algorithm monogenic inflammatory bowel disease. Patients with endoscopically and histologically confirmed diagnosis of inflammatory bowel disease are assessed for red flag signs that might raise suspicion for a monogenic IBD (age of onset, family history, clinical presentation, and laboratory results; Tables 3 and 4). In patients with suspected monogenic IBD (either <2 year of onset of IBD or >2 year of IBD-onset with additional red flag features) a multidisciplinary team assessment will help to establish a diagnostic and therapeutic care plan. After appropriate counselling of the family, clinical sequencing will be performed based on availability of the technologies, the expected coverage of likely monogenic IBD candidate genes and urgency to have results available. The role of suspected genetic variants will be functionally validated und results will be discussed by a multidisciplinary team to assess the therapeutic consequences and to amend the diagnostic and therapeutic care plan of the patient. IBD = inflammatory bowel disease.
Qualitative reports and reviews describe how clinical genomics pathways for patients with suspected monogenic IBD are best organised (10,11,107). Multidisciplinary care of patients with expected or confirmed monogenic IBD is key to deliver state-of-the-art patient-centred care. Setup of dedicated clinics with focus on VEO-IBD, monogenic IBD, and genomic medicine can help to implement such interdisciplinary care (107). This supports provision of specialized paediatric gastroenterologist service care jointly with immunology and genetic services, as well as additional disciplines, such as radiology, surgery, rheumatology, dermatology, dietetics, pharmacists (potential use of off licence treatments), psychology (severe chronic disorders, family support), primary care clinicians, as well as ethicists (implications for other family members, novel treatment options, families who consider prenatal screening), and research scientists. Individual specialities such as paediatric radiology (108) have highlighted the specific needs of patients with VEO-IBD and monogenic IBD within their diagnostic algorithms. Virtual multidisciplinary case discussions can be helpful to connect not only local but national and international specialists.
Before genetic investigations, it is important to discuss indication, previous diagnostic findings, the genetic screening process, the likelihood to identify incidental findings, and the potential therapeutic consequences (Fig. 1). Up-to-date standards and guidelines and practice resource for clinical genomics are available via specialist societies, such as the European Society of Human Genetics (109) or the American College of Medical Genetics (https://www.nature.com/gim/). There are country-specific rules on handling patient consent, genetic data storage, and data protection as well as incidental findings. Children should always be encouraged to actively engage in the consent/assent process, respecting their rights in light of their age-related cognitive development (110). After the genetic investigations, a multidisciplinary team meeting helps to assess the genetic and functional validation results, to assess the need of additional diagnostic tests and therapeutic implications, to discuss incidental findings, and to prepare communicating genetic findings to patients (Fig. 1). Given the complexity of the findings, it is important to communicate the genetic test results to patients and their families as well as nonspecialists in a understandable way (111). How to communicate information about less well-understood genetic variants is a matter of debate. In light of the large number of variants of unknown significance, there is good reason not to report variants of unknown significance (112). Pre- and post-test counselling on variants of unknown significance and their spectrum of potential consequences can help to reduce misinterpretations, anxiety, as well as decisional regret (113).
Although centralised sequencing and analysis has several advantages, hospital-based analysis was associated with higher diagnostic yield compared with centralised exome analysis suggesting that local expert knowledge (on patient and phenotype) contributed significantly to the increased diagnostic yield (22).
The urgency of genetic testing is influenced by the clinical condition of the patient and the therapeutic implications of the genetic results. The former can change and the latter are difficult to predict before sequencing results are received, however. This is illustrated by a patient with VEO-IBD who was in stable condition at the time of study participation but died after an unexpected EBV infection response and liver failure because of X-linked lymphoproliferative type 1 disorder before the exome sequencing results became available (8). An ‘‘emergency’’ sequencing is rarely required but technically feasible in critically ill infants (114). In a patient with infantile-onset Crohn’s disease, such an emergency 24-hour trio-exome sequencing was performed revealing compound heterozygous IL10RA defects allowing to proceed rapidly with curative haematopoietic stem cell transplantation (114).
For young adult patients with suspected or confirmed monogenic monogenic IBD, defined arrangements for transitional care from paediatric to adult care is important. In some patients, the monogenic IBD diagnosis has been made only when transitioned to adult gastroenterology care emphasizing decades of diagnostic delay some patients face (37,91).
Recommendations and Position Statement
There is sufficient evidence to recommend the use of next-generation DNA sequencing technologies to diagnose known monogenic causes of IBD in routine clinical practice. We propose a diagnostic pathway for monogenic IBD that complements the standard IBD guideline workup, facilitates multidisciplinary team assessment of patients with suspected monogenic IBD, supports genetic counselling and consent to research if appropriate, and implements next-generation sequencing technologies as well as multidisciplinary team assessment of genetic results (Fig. 1). This diagnostic pathway involves the full assessment of the patients clinical phenotype, the family history, the test results from previous immunological investigations (Table 4), as well as interpretation of results and incidental findings and its implications for prognosis and therapies. We formulated 9 statements on the use of genomic technologies in routine clinical care (Table 6). Special considerations for low-resource countries are formulated to address the fact that specialised clinics with focus on monogenic IBD and clinical genetics next-generation sequencing technologies are difficult to access and that socioeconomic conditions, such as lack of health insurance coverage prevent access to those services (Box 3).
TABLE 6.
Statements
| Statements | Consensus rate |
|---|---|
| 1. The diagnostic process and care of a patient with suspected or confirmed monogenic IBD is best coordinated by a multidisciplinary team of specialists, including gastroenterologists, geneticists, immunologists, and other subspecialists contingent on the individual gene defect, comorbidities and extraintestinal manifestations | 32/32 (100%) |
| 2. Next-generation DNA sequencing technologies are recommended to diagnose known monogenic causes of IBD in routine clinical practice | 32/32 (100%) |
| 3. Genetic screening for monogenic IBD is recommended in all patients with infantile-onset IBD (<2 years) and should be considered in patients with very early-onset IBD (<6 years), in particular, in those patients with relevant comorbidity, extraintestinal manifestations, and/or family history | 31/32 (97%) |
| 4. Although a rare or very rare diagnosis, a monogenic form of IBD should be considered in patients with any paediatric or adult age IBD-onset if they present with relevant comorbidity, extraintestinal manifestations, and/or family history | 27/32 (84%) |
| 5. Routine genetic screening for all IBD patients is not recommended since a monogenic cause of IBD in patients with IBD onset over 6 year of age, especially those with adolescent or adult age onset of IBD is exceptional in the absence of relevant comorbidity | 32/32 (100%) |
| 6. Genetic investigations to establish monogenic IBD are recommended in advance of hematopoietic stem cell transplantation unless the bowel inflammation can be clearly explained (eg, drug-induced colitis) | 32/32 (100%) |
| 7. Panel sequencing, exome, and genome sequencing technologies have complementary diagnostic strength; the first-line technology should be guided by availability and degree of diagnostic suspicion | 30/32 (94%) |
| 8. Functional assessment of novel gene defects and variants of unknown significance is necessary to establish causality | 32/32 (100%) |
| 9. Patients and their families with suspected or confirmed monogenic IBD should be offered the opportunity to participate in research studies. A therapeutically relevant genetic result established in a research setting should be confirmed in a clinical genetics setting | 30/32 (94%) |
IBD = inflammatory bowel disease.
Comments: relevant comorbidities and extraintestinal manifestations and family history are summarised in Box 2.
Box 3. Specific considerations for low-resource countries.
To support patients and families with suspected monogenic inflammatory bowel disease (IBD) within low-resource settings, knowledge on local health care pathways, health care insurance provision, ethnic and religious considerations are important to choose effective and pragmatic diagnostic pathways and to plan subsequent provision of relevant therapies
Socioeconomic conditions and regional environmental factors, such as infections that can mimic IBD and primary immunodeficiencies within different parts of the world (environmental enteropathy, intestinal tuberculosis, etc) and are therefore, important considerations to assess the pre-test risk of monogenic IBD
Access to multidisciplinary teams might be facilitated by centralised care of children with suspected monogenic IBD within each country and via international collaboration
Access to multidisciplinary teams might be facilitated via virtual clinics
Sanger sequencing can be a cost-effective test strategy in particular in regions with enrichment of known genetic variants because of founder effects
Participation in international research projects can provide genetic testing and functional validation in settings were routine genetic and immunological clinical resources and health care utilisation are not available
Opportunities, Challenges, and the Need for Education and Research
A prerequisite for the implementation of genomic diagnostics for monogenic IBD in routine clinical practice is education. Rapid developments in genomic technologies and its ethical implications requires an updated training syllabus for specialty training as well as continued professional education of paediatric and adult gastroenterologists. In a 2017 web-based nationwide survey of UK gastroenterology specialty trainees, 91% of trainees regard their local training program not adequate in regard to genomic medicine (115). In paediatric gastroenterology, the identification and understanding of genetic conditions is part of the ESPGHAN and NASPGHAN training syllabus but genomic medicine is not currently specified.
Studies on the perspective of patients and patient-reported outcome are lacking in the field of monogenic IBD.
Technologies, such as RNA sequencing, single cell RNA sequencing, proteomic and metabolomic technologies do complement current clinical genomic technologies. Defined applications will soon reach clinical practice to aid splice defect analysis and understand isoform usage, cellular mosaicism, and posttranslational modification.
The field of precision medicine and genomics in rare diseases is heavily research-driven. One challenge is to combine clinical genetic practice and research. The clinical genetic perspective is focused to identify known disorders with a strong previously published or accessible evidence (identification of pathogenic variants or likely pathogenic variants based on literature or established databases) whereas translational science aims to identify novel causative genetic variants that are relevant for disease pathogenesis and treatments. Expectations in regard to timely reporting of known and novel genetic results can be challenging as it might take months or years to validate novel findings. Di- and potentially oligogenic forms of IBD are not yet robustly defined.
Open access to research genetic data is challenging in times of increasing scrutiny towards personal data protection and genetic confidentiality on the one side and opportunities to identify individuals based on ancestry analysis on the other (116). The intimate interaction between clinical genetics and translational science from using genetic technologies and functional validation in the research setting to applying genetically informed treatments off-label is only partially sanctioned by law and regulators.
Another emerging challenge is the use of direct-to-consumer genetics (117). Genetic tests initiated and paid for by patients via commercial sequencing facilities can provide IBD-relevant genetic results (for instance by providing direct to consumer genome sequencing Nebula Genomics, https://nebula.org/whole-genome-sequencing/). In the absence of a multidisciplinary team assessment and disease specific specialist input, however, interpretation of direct-to consumer genetics test results will be challenging for patients and clinicians alike
A formal health economic assessment for monogenic IBD is required. This requires assessment of the diagnostic costs in relation with treatment and procedure costs in a group of very diverse monogenic disorders with only gradually emerging standards of care in a setting of centralized and specialized multidisciplinary care. For many disorders, medications are not formally licenced, novel medications that are provided as part of research (no costs can be affiliated yet), and patients with extremely expensive forms of health care utilisation, such as multiple operations, parental nutrition, and stem cell transplantation will strongly influence the analysis.
CONCLUSIONS
The use of genomic medicine offers essential diagnostic opportunities and has complex medical, and scientific as well as ethical, legal, financial, and social implications. Implementation of appropriate genomics into clinical practice requires, therefore, not just the use of evolving clinical genetics technologies but a patient-centred multidisciplinary approach.
Supplementary Material
Acknowledgments
The Genius group and the COLORS in IBD project were supported via ESPGHAN network grants (F.M.R., H.H.U., and D.W.). H.H.U., and S.T. are funded by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). R.K.R. is supported by an NHS Research Scotland Career Researcher Clinician award. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. H.H.U., S.S., C.K., A.M. are supported by the Leona M. and Harry B. Helmsley Charitable Trust (VEO-IBD consortium).
For the last 3 years, H.H.U. received research support or consultancy fees from UCB Pharma, Eli Lilly, Pfizer, AbVie, Celgene, OMass Therapeutics, and MiroBio. For the last 3 years, D.T. received consultation fee, research grant, royalties, or honorarium from Janssen, Pfizer, Hospital for Sick Children, Ferring, Abbvie, Takeda, Atlantic Health, Shire, Celgene, Lilly, Roche, ThermoFisher, BMS. For the last 3 years, R.K.R. received consultation fee, research grant, royalties, or honorarium from Janssen, Abbvie, Takeda, Celgene, Lilly, Nestle Health Sciences, Vifor, Celltrion, Therakos, Pharmacosmos and Tillots. S.P.L.T. has been adviser to, in receipt of educational or research grants from, or invited lecturer for AbbVie, Amgen, Asahi, Biogen, Boehringer Ingelheim, BMS, Cosmo, Elan, Enterome, Ferring, FPRT Bio, Genentech/Roche, Genzyme, Glenmark, GW Pharmaceuticals, Janssen, Johnson & Johnson, Lilly, Merck, Novartis, Novo Nordisk, Ocera, Pfizer, Shire, Santarus, SigmoidPharma, Synthon, Takeda, Tillotts, Topivert, Trino Therapeutics with Wellcome Trust, UCB Pharma, Vertex, VHsquared, Vifor, Warner Chilcott, and Zeria. A.M.G. has served as a speaker or consultant or advisory board member for Abbvie, Amgen, Bristol Meyers Squibb, Celgene, Janssen, Lilly, Merck, Nestle, Pfizer, Roche, and has received research grant from Abbvie. For the last 3 years, Ld.R. received consultation fee, research grant, or honorarium from Pfizer, Abbvie, Roche, Celgene, and Nestle. For the last 3 years, T.S. received speaker’s fees from Nutricia. For the last 3 years, D.S.S. received consultation fee and research grant from AbbVie and Takeda. In the last 3 years, D.C.W. has received consultancy fees, speaker fees, and/or travel support from Abbvie, Nestle Health Sciences, Roche, Ferring, and Predictimmune. J.V.L. reports consulting, travel and/ or speaker fees, and research support from AbbVie, Janssen, Nestlé Health Science, Novalac, Pfizer, Merck, P&G, GSK, Illumina, Otsuka. For the last 3 years, F.M.R. received consultation fees, honorarium from Johnson & Johnson, Centocor, AbbVie, MSD France, NestléNutrition Institute, Nestlé Health Science, Danone, Mead Johnson, Takeda, Celgene, BioGen, and Arkopharma, AMGEN; F.M.R. received research grants from AbbVie. In the last 3 years, S.B.S. has received grant support from Novartis, Pfizer, Janssen, Merck, Regeneron; consultancy and/or advisory board related activities for Pfizer, Janssen, Merck, Takeda, Lilly, Celgene, BMS, IFM therapeutics, Amgen, Pandion, Hoffman La-Roche, Applied Molecular Transport, Cosmo Pharmaceuticals.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text, and links to the digital files are provided in the HTML text of this article on the journal’s Web site (www.jpgn.org).
Disclaimer and qualifying statement: ESPGHAN is not responsible for the practices of physicians and provides guidelines and position papers as indicators of best practice only. Diagnosis and treatment is at the discretion of physicians. This guidance may be revised as necessary to account for changes in technology, new data, or other aspects of clinical practice. This guidance is intended to be an educational device to provide information that may assist clinicians in providing care to patients. They are not a rule and should not be construed as establishing a legal standard of care or as encouraging, advocating, requiring, or discouraging any particular treatment. Clinical decisions in any particular case involve a complex analysis of the patient’s condition and available courses of action. Therefore, clinical considerations may require taking a course of action that varies from the suggestions made as part of this guidance.
REFERENCES
- 1.Graham DB, Xavier RJ. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020;578:527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhlig HH, Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu Rev Immunol 2018;36:755–81. [DOI] [PubMed] [Google Scholar]
- 3.Cleynen I, Boucher G, Jostins L, et al. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet 2016;387:156–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Lange KM, Moutsianas L, Lee JC, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 2017;49:256–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015;47:979–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Y, de Lange KM, Jostins L, et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat Genet 2017;49:186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serra EG, Schwerd T, Moutsianas L, et al. Somatic mosaicism and common genetic variation contribute to the risk of very-early-onset inflammatory bowel disease. Nat Commun 2020;11:995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Splinter K, Adams DR, Bacino CA, et al. , Undiagnosed Diseases Network. Effect of genetic diagnosis on patients with previously undiagnosed disease. N Engl J Med 2018;379:2131–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan KE, Conrad M, Kelsen JR. Very early-onset inflammatory bowel disease: an integrated approach. Curr Opin Allergy Clin Immunol 2018;18:459–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlig HH, Muise AM. Clinical genomics in inflammatory bowel disease. Trends Genet 2017;33:629–41. [DOI] [PubMed] [Google Scholar]
- 12.Ouahed J, Spencer E, Kotlarz D, et al. Very early onset inflammatory bowel disease: a clinical approach with a focus on the role of genetics and underlying immune deficiencies. Inflamm Bowel Dis 2020;26:820–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pazmandi J, Kalinichenko A, Ardy RC, et al. Early-onset inflammatory bowel disease as a model disease to identify key regulators of immune homeostasis mechanisms. Immunol Rev 2019;287:162–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelsen JR, Sullivan KE, Rabizadeh S, et al. NASPGHAN Position Paper on The Evaluation and Management for Patients with Very Early-Onset Inflammatory Bowel Disease (VEO-IBD). J Pediatr Gastroenterol Nutr 2020;70:389–403. [DOI] [PubMed] [Google Scholar]
- 15.Kim KY, Lee EJ, Kim JW, et al. Higher morbidity of monogenic inflammatory bowel disease compared to the adolescent onset inflammatory bowel disease. Pediatr Gastroenterol Hepatol Nutr 2018;21:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kammermeier J, Dziubak R, Pescarin M, et al. Phenotypic and genotypic characterisation of inflammatory bowel disease presenting before the age of 2 years. J Crohns Colitis 2017;11:60–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sokol H, Mahlaoui N, Aguilar C, et al. Intestinal dysbiosis in inflammatory bowel disease associated with primary immunodeficiency. J Allergy Clin Immunol 2019;143:775.e6–8.e6. [DOI] [PubMed] [Google Scholar]
- 18.Huang C, De Ravin SS, Paul AR, et al. , NIDDK IBD Genetics Consortium. Genetic risk for inflammatory bowel disease is a determinant of Crohn’s disease development in chronic granulomatous disease. Inflamm Bowel Dis 2016;22:2794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen BS, Fredrich B, Hoeppner MP, et al. Opportunities and challenges of whole-genome and -exome sequencing. BMC Genet 2017;18:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Z, Zhu L, Roberts R, et al. Toward clinical implementation of next-generation sequencing-based genetic testing in rare diseases: where are we? Trends Genet 2019;35:852–67. [DOI] [PubMed] [Google Scholar]
- 21.Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet 2015;47:717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med 2018;3:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belkadi A, Bolze A, Itan Y, et al. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc Natl Acad Sci U S A 2015;112:5473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim SZ, Chua EW. Revisiting the role of thiopurines in inflammatory bowel disease through pharmacogenomics and use of novel methods for therapeutic drug monitoring. Front Pharmacol 2018;9:1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heap GA, Weedon MN, Bewshea CM, et al. HLA-DQA1-HLA-DRB1 variants confer susceptibility to pancreatitis induced by thiopurine immunosuppressants. Nat Genet 2014;46:1131–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang SK, Hong M, Baek J, et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat Genet 2014;46:1017–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker GJ, Harrison JW, Heap GA, et al. , IBD Pharmacogenetics Study Group. Association of genetic variants in NUDT15 with thiopurine-induced myelosuppression in patients with inflammatory bowel disease. JAMA 2019;321:773–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sazonovs A, Kennedy NA, Moutsianas L, et al. HLA-DQA1 *05 carriage associated with development of anti-drug antibodies to infliximab and adalimumab in patients with Crohn’s disease. Gastroenterology 2020;158:189–99. [DOI] [PubMed] [Google Scholar]
- 29.Uhlig HH, Schwerd T, Koletzko S, et al. , COLORS in IBD Study Group and NEOPICS. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014;147:990.e3–1007.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tegtmeyer D, Seidl M, Gerner P, et al. Inflammatory bowel disease caused by primary immunodeficiencies-clinical presentations, review of literature, and proposal of a rational diagnostic algorithm. Pediatr Allergy Immunol 2017;28:412–29. [DOI] [PubMed] [Google Scholar]
- 31.Charbit-Henrion F, Parlato M, Hanein S, et al. Diagnostic yield of next-generation sequencing in very early-onset inflammatory bowel diseases: a multicenter study. J Crohns Colitis 2018;12:1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis 2011;17:1314–21. [DOI] [PubMed] [Google Scholar]
- 33.Richards S, Aziz N, Bale S, et al. , ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang YH, Luo YY, Yu JD, et al. Phenotypic and genotypic characterization of inflammatory bowel disease in children under six years of age in China. World J Gastroenterol 2018;24:1035–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lega S, Pin A, Arrigo S, et al. Diagnostic approach to monogenic inflammatory bowel disease in clinical practice: a ten-year multicentric experience. Inflamm Bowel Dis 2020;26:720–7. [DOI] [PubMed] [Google Scholar]
- 36.Crowley E, Warner N, Pan J, et al. Prevalence and clinical features of inflammatory bowel diseases associated with monogenic variants, identified by whole-exome sequencing in 1000 children at a single center. Gastroenterology 2020;158:2208–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quaranta M, Wilson R, Goncalves Serra E, et al. Consequences of identifying XIAP deficiency in an adult patient with inflammatory bowel disease. Gastroenterology 2018;155:231–4. [DOI] [PubMed] [Google Scholar]
- 38.Amininejad L, Charloteaux B, Theatre E, et al. , International IBD Genetics Consortium. Analysis of genes associated with monogenic primary immunodeficiency identifies rare variants in XIAP in patients with Crohn’s disease. Gastroenterology 2018;154:2165–77. [DOI] [PubMed] [Google Scholar]
- 39.Fiskerstrand T, Arshad N, Haukanes BI, et al. Familial diarrhea syndrome caused by an activating GUCY2C mutation. New Engl J Med 2012;366:1586–95. [DOI] [PubMed] [Google Scholar]
- 40.Aguilar C, Lenoir C, Lambert N, et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol 2014;134:1131.e9–41.e9. [DOI] [PubMed] [Google Scholar]
- 41.Gambineri E, Ciullini Mannurita S, Hagin D, et al. Clinical, immunological, and molecular heterogeneity of 173 patients with the phenotype of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Front Immunol 2018;9:2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001;411:599–603. [DOI] [PubMed] [Google Scholar]
- 43.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001;411:603–6. [DOI] [PubMed] [Google Scholar]
- 44.Habibi S, Zaki-Dizaji M, Rafiemanesh H, et al. Clinical, immunologic, and molecular spectrum of patients with LPS-responsive beige-like anchor protein deficiency: a systematic review. J Allergy Clin Immunol Pract 2019;7:2379.e5–86.e5. [DOI] [PubMed] [Google Scholar]
- 45.Cagdas D, Halacli SO, Tan C, et al. A spectrum of clinical findings from ALPS to CVID: several novel LRBA defects. J Clin Immunol 2019;39:726–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Egg D, Schwab C, Gabrysch A, et al. Increased risk for malignancies in 131 affected CTLA4 mutation carriers. Front Immunol 2018;9:2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kammermeier J, Drury S, James CT, et al. Targeted gene panel sequencing in children with very early onset inflammatory bowel disease–evaluation and prospective analysis. J Med Genet 2014;51:748–55. [DOI] [PubMed] [Google Scholar]
- 48.Kelsen JR, Dawany N, Moran CJ, et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 2015;149:1415–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashton JJ, Andreoletti G, Coelho T, et al. Identification of variants in genes associated with single-gene inflammatory bowel disease by whole-exome sequencing. Inflamm Bowel Dis 2016;22:2317–27. [DOI] [PubMed] [Google Scholar]
- 50.Ostrowski J, Paziewska A, Lazowska I, et al. Genetic architecture differences between pediatric and adult-onset inflammatory bowel diseases in the Polish population. Sci Rep 2016;6:39831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao Y, Wang XQ, Yu Y, et al. Comprehensive mutation screening for 10 genes in Chinese patients suffering very early onset inflammatory bowel disease. World J Gastroenterol 2016;22:5578–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petersen BS, August D, Abt R, et al. Targeted gene panel sequencing for early-onset inflammatory bowel disease and chronic diarrhea. Inflamm Bowel Dis 2017;23:2109–20. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki T, Sasahara Y, Kikuchi A, et al. Targeted sequencing and immunological analysis reveal the involvement of primary immunodeficiency genes in pediatric IBD: a Japanese Multicenter Study. J Clin Immunol 2017;37:67–79. [DOI] [PubMed] [Google Scholar]
- 54.Ashton JJ, Mossotto E, Stafford IS, et al. Genetic sequencing of pediatric patients identifies mutations in monogenic inflammatory bowel disease genes that translate to distinct clinical phenotypes. Clin Transl Gastroenterol 2020;11:e00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kelsen JR, Dawany N, Martinez A, et al. A de novo whole gene deletion of XIAP detected by exome sequencing analysis in very early onset inflammatory bowel disease: a case report. BMC Gastroenterol 2015;15:160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meeths M, Entesarian M, Al-Herz W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood 2010;116:2635–43. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Xia X, Zhang J, et al. Haemophagocytic lymphohistiocytosis in inflammatory bowel disease with virus infection. Prz Gastroenterol 2015;10:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwab C, Gabrysch A, Olbrich P, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol 2018;142:1932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fabre A, Marchal S, Barlogis V, et al. Clinical aspects of STAT3 gain-of-function germline mutations: a systematic review. J Allergy Clin Immunol Pract 2019;7:1958.e9–69.e9. [DOI] [PubMed] [Google Scholar]
- 60.Toubiana J, Okada S, Hiller J, et al. , International STAT1 Gain-of-Function Study Group. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016;127:3154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ballew BJ, Joseph V, De S, et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet 2013;9:e1003695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Artac H, Emsen A, Ucaryilmaz H, et al. Infliximab therapy for inflammatory colitis in an infant with NEMO deficiency. Immunol Res 2019;67:450–3. [DOI] [PubMed] [Google Scholar]
- 63.Hartley JL, Zachos NC, Dawood B, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology 2010;138:2388.e1–98.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fabre A, Charroux B, Martinez-Vinson C, et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet 2012;90:689–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neven B, Mamessier E, Bruneau J, et al. A Mendelian predisposition to B-cell lymphoma caused by IL-10R deficiency. Blood 2013;122:3713–22. [DOI] [PubMed] [Google Scholar]
- 66.Bratanic N, Kovac J, Pohar K, et al. Multifocal gastric adenocarcinoma in a patient with LRBA deficiency. Orphanet J Rare Dis 2017;12:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Avitzur Y, Guo C, Mastropaolo LA, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 2014;146:1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kotlarz D, Beier R, Murugan D, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology 2012;143:347–55. [DOI] [PubMed] [Google Scholar]
- 69.Kotlarz D, Marquardt B, Baroy T, et al. Human TGF-beta1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat Genet 2018;50:344–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 2011;13:255–62. [DOI] [PubMed] [Google Scholar]
- 71.Freeman EB, Koglmeier J, Martinez AE, et al. Gastrointestinal complications of epidermolysis bullosa in children. Br J Dermatol 2008;158:1308–14. [DOI] [PubMed] [Google Scholar]
- 72.Williams KW, Milner JD, Freeman AF. Eosinophilia associated with disorders of immune deficiency or immune dysregulation. Immunol Allergy Clin North Am 2015;35:523–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Warnatz K, Bossaller L, Salzer U, et al. Human ICOS deficiency abrogates the germinal center reaction and provides a monogenic model for common variable immunodeficiency. Blood 2006;107:3045–52. [DOI] [PubMed] [Google Scholar]
- 74.Roifman CM. The case for adding X-linked agammaglobulinemia to newborn screening. LymphoSign J 2015;2:53–5. [Google Scholar]
- 75.Uhlig HH. Mendelian diseases and inflammatory bowel disease-data mining for genetic risk and disease-associated confounders. Inflamm Bowel Dis 2018;24:467–70. [DOI] [PubMed] [Google Scholar]
- 76.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 2009;361:2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barzaghi F, Amaya Hernandez LC, Neven B, et al. , Primary Immune Deficiency Treatment Consortium (PIDTC) and the Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT). Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol 2018;141:1036.e5–49.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lien R, Lin YF, Lai MW, et al. Novel mutations of the tetratricopeptide repeat domain 7A gene and phenotype/genotype comparison. Front Immunol 2017;8:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miot C, Imai K, Imai C, et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG /NEMO mutations. Blood 2017;130:1456–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lehle AS, Farin HF, Marquardt B, et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology 2019;156:275–8. [DOI] [PubMed] [Google Scholar]
- 81.Li Y, Fuhrer M, Bahrami E, et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc Natl Acad Sci U S A 2019;116:970–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cuchet-Lourenco D, Eletto D, Wu C, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 2018;361:810–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shouval DS, Ebens CL, Murchie R, et al. Large B-cell lymphoma in an adolescent patient with interleukin-10 receptor deficiency and history of infantile inflammatory bowel disease. J Pediatr Gastroenterol Nutr 2016;63:e15–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shouval DS, Biswas A, Kang YH, et al. Interleukin 1beta mediates intestinal inflammation in mice and patients with interleukin 10 receptor deficiency. Gastroenterology 2016;151:1100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Levy M, Arion A, Berrebi D, et al. Severe early-onset colitis revealing mevalonate kinase deficiency. Pediatrics 2013;132:e779–83. [DOI] [PubMed] [Google Scholar]
- 86.Canna SW, de Jesus AA, Gouni S, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014;46:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Canna SW, Girard C, Malle L, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol 2017;139:1698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lo B, Zhang K, Lu W, et al. Autoimmune disease patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015;349:436–40. [DOI] [PubMed] [Google Scholar]
- 89.Ye Z, Hu W, Wu B, et al. Predictive prenatal diagnosis for infantile-onset inflammatory bowel disease due to interleukin-10 signalling defects. J Pediatr Gastroenterol Nutr 2020. [Online ahead of print]. [DOI] [PMC free article] [PubMed]
- 90.Tangye SG, Al-Herz W, Bousfiha A, et al. Human Inborn errors of immunity: 2019 update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol 2020;40:24–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bolton C, Burch N, Morgan J, et al. Remission of inflammatory bowel disease in glucose-6-phosphatase 3 deficiency by allogeneic haematopoietic stem cell transplantation. J Crohns Colitis 2020;14:142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schwerd T, Bryant RV, Pandey S, et al. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal Immunol 2018;11:562–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rehm HL, Berg JS, Brooks LD, et al. ClinGen–the clinical genome resource. N Engl J Med 2015;372:2235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gifford CE, Weingartner E, Villanueva J, et al. Clinical flow cytometric screening of SAP and XIAP expression accurately identifies patients with SH2D1A and XIAP/BIRC4 mutations. Cytometry B Clin cytometry 2014;86:263–71. [DOI] [PubMed] [Google Scholar]
- 96.Kanegane H, Hoshino A, Okano T, et al. Flow cytometry-based diagnosis of primary immunodeficiency diseases. Allergol Int 2018;67:43–54. [DOI] [PubMed] [Google Scholar]
- 97.Dhillon SS, Fattouh R, Elkadri A, et al. Variants in NADPH oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology 2014;147:680–9. [DOI] [PubMed] [Google Scholar]
- 98.Uhlig HH, Booth C. A spectrum of genetic variants contributes to immune defects and pathogenesis of inflammatory bowel diseases. Gastroenterology 2018;154:2022–4. [DOI] [PubMed] [Google Scholar]
- 99.Forbester JL, Lees EA, Goulding D, et al. Interleukin-22 promotes phagolysosomal fusion to induce protection against Salmonella enterica Typhimurium in human epithelial cells. Proc Natl Acad Sci U S A 2018;115:10118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bigorgne AE, Farin HF, Lemoine R, et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J Clin Invest 2013;124:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goettel JA, Biswas S, Lexmond WS, et al. Fatal autoimmunity in mice reconstituted with human hematopoietic stem cells encoding defective FOXP3. Blood 2015;125:3886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schwarze K, Buchanan J, Taylor JC, et al. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet Med 2018;20:1122–30. [DOI] [PubMed] [Google Scholar]
- 103.van Nimwegen KJ, van Soest RA, Veltman JA, et al. Is the $1000 genome as near as we think? A cost analysis of next-generation sequencing. Clin Chem 2016;62:1458–64. [DOI] [PubMed] [Google Scholar]
- 104.Schwarze K, Buchanan J, Fermont JM, et al. The complete costs of genome sequencing: a microcosting study in cancer and rare diseases from a single center in the United Kingdom. Genet Med 2020;22:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stark Z, Schofield D, Alam K, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med 2017;19:867–74. [DOI] [PubMed] [Google Scholar]
- 106.Levine A, Koletzko S, Turner D, et al. , European Society of Pediatric Gastroenterology, Hepatology, and Nutrition. ESPGHAN revised porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr 2014;58:795–806. [DOI] [PubMed] [Google Scholar]
- 107.Lazaridis KN, Schahl KA, Cousin MA, et al. , Individualized Medicine Clinic Members. Outcome of whole exome sequencing for diagnostic odyssey cases of an individualized medicine clinic: the Mayo Clinic Experience. Mayo Clin Proc 2016;91:297–307. [DOI] [PubMed] [Google Scholar]
- 108.Watson TA, Petit P, Augdal TA, et al. European Society of Paediatric Radiology abdominal imaging task force: statement on imaging in very early onset inflammatory bowel disease. Pediatr Radiol 2019;49:841–8. [DOI] [PubMed] [Google Scholar]
- 109.Matthijs G, Souche E, Alders M, et al. , EuroGentest, European Society of Human Genetics. Guidelines for diagnostic next-generation sequencing. Eur J Hum Genet 2016;24:2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bush LW, Bartoshesky LE, David KL, et al. , ACMG SELI committee. Pediatric clinical exome/genome sequencing and the engagement process: encouraging active conversation with the older child and adolescent: points to consider-a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2018;20:692–4. [DOI] [PubMed] [Google Scholar]
- 111.Farmer GD, Gray H, Chandratillake G, et al. Recommendations for designing genetic test reports to be understood by patients and nonspecialists. Eur J Hum Genet 2020;28:885–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vears DF, Senecal K. Borry P Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet 2017;60:553–8. [DOI] [PubMed] [Google Scholar]
- 113.Clift K, Macklin S, Halverson C, et al. Patients’ views on variants of uncertain significance across indications. J Community Genet 2020;11:139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang H, Qian Y, Lu Y, et al. Clinical utility of 24-h rapid trio-exome sequencing for critically ill infants. NPJ Genom Med 2020;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Al Bakir I, Sebepos-Rogers GM, Burton H, et al. Mainstreaming of genomic medicine in gastroenterology, present and future: a nationwide survey of UK gastroenterology trainees. BMJ Open 2019;9:e030505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kim J, Edge MD, Algee-Hewitt BFB, et al. Statistical detection of relatives typed with disjoint forensic and biomedical loci. Cell 2018;175:848.e6–58.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Thiebes S, Toussaint PA, Ju J, et al. Valuable genomes: taxonomy and archetypes of business models in direct-to-consumer genetic testing. J Med Internet Res 2020;22:e14890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Uchida T, Suzuki T, Kikuchi A, et al. Comprehensive targeted sequencing identifies monogenic disorders in patients with early-onset refractory diarrhea. J Pediatr Gastroenterol Nutr 2020;71:333–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.