

Abstract
Plasmacytoid dendritic cells (pDCs) can rapidly produce interferons and other soluble factors in response to extracellular viruses or virus mimics such as CpG-containing DNA. pDCs can also recognize live cells infected with certain RNA viruses, but the relevance and functional consequences of such recognition remain unclear. We studied the response of primary DCs to the prototypical persistent DNA virus, human cytomegalovirus (CMV). Human pDCs produced high amounts of IFN-I when incubated with live CMV-infected fibroblasts, but not with free CMV; the response involved integrin-mediated adhesion, transfer of DNA-containing virions to pDCs and the recognition of DNA through TLR9. Compared to transient polyfunctional responses to CpG or free influenza virus, pDC response to CMV-infected cells was long-lasting, dominated by the production of type I (IFN-I) and type III (IFN-III) interferons, and lacked diversification into functionally distinct populations. Similarly, pDC activation by influenza-infected lung epithelial cells was highly efficient, prolonged and dominated by interferon production. Prolonged pDC activation by CMV-infected cells facilitated the activation of natural killer cells critical for CMV control. Finally, patients with CMV viremia harbored phenotypically activated pDCs and increased circulating IFN-I and IFN-III. Thus, recognition of live infected cells is a mechanism of virus detection by pDCs that elicits a unique antiviral immune response.
One-sentence Summary
pDCs recognize cells infected with human cytomegalovirus and respond with a unique prolonged activation program
Introduction
Dendritic cells (DCs) recognize pathogens through pattern recognition receptors such as Toll-like receptors (TLRs), produce cytokines and chemoattractants, and present antigens (Ag), all to orchestrate an immune response. Classical or conventional DCs (cDCs) are specialized Ag-presenting cells that comprise two distinct subsets termed cDC1 and cDC2, which preferentially engage in CD8+ T cell cross-priming and CD4+ T cell activation, respectively (1). Plasmacytoid DCs (pDCs) mediate rapid production of type I (IFN-α/β, IFN-I) and type III (IL-28/29, IFN-λ, IFN-III) interferon, as well as other cytokines such as TNF-α upon activation through DNA- and RNA-sensing endosomal TLR9 and TLR7, respectively (2, 3). Accordingly, pDCs are involved in controling acute and persistent viral infections (4–7).
Although certain viruses can infect pDCs and elicit IFN-I responses in a replication-dependent manner (8, 9), pDCs are resistant to infection by many (if not most) viruses. For some viruses such as influenza A (Flu) virus, pDCs can endocytose free virions and recognize them through endosomal TLRs in a replication-independent way (4, 10). However, these recognition mechanisms would be ineffective against viruses that replicate slowly and/or are present primarily inside cells. In that respect, pDCs can recognize cells infected with several RNA viruses rather than free viruses themselves, in a process that requires the transfer and TLR7-mediated recognition of viral RNA (11–16). This recognition involves cell-cell interactions mediated by the integrin LFA-1 (15), which may promote the formation of a polarized structure at the pDC-infected cell interface termed “interferogenic synapse” (17). However, it remains unclear whether this mechanism is broadly operational in responses to both RNA and DNA viruses, and whether the pDC activation by virus-infected cells has the same features and outcome as activation by solution-born stimuli such as free viruses or synthetic TLR ligands.
Human cytomegalovirus (CMV) is a large, enveloped DNA herpesvirus that latently infects the majority (60–90%) of adult human population and may become reactivated in immunodeficient subjects such as immunosuppressed transplantation recipients (18). The lifelong control of CMV in immunocompetent subjects is mediated by NK and T cell responses, which require Ag presentation and cytokine production by DCs (19). Culture-derived cDCs and macrophages as well as primary pDCs and cDC2 can recognize CMV in vitro and produce IFN-I and/or other cytokines (20–22). Similarly, murine pDCs produce IFN-I in response to free murine cytomegalovirus (MCMV) in vitro, and pDCs and cDCs facilitate NK cell activation, T cell cross-priming, and MCMV control in vivo (5, 6, 23). However, it is unclear how DCs sense CMV in vivo, given that little virus is produced extracellualrly during CMV persistence.
Here, we showed that pDCs efficiently responded to CMV-infected cells, and the resulting response was distinguished by its sensitivity, long duration, and enhanced production of IFN-I and IFN-III. These results establish the recognition of virus-infected cells by pDCs as a mechanism that elicits a unique antiviral response.
Results
pDCs preferentially responded to live CMV-infected cells
To evaluate the response of DC subsets to CMV, we stimulated primary DCs and monocytes (Fig. S1A) with CMV strain TB40E encoding green fluorescence protein (CMV-GFP). Purified monocytes and cDC2 were readily infected with CMV as revealed by high GFP fluorescence after 36 hours, whereas pDCs showed no productive CMV infection (Fig. 1A). pDCs produced little IFN-α when stimulated with free CMV, even at a multiplicity of infection (MOI) of up to 80 (Fig. S1B). Next, we infected the human lung fibroblast cell line MRC5 with CMV-GFP (CMV-MRC5, Fig. S1C) and cocultured it with enriched primary DCs. CMV-MRC5, but not uninfected MRC5, elicited intracellular IFN-α and TNF-α accumulation in pDCs (Fig. 1B). The response was comparable to that elicited by unmethylated CpG oligonucleotide (CpG), a synthetic TLR9 ligand commonly used for pDC activation, but was biased towards IFN-α versus TNF-α production (Fig. 1B). cDC1 and cDC2 failed to respond to CMV-MRC5, although the TNF-α response of cDCs to TLR3 and TLR4 ligands was detectable (Fig. 1B). As with the free CMV, pDCs remained GFP-negative, whereas both cDC subsets showed GFP fluorescence (Fig. 1C). When total PBMC were cocultured with CMV-MRC5, IFN-α production was detected only among pDCs (Fig. 1D). IFN production by pDCs was accompanied by the induction of CD69 (Fig. S1D), whereas CD40 was upregulated on both pDCs and cDCs after 24 hours of coculture with CMV-MRC5 (Fig. S1E). We measured IFN-α protein secretion by purified pDCs incubated with free CMV at 10 MOI or with CMV-MRC5. IFN-α was robustly produced only in the latter condition, exceeding the levels induced by CpG (Fig. 1E). Cytokine IL-3 facilitates survival and activation of pDCs in vitro (24, 25) and was used in previous studies looking at pDC responses to CMV (22). The addition of IL-3 resulted in detectable IFN-α responses to free CMV; however, pDCs cocultured with CMV-MRC5 still produced ~6-fold more IFN-α (Fig. 1F). Thus, pDCs mounted a specific IFN response to CMV-infected fibroblasts more efficiently than the responses to free CMV.
Fig. 1. pDCs produced interferon upon contact with live CMV-infected cells.
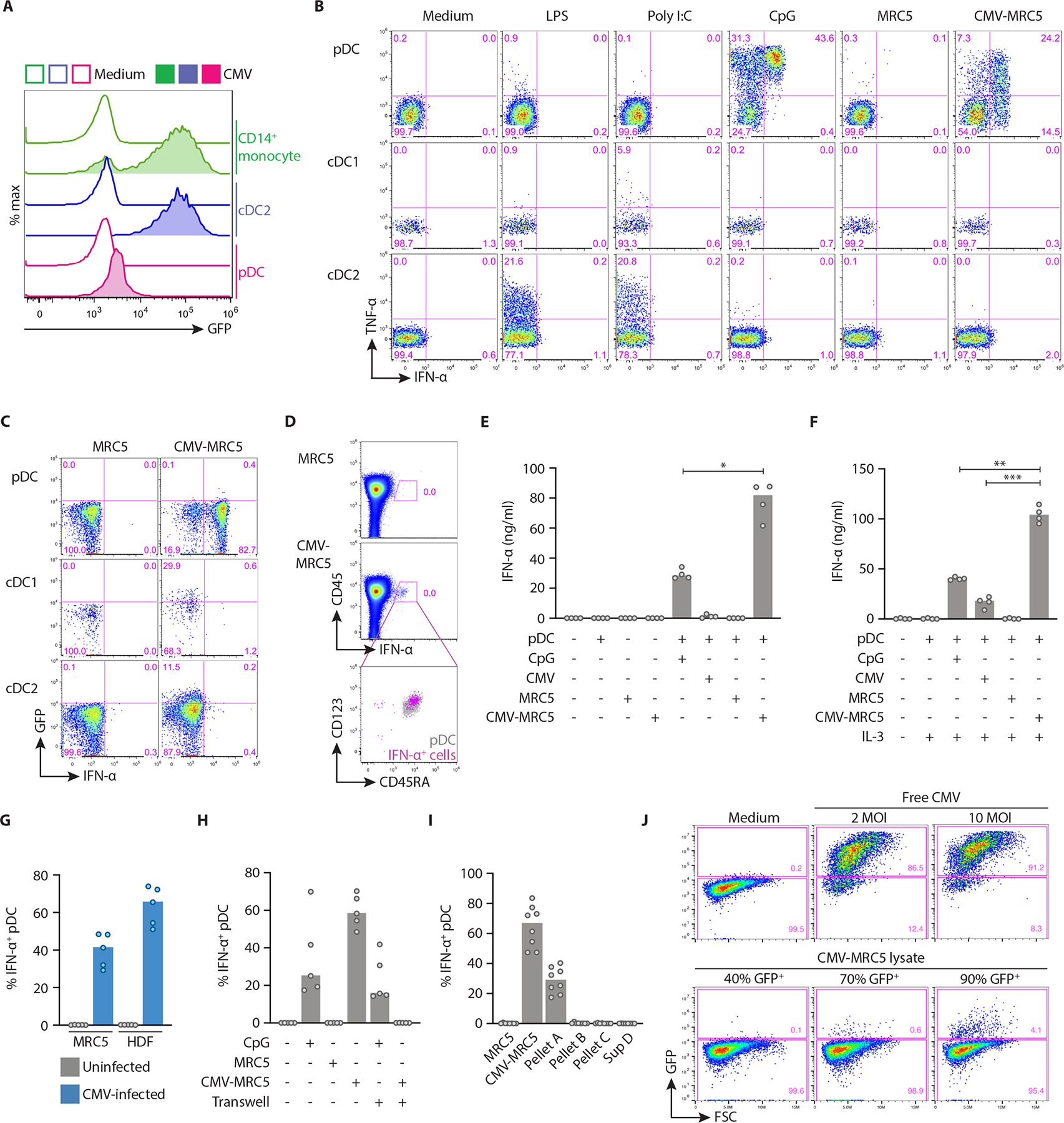
(A) The expression of GFP in purified human pDCs, cDC2 or CD14+ monocytes incubated with free CMV-GFP at 10 MOI for 36 hours.
(B) The expression of IFN-α and TNF-α in DC subsets stimulated with the indicated stimuli including MRC5 cells alone or infected with CMV (CMV-MRC5). Enriched DCs were cocultured with MRC5, and each plot was gated for pDC (CD123+ CD45RA+), cDC1 (CD11c+ CD141+) and cDC2 (CD11c+ CD1c+).
(C) The expression of IFN-α and GFP (as an indicator of CMV infection) in DC subsets cocultured with or without CMV-MRC5 and gated as in panel A. Representative of two experiments.
(D) The expression of IFN-α in total PBMC cocultured with or without CMV-MRC5. Right panel shows gated IFN-α+ cells (pink) overlaid onto gated CD123+ CD45RA+ total pDCs (grey).
(E-F) The concentration of IFN-α protein measured by ELISA in the supernatant of enriched pDCs stimulated with CpG, free CMV (10 MOI), MRC5 or CMV-MRC5 for 24 hours in the absence of IL-3 (E) or presence of IL-3 (F).
(G) Percentage of IFN-α+ pDCs cocultured with CMV-MRC5 or primary human dermal fibroblasts (HDF), infected at 0.5 MOI and cultured for 2 or 5 days, respectively.
(H) Percentage of IFN-α+ pDCs cocultured with CMV-MRC5 or CpG in transwell cultures or in the same wells as controls.
(I) Percentage of IFN-α+ pDCs cocultured with different fractions of CMV-infected cells. CMV-MRC5 were centrifuged to generate pellet A (live cells), pellet B (dead cells), pellet C (cell debris) and supernatant D (virus, exosomes and microparticles).
(J) Titration of infectious CMV particles from lysates of CMV-MRC5. Lysates from CMV-MRC5 cultures or free CMV were used to infect MRC5. Shown is the fraction of GFP+ cells (as a measure of CMV replication) on the day 2 post infection.
In panels E-I, symbols indicate values from individual donors; bars indicate median.
The response of pDCs correlated with the fraction of CMV-infected cells within MRC5 cultures and was detectable when only 10% of cells were infected (Fig. S1F). In addition to MRC5, pDCs also mounted IFN response to CMV-infected primary human dermal fibroblasts (Fig. 1G). In addition to the cloned TB40E strain of CMV, pDCs responded to native CMV strain AD169 and to TB40E strain with a deletion in the region responsible for virus latency (Fig. S1G). Notably, pDCs failed to respond to CMV-MRC5 in transwell cultures (Fig. 1H), or when incubated with subcellular fractions of CMV-MRC5 culture supernatant (Fig. 1I). The response was nearly abolished when CMV-MRC5 were killed by UV irradiation or repeated freezing (Fig. S2A), or when they were fixed with formaldehyde (Fig. S2B). Finally, we estimated the amount of virus present in CMV-MRC5 by lysing these cells and testing infectivity of the lysate. CMV-MRC5 cells at 40%−70% infection rate contained very little infectious virus as determined by GFP expression, and even those at 90% contained ~20-fold less virus than purified virus preparations at the MOI of 2 (Fig. 1J). Thus, IFN-I production by pDCs required a direct interaction with live CMV-infected cells, and was triggered by very few viral particles within infected cells.
pDCs response to CMV-infected cells required cell adhesion and TLR9-mediated recognition
Immunofluorescent staining of cultured pDCs confirmed the production of IFN-I in response to CMV-MRC5 (Fig. 2A) but not to uninfected MRC5 (Fig. S3A). Notably, ~85% of IFN-α-producing pDCs were in contact with CMV-MRC5, in contrast to only ~50% of IFN-α-negative pDCs (Fig. 2A and S3B). We also observed that IFN-α+ pDCs expressed increased levels of the activated integrin LFA-1 (Fig. 2B), although polarized distribution of activated LFA-1 (17) was not observed (Fig. S3C). The percentage of IFN-α+ pDCs in CMV-MRC5 cocultures was significantly decreased after antibody-mediated blockade of LFA-1 or of its ligand ICAM-1 (Fig. 2C), whereas pDC response in cocultures with CpG and uninfected MRC5 was not affected. No effect was observed after the blockade of other homologous LFA-1 ligands ICAM-2 or ICAM-3, or of the adhesion molecule E/P-selectin (Fig. S3D and E). Pre-incubation of CMV-MRC5, but not of pDCs, with anti-ICAM-1 prior to coculture reduced the cytokine response (Fig. 2D), suggesting that the activation of LFA-1 on pDCs by ICAM-1 on CMV-infected cells facilitated the interaction-dependent pDC response.
Fig. 2. pDC response to CMV-infected cells required LFA-1-mediated adhesion and TLR9.
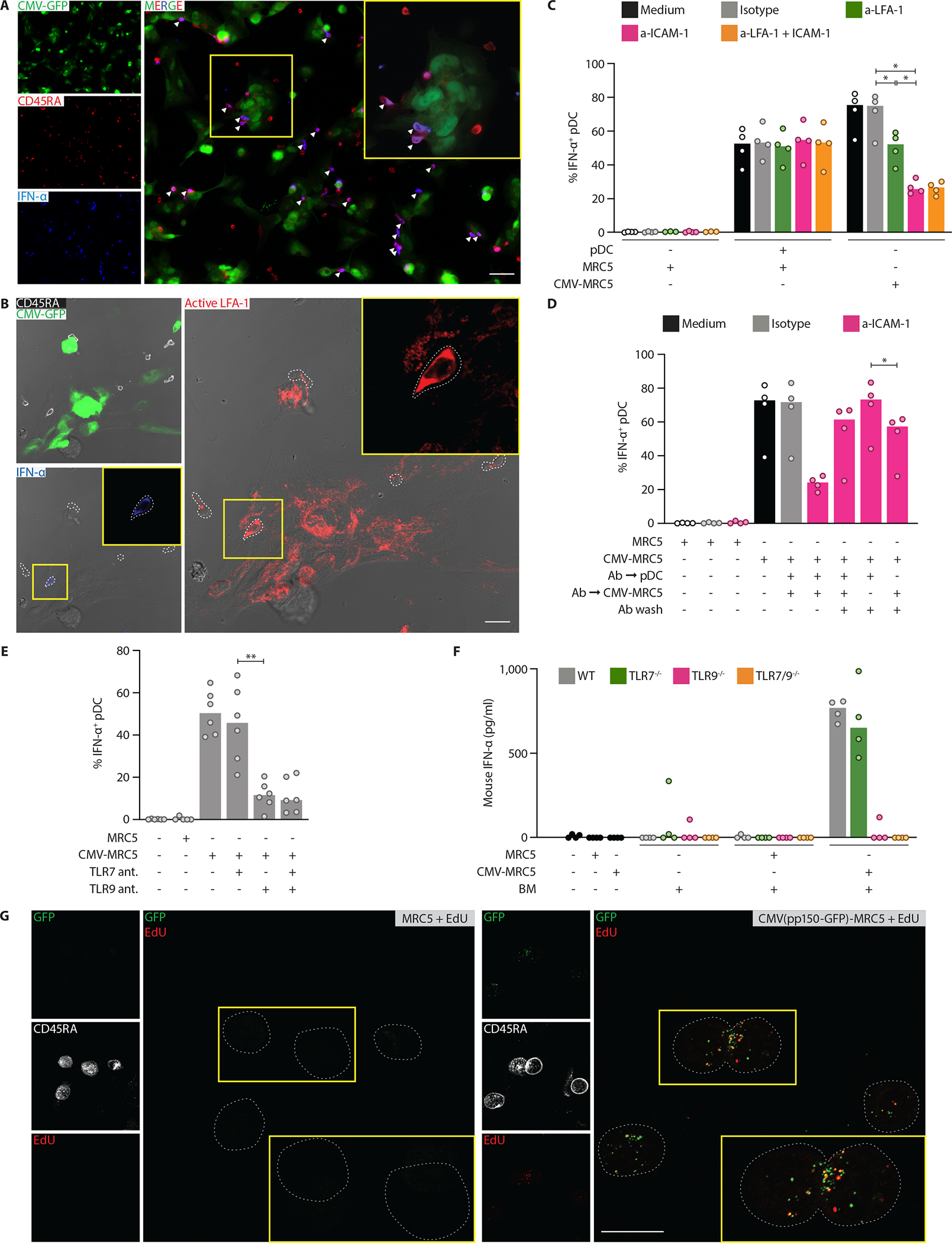
(A) Representative localization of pDCs double positive (arrowhead) for CD45RA (red) and IFN-α (blue) in close contact with CMV-MRC5 (green) after 8 hours of coculture. The area in the yellow box is zoomed in the inset. Scale bar, 50 μm.
(B) Representative pDCs double positive for active LFA-1 (red) and IFN-α (blue). pDCs cocultured with CMV-MRC5 (green) for 8 hours were detected by CD45RA (white). The area in the yellow box is zoomed in the inset. White dotted lines depict cell borders defined by CD45RA. Images are merged with brightfield. Scale bar, 20 μm.
(C) The expression of IFN-α in pDCs stimulated with CpG or cocultured with CMV-MRC5 in the continuous presence of blocking antibodies against LFA-1 and/or ICAM-1.
(D) The expression of IFN-α in pDCs cocultured with CMV-MRC5 after washing off the blocking antibody against ICAM-1. pDCs, CMV-MRC5 or both were preincubated with anti-ICAM-1 for 30 minutes and washed before coculture.
(E) The expression of IFN-α in pDCs cocultured with CMV-MRC5 in the presence of TLR7 or TLR9 oligonucleotide antagonists.
(F) Production of IFN-α by murine primary bone marrow cells (BM) in response to CMV-infected human cells. Total murine BM was cocultured with CMV-MRC5 for 24 hours, and murine IFN-α protein was measured in the supernatant by ELISA. Mouse strains included wild-type controls (WT) and those deficient in TLR7 and/or TLR9.
(G) The transfer of DNA-containing CMV virions to pDCs. Representative images of pDCs after coculture with MRC5 infected with CMV carrying GFP in the virion (CMV (pp150-GFP)). MRC5 were infected with CMV(pp150-GFP), pulsed with EdU and cocultured with pDCs for 8 hours. pDCs were stained for CD45RA, sorted and stained for GFP (green) and EdU (red). The area in the yellow box is zoomed in the inset. White dotted lines depict pDC cell borders defined by CD45RA (white). Scale bar, 20 μm.
In panels C–F, symbols indicate values from individual donors or mice; bars indicate median.
The fraction of IFN-α+ pDCs was significantly decreased by the oligonucleotide antagonist of TLR9, whereas TLR7 antagonist had no independent or additive effect (Fig. 2E). To confirm the observed TLR9 dependence by genetic means, we tested the response of murine pDCs to CMV-MRC5. Human CMV does not replicate in murine cells (19). Nevertheless, total bone marrow cells and pDC-containing cultures from the bone marrow of wild-type mice produced IFN-α in response to CMV-MRC5; the response was intact in Tlr7−/− mice but nearly abolished in Tlr9−/− and Tlr7−/−Tlr9−/− mice (Fig. 2F and S4A). These data confirmed that the activation of pDCs by CMV-infected cells did not involve the infection of pDCs and is mediated through TLR9.
Because the dependence of pDC activation on TLR9 implied the recognition of viral DNA, we tested how the DNA becomes available to pDCs. Whereas the activation of pDCs by CpG was abolished by DNASE1 treatment, the activation of pDCs by CMV-MRC5 was not, suggesting that the viral DNA is sequestered (Fig. S4B). To test the transfer of CMV DNA, we utilized the TB40-UL32-GFP strain expressing GFP fused to the tegument protein pp150; unlike the CMV-GFP used above, the fluorescence of CMV(pp150-GFP) marks cell-associated virions rather than infected cells. We also added thymidine analogue 5-ethynyl-2’-deoxyuridine (EdU) to infected MRC5 to label newly synthesized DNA (both cellular and viral). pDCs were cocultured with CMV(pp150-GFP)-infected EdU-labeled MRC5 cells and sorted (Fig. S4C), followed by staining for GFP and EdU. Fluorescent microscopy showed that these pDCs contained multiple GFP+ speckles, many of which were also EdU+, consistent with DNA-containing virions (Fig. 2G). No EdU transfer from uninfected MRC5 was observed, suggesting (albeit not proving) that EdU labeling represents viral DNA. The GFP fluorescence of CMV(pp150-GFP) was also detected in pDCs by flow cytometry as early as 2 hours after the coculture, and over time was predominant in the activated CD69+ pDCs (Fig. S4D). Finally, we sorted CMV(pp150-GFP)-positive CD69+ pDCs (Fig. S4E) and confirmed the presence CMV DNA in them by qPCR (Fig. S4F). These results documented the transfer of DNA-containing virions from CMV-infected cells to pDCs, likely triggering the TLR9-mediated response in pDCs.
pDC responses to CMV-infected cells were prolonged and biased towards IFN production
The production of TNF-α and IFN-α by pDCs stimulated with CpG or Flu was maximal at 4–8 hours and rapidly decreased thereafter to almost undetectable levels by 24 hours (Fig. S5A). In contrast, IFN-α production by pDCs stimulated with CMV-MRC5 was prominent at 18 hours and still readily detectable by 30 hours (Fig. 3A); at these late time points, little TNF-α was produced (Fig. 3B). Cytokine production in response to free CMV+IL-3 was low as noted above (Fig. 1F) but peaked at 8–12 hours (Fig. S5A), suggesting that the virus itself may have partially contributed to the prolonged pDC response. The observed differences in the pDC response were not due to differential cell survival, which was increased by all tested stimuli (Fig. S5B).
Fig. 3. Kinetics and magnitude of pDC activation by CMV-infected cells.
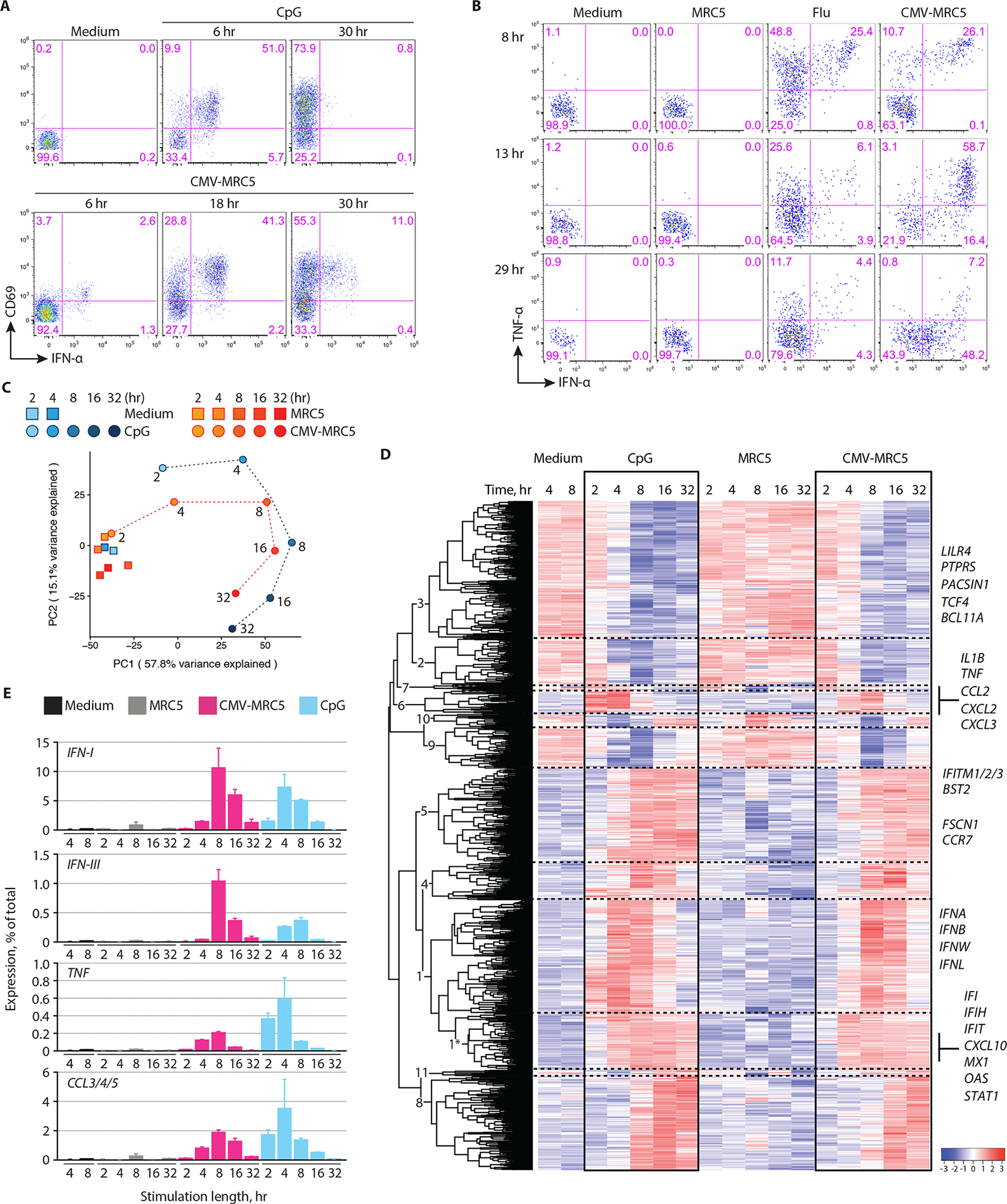
(A) The expression of IFN-α and CD69 in pDCs that were incubated with CpG or cocultured with CMV-infected MRC5 cells (CMV-MRC5) for the indicated time periods.Representative of 4 experiments.
(B) The expression of IFN-α and TNF-α in pDCs that were incubated with influenza virus (flu) or cocultured with uninfected or CMV-infected MRC5 cells for the indicated time periods. Representative of two experiments.
(C-E) Whole-transcriptome analysis of pDC activation. pDCs were incubated with MRC5, CMV-MRC5 or CpG for 2, 4, 8, 16 or 32 hours or in medium only for 4 or 8 hours, sorted and analyzed by RNA-Seq. All data represent averages of three biological replicates.
(C) Principal component (PC) analysis of expression profiles in each condition and time point (different time points of each activation conditions are connected by dashed lines). The fraction of variance captured by each PC is indicated.
(D) Expression dynamics of genes that were differentially expressed during activation by both CMV-MRC5 and CpG. Shown is the hierarchically clustered heatmap of mean relative expression intensities, with cluster numbers indicated (see Table S1). A subcluster of cluster 1 that shows early stable upregulation upon activation is indicated with an asterisk. Select genes within each cluster are listed.
(E) The fraction of transcripts encoding soluble factors in activated pDCs. Shown are cumulative percentages of transcripts for the indicated cytokines and chemokines out of the total transcriptome in the indicated stimulation conditions and time points (mean ± S.D. of triplicate samples).
To confirm the observed kinetic differences at the transcriptome level, we performed global mRNA sequencing (RNA-Seq) using the high-throughput PLATE-Seq approach (26) adapted for low-input samples (27). Enriched primary pDCs from three donors were separately incubated with CpG or CMV-MRC5 (or with medium or uninfected MRC5 as respective controls) for (2, 4, 8, 16 or 32 hours. Activated pDCs were sorted to separate them from MRC5 cells based on size (Fig. S6A) and analyzed by PLATE-Seq, yielding normalized transcript values (counts per million). Principal component analysis (PCA) separated activated from control pDCs (PC1), and pDCs at late activation time points from those at early time points (PC2) (Fig. 3C). Notably, pDCs incubated with CMV-MRC5 clustered with CpG-activated pDCs from the preceding time points (e.g. CMV-MRC5 4 hours with CpG 2 hours, etc). Next, we defined genes that were significantly changed in each activation conditions (Fig. S6B), created an overlapping set of 1,090 genes and analyzed them by hierarchical clustering which yielded 11 clusters (Fig. 3D and Table S1). The data revealed dynamic transcriptome changes, including transient (cluster 9) and permanent (clusters 2–3) downregulation and several waves of upregulation. Among the upregulated genes, a small fraction (cluster 5 and a sub-cluster of cluster 1 designated 1*) showed permanent induction with comparable kinetics in both stimulation conditions; most of these corresponded to IFN-inducible genes such as MX, OAS and IFI family members, STAT1 and CXCL10 (Fig. 3D). Transient gene induction included an early wave including inflammatory cytokines and chemokines (cluster 6), and a delayed transient induction that included all IFN-I and IFN-III genes (cluster 1) (Fig. 3D). Consistent with PCA, all these and other dynamic clusters showed delayed dynamics in CMV-MRC5 compared to CpG (Fig. 3D). Thus, while some genes and clusters showed comparable kinetics, the overall program of pDC activation by CMV-infected cells appeared delayed and prolonged.
We took advantage of the quantitative nature of PLATE-Seq to estimate the fraction of key transcripts within the transcriptome of each sample. During activation by CMV-MRC5, all IFN-I gene transcripts comprised ~10% of the entire pDC transcriptome at the peak (8 hours) and >1% at 32 hours, compared to ~7% and <0.1% for CpG (Fig. 3E). This corresponds to up to ~20% of activation-induced new transcripts in pDCs, comparable with (albeit lower than) previous semi-quantitative estimates (~60% of new transcripts, (28)). IFN-III gene transcripts comprised ~1% at the peak and were still detectable at 32 hours, whereas they were ~3-fold lower in CpG at the peak and absent by 32 hours. Conversely, the expression of TNF and of related chemokines CCL3/4/5 was higher in CpG, the latter comprising >3% of the transcriptome (Fig. 3E). The expression of key IFN-inducible genes was comparable in all activation conditions, reaching at least 3% of the transcriptome, while the expression of MHC class I and II genes was relatively constant and accounted for 1–2% (Fig. S6C). These data confirmed that the pDC response to CMV-infected cells was prolonged and biased towards IFN-I/IFN-III production at the expense of other inflammatory mediators.
Gene cluster 3 that was permanently downregulated during activation (Fig. 3D) included genes that define the pDC lineage, such as those encoding specific surface markers (LILRA4, PTPRS), endocytic adaptors (PACSIN1) and transcription factors (TCF4, BCL11A). Indeed, gene set enrichment analysis (GSEA) showed that the transcriptomic signature of pDCs (29) was significantly downregulated at 32 hours in both stimulation conditions (Fig. S7A). Conversely, the common signature of cDCs was significantly upregulated in pDCs activated by CMV-infected cells, but not in CpG-activated pDCs (Fig. S7A). To characterize transcriptional regulation of the process, we used ARACNe-AP to construct a co-expression network (30) and the VIPER algorithm to infer transcription factor activity (31). TCF4 was downregulated at 16–32 hours (Fig. S7B) with the highest significance of the normalized enrichment score (NES), and therefore was identified by VIPER as the potential top driver of the process. TCF4 is a transcriptional master regulator of pDC development and maintenance, and its deletion from mature pDCs induces their transdifferentiation into cDC-like cells (32). Other transcriptional drivers of pDC lineage (SPIB, RUNX2, IKZF1, CXXC5, MEF2C/D) represent direct targets of TCF4 (33, 34) and were similarly identified as downregulated by VIPER (Fig. S7B). Furthermore, essential cofactors of TCF4 such as BCL11A (32, 35) and CBFA2T3 (33) were downregulated whereas TCF4 antagonist TAL1 (36) was strongly induced (Fig. S7C). Indeed, direct transcriptional targets of TCF4 (34) were significantly downregulated in activated pDCs, reaching maximum at 16 hours (Fig. S7D). Conversely, upregulated transcription factors included regulators of cell activation state (ATF4, NFKB1), IFN/cytokine signaling (STAT1, STAT2, STAT5A, STAT6) and cDC differentiation (IRF4, BATF3, BATF (37)) (Fig. S7B). Thus, the activation of human pDCs by CMV-infected cells triggered the downregulation of TCF4-mediated lineage-specific expression program and the induction of cDC expression features.
pDC response to CMV-infected cells did not involve phenotypic diversification
To characterize the response of primary pDCs and other DC subsets to different stimuli, we used mass cytometry with a panel of 36 antibodies encompassing surface markers of lineage and activation state, cytokines and chemokines. Enriched pan-DCs (Fig. S1A) were cultured with CpG, Flu, CMV-MRC5 or control uninfected MRC5 for 9–11 hours or 24 hours, fixed and analyzed by mass cytometry. We used surface markers to define DC subsets including pDCs, cDC1 and cDC2, the latter including into two functionally distinct subsets (CD5lo and CD5hi) as described recently (38, 39) (Fig. 4A–C). We also identified the CD123+ SIGLEC1+ CD5+ subset corresponding to AXL+ DCs or transitional DCs (tDC) (Fig. 4B–C), a distinct DC subset with features of pDCs and cDC2 (38–41). Consistent with the data in Figure 3, at the early time point pDCs produced IFN-α in response to all stimuli, IFN-λ in response to CMV-MRC5 and CpG, and TNF-α and other mediators predominantly in response CpG and/or Flu (Fig. 4D). In contrast, cDC1 and cDC2 produced hardly any soluble mediators except CXCL10, but showed induction of CD38, CD83, CD86, HLA-DR, CD40 and (for cDC2) SIGLEC1 at the late time point (Fig. 4D).
Fig. 4. Distinct nature of the pDC response to CMV-infected cells.
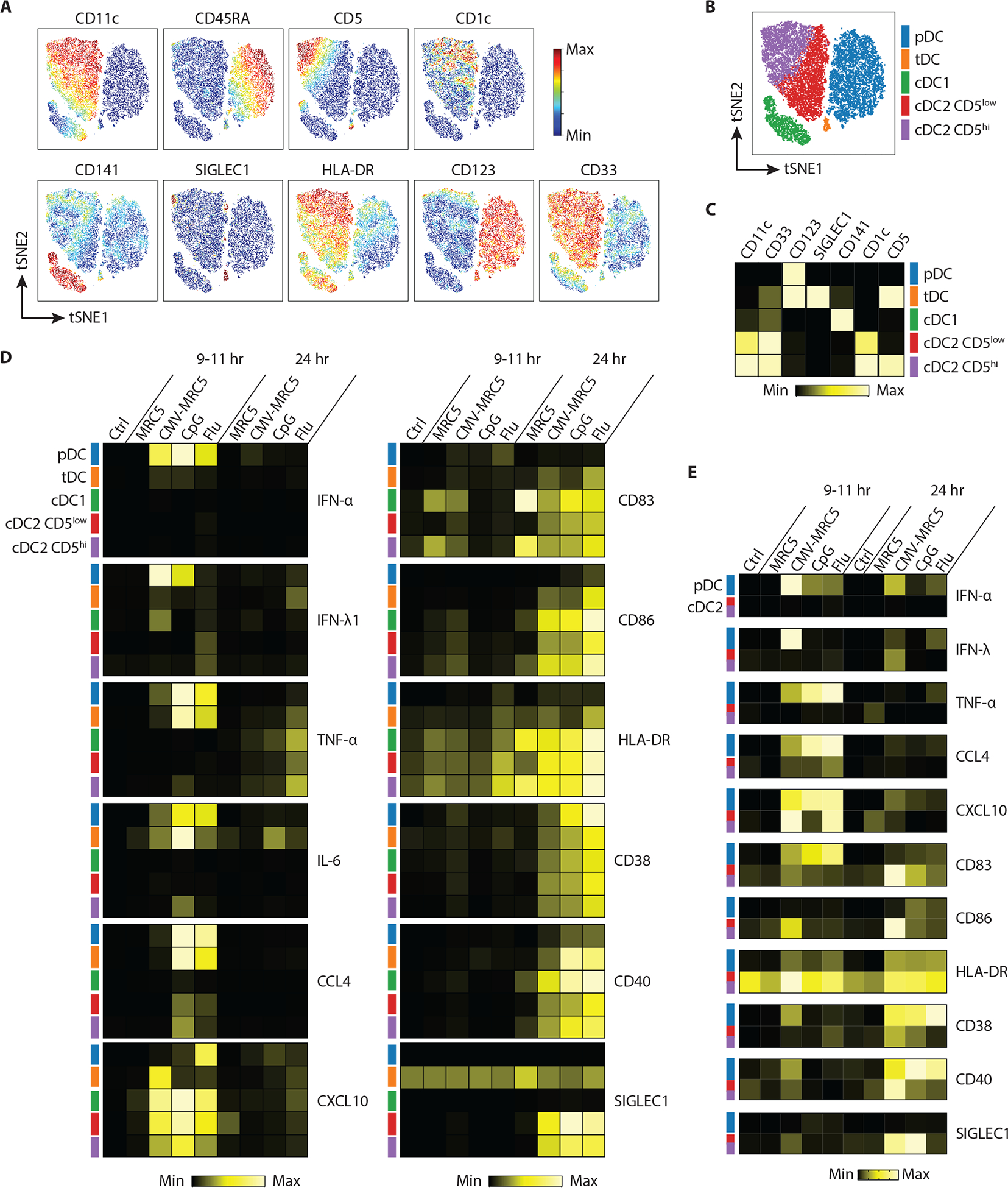
(A–D) Total DC-enriched fraction of normal PBMC was left unstimulated (control) or stimulated with uninfected or CMV-infected MRC5 cells, CpG or the Flu virus, and analyzed by mass cytometry at early (9–11 hours) or late (24 hours) time points.
(A) Relative expression of indicated subset markers within the total DC population visualized on a tSNE plot.
(B) tSNE plot of the control DC population showing five manually gated DC subsets highlighted by distinct colors.
(C) Corresponding heatmap showing the relative marker expression across each of the defined subsets from panel A.
(D) Heatmaps of cytokines (left panel) and activation marker (right panel) expression within the five DC subsets after stimulation.
(E) Sorted pDCs or cDC2 (both CD5low and CD5high) were stimulated as above and analyzed by flow cytometry. Shown are heatmaps of indicated cytokine and activation marker expression after stimulation.
In panels C–E, highest and lowest positive cell fractions (for cytokines) or median intensities (for surface markers) were set to the maximum and minimum, respectively.
We confirmed the results of mass cytometry using sorted pDCs (separated from tDCs, Fig. S8A) and cDC2 (encompassing both CD5lo and CD5hi subsets) in similar activation conditions (Fig. S8). The elevated expression of IFN-α and IFN-λ in response to CMV-MRC5 versus CpG or Flu was more pronounced in isolated pDCs (Fig. 4E), consistent with RNA-Seq results (Fig. 3E) and highlighting the cell-intrinsic nature of these differences. On the other hand, the induction of some activation markers on cDC2s was lower, suggesting that this induction may have represented a bystander effect of pDC activation (Fig. 4E). Phenotypic changes in pDCs were not due to contamination with tDCs (defined as AXL+ SIGLEC6+), which comprised ~1% of enriched pDCs (Fig. S9A). We also directly compared the response of sorted pDCs and tDCs to CMV-MRC5 and CpG using cytokine bead array (Fig. S9B). Consistent with Fig. 4D, only pDCs produced IFN-I and IFN-III, whereas both subsets produced IP-10 and IL-6 (Fig. S9B), emphasizing the distinct and specific nature of tDC response.
We used mass cytometry data to gate the pDC cluster (Fig. 4B), combine pDCs from the early time point of activation and analyze them for cytokine/chemokine production. Dimensionality reduction revealed 5 functionally distinct pDC clusters characterized by different combinations of intracellular cytokine expression (Fig. 5A and B). These clusters largely segregated with stimulation conditions (Fig. 5, C to E): thus, pDCs from the CMV-MRC5 samples showed prominent co-expression of IFN-I and IFN-III (cluster 3), whereas pDCs activated by CpG and/or Flu predominantly produced TNF-α, IL-6, IL-8 CCL4 and/or CXCL10 (clusters 1, 4 and 5). We next examined the diversification of pDCs into distinct functional populations following activation (42). These populations, termed P1 and P3, can be distinguished by the expression PD-L1 and CD80 and preferentially mediate IFN-I production and Ag presentation, respectively. pDCs activated by CpG or by TLR7 ligand R848 for 24 hours comprised P1 (PD-L1+ CD80−), P3 (PD-L1− CD80+) and the intermediate P2 (PD-L1+ CD80+) populations (Fig. 5F). However, pDCs activated by CMV-MRC5 showed little CD80 expression and comprised almost exclusively P1 for up to 36 hours (Fig. 5F). Using transcriptional signatures of P1 and P3 populations (42), we found that pDCs activated by CMV-MRC5 for 32 hours showed a significant enrichment of the P1 signature and depletion of the P3 signature compared to CpG-activated pDCs (Fig. 5G). Thus, the response of pDCs to CMV-infected cells was focused on the production of IFN-I and IFN-III at the expense of other soluble mediators, and lacked phenotypic diversification.
Fig. 5. CMV-infected cells elicited a distinct IFN-focused pDC response.
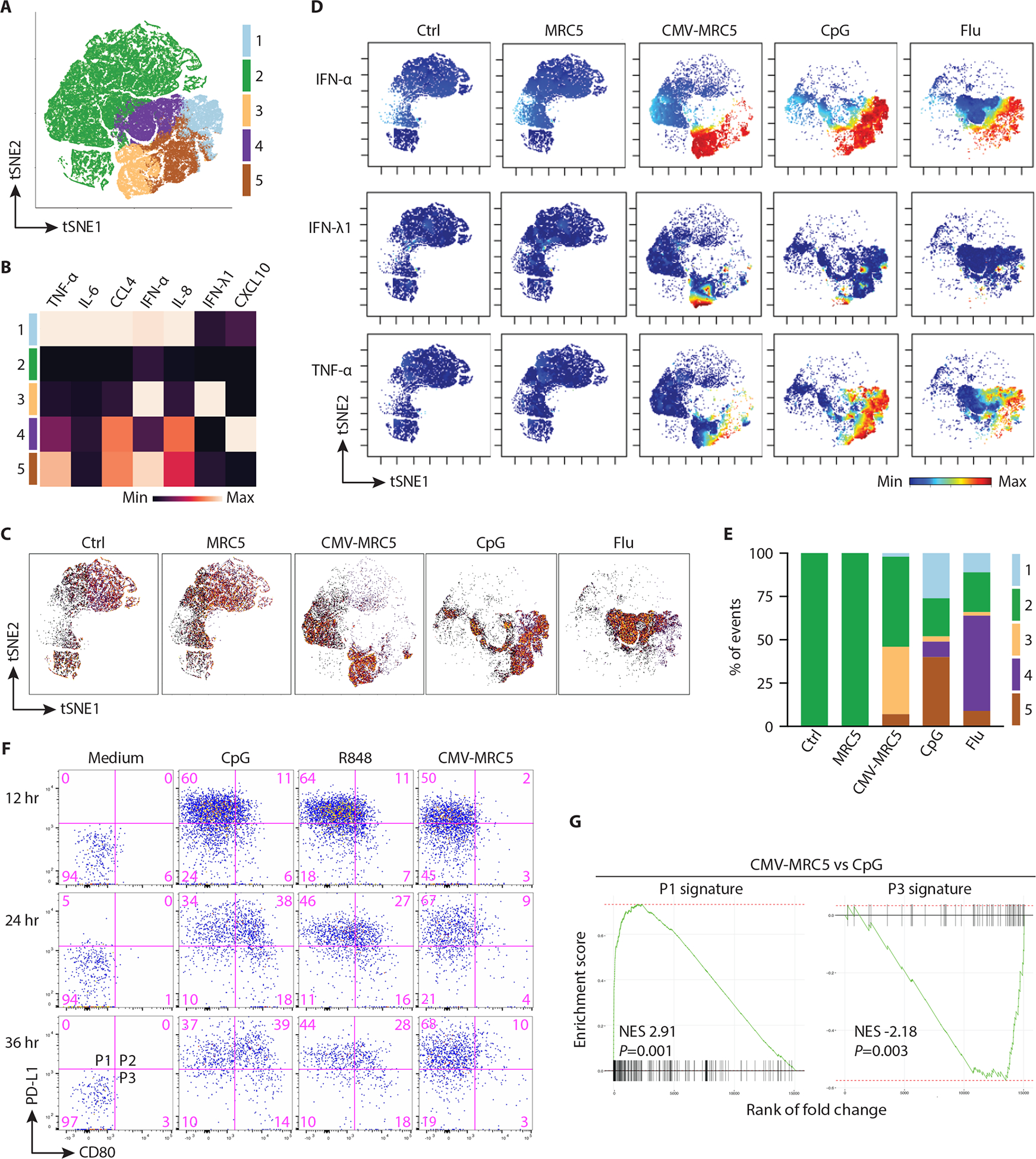
(A) tSNE plot showing equal numbers of pDC pooled from each of the early (9–11 hours) time point stimulation conditions and clustered based on cytokine expression alone; the resulting clusters are indicated bby colors.
(B) Heatmap of cytokine expression in pDC clusters defined in panel A. Highest and lowest positive cell fractions for each cytokine were set to the maximum and minimum, respectively.
(C) tSNE plots showing the relative distribution of cells under each stimulation condition.
(D) Relative expression of indicated cytokines within the pDC population visualized on a tSNE plot.
(E) The relative frequency of cell clusters in each stimulation condition.
(F) Phenotypic diversification of pDCs in response to CpG, R848 or CMV-MRC5. Enriched pDCs were stimulated with TLR ligands or CMV-MRC5 for the indicated times and analyzed by flow cytometry to define the highlighted P1–P3 populations. Representative of 4 experiments.
(G) Transcriptional signatures of pDC subpopulations in activated pDCs. Gene set enrichment analysis (GSEA) was performed on pDCs activated by CMV-MRC5 compared to CpG for 32 hours. Shown is the normalized enrichment score (NES) and enrichment p-value of P1 and P3 subpopulation signatures derived by the analysis of RNA-Seq data from (42).
A distinct pDC activation program in response to virus-infected cells
To test whether the observed distinct response to CMV-infected cells was specific to CMV or was generalizable to cells infected with other viruses, we tested the response of pDCs to the A549 lung epithelium carcinoma cell line infected with the Flu virus encoding GFP (Flu-GFP). Both the free Flu-GFP virions and A549 cells infected with Flu-GFP at ~50% efficiency (Fig. 6A) induced strong activation of primary human pDCs (Fig. 6B). Notably, Flu-GFP-infected A549 cells (Flu-A549) induced significantly more IFN-α2 and IFN-λ1, but almost no TNF-α, compared to free Flu-GFP (Fig. 6B). Intracellular staining showed that the increased production of IFN-α and IFN-λ by pDCs activated with Flu-A549 persisted for >24 hours (Fig. 6C and S10). pDCs activated with Flu-A549 showed little phenotypic diversification and comprised primarily the P1 population, in contrast to the full P1–P3 spectrum of Flu-activated pDCs (Fig. 6D). Finally, the lysate of Flu-A549 contained very few infectious virus particles (Fig. 6E), revealing that pDC activation by Flu-infected cells was highly efficient on a per virion basis. Thus, compared to the free Flu virus, pDC activation by Flu-infected cells was efficient, prolonged, biased towards interferon production and lacked phenotypic diversification. This activation program resembled the one elicited by CMV-infected cells, suggesting that it may be a more general characteristic of pDC activation by virus-infected cells.
Fig. 6. Influenza virus-infected cells elicited a prolonged IFN-focused pDC response.
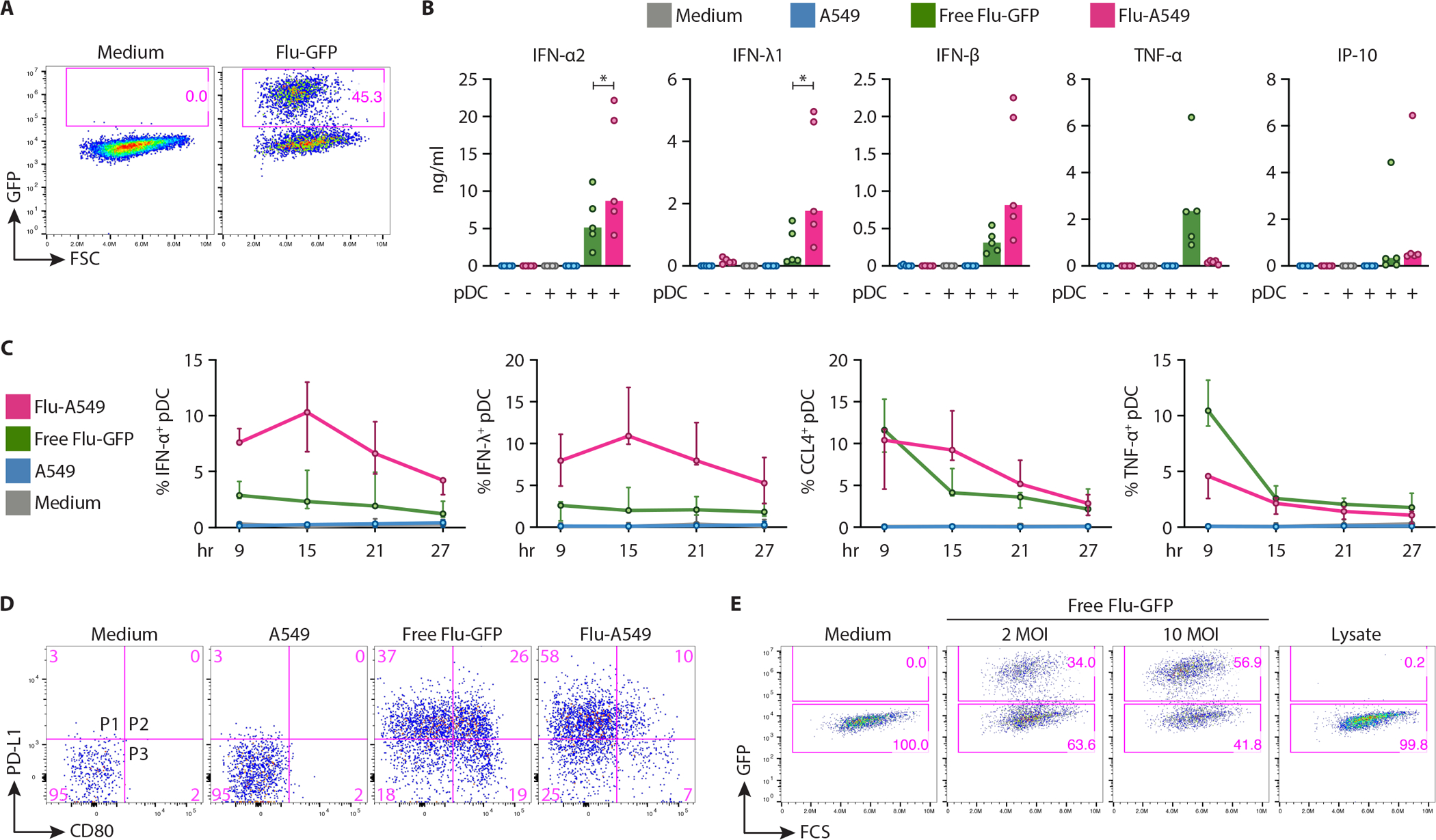
(A–D) Human pDCs were enriched from donor PBMC and incubated with free Flu-GFP virus (10 MOI) or with A549 cells infected with Flu-GFP (Flu-A549) to 40–60% efficiency, or control uninfected A549 cells.
(A) Flow cytometry analysis of A546 cells infected with Flu-GFP at 0.5 MOI for 36 hours. Representative of 6 experiments.
(B) Cytokines in the supernatants of pDCs cocultured with the indicated stimuli for 24 hours, as determined by multiplex bead array. Symbols indicate values from individual PBMC donors (n=5); bars indicate median.
(C) Fractions of cytokine-positive cells among pDCs as determined by intracellular staining at the indicated time points of coculture. Data show median ± range of 3 donors.
(D) Phenotypic diversification of pDCs in response to Flu-A549 vs free Flu, as determined by flow cytometry at 24 hours of coculture. Representative of 4 experiments.
(E) Titration of infectious Flu particles from the lysate of Flu-A549. Flu-A549 cultures (panel A) were freeze-thawed twice and used to infect A549 cells, along with free Flu at the indicated MOI. Shown is the fraction of GFP+ cells 36 hours post infection.
pDC activation by CMV-infected cells facilitated NK cell activation
To test the functional outcome of pDC activation by CMV-infected cells, we analyzed the crosstalk of pDCs with NK cells, the key cell type in the control of CMV (19). pDCs were activated by CMV-MRC5 or Flu for 18 hours, NK cells were added and analyzed after 60 hours of coculture (Fig. S11A). We noticed that fewer GFP+ CMV-MRC5 cells remained after coculture of activated pDCs with NK cells, suggesting increased killing by NK cells (Fig. S11B–C). Accordingly, pDCs activated by CMV-MRC5 caused upregulation of the NK cell activation marker CD69 (Fig. 7A–B) and enhanced NK cell secretion of soluble Fas ligand (sFasL), perforin and IFN-γ (Fig. 7C). The induction of sFasL and IFN-γ was significantly higher when pDCs were activated by CMV-MRC5 compared to Flu (Fig. 7C). All readouts of NK cell activation (Fig. 7D–E) and enhanced killing of MRC5-CMV (Fig. S11B–C) were reduced by IFN-I blockade, suggesting that pDCs facilitate NK cell activation via IFN-I. To test the function of pDCs that underwent initial activation with CMV-MRC5 or Flu, pDCs were activated for 18 hours, purified, washed and cocultured with NK cells for 60 hours. In this setting, pDCs activated with CMV-MRC5 elicited more NK cell activation than pDCs activated with Flu (Fig. 7F,G). Thus, the prolonged program of pDC activation in response to CMV-infected cells, as opposed to a transient activation by a free virus, was optimal for NK cell activation.
Fig. 7. pDCs activated with CMV-infected cells facilitated NK cell activation.
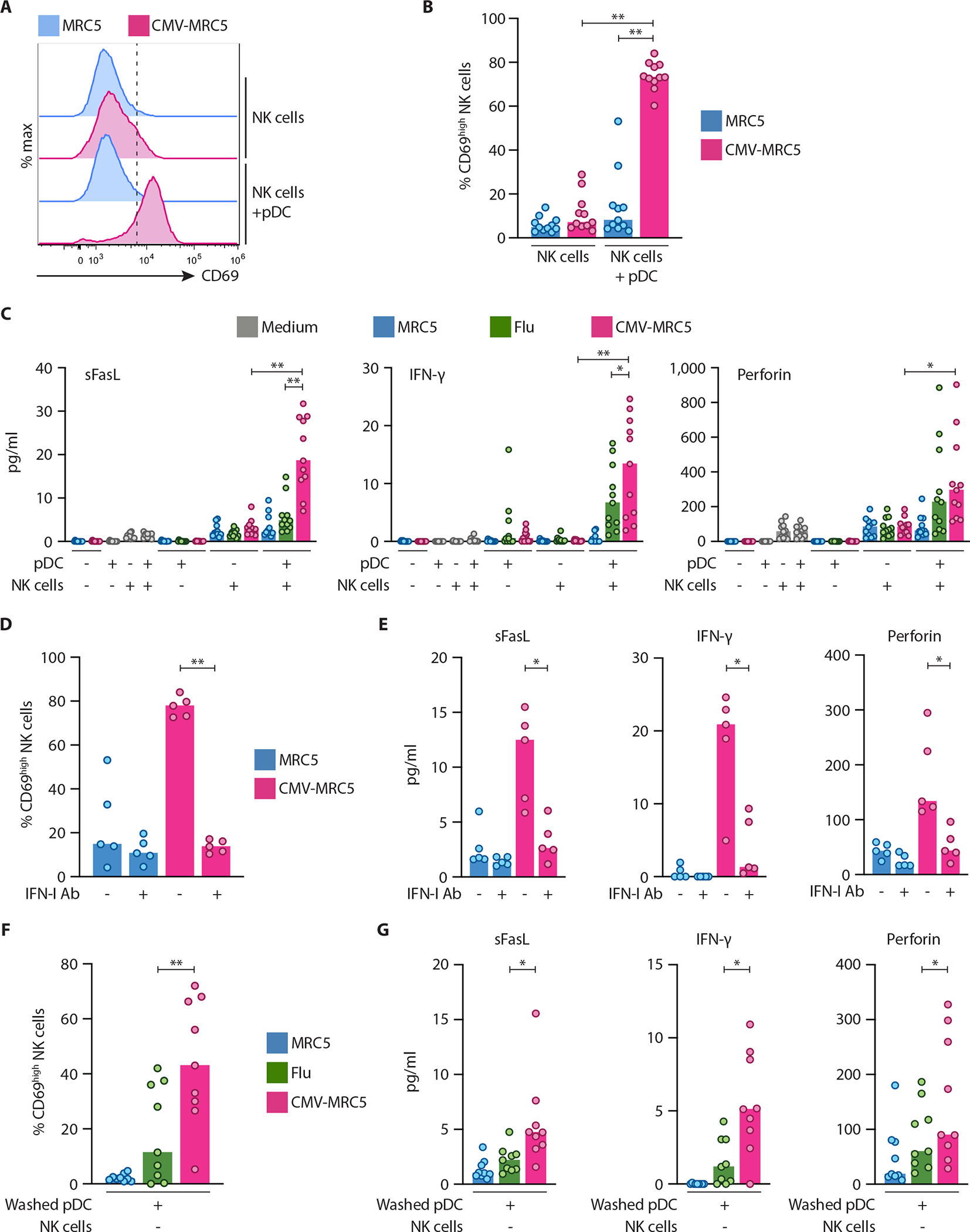
Human pDCs were enriched from donor PBMC, incubated in medium, with control MRC5 cells or with CMV-MRC5 or free Flu virus for 18 hours, and then cocultured with enriched NK cells directly (A to E) or after wash/live cell purification (F and G) for 60 hours.
(A–B) Representative histograms of CD69 expression on NK cells (A) and the fraction of CD69+ NK cells (B) after coculture with pDCs.
(C) NK cell-derived soluble factors in the supernatants of pDC-NK cell cocultures as determined by multiplex bead array.
(D–E) Percentage of CD69+ NK cells (D) and NK cell-derived soluble factors in the supernatants (E) in the absence or presence of IFN-I/IFNAR2 neutralizing antibody mixture (IFN-I Ab).
(F–G) Percentage of CD69+ NK cells (F) and NK cell-derived soluble factors in the supernatants (G) after coculture with washed/purified pDC.
In panels A–G, symbols indicate values from individual PBMC donors; bars indicate median.
pDC activation and interferon production in patients with CMV viremia
To test whether DCs responded to the presence of CMV in vivo, we analyzed transplantation recipients with reactivated CMV replication due to immunosuppression. Ten of 13 viremic patients were completely asymptomatic, despite a broad range of CMV titers in the peripheral blood (PB) (Table S2). A comparable group of transplantation recipients without CMV viremia was used as a control. The fractions of pDCs and cDC2 among peripheral blood mononuclear cells (PBMC) (Fig. S12A) were significantly reduced in viremic patients compared to non-viremic controls (Fig. 8A). In addition, the levels of activation-induced costimulatory molecule CD40 and chemokine receptor CCR7 were significantly increased on all DC subsets (but not on B cells) in viremic patients (Fig. 8B and C). The expression of immediate-early activation marker CD69 was significantly induced only on cDC2 (Fig. 8B and C), whereas the expression of other activation markers (CD80, HLA-DR, CCR5) was not altered on any DC subset (Fig. S12B).
Fig. 8. The DC compartment and cytokines in patients with CMV viremia.
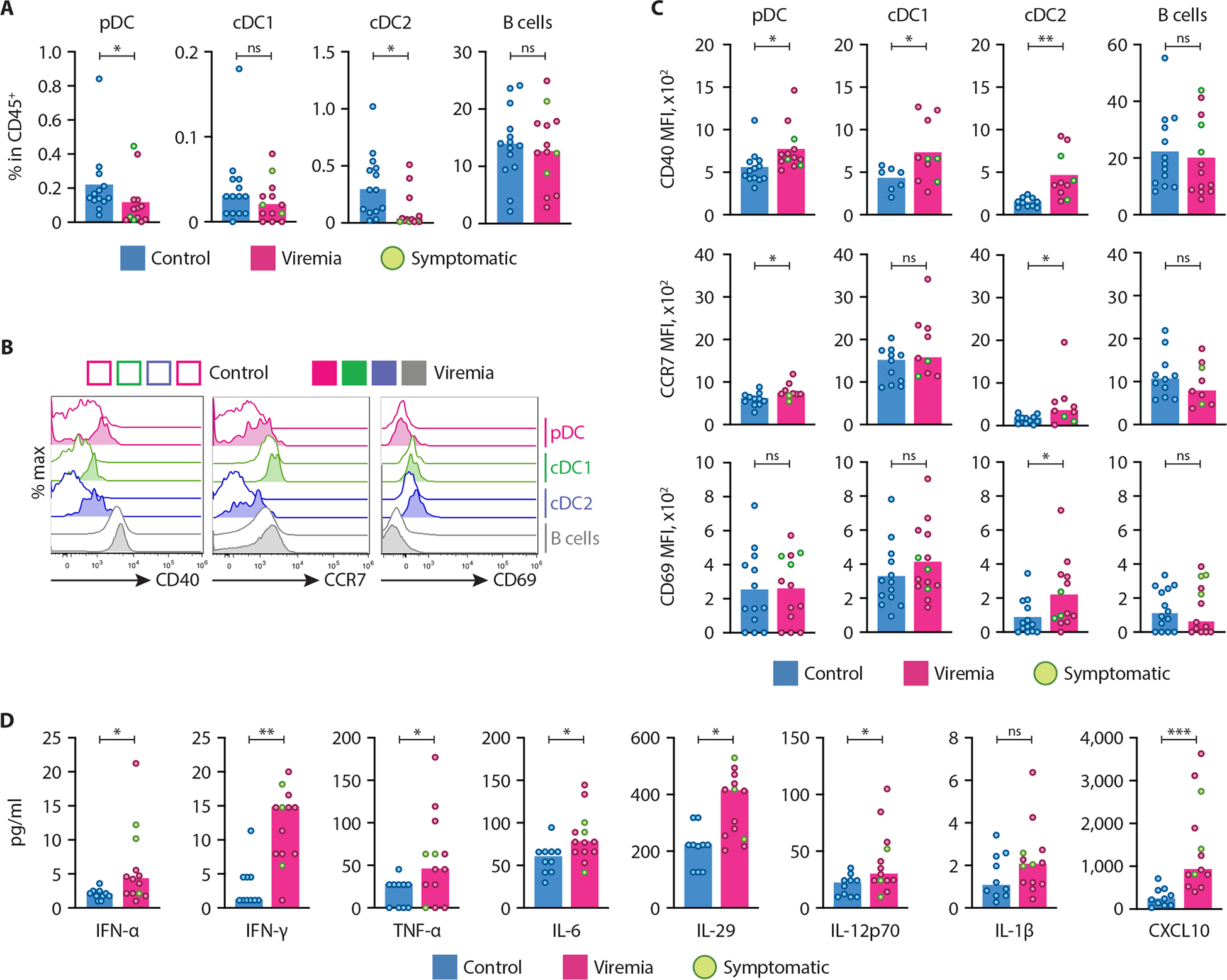
(A–D) PBMC and plasma from organ transplant recipients with (viremia) or without (control) CMV viremia were analyzed by flow cytometry and multiplexed bead array (n=13 in each group). Three patients with clinical symptoms are marked green. The pDCs, cDC1, cDC2 and B cells were defined as shown in Fig. S11A.
(A) The fraction of indicated cell types among total CD45+ PBMC.
(B) Representative histograms of CD40, CCR7 and CD69 expression on the indicated cell types from control and viremic patients.
(C) Median fluorescence intensity (MFI) of CD40, CCR7 and CD69 on the indicated cell types from control and viremic patients.
(D) The concentration of cytokines and chemokines measured by multiplexed bead array in the plasma of control and viremic patients.
In panels A, C and D, symbols indicate values from individual patients; bars indicate median.
We also measured the levels of 7 cytokines (IL-6, TNF-α, IL-12p70, IL-1β, IFN-γ, IFN-α and IL-29) and an interferon-inducible chemokine CXCL10 in the plasma of all patients. We observed a significant increase of all cytokines (except for IL-1β) in viremic patients, again irrespectively of the clinical symptoms (Fig. 8D). The levels of TNF-α or any other cytokine showed no correlation with DC depletion (Fig. S13A), arguing against the causal relationship between them. The only significant correlation between cytokine levels and CMV titers was the inverse correlation in the case of IFN-α (Fig. S13B), suggesting that patients with high IFN-α levels show better virus control. Collectively, these data suggested that active CMV replication, independently of clinical disease, induced phenotypic activation and depletion of select DC subsets including pDCs from the blood, as well as the production of multiple cytokines including IFN-I and IFN-III.
Discussion
This study reports that human pDCs recognized cells infected with CMV and mount a specific cytokine response. This response was distinct from the previously characterized response to solution-born stimuli as pDCs responded minimally to free CMV and only in the presence of IL-3 (22, 43); the latter study also noted an even lower pDC response to CMV-infected retinal pigment epithelial cells. The relatively low response of pDCs to CMV in our hands may also have reflected the high purity of CMV preparation. In contrast to the low IFN response pf pDCs to free CMV, we observed a potent response to CMV-infected fibroblasts (40–60% of IFN-α+ pDCs); the response required a direct contact between live cells that was mediated in part by the integrin LFA-1 on pDCs and its ligand ICAM-1 on infected cells. LFA-1 has recently emerged as an important regulator of IFN production by pDCs (44) that facilitates homotypic (45) or heterotypic (15, 17) cell-cell interactions. While the transfer of viral marker protein or RNA from infected cells to pDCs has been observed for other herpesviruses (46, 47), we identified that viral DNA is the relevant CMV-derived stimulus and TLR9 is its sensor in pDCs. The exact mechanism of how transferred viral DNA activates pDCs remains unclear, but likely reflects the uncoating of virions in the early endosomal compartment to release DNA for TLR9 binding. This idea was supported by the fact that pDCs recognized human CMV-infected cells in a TLR9-dependent manner across the species barrier. These results support the recognition of cells infected with RNA and DNA viruses as a common mechanism of pDC activation.
Cytokine production by pDCs activated by free viruses or TLR ligands peaks after a few hours and is rapidly extinguished (28, 48), whereas pDC response to CMV-infected cells peaked at 8–16 hours and proceeded for >30 hours. The pDC response to solution-born stimuli involved the production of IFN-I and several other cytokines and chemokines including TNF-α, as well as the emergence of a CD80+ Ag-presenting population (42). In contrast, pDC response to CMV-infected cells was focused on the production of IFN-I and IFN-III at the expense of TNF-α and other factors. The diversification into CD80+ cells was largely absent, consistent with the role of TNF-α in the process (42). Finally, the response to CMV-infected cells was extremely efficient, as it required a small fraction of infected cells and very little virus within them. Some of these features such the prolonged activation kinetics may reflect the specific nature of CMV as a stimulus. Nevertheless, the prolonged, interferon-focused program of pDC activation by virus-infected cells was confirmed for Influenza, an RNA virus that (unlike CMV) could also activate pDCs directly. Collectively, these results revealed a previously unappreciated flexibility of pDC responses and highlighted the distinct nature of virus-infected cells as innate immune stimuli.
pDC activation by CMV-infected cells enhanced activation of NK cells, revealing an apparent advantage of the prolonged activation program elicited by CMV-infected cells. pDC activation by CMV-infected cells coordinated a reduction of the TCF4-driven pDC expression program and the acquisition of transcriptomic features of cDCs, independently of phenotypic diversification. This activation-induced conversion into a cDC-like state appeared to be distinct from IFN-driven conversion of immature pDCs into cDC2 (49); on the other hand, this conversion was consistent with original observations on pDC activation in culture (24, 50) and in MCMV-infected mice (51). The acquisition of cDC-like features by activated pDCs may facilitate the presentation of virus antigens to T cells and/or local bystander T cell activation, both of which need further study.
We found that pDC and cDC2 were reduced in the PB of CMV viremic patients even in the absence of clinical disease, likely reflecting the response to CMV rather than a secondary effect of inflammation proposed for the clinical CMV infection (52). The depletion of pDCs was also consistent with the age-dependent reduction of pDCs in CMV-seropositive human subjects (53). Persistent activation of pDCs was further supported by their phenotypic activation and the upregulation of key cytokines including IFN-I and IFN-III, again irrespectively of the clinical disease. Notably, the observed inverse correlation of IFN-α with CMV titers suggested that the former may inhibit the latter, implicating IFN-I as a mechanism of CMV infection control. These data suggest an involvement of primary DCs in CMV infection and emphasize the role of IFN-I and pDCs as a source of control in CMV persistence.
Collectively, our results highlighted the unique capacity of pDCs to sense a broad range of viruses, including persistent DNA viruses such as CMV, even in their absence in extracellular space. The response to CMV-infected cells was very sensitive and resistant to the highly efficient immunoevasion mechanisms of CMV, including its capacity to inhibit intracellular DNA sensing and IFN production (54). Our results elucidated an innate immunity mechanism that likely maintains constant pressure on persistent viruses and prevents even low-level replication within cells. These data also reveal the previously unknown diversity of pDC responses, which appear tailored to specific stimuli such as solution-born acute viruses or cells infected with persistent viruses.
Materials and Methods
Study design.
The bulk DC population or individual DC subsets were isolated from PBMC of healthy blood donors, incubated with various stimuli in vitro, and analyzed for their cytokine response and differentiation; replicates are indicated for each experiment. In Fig. 7, isolated pDCs were activated and cocultured with NK cells isolated from the same donor. In Fig. 8, transplant recipients with or without CMV viremia were analyzed for cytokine levels in the plasma and for DC numbers among PBMC; the analysis was blinded to sample identity.
Human samples.
We collected PBMC from consenting healthy human donors at New York University Langone Health (NYULH) as approved by the NYULH Institutional Review Board (IRB). Buffy coats were obtained from New York Blood Center (NYBC) under the waiver from IRB. The control and CMV viremia patient samples were obtained from the BioBank at Columbia University Medical Center. All outliers were included in this study.
Mice.
Female mice (13–15 weeks of age) were used for experiments with mouse cells. Female mice C57BL/6J [#000664], TLR7−/− [B6.129S1-Tlr7tm1Flv/J, #008380] and TLR9−/− [C57BL/6J-Tlr9M7Btlr/Mmjax, #34329-JAX] were purchased from Jackson Laboratories and maintained under the approval of NYULH Institutional Animal Care and Use Committee (IACUC).
Viruses.
Bacterial artificial chromosome (BAC) stocks of TB40/E WT virus was engineered to express GFP from a SV40-promoter (55). CMV(pp150-GFP) was a gift from Christian Sinzger (56). AD169 strains was acquired from ATCC. Virions were purified by density gradient centrifugation through a D-sorbitol cushion, resuspended in Iscove-modified Dulbecco medium (IMDM) supplemented with 2% FBS and stored at −80°C. Before infection, virus was thawed, sonicated, and centrifuged to pellet debris. Infectious virus yields were assayed on human fibroblasts. Influenza virus H1N1 (PR8) was prepared as described previously (57). Influenza virus NS1-GFP (NS1-GFP (a generous gift of A. Garcia-Sastre) (58) was prepared and kindly provided by Kamal Khanna, NYU (59).
Cell culture and infection.
All cells were incubated at 37°C/5% CO2. Human lung fibroblast cell line MRC5 (control or infected with CMV) or human lung adenocarcinoma cell line A549 (control or infected with influenza virus) were cocultured with PBMC fractions in complete RPMI-1640 medium (herein referred to as cRPMI) containing 2 mM L-Glutamine, 1 mM sodium pyruvate, 1X MEM non-essential amino acids, 100 units/ml penicillin-streptomycin (all from Gibco) and 10% FBS (Sigma). Mouse bone marrow (BM) cells were cocultured with MRC5 or CMV-MRC5 in cRPMI with 5 μM 2-Mercaptoethanol or cultured for 7 days in cRPMI supplemented with 5 μM 2-Mercaptoethanol and 10% supernatant of the Flt3L-producing B16 cell line to generate mouse DCs, followed by coculture with MRC5 cells. Uninfected neonatal primary human dermal fibroblasts (HDF) were cultured in fibroblast basal medium with fibroblast growth kit – low serum (all from ATCC) and cocultured with pDCs in cRPMI.
MRC5 and HDF were infected with 0.5 MOI of CMV-GFP and cultured for 2–5 days to yield 40–70% GFP+ cells by flow cytometry. A549 were infected with 0.5. MOI of influenza NS1-GFP and cultured for ~36 hours to yield 40–60% GFP+ cells. Infected cells were trypsinized and frozen in FBS containing 10% DMSO. Upon thawing, cells were cultured overnight, plated at 6×104 cells/cm2 in 96, 48 or 24 well plates for at least 8 hours, and cocultured with 5×103-4×104 pDCs. For EdU incorporation, MRC5 (uninfected or infected with 0.5 MOI of CMV(pp150-GFP) for 1 day) were cultured with 5 μM EdU (Life Technologies) for 2 days, trypsinized and frozen as above. Thawed EdU-labeled cells were cultured overnight with 5 μM EdU, washed and used for coculture with pDCs. For UV irradiation, uninfected or CMV-infected MRC5 cells were irradiated with 4.5 Joules of 254 nm UV-C using UV Stratalinker 1800 (Stratagene) and cultured for 2 days. For freeze-thawing, cells were frozen at −80°C and thawed at 37°C twice. For fixation, cells were fixed by 4% formaldehyde in PBS for 20 min at room temperature, washed, quenched with 0.1 M Glycine in PBS for 30 min and washed with cRPMI five times. For differential centrifugation, infected MRC5 cells were collected by gentle pipetting in the culture supernatant and centrifuged at 4°C sequentially at 300 g/10 min (pellet A, live cells), 2,000 g/10 min (pellet B, dead cells) and 10,000 g/30 min (pellet C, cell debris and supernatant D, virus and subcellular particles). For transwell cultures, MRC5 cells were cultured at 6×104 cells/cm2 in 0.9 ml of cRPMI in 24-well plate for 8 hours, followed by insertion of Transwells (Corning, 1.0 μm pore size) and addition of pDCs in 0.1 ml cRPMI for another 11 hours.
PBMC, pDCs, pan-DC and monocytes enrichment and sorting.
PBMC from donors’ blood were isolated by Ficoll gradient (GE Healthcare). pDCs, monocytes and pan-DCs were enriched from fresh PBMC by negative selection using EasySep Human Plasmacytoid DC Enrichment Kit, Monocyte Enrichment kit and pan-DC Pre Enrichment Kit (all Stemcell Technologies) to ~80–90% purity. For sorting, pDC and pan-DCs were enriched as above, stained using Zombie Aqua Fixable Viability Kit (Biolegend) for 20 min at 4°C, washed in cRPMI once and stained with fluorophore-labeled mAbs for CD11c (PerCP/Cy5.5, clone: Bu15), CD1c (BV421, clone: L161), CD123 (PE or BV421, clone: 6H6), CD141 (PE/Cy7, clone: M80, all from Biolegend), AXL (PE, clone: 108724) or Siglec6 (APC, clone: 767329, all from R&D) for 20 min at 4°C, followed by two cRPMI washes and resuspension in cRPMI. Cells fractions including CD11c−CD123+ pDCs, CD123+AXL−Siglec6− pDC, CD123+AXL+Siglec6+ tDC, CD11c+CD123−CD1c+ cDC1 and CD11c+CD123−CD141+ cDC2 were sorted on SY3200 or SH800Z sorters using built-in instrument software (SONY Biotechnology) and analyzed using FlowJo software (FlowJo, LLC). Population compositions and/or gating strategies for sorting are illustrated in Fig. S1A, S4C, S4E, S6A, S8A, S9A and S11A.
Blocking antibodies and antagonists.
Cells were preincubated with 10 μg/ml isotype controls or blocking antibodies against LFA-1 (clone: HI111, Biolegend), ICAM-1 (clone: HCD54, Biolegend), ICAM-2 (clone: CBR-IC2/2, eBioscience), ICAM-3 (clone: CBR-IC3/1, eBioscience), CD62 E/P-selectin (clone: BBIG-E6 (13D5), R&D) and IFN-I/IFNAR2 (39000-1, PBL) for 30 minutes, or with 3 μM single-stranded oligodeoxynucleotides ODN 20958 (TLR7 antagonist, Miltenyi) or ODN 2088 (TLR9 antagonist, Invivogen) for 5 hours, followed by stimulation in the presence of blocking reagents unless indicated otherwise.
Cell stimulation.
Cells were stimulated with 5 μg/ml R848 for 4.5 hours; 2.5 μM CpG class A (ODN 2216), 0.1 μg/ml LPS or 25 μg/ml Poly-I:C for 6 hours (all ligands from Invivogen); or free CMV, MRC5 or CMV-MRC5 for 11 hours, unless otherwise indicated. For intracellular staining, Brefeldin A (BFA, 10 μg/ml) was added for the last 3–5 hour of incubation. For flow cytometry, cells were stained for viability and surface markers as above, washed in cRPMI and fixed using Foxp3 staining buffer set (eBioscience), followed by staining for intracellular IFN-α (PE or APC, clone: LT27:295, Miltenyi), IFN-λ (biotin, clone: 247801), CCL4 (PE, clone: 24006, all R&D), CXCL10 (PE, clone: J034D6) or TNF-α (PE/Dazzle 594, clone: Mab11, all Biolegend). Cells were acquired on the Attune NxT flow cytometer (ThermoFisher) and analyzed using FlowJo software (FlowJo, LLC).
For stimulation of NK cells, both pDC and autologous NK cells were enriched from frozen PBMC by negative isolation using EasySep Human pDC Enrichment Kit and NK cell isolation kit (all Stemcell Technologies). Enriched pDC (50,000 cells/mL) were stimulated with indicated stimuli for 18 hours, while enriched NK cells were cultured in cRPMI. To wash and purify pDCs, stimulated pDCs were incubated with Dead Cell Removal Kit (Miltenyi) and washed with cRPMI to remove stimuli and activation products. The recovered NK cells (500,000 cells/mL) were added to the original pDC cultures or washed pDC and cocultured for 60 hours.
Immunofluorescent microscopy.
Uninfected or infected MRC5 cells were plated at 6×104 cells/cm2 onto 12 mm micro coverglass for 8 hours. pDCs (2×104) were added for another 8 hours, followed by fixation with 4% formaldehyde for 20 min, permeabilization with 0.5% Triton X-100 in PBS for 20 min and blocking with 10% goat serum containing 0.2 M glycine for 1 hour. Fixed cells were washed with PBS 3 times between steps. Cells were stained for GFP (AF488, Invitrogen), CD45RA (biotin, clone: HI100, Biolegend) followed by streptavidin (AF555, Invitrogen), and IFN-α (APC, clone: LT27:295, Miltenyi) followed by anti-mouse IgG1 (HRP, clone: SB77e, Southern Biotech) for anti-IFN-α-APC with Alexa Fluor 647 Tyramide SuperBoost Kit (Invitrogen). Images were collected using BZ-X710 microscope with 4x objective lens and analyzed with Keyence Analysis software (all Keyence). For activated LFA-1, cells were stained for GFP (AF488, Invitrogen), CD45RA (BV421, clone: HI100, Biolegend), active LFA-1 (biotin, clone: m24, Biolegend) and IFN-α (APC, clone: LT27:295, Miltenyi), followed by secondary antibodies staining using a-mouse IgG2b (BV421, clone: R12–3, BD Biosciences) for anti-CD45RA-BV421, streptavidin (AF555, Invitrogen) for active-LFA-1-biotin and anti-mouse IgG1 (APC, clone: RMG1-1, Biolegend) for a-IFN-α-APC. Images were collected using an LSM 880 microscope with 63x objective lens and analyzed with Zen software (all Zeiss).
pDCs cocultured with MRC5 infected with CMV(pp150-GFP) were sorted, subjected to cytospin, fixed and permeabilized as above. Cells were stained for GFP and CD45RA with anti-GFP-AF488 and anti-CD45RA-BV421, respectively, followed by labeling with secondary antibodies using anti-mouse IgG2b-BV421 for anti-CD45RA-BV421. EdU was detected using Click-iT Plus EdU AF594 Imaging Kit (ThermoFisher). Images were collected using an LSM 880 microscope with 100x objective lens and analyzed with Zen software (all Zeiss).
Cytokine measurement.
IFN-α and TNF-α in cell supernatants were measured by ELISA using Human IFN-α Pan ELISA kit (Mabtech), Human TNF-alpha DuoSet ELISA Kit (R&D) or Mouse IFN Alpha ELISA kit (PBL Assay Science). Cytokines (IL-6, TNF-α, IL-12p70, IFN-γ, IFN-α, IL-29, CXCL10 and IL-1β) in frozen plasma samples were measured using a custom ProcartaPlex Multiplex Immunoassay (ThermoFisher) on a Luminex 200 reader. Cytokines from CMV- or influenza-activated pDCs and from pDC-NK cell cocultures were measured using LEGENDplex human anti-virus panel and human CD8/NK panel (Biolegend), respectively.
CyTOF.
Enriched pan-DCs from one donor were co-cultured with indicated stimuli for 9–11 or 24 hours and fixed. Samples were barcoded, pooled, stained with antibodies against surface markers and cytokines, and analyzed using Helios mass cytometer (Fluidigm). Further details of cell processing and analysis are provided in Supplemental Methods.
qPCR.
DNA from sorted pDCs cocultured with CMV(pp150-GFP)-MRC5 was isolated using QIAamp DNA Blood kit (Qiagen) and measured using Nanodrop 8000 (ThermoFisher). qPCR measurements of CMV UL132 gene were setup using Taqman assay (Vi03453400_s1, FAM, Thermo Scientific) with Quantifast Pathogen kit (Qiagen). CMV copy number was quantified by a standards curve of 101–107 copies per reaction. Samples were amplified in duplicate using CFX96 Touch Real-Time PCR Detection System (Bio-Rad) and analyzed using the CFX manager software according to the ΔΔCT method. DNA input was normalized by the 18S amplicon (Hs99999901_s1, VIC, Thermo Scientific).
RNA-seq sample preparation, processing, and analysis.
Enriched pDCs from 3 different donors were cocultured with the indicated stimuli for 2–32 hours as indicated, harvested by gentle pipetting and stained for viability as above. Live pDCs were sorted using scatter parameters to eliminate MRC5 cells using the SH800Z sorter (SONY Biotechnology), and total RNA was isolated by RNeasy Mini Kit (Qiagen). RNA-seq was conducted using a low-input protocol as described (27); barcoded cDNA libraries were pooled and sequenced on an Illumina NextSeq 500 instrument. The resulting sequences were aligned to the genome, demultiplexed using sample-specific barcodes and used to generate a digital matrix of molecular counts for each sample. Further details are provided in Supplemental Methods.
Statistical Analysis.
Graphing and statistical analysis was performed using Prism software (GraphPad), using paired Student’s t-test except in Fig. 8 and S12B, which used unpaired Mann-Whitney test. Correlation in Fig. S13 was estimated using Spearman method with a two-tailed p value for significance. Statistical significance is indicated as follows: *p ≤ 0.05; **p ≤ 0.005; ***p ≤ 0.0005; ****p ≤ 0.0001; ns, not significant.
Supplementary Material
Acknowledgements
We thank Ioanna Tiniakou and Chetna Soni for help with murine pDC stimulation, Kamal Khanna for the Flu-GFP virus and Donna Collins-McMillen for a batch of CMV.
Funding
Supported by the NIH Human Immunology Project Consortium grant AI128949 (D.L.F., F.G., P.A.S., B.R.) and NIH grant AI072571 (B.R.). Mass cytometry instrumentation at the Mt. Sinai Human Immune Monitoring Center was supported by NIH grant S10 OD023547-01.
Footnotes
Competing Interests
The authors declare no competing financial interest.
Data and materials availability
Sequencing data were deposited in the NCBI GEO database under accession number GSE163707. All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials, and/or available from the authors upon request.
References and notes
- 1.Guilliams M et al. , Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14, 571–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swiecki M, Colonna M, The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol 15, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reizis B, Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 50, 37–50 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A, Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198, 513–520 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB, Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 4, 374–386 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M, Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity 33, 955–966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cervantes-Barragan L et al. , Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci U S A 109, 3012–3017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornung V et al. , Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol 173, 5935–5943 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A, Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Asselin-Paturel C et al. , Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol 2, 1144–1150 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA, HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology 343, 256–266 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Takahashi K et al. , Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc Natl Acad Sci U S A 107, 7431–7436 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dreux M et al. , Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 12, 558–570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wieland SF et al. , Human plasmacytoid dendritic cells sense lymphocytic choriomeningitis virus-infected cells in vitro. J Virol 88, 752–757 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia-Nicolas O et al. , Sensing of Porcine Reproductive and Respiratory Syndrome Virus-Infected Macrophages by Plasmacytoid Dendritic Cells. Front Microbiol 7, 771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sinigaglia L et al. , Immature particles and capsid-free viral RNA produced by Yellow fever virus-infected cells stimulate plasmacytoid dendritic cells to secrete interferons. Sci Rep 8, 10889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Assil S et al. , Plasmacytoid Dendritic Cells and Infected Cells Form an Interferogenic Synapse Required for Antiviral Responses. Cell Host Microbe 25, 730–745 e736 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Haidar G, Boeckh M, Singh N, Cytomegalovirus Infection in Solid Organ and Hematopoietic Cell Transplantation: State of the Evidence. J Infect Dis 221, S23–S31 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picarda G, Benedict CA, Cytomegalovirus: Shape-Shifting the Immune System. J Immunol 200, 3881–3889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kvale EO et al. , CD11c+ dendritic cells and plasmacytoid DCs are activated by human cytomegalovirus and retain efficient T cell-stimulatory capability upon infection. Blood 107, 2022–2029 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Varani S et al. , Human cytomegalovirus differentially controls B cell and T cell responses through effects on plasmacytoid dendritic cells. J Immunol 179, 7767–7776 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Paijo J et al. , cGAS Senses Human Cytomegalovirus and Induces Type I Interferon Responses in Human Monocyte-Derived Cells. PLoS Pathog 12, e1005546 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zucchini N et al. , Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol 180, 5799–5803 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Grouard G et al. , The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med 185, 1101–1111 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadowaki N, Liu YJ, Natural type I interferon-producing cells as a link between innate and adaptive immunity. Hum Immunol 63, 1126–1132 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Bush EC et al. , PLATE-Seq for genome-wide regulatory network analysis of high-throughput screens. Nature communications 8, 105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Snyder ME et al. , Generation and persistence of human tissue-resident memory T cells in lung transplantation. Science immunology 4, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito T, Kanzler H, Duramad O, Cao W, Liu YJ, Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 107, 2423–2431 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Heidkamp GF et al. , Human lymphoid organ dendritic cell identity is predominantly dictated by ontogeny, not tissue microenvironment. Science immunology 1, (2016). [DOI] [PubMed] [Google Scholar]
- 30.Lachmann A, Giorgi FM, Lopez G, Califano A, ARACNe-AP: gene network reverse engineering through adaptive partitioning inference of mutual information. Bioinformatics 32, 2233–2235 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez MJ et al. , Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet 48, 838–847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B, Continuous expression of the transcription factor e2–2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity 33, 905–916 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh HS et al. , ETO family protein Mtg16 regulates the balance of dendritic cell subsets by repressing Id2. J Exp Med 211, 1623–1635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ceribelli M et al. , A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell 30, 764–778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ippolito GC et al. , Dendritic cell fate is determined by BCL11A. Proc Natl Acad Sci U S A 111, E998–1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka A, Itoh F, Itoh S, Kato M, TAL1/SCL relieves the E2–2-mediated repression of VEGFR2 promoter activity. J Biochem 145, 129–135 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Murphy TL et al. , Transcriptional Control of Dendritic Cell Development. Annu Rev Immunol 34, 93–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alcantara-Hernandez M et al. , High-Dimensional Phenotypic Mapping of Human Dendritic Cells Reveals Interindividual Variation and Tissue Specialization. Immunity 47, 1037–1050 e1036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villani AC et al. , Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.See P et al. , Mapping the human DC lineage through the integration of high-dimensional techniques. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leylek R et al. , Integrated Cross-Species Analysis Identifies a Conserved Transitional Dendritic Cell Population. Cell Rep 29, 3736–3750 e3738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alculumbre SG et al. , Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat Immunol 19, 63–75 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Becker J et al. , Human monocyte-derived macrophages inhibit HCMV spread independent of classical antiviral cytokines. Virulence 9, 1669–1684 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomasello E et al. , Molecular dissection of plasmacytoid dendritic cell activation in vivo during a viral infection. EMBO J 37, e98836 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitoh SI et al. , TLR7 mediated viral recognition results in focal type I interferon secretion by dendritic cells. Nature communications 8, 1592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Megjugorac NJ et al. , Image-based study of interferongenic interactions between plasmacytoid dendritic cells and HSV-infected monocyte-derived dendritic cells. Immunol Invest 36, 739–761 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Baglio SR et al. , Sensing of latent EBV infection through exosomal transfer of 5’pppRNA. Proc Natl Acad Sci U S A 113, E587–596 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coccia EM et al. , Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 34, 796–805 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB, Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol 5, 1227–1234 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cella M, Facchetti F, Lanzavecchia A, Colonna M, Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol 1, 305–310 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Abbas A et al. , The activation trajectory of plasmacytoid dendritic cells in vivo during a viral infection. Nat Immunol 21, 983–997 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varani S et al. , High TNF-alpha and IL-8 levels predict low blood dendritic cell counts in primary cytomegalovirus infection. J Clin Virol 53, 360–363 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Puissant-Lubrano B et al. , Distinct effect of age, sex, and CMV seropositivity on dendritic cells and monocytes in human blood. Immunol Cell Biol 96, 114–120 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Dell’Oste V et al. , Tuning the Orchestra: HCMV vs. Innate Immunity. Front Microbiol 11, 661 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Umashankar M et al. , A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7, e1002444 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sampaio KL, Cavignac Y, Stierhof YD, Sinzger C, Human cytomegalovirus labeled with green fluorescent protein for live analysis of intracellular particle movements. J Virol 79, 2754–2767 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL, Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper-independent mechanisms. J Virol 84, 9217–9226 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Manicassamy B et al. , Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A 107, 11531–11536 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ural BB et al. , Identification of a nerve-associated, lung-resident interstitial macrophage subset with distinct localization and immunoregulatory properties. Science immunology 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.